

Abstract
Human rhinovirus (HRV) infections trigger exacerbations of lower airway diseases. HRV infects human airway epithelial cells and induces proinflammatory and antiviral molecules that regulate the response to HRV infection. Interferon (IFN)-stimulated gene of 15 kDa (ISG15) has been shown to regulate other viruses. We now show that HRV-16 infection induces both intracellular epithelial ISG15 expression and ISG15 secretion in vitro. Moreover, ISG15 protein levels increased in nasal secretions of subjects with symptomatic HRV infections. HRV-16-induced ISG15 expression is transcriptionally regulated via an IFN regulatory factor pathway. ISG15 does not directly alter HRV replication but does modulate immune signaling via the viral sensor protein RIG-I to impact production of CXCL10, which has been linked to innate immunity to viruses. Extracellular ISG15 also alters CXCL10 production. We conclude that ISG15 has a complex role in host defense against HRV infection, and that additional studies are needed to clarify the role of this molecule.
Introduction
Human rhinovirus (HRV) is the dominant viral pathogen linked to acute exacerbations of lower airway diseases such as asthma, chronic obstructive pulmonary disease, and cystic fibrosis.1, 2, 3 The airway epithelial cell is the primary site of HRV infection and it is believed that viral alterations in epithelial cell biology regulate the outcomes to HRV infection in the airways. In support of this, HRV infection, in vivo and in vitro, induces epithelial expression of a wide range of proinflammatory and antiviral host defense genes.4, 5, 6, 7 Several studies have focused on the HRV-induced epithelial production of proinflammatory cytokines and chemokines.8, 9, 10, 11 By contrast, although nitric oxide and viperin have been shown to inhibit HRV replication,4, 12 little is known about the role of other host defense molecules in limiting HRV infections.
Interferon (IFN)-stimulated gene of 15 kDa (ISG15) is an ubiquitin-like modifier that is highly induced upon HRV infection of airway epithelial cells, both in vitro and in vivo.4, 6 ISG15 can be covalently coupled to many host cellular proteins (a process known as ISGylation), often modulating their functions.13 Moreover, ISG15 can conjugate to some viral proteins, such as the NS1 protein of influenza, resulting in antiviral activity.14 Unlike most proteins involved in post-translational modification of proteins, ISG15 can be secreted from some cell types and exert immunomodulatory effects, including stimulation of T-cell proliferation, and expansion and activation of natural killer (NK) cells.15, 16 Despite its effects in other systems, the regulation and role of ISG15 during HRV infection has not been examined.
We hypothesized that HRV infection would induce epithelial expression and secretion of ISG15, and that ISG15 would exert modulatory effects on epithelial responses to HRV infection. We now demonstrate that HRV-induced epithelial expression of ISG15 is regulated transcriptionally via an IFN regulatory factor (IRF) binding site(s) in the gene promoter, and that HRV infection induces epithelial expression and extracellular secretion of ISG15 both in vitro and in vivo. Although ISG15 does not directly affect replication of HRV in epithelial cells, it regulates HRV-induced epithelial production of CXCL10 but not CXCL8. Thus, our data suggest that ISG15 exerts a complex immune regulatory role during HRV infections.
Results
HRV-16 infection of human bronchial epithelial cells induces time-dependent expression and release of ISG15
HRV-16 infection induced a time-dependent (P<0.05) increase in ISG15 mRNA levels compared with medium (Figure 1a). Constitutive expression of ISG15 mRNA was observed but did not significantly change with time. Compared with medium, HRV-16 infection significantly increased ISG15 mRNA levels at both 24 (P<0.05) and 48 h (P<0.001) post infection (Figure 1a). Similar data were observed using the human bronchial epithelial (HBE) cell line, BEAS-2B, with maximal expression at 48 h (data not shown). HRV infection increased ISG15 protein expression in HBE cells in a time-dependent manner, with protein detectable at 24 h and peak expression at 48 h (Figure 1b). Similar data were observed using BEAS-2B cells (data not shown). Release of ISG15 protein into HBE supernatants was measured at 24 and 48 h. Constitutive secretion was observed at both time points, but this was increased following HRV-16 infection, with the difference reaching statistical significance (P<0.05) at 48 h (Figure 1c).
Figure 1.

Human rhinovirus (HRV)-16 infection induces the expression of interferon-stimulated gene 15 kDa (ISG15) mRNA and protein in human bronchial epithelial (HBE) cells. (a) Induction of ISG15 mRNA expression after HRV-16 infection in HBE cells assessed by quantitative reverse transcriptase-PCR (femtograms±s.e.m.; n=6). (b) Immunoblot detection of intracellular ISG15 protein production following HRV-16 infection of HBE cells (representative of n=3). (c) Levels of released ISG15 into the supernatants of HRV-16-infected HBE (μg ml−1±s.e.m.; n=6). M, medium control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HRV, HRV-16. GAPDH was used as an internal control. Asterisk indicates significant difference between medium and HRV-16-infected cells at the respective time point (P<0.05).
ISG15 protein release into nasal secretions correlates with viral load during naturally acquired HRV infections
We measured ISG15 protein levels in nasal secretions from 10 different healthy subjects with confirmed naturally acquired HRV infections (Figure 2). Nasal lavages were obtained on 3 consecutive days when subjects were symptomatic and were shedding HRV (visits 1–3), and again at least 2 weeks after the resolution of cold symptoms when no HRV could be detected (visit 4). ISG15 protein levels were highest in nasal secretions obtained at visit 1 and displayed a time-dependent decrease (P<0.05), with ISG15 protein levels significantly higher at visit 1 (100.1±46.1 ng ml−1) compared with visit 4 (5.2±2.5 ng ml−1; Figure 2). Moreover, levels of secreted ISG15 significantly correlated (r=0.5, P<0.005) with HRV viral titers and with symptom scores (rs=0.43, P<0.01).
Figure 2.

Healthy volunteers with naturally acquired human rhinovirus (HRV) colds have increased levels of interferon-stimulated gene of 15 kDa (ISG15) protein in nasal lavages. ISG15 protein (ng ml−1) and corresponding viral titers (log TCID50 U ml−1) in samples from HRV-infected individuals at the onset of cold symptoms (visit 1), 2 subsequent days (visits 2 and 3), and 4 weeks after onset of symptoms and clearance of viral infection (visit 4). Data represent mean±s.e.m. (n=10). Asterisks indicate significant (P<0.05) difference between visit 1 and visit 4 (within each respective group).
HRV-16-induced ISG15 expression is transcriptionally regulated through a specific IRF DNA-binding site
To examine transcriptional regulation of ISG15, we compared responses of a 634-bp ISG15 promoter construct with various truncated forms of the promoter, as illustrated in Figure 3. Data are expressed both as fold increase above that induced by appropriate medium control and as relative light units (RLUs) so that differences in basal level of induction between constructs can be seen. Constructs were transfected into BEAS-2B cells and stimulated for 24 h. HRV-16 infection caused a significant (P<0.001) drive of the 634-bp construct compared with medium. Truncation of the ISG15 promoter to 308-bp, which excluded the IRF-C and overlapping signal transducer and activator of transcription (STAT)/nuclear factor-κB (NF-κB)-2-binding sites did not significantly alter HRV-16-induced promoter drive compared with the 634-bp construct. Further truncation to a 215-bp construct, which excluded the proximal NF-κB1 site but retained the proximal IRF-A, IRF-B, and a cyclic adenosine monophosphate response element (cAMP response element) site, significantly (P<0.05) reduced HRV-16-induced promoter drive when analyzed as fold induction, but failed to achieve significance when expressed in RLUs. Analysis of a 155-bp construct that deleted the IRF-A- and IRF-B-binding sites abrogated virus-induced ISG15 promoter drive to near-basal levels either when analyzed as fold induction or as RLUs.
Figure 3.

Human rhinovirus (HRV)-16-induced interferon-stimulated gene of 15 kDa (ISG15) expression is controlled at the level of transcription. A 634-bp ISG15 promoter sequence and various truncated forms were cloned upstream into a pGL4.10 firefly luciferase plasmid and transiently transfected into BEAS-2B cells using lipid-mediated techniques. Cells were exposed to medium (M) or infected with HRV-16 (V) for 24 h and luciferase activity was measured. Data are presented as (a) fold increase over medium control and (b) relative light units (RLUs), and represent mean±s.e.m. (n=6). Asterisk indicates significant difference between 634-bp vs. truncated forms in HRV-16-infected cells (P<0.05). Transcription factor binding sites in the ISG15 promoter: CRE, cAMP response element; IRF, interferon regulatory factor; κB, nuclear factor-κB; STAT, signal transducer and activator of transcription.
Specific point mutations were introduced into the putative transcription factor-binding sites found in the 634-bp promoter construct. Consistent with truncation mutant studies, separate point mutations of the IRF-C or the overlapping STAT /NF-κB2 sites did not significantly alter HRV-16-induced promoter activity compared with the wild-type construct (data not shown). As shown in Figure 4a, individual mutations were introduced into the remaining NF-κB1, IRF-A, IRF-B, and cAMP response element sites. For each of these mutated constructs, basal drive was lower than that observed for the wild-type construct, such that interpretation of results is complex and varies depending on whether fold induction or RLUs are compared. On the basis of fold induction values, only mutation of the IRF-B-binding sequence significantly (P<0.05) reduced HRV-induced promoter drive compared with the wild-type sequence. Consistent with these data, simultaneous mutation of both the IRF-A and the NF-κB1 sites did not decrease promoter activity compared with wild type (Figure 4c). Double mutation of IRF-B and IRF-A or IRF-B and NF-κB1 sites significantly (P<0.05) inhibited HRV-induced promoter drive compared with wild type in each case (Figure 4c), although the effect of these double mutations was not greater than that observed on mutation of the IRF-B site alone. By contrast, when data were expressed as RLUs, mutation of each of the sites examined results in a significant reduction of viral drive. Of note, however, mutation of the putative IRF-binding sites cause the greatest abrogation of viral drive (Figure 4b). Any double mutant that included mutation of an IRF site showed significant reduction of viral drive relative to the wild-type construct (Figure 4d). Thus, although data need to be interpreted with caution, putative IRF-binding sites appear to have a significant role in HRV-induced ISG15 gene activation.
Figure 4.

Human rhinovirus (HRV)-16-induced interferon-stimulated gene of 15 kDa (ISG15) transcriptional upregulation is mediated specifically through the interferon regulatory factor (IRF)-B DNA-binding site. Specific point mutations were introduced into either individual (a) or multiple (b) DNA-binding sites in the 634-bp ISG15 promoter sequence using site-directed mutagenesis techniques. BEAS-2B cells were transiently transfected using lipid-mediated techniques. Cells were infected with HRV-16 for 24 h and luciferase activity was measured in cell lysates. Data are presented as (a,c) fold increase over medium control and (b,d) as relative light units (RLUs), and represent mean±s.e.m. (n=6). Asterisk indicates significant difference between wild type and mutated forms in HRV-16-infected cells (P<0.05). Transcription factor binding sites in the ISG15 promoter: CRE, cAMP response element; κB, nuclear factor-κB; STAT, signal transducer and activator of transcription; M, medium control; V, infection with HRV-16.
We have previously shown that HRV-16 infection induces epithelial expression of the transcription factor, IRF-1.17, 18 Given the putative role of the IRF-binding site(s) in the regulation of HRV-16-induced ISG15 expression, we used two separate IRF-1-specific short-interfering RNA (siRNA) duplexes to examine the role of this transcription factor in ISG15 expression. Both duplexes markedly attenuated HRV-16-induced intracellular ISG15 protein production as assessed by immunoblotting (Figure 5).
Figure 5.
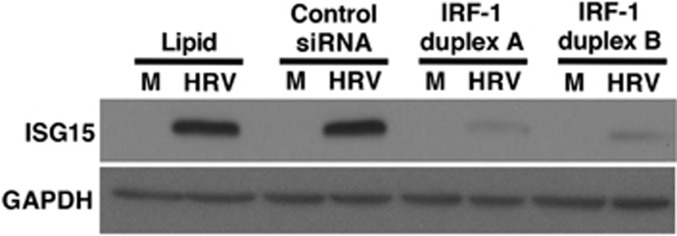
Interferon (IFN) regulatory factor (IRF)-1 knockdown reduces human rhinovirus (HRV)-16-induced IFN-stimulated gene of 15 kDa (ISG15) protein production in human bronchial epithelial (HBE) cells. HRV-16-induced ISG15 protein (48 h after infection) was assessed by immunoblotting following lipid-mediated transfection (24 h) of 10 nM IRF-1 short-interfering RNA (siRNA) duplexes A or B compared with control, nontargeting siRNA. ISG15 blot is representative of three separate experiments. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. M, medium control; HRV, HRV-16.
ISG15 siRNA knockdown does not directly alter HRV-16 replication in HBE cells
Previous reports have shown that ISG15 exerts antiviral actions against various viruses,19 including influenza virus.14 To determine whether ISG15 has a direct antiviral role against HRV in epithelial cells, HBE cells were transfected with two separate ISG15 siRNA duplexes (10 nM each). Effective knockdown of ISG15 protein was demonstrated compared with control siRNA at 48 h following HRV-16 infection (Figure 6a). Given that peak ISG15 protein expression occurs at 48 h following HRV infection, we assessed the effect of ISG15 knockdown on viral replication at times of peak ISG15 protein production (48–72 h time point). Thus, HBE cells were transfected with either one of two ISG15 siRNA duplexes or a control, nontargeting siRNA, and exposed to HRV-16 for 3 h. Cells were then washed thoroughly to remove noninternalized viral particles and fresh medium was added. Every 24 h, supernatants were collected and fresh medium was replaced for a total of 72 h. At the 48- to 72-h collection time point following peak ISG15 expression, HRV-16-infected HBE supernatants and cell lysates were assessed for levels of HRV-16 RNA by real-time PCR (Figure 6b). No significant difference in virus levels was found in supernatants between HRV-16-infected cells transfected with control siRNA or either of the ISG15 siRNA duplexes. Furthermore, loss of ISG15 using either duplex did not significantly alter HRV-16 RNA levels in whole cell lysates compared with control siRNA cells (Figure 6b). HRV-16 RNA was not detected in uninfected HBE cells (not shown). We also monitored the effect of ISG15 siRNA knockdown on the release of intact, infective virions using the same HRV-16-infected HBE supernatants by measuring viral titers (TCID50 ml−1). In agreement with our reverse transcriptase PCR (RT-PCR) studies, ISG15 knockdown did not significantly alter the release of infectious HRV-16 compared with control siRNA-infected cells (Figure 6c). Similar to our findings in HBE cells, knockdown of ISG15 (duplex A) in BEAS-2B cells did not significantly affect levels of HRV-16 viral titers compared with control siRNA-treated cells (not shown).
Figure 6.

Interferon-stimulated gene of 15 kDa (ISG15) knockdown markedly inhibits human rhinovirus (HRV)-16-induced ISG15 protein expression, but does not alter HRV-16 replication in human bronchial epithelial (HBE) cells. (a) HRV-16-induced ISG15 protein expression (48 h after infection) is reduced after transfection of 10 nM ISG15 short-interfering RNA (siRNA) duplexes A or B compared with control, nontargeting siRNA (n=3). HBE cells were initially transfected with either ISG15-specific siRNA duplex or control siRNA followed by HRV-16 infection for 3 h, washed with Hank's balanced salt solution (HBSS) three times, and fresh medium was added and collected every 24 h (72 h total). (b) HRV-16 RNA levels were measured (72 h after infection) in HBE lysates (white bars) and supernatants (black bars) using 5′-untranslated region (UTR)-specific primers and probe (cycle threshold counts±s.e.m.; n=3). (c) HRV-16 titers from HBE were measured (72 h after infection) from supernatants using WI-38 lung fibroblast viral titer assay (log TCID50 U ml−1±s.e.m.; n=3).
HRV-16 infection induces the binding of ISG15 to RIG-I
In HeLa cells exposed to IFN, ISG15 modulates activity of the double-stranded RNA pattern recognition receptor, RIG-I,20 which has been reported, along with melanoma differentiation associated gene-5 (MDA5), to regulate HRV-induced epithelial expression of downstream genes.21 We determined whether ISG15 regulates HRV-induced expression of RIG-I and/or MDA5. Transfection of HBE cells with either of the two ISG15-selective siRNA duplexes had no effect on intracellular expression of either RIG-I or MDA5 protein 48 h after HRV-16 infection (Figure 7a).
Figure 7.

Interferon-stimulated gene of 15 kDa (ISG15) does not alter human rhinovirus (HRV)-16-induced RIG-I protein expression, but ISG15 binding to RIG-I is increased following HRV infection. (a) HRV-16-induced RIG-I and MDA-5 protein levels in human bronchial epithelial (HBE) cell lysates (48 h after infection) were assessed by western blotting after lipid-mediated transfection (24 h) of 10 nM ISG15 short-interfering RNA (siRNA) duplexes A or B, compared with control, nontargeting siRNA. (b) HRV-16 infection induces ISG15 binding to RIG-I in BEAS-2B cells. A pTIS plasmid encoding FLAG-tagged RIG-I was transiently transfected using lipid-mediated techniques. Cells were infected with HRV-16 for 48 h and cells were lysed. (c) HRV-16-induced ISG15 and RIG-I protein levels (24 h after infection) in HBE cells were reduced after lipid-mediated transfection (24 h) of 10 nM RIG-I siRNA duplexes A or B compared with control, nontargeting siRNA. Blots are representative of three separate experiments. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. M, medium control; HRV, HRV-16.
We transfected BEAS-2B cells with a previously described pTIS vector expressing FLAG-tagged RIG-I (pTIS-FLAG-RIG-I) or empty pTIS vector as a negative control.22 Expression of RIG-I was assessed by immunoprecipitation followed by immunoblot detection of FLAG protein. As expected, BEAS-2B cells transfected with the control pTIS plasmid did not contain detectable RIG-I. Similarly, immunoprecipitation with IgG control antibody did not result in detectable FLAG-labeled RIG-I protein in cells transfected with the RIG-I vector (data not shown). By contrast, immunoprecipitation and blotting with anti-FLAG showed clear expression of RIG-I in BEAS-2B cells and the levels of expressed FLAG-labeled RIG-I were not different in cells exposed to medium or HRV-16 for 48 h (Figure 7b, upper panel). ISG15 protein was not detectable in cells transfected with either the pTIS or RIG-I plasmids and exposed to medium, but ISG15 was induced on HRV-16 infection of cells transfected with either the RIG-I or control vectors (Figure 7b, lower panel). To assess binding of ISG15 to RIG-I, cells were transfected with the RIG-I or control vectors and then exposed to medium alone or HRV-16. Cellular proteins were immunoprecipitated with anti-FLAG and blots were probed with anti-ISG15. No binding was detected in cells transfected with the control pTIS vector and exposed to either medium alone or HRV-16. In RIG-transfected cells, however, HRV-16 infection, but not exposure to medium, resulted in ISG15 binding to RIG-I (Figure 7b, middle panel).
RIG-I protein is detected in epithelial cells within 8 h of HRV infection,21 which is before the maximal intracellular ISG15 protein production. We therefore examined whether HRV-induced ISG15 expression is dependent on the presence of RIG-I. Transfection of HBE with two separate RIG-I siRNA duplexes abrogated HRV-16-induced ISG15 protein levels (Figure 7c), suggesting a potential feedback loop in the regulation of ISG15.
ISG15 selectively regulates HRV-16-induced CXCL10 expression via intracellular and extracellular pathways
As RIG-I has been reported to regulate HRV-induced epithelial expression of downstream genes, including CXCL10,21 we examined whether ISG15 regulated HRV-16-induced CXCL10 expression. Transfection of HBE with ISG15-selective siRNA duplexes resulted in a significant (P<0.01) enhancement of CXCL10 protein release 48 h post HRV-16 infection (Figure 8a). Given that ISG15 can also be secreted from HRV-infected HBE cells, we evaluated effects of extracellular application of recombinant active and inactive (precursor) forms of ISG15. Human recombinant active ISG15 caused a concentration-dependent (P<0.05) inhibition of HRV-16-induced CXCL10 release compared with virus alone, with significant inhibition observed at both 30 and 10 μg ml−1 active ISG15 (Figure 8b), whereas inactive pro-ISG15 had no significant effect. Incubation of HBE cells with ISG15 alone did not induce CXCL10 protein release at any concentration used (data not shown). HRV-16-induced epithelial CXCL10 expression is transcriptionally regulated.11, 17 To determine whether ISG15 affected CXCL10 transcription, BEAS-2B cells were transiently transfected with a 972-bp CXCL10 promoter luciferase construct followed by incubation with HRV-16 alone, 30 μg ml−1 of active or pro-ISG15 alone, and active or pro-ISG15 in combination with HRV-16 for 24 h (Figure 8c). HRV-induced CXCL10 promoter luciferase activity was significantly (P<0.05) inhibited in the presence of 30 μg ml−1 active ISG15, whereas the inactive form of ISG15 protein (30 μg ml−1) did not alter HRV-16-induced CXCL10 promoter drive. Neither form of recombinant ISG15 protein caused CXCL10 promoter drive by itself (Figure 8c). The effects of ISG15 on HRV-16-induced CXCL10 were selective, as neither the active or inactive form of ISG15 inhibited HRV-16-induced CXCL8 release (Figure 8d).
Figure 8.

Human rhinovirus (HRV)-16-induced CXCL10 protein production from human bronchial epithelial (HBE) cells is modulated in the presence of interferon-stimulated gene of 15 kDa (ISG15) short-interfering RNA (siRNA) and recombinant, active ISG15 protein. (a) HRV-16-induced CXCL10 protein release in the presence of ISG15-specific or control, nontargeting siRNA was assessed by enzyme-linked immunosorbent assay (ELISA) at 48 h after infection. Asterisk indicates significant difference between HRV-16-induced CXCL10 production in ISG15 siRNA vs. control siRNA-treated cells (P<0.05). HRV-16-induced (b) CXCL10 or (d) CXCL8 protein levels in supernatants were assessed using ELISA in the presence of various concentrations of either active or inactive ISG15 protein after 24 h incubation. (c) BEAS-2B cells were transiently transfected with a full-length 972-bp CXCL10 promoter-luciferase construct and incubated with either active or inactive ISG15 (30 μg ml−1) in the presence or absence of HRV-16 for 24 h. Luciferase activity was measured in cell lysates. Data are presented as fold increase over medium control and represent mean±s.e.m. (n=6). CXCL10 and CXCL8 protein data are means±s.e.m. (n=3–5). Asterisk indicates significant difference between HRV-16 alone and HRV-16+active ISG15 (P<0.05).
HRV-induced epithelial cell expression of ISG15 and CXCL10 is not dependent on IFNs
Given that ISG15 and CXCL10 are both IFN-inducible genes, it is possible that the ability of HRV-infection to induce epithelial expression of one or both of these genes may occur secondary to viral induction of IFNs. We previously showed that HRV-induced epithelial CXCL10 expression is not dependent on type I IFNs, as blockade of the type I IFN receptor blocked induction of CXCL10 by IFN-β but not by HRV infection.11 Moreover, although HRV-induced IFN-β secretion from epithelial cells has been described,23 we, and others, have been unable to detect measurable type I IFN protein secretion from HRV-infected epithelial cells.6, 11, 24, 25 By contrast, HRV-induced IFN-λ1 (interleukin (IL)-29) secretion has been consistently observed from human airway epithelial cells.6, 26 To determine whether viral induction of IL-29 was necessary for HRV-induced ISG15 and CXCL10 expression, we initially tried using siRNA approaches in primary HBE cells. Despite using multiple siRNA duplexes, we were unable to achieve substantial knockdown of IL-29. Therefore, as an alternative approach we exposed BEAS-2B cells, which do not produce IL-29 on HRV infection (unpublished data), to HRV or 1 ng ml−1 of IL-29 to examine the role of IL-29 in HRV-induced epithelial expression of ISG15 and CXCL10 without potential interference from endogenous IFN production. The concentration of IL-29 used (1 ng ml−1) is the maximal level we have ever detected from primary HBE cells on HRV infection.6 As expected, HRV infection caused robust induction of both ISG15 and CXCL10 at 24 and 48 h post infection, but did not stimulate measurable IL-29 production (Figure 9). By contrast, stimulation with IL-29 induced only weak expression of ISG15 and low levels of CXCL10 secretion. Greater than 80% of the exogenously administered IL-29 was recovered even at 48 h after application, indicating that the lack of measurable production of IL-29 from HRV-infected BEAS-2B cells is unlikely to be due to degradation. Thus, HRV-induced ISG15 and CXCL10 production is not dependent on IL-29 production.
Figure 9.

Human rhinovirus (HRV)-16-induced interferon (IFN)-stimulated gene of 15 kDa (ISG15) expression and CXCL10 production is not dependent on interleukin (IL)-29 (type III IFN). BEAS-2B cells were infected with HRV-16 (HRV) or exposed to medium (M) or exogenous IL-29 (1 ng ml−1). (a) Western blotting for ISG15 expression (representative of n=3). (b) Enzyme-linked immunosorbent assay (ELISA) to determine levels of IL-29 in supernatants at 24 and 48 h post stimulation (data are mean±s.e.m. from three experiments). (c) Levels of CXCL10 secretion in supernatants at 24 and 48 h post stimulation (data are mean±s.e.m. from three experiments).
Discussion
ISG15 can be covalently attached to both host and viral proteins via the action of a unique intracellular ligase cascade that parallels that involved in ubiquitination.27 Conjugation (ISGylation) is postulated to alter the function of viral and host proteins leading to antiviral activity.19, 28 Although ISG15 expression has been described in epithelial cells infected with HRV,4, 6 the regulation of its expression and its potential role in antiviral immunity to HRV has not been previously examined.
We confirmed that HRV infection induces epithelial expression of ISG15 mRNA and protein, and provided the first demonstration that ISG15 is secreted from HRV-infected primary human airway epithelial cells. Expression and secretion of ISG15 was not observed until 24–48 h post infection, suggesting that induction is dependent on late, virus replication-dependent signaling pathways. We have previously shown that HRV-16 infection does not alter cell viability;29, 30 thus, release must be due to active secretion rather than cell lysis. As ISG15 does not contain a secretory leader sequence,16 it has been proposed that it is released through a noncanonical pathway independent of the endoplasmic reticulum and Golgi complex, similar to IL-1β or fibroblast growth factors 1 and 2.16, 31 Although it is feasible that ISG15 could be secreted as a complex with another protein, there are no reports of ISG15 conjugate secretion, and the secreted ISG15 we measure dilutes appropriately in the enzyme-linked immunosorbent assay, indicating that if a conjugate is formed it must be with a protein small enough to not interfere with antibody recognition. As noted, ISG15 is an ubiquitin-like protein; thus, it is of interest that both ubiquitin and SUMO-3 can also be secreted from some cell types in humans.31 We also provide the first demonstration that ISG15 is released into nasal secretions of subjects with naturally acquired HRV infections, and that levels of ISG15 in secretions significantly correlate with viral load and symptom scores. As ISG15 in the extracellular milieu can exert immunomodulatory activities, including inducing proliferation of T lymphocytes, expansion and activation of NK cells, and recruitment of neutrophils,15, 32 secretion of ISG15 into nasal secretions could regulate antiviral immunity during HRV infections.
HRV-induced epithelial expression of ISG15 is regulated, at least in part, at the level of transcription. Our data show that activation of the ISG15 promoter upon HRV infection is dependent, at least in part, on interaction of a transcription factor(s) of the IRF family with a specific recognition sequence(s). Our data suggest a clear role for the IRF-B recognition sequence, while the role of the IRF-A site is not entirely clear, as data can be interpreted differently depending on whether expressed as fold induction above respective basal drive or as raw RLUs. Interestingly, the IRF-B site has been shown to be important for transcriptional regulation of ISG15 induction in response to IFN-β.33 Although viral induction of genes is often linked to activation of IRF-3,34 our prior studies have indicated no role for IRF-3 in induction of epithelial expression of CXCL10 on HRV infection, but an essential role for IRF-1.17, 18 Consistent with our earlier observations, selective knockdown of IRF-1 markedly inhibited HRV-induced ISG15 expression, suggesting that interactions of IRF-1 with the IRF recognition sequence(s) in the ISG15 promoter contributes to ISG15 gene regulation.
Although ISG15 inhibits the replication of a number of viruses, either via direct ISGylation of viral proteins or via conjugation to host cell proteins, or both,14, 35 highly effective knockdown of ISG15 using each of two different siRNA duplexes targeting ISG15 had no effect on HRV replication in airway epithelial cells. We have previously shown that viral replication in cultured epithelial cells remains constant over at least a 3-day period;8 hence, we were able to assess the effects of siRNA knockdown of ISG15 at a time after it was normally maximally induced. Consistent with these data, showing no direct role of ISG15 in regulating replication of HRV, replication of other viruses, including herpes simplex virus, is unaltered in cells derived from patients with inherited ISG15 deficiency,36 and ISG15-deficient mice show increased susceptibility to some, but by no means all, of the viruses examined.35
This lack of direct effect on HRV replication does not rule out a potential modulatory role of ISG15 on antiviral immunity via effects on host proteins. The RNA helicases, RIG-I and MDA5, serve as pattern recognition receptors for viral double-stranded RNA and signaling via both of these molecules has been linked to HRV-induced expression of a number of downstream gene products.21, 37 Knockdown of ISG15 did not alter expression of RIG-I or MDA5 upon HRV infection, but this did not rule out effects on RIG-I function, as conjugation of ISG15 to RIG-I has previously been reported to regulate downstream signaling.38
Although RIG-I has been reported to be a target for ISGylation in HeLa cells stimulated with IFN,20 this has not been examined in response to HRV infection of bronchial epithelial cells. Using a FLAG-tagged RIG-I overexpression plasmid and immunoprecipitation, we showed that when ISG15 expression was induced in cells infected with HRV-16, ISG15 binding to RIG-I occurred. These data are consistent with the concept that ISGylation of RIG-I occurs in HRV-infected epithelial cells and, presumably, is involved in downregulating RIG-I function.
As induction of RIG-I occurs within 8 h in HRV-infected cells,21 we used siRNA knockdown to determine whether RIG-I may also mediate HRV-induced ISG15 expression, and found that preventing RIG-I induction did indeed inhibit HRV-induced ISG15 expression. Thus, although HRV-16-induced ISG15 conjugates to RIG-I, the expression of ISG15 following HRV infection is also dependent on RIG-I-mediated signaling, forming a potential negative feedback loop.
RIG-I-mediated signaling has been linked to downstream production of several proteins in HRV-infected epithelial cells,21 including CXCL10, a chemokine linked to antiviral immunity via its ability to recruit activated type 1 T lymphocytes and NK cells to sites of infection. CXCL10-deficient mice have decreased ability to control viral infections, and impaired T-cell recruitment and activation, whereas CXCL10 transgenic mice show improved control of infection and enhanced NK cell responses.39, 40 The demonstration that ISG15 knockdown led to enhanced HRV-16-induced secretion of CXCL10 implies that ISG15 has a negative regulatory role on CXCL10 secretion. Consistent with the siRNA knockdown studies, we found that exogenous application of active, but not an inactive precursor form of, ISG15 inhibited HRV-induced CXCL10 production. Exogenous ISG15 also was able to suppress transcriptional activation of the CXCL10 gene. This effect appears to have some selectivity, as CXCL8 production in response to HRV-16 infection was not altered. CXCL8 is induced early after HRV infection and can occur even with virus-rendered replication deficient via direct receptor-mediated signaling.41, 42, 43 The lack of effect on CXCL8 may reflect the fact that its induction, unlike CXCL10, does not require the interaction of viral replication intermediates with RNA helicases, such as RIG-I, or induction of IRF-1. Given that ISG15 does not inhibit viral replication and suppresses production of CXCL10, which has been linked to antiviral immunity, it may be tempting to speculate that ISG15 actually suppresses antiviral immunity to HRV infection. However, such speculation may be premature, as a comprehensive assessment of the effects of ISG15 on other genes with potential antiviral effects has not yet been performed. In addition, the ability of ISG15 itself to trigger expansion and activation of NK cells implies that the role of this molecule in the antiviral response to HRV infection is likely to be complex. Furthermore, the putative receptor(s) for extracellular ISG15 has not been identified and it is unknown how many other cell types may be regulated via extracellular ISG15.
Both ISG15 and CXCL10 are considered members of the so-called “IFN-inducible transcriptome”. However, although these genes are IFN-inducible, there is precedent that many inducible genes can also be induced by IFN-independent mechanisms. Human airway epithelial cells do not produce IFN-γ and the induction of type I IFNs in HRV-infected epithelial cells is controversial. There are reports of HRV-induced epithelial production of IFN-β, and it has been suggested that deficient HRV-induced production of IFN-β is a characteristic of asthma.23 By contrast, several groups, including us, have been unable to measure secretion of type I IFNs from HRV-infected airway epithelial cells.6, 11, 24, 25 Moreover, we have previously shown that blockade of the type I IFN receptor inhibits IFN-β-induced airway epithelial cell production of CXCL10, but does not modulate HRV-induced CXCL10 production.11 Recent data suggest that the predominant IFN produced on HRV infection of primary cultures of human airway epithelial cells is IFN-λ1 (IL-29).6, 25 We demonstrated, however, that HRV infection of BEAS-2B cells (which do not produce IL-29 in response to infection) generated substantial amounts or ISG15 and CXCL10. By contrast, exogenous administration of IL-29, at the maximal level we have observed from HRV-infected primary airway epithelial cultures, was a much weaker stimulus for induction of ISG15 and CXCL10. Taken together, therefore, our data indicate that HRV infection induces both ISG15 and CXCL10 independently of type I or type III IFNs. This is consistent with a recent study demonstrating that influenza infection of cells lacking both type I and type III IFN receptors still induced a full “IFN-like transcriptome,” including both ISG15 and CXCL10.44
In summary, HRV infection of epithelial cells induces expression and secretion of ISG15. Induction of ISG15 is independent of IFN production but depends on transcriptional regulation via IRF recognition sequence(s) in the ISG15 promoter. Knockdown of IRF-1 suppresses HRV-induced ISG15 production. ISG15 does not directly regulate replication of HRV in airway epithelial cells but can modulate immune responses via effects on the double-stranded RNA pattern recognition receptor, RIG-I, and by regulating CXCL10 production. Additional studies are required to more fully define the complex role of ISG15 in host antiviral defenses against HRV infection.
Methods
Reagents and antibodies. The following reagents were purchased: Eagle's minimal essential medium, Hank's balanced salt solution, penicillin–streptomycin–amphotericin B, and L-glutamine, TRIzol reagent, sodium pyruvate, nonessential amino acids, gentamicin, and fetal bovine serum (Invitrogen, Burlington, ON, Canada); bronchial epithelial cell basal medium, and additives to create bronchial epithelial cell growth medium (BEGM; Lonza, Walkersville, MD); TaqMan Master Mix, 20 × glyceraldehyde-3-phosphate dehydrogenase, RNase inhibitor, and reverse transcriptase (Applied Biosystems, Streetsville, ON, Canada); TransIT LTI (Mirius, Madison, WI); firefly luciferase reporter plasmid pGL4.10[luc2], and 5 × passive lysis buffer (Promega, Madison, WI); firefly luciferase assay kit (Biotium, Hayward, CA); protease inhibitor tablets (Roche, Mississauga, ON, Canada); horseradish peroxidase-conjugated anti-rabbit Ig Ab and enhanced chemoluminescent substrate reagent (GE Healthcare Bio-Sciences, Piscataway, NJ); specific antibodies: ISG15 and RIG-I (Cell Signaling Technology, Danvers, MA), IRF-1 (Santa Cruz Biotechnology, Santa Cruz, CA), MDA5 (Enzo Life Sciences, Plymouth Meeting, PA); FLAG (Sigma-Aldrich, Oakville, ON, Canada); recombinant active ISG15 and pro-ISG15 (Boston Biochem, Boston, MA). All other chemicals were purchased from Sigma-Aldrich.
Viruses and cell lines. The BEAS-2B cell line was a gift from Dr Curtis Harris (National Cancer Institute, Bethesda, MD). This cell line shows similar responses to HBE on HRV infection.17, 18, 29, 45 WI-38 cells were purchased from the American Type Culture Collection (Manassas, VA). HRV-16 viral stocks were propagated in WI-38 cells and purified by centrifugation through sucrose to remove ribosomes and soluble factors of WI-38 origin as previously described.12, 46
Epithelial cell culture. Normal human lungs not used for transplantation were obtained from a tissue retrieval service (International Institute for the Advancement of Medicine, Jessup, PA). Approval to use recovered organs for these studies was obtained from the Conjoint Health Research Ethics Board of the University of Calgary. Primary HBE cells were obtained by protease digestion of dissected airways as previously described.47 Primary HBE cells from a total of 12 donors (10 male; age range 18–62 years) were used for the current studies. All donors died from head trauma or cerebrovascular causes, and none of the donors had any inflammatory lung disease. Both BEAS-2B and HBE cells were grown on six-well culture plates in BEGM. Before stimulation, cells were cultured overnight in BEGM from which hydrocortisone had been removed after washing with Hank's balanced salt solution, and this hydrocortisone-free medium was used for all experiments.
Viral infection of epithelial cells. BEAS-2B cells were infected with 104.5 50% tissue culture-infective dose (TCID50) U ml−1 (multiplicity of infection of ∼0.1) HRV-16, whereas HBE cells were infected with 105.5 TCID50 U ml−1 (multiplicity of infection of ∼1.0) HRV-16. A higher dose was necessary for HBE cells, as only up to 10% of cells are infected, even with high doses of HRV.48
siRNA knockdown of ISG15, RIG-I, and IRF-1. Subconfluent BEAS-2B or HBE cells were transfected with 10 nM of specific siRNAs targeting each molecule of interest, or an appropriate control, nontargeting siRNA (Life Technologies, Burlington, ON, Canada) for 24 h at 37 °C using Lipofectamine RNAiMAX (Life Technologies) in BEGM without antibiotics. For each molecule of interest, two different siRNAs were tested to confirm selectivity. After transfection, medium was changed and cells were allowed to recover for 48 h, with media changed to BEGM without hydrocortisone for the second 24-h period. Cells were then infected with HRV-16, and supernatants were collected 48 h post infection. Whole-cell lysates from BEAS-2B and HBE cells were collected 24 or 48 h post infection. The specific forward siRNA sequences used were as follows: ISG15 duplex A, 5′-GCAUCCUGGUGAGGAAUAACAAGGG-3′ ISG15 duplex B, 5′-GAGCUGAAGGCGCAGAUCATT-3′ RIG-I duplex A, 5′-AAGCTTTACAACCAGAATTTA-3′ RIG-I duplex B, 5′-TTCTACAGATTTGCTCTACTA-3′ IRF-1 duplex A, 5′-UCCCAAGACG UGGAAGGCCAACUUU-3′ and IRF-1 duplex B, 5′-CGGACAGCACCAGUGAUCUGUACAA-3′.
Assessment of CXCL10, IRF-1, RIG-I, MDA5, and ISG15 protein. CXCL10 protein levels were assayed by enzyme-linked immunosorbent assay using matched antibody pairs (R&D Systems, Minneapolis, MN). CXCL8 levels were measured using enzyme-linked immunosorbent assay as previously described.8 ISG15 protein levels from HBE supernatants and HRV-infected individuals were measured using a commercial enzyme-linked immunorsorbent assay kit (Cusabio, Wuhan, China). Whole-cell lysates from HRV-16-infected cells were isolated and separated on SDS-polyacrylamide gel electrophoresis as previously described.41 Membranes were probed with IRF-1, RIG-I, MDA5, or ISG15 specific antibody overnight at 4 °C. Membranes were washed and incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit or mouse IgG, and visualized with enhanced chemoluminescent substrate reagent. Membranes were stripped and re-probed with antibody to glyceraldehyde-3-phosphate dehydrogenase to ensure equal loading.
Assessment of ISG15 mRNA. Total cellular RNA from HRV-16-infected BEAS-2B or HBE cells was isolated with TRIzol and treated with DNase (Ambion, Austin, TX). ISG15 mRNA expression was assessed by real time RT-PCR using intron-spanning specific primers and a Taqman probe (Applied Biosystems): ISG15 forward primer, 5′-GCTGGGACCTGACGGTGA-3′, ISG15 reverse primer, 5′-TGGAGCTGCTCAGGGACAC-3′ ISG15 probe, 5′-FAM-ATGCTGGCGGGCAACGAATTCC-MGB-3′. To permit absolute quantification, a first-strand cDNA standard encompassing the sequences bound by ISG15 primers and probe was synthesized (University of Calgary DNA Services, Calgary, AB, Canada). Expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase was also assayed using reagents obtained from Applied Biosystems. Data were expressed as femtograms (fg) calculated from the standard curve after correction for variations in housekeeping gene levels.
Overexpression of FLAG-tagged RIG-I and immunoprecipitation. The pTIS and pTIS-FLAG-RIG-I plasmids22 were kindly provided by Dr Kui Li (University of Tennessee Health Sciences Center). BEAS-2B cells were transfected with either overexpression plasmid for 6 h. Cells were then allowed to recover overnight and either treated with media or purified HRV-16 for 48 h. Cells were washed with cold phosphate buffered saline and then scraped in lysis buffer (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1 × mini complete, 1 mM phenylmethylsulfonyl fluoreide and 1% Triton X-100). Lysates were incubated on ice for 10 min, sonicated, and centrifuged at 10,000 × g for 10 min at 4 °C. One milligram of protein was incubated with 10 μg ml−1 anti-FLAG M2 antibody (Sigma, Oakville, ON, Canada) overnight at 4 °C. Protein G-Sepharose beads (GE Heathcare) were added to the sample and incubated for 30 min at 4 °C. Beads were washed 4 × in immunoprecipitation buffer (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl) and Laemmli's buffer was added. Samples were subsequently run on SDS-polyacrylamide gel electrophoresis.
ISG15 promoter constructs. A 634-bp ISG15 promoter–luciferase construct, corresponding to the sequence from −513 to +121 (relative to the transcriptional start site) of the 5′-flanking region of the human ISG15 gene, was amplified from human genomic DNA and cloned into a pGL4.10 firefly luciferase vector. Point mutations in the putative cAMP response element, proximal NF-κB1 (κB1), and IRF promoter sites were generated in the truncated construct using standard site-directed mutagenesis techniques. Boldface, lowercase letters in the following sequences denote mutation sites in forward primers. For cAMP response element, 5′-ACGCCCtCTGAtGTGTGTGCCTCA-3′ for IRF-A, 5′-TCGGGAAAGGGAAACCttAACTGAAGC-3′ for IRF-B, 5′-TCGGGAAAGttAAACCGAA ACTGAAGC-3′ and for κB1, 5′-GTGGtGATaCCGTCCGCTGTCCG-3′. Generation of the 972-bp CXCL10 full-length promoter-luciferase construct has been previously described.11 Successful generation of all constructs was confirmed by sequencing (University of Calgary DNA Services).
Lipid transfection and luciferase assay. Subconfluent (40–50%) monolayers of BEAS-2B cells were transiently transfected with 0.1 μg of each ISG15 promoter construct for 6 h at 37 °C using TransIT LT1 (Mirius) in basal medium as per the manufacturer's protocol. Alternatively, full-length 972-bp CXCL10 promoter luciferase construct (0.01 μg) was transfected as previously described.11, 30 After 6 h, medium was aspirated and cells were allowed to recover overnight in BEGM (without hydrocortisone) containing 5% fetal bovine serum. Cells were infected with HRV-16 (1 × 105 TCID50 U ml−1) for 24 h at 34 °C and then lysed in passive lysis buffer. Firefly luciferase activity was measured following the addition of D-luciferin substrate. Data were expressed as fold increase of stimulated cells over control, or as luminometer readings in the form of RLUs. Within each experiment, results were averaged from triplicate wells.
Natural cold study samples. Samples were taken from a previously conducted, prospective, cross-sectional study in which otherwise healthy, nonsmoking, nonallergic subjects reported to the research clinic within 36 h after the onset of cold symptoms.49 Subjects completed a validated symptom questionnaire and underwent nasal lavage, as described previously.4, 50 Symptom scores and nasal lavage were repeated on the two subsequent days. At least two full weeks after the resolution of cold symptoms, subjects returned to the clinic for a retrospective baseline visit (visit 4), at which time symptom scores and nasal lavage were repeated. Collection and processing of nasal lavage samples has been previously described.50 All subjects provided written informed consent, and the protocol was approved by the Conjoint Health Research Ethics Board of the University of Calgary.
Viral RNA from nasal lavage samples was isolated using the QIAamp Viral RNA Mini Kit (Qiagen, Mississauga, ON, Canada). RNA was reverse transcribed using oligo(dT) and Superscript II reverse transcriptase. Quantitative analysis of HRV was performed using a previously described real-time RT-PCR technique that detects all known species of HRV.49 To quantify HRV genomic material in nasal lavages, a purified HRV-16 preparation of known titer was used to establish a standard curve that was run in each assay. The standard curve permitted quantification of HRV over the range of 1 to 105 TCID50 ml−1 of original nasal lavage fluid.49 Samples were obtained from 10 subjects (mean age, 38 years; range, 22–67 years; 5 women) during established HRV infections and ∼4 weeks later, when they had been symptom-free for more than 2 weeks. These latter samples were confirmed to be negative for HRV by real-time RT-PCR.
Measurement of HRV-16 viral mRNA and titers. HBE cells were plated in six-well plates and infected for 3 h at 34 °C followed by washing of cells with Hank's balanced salt solution three times. Fresh medium was added to cells and further incubated for a total of 72 h at 34 °C with medium collected and replaced every 24 h. At 72 h post infection, cells were scraped in fresh medium, sonicated for 10 s, and centrifuged for 10 min at 20,000 × g at 4 °C. Viral RNA was isolated as above from cell supernatants and lysates, and subjected to quantitative RT-PCR using primers and probe directed to the 5′-untranslated regions of HRV-16. Forward primer: 5′-TCCTCCGGCCCCTGAA-3′, reverse primer: 5′-ACTGGATTGTGTGCACTGGCT-3′ probe: 5′-FAM-TGGCTAACCTTAAACCT-MGB-3′. This system did not detect other rhinovirus strains, including HRV-14, HRV-39, and HRV-1A. HRV-16 viral titers were assessed in HBE supernatants using WI-38 cells, as previously described.12
Statistical analysis. For normally distributed data, between-group comparisons were made by appropriate one-way or two-way analysis of variance, with post-hoc analysis using Neuman–Keuls post-hoc multiple comparison testing for statistical significance. For nonparametric data, Kruskal–Wallis analysis of variance was used, followed by Wilcoxon matched-pairs signed-rank test. Correlations between continuous variables were assessed using Pearson's correlation coefficient. Correlations involving nonparametric data were assessed using Spearman's rank correlation coefficient. For all statistical tests, a two-tailed P-value of <0.05 was assumed to be significant.
Acknowledgments
We thank Dr Kui Li for providing the pTIS-FLAG-RIG-I and pTIS control plasmids. Grant number 43923 from the Canadian Institutes of Health Research supported this work. D.P. holds a Canada Research Chair in Inflammatory Airway Disease. R.L. holds the GlaxoSmithKline/Canadian Institutes of Health Research Professorship in Inflammatory Lung Diseases. M.H. is the recipient of a studentship from Alberta Innovates-Health Solutions.
The authors declared no conflict of interest.
References
- Proud D, Chow C.-W. Role of viral infections in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2006;35:513–518. doi: 10.1165/rcmb.2006-0199TR. [DOI] [PubMed] [Google Scholar]
- Traves S.L., Proud D. Viral-associated exacerbations of asthma and COPD. Curr. Opin. Pharmacol. 2007;7:252–258. doi: 10.1016/j.coph.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat D., et al. The role of respiratory viruses in cystic fibrosis. J. Cyst. Fibros. 2008;7:320–328. doi: 10.1016/j.jcf.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud D., et al. Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am. J. Respir. Crit. Care Med. 2008;178:962–968. doi: 10.1164/rccm.200805-670OC. [DOI] [PubMed] [Google Scholar]
- Bochkov Y.A., Hanson K.M., Keles S, Brockman-Schneider R.A., Jarjour N.N., Gern J.E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud D., et al. Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS One. 2012;7:e40762. doi: 10.1371/journal.pone.0040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., et al. Rhinovirus induces airway epithelial gene expression through double-stranded RNA and IFN-dependent pathways. Am. J. Respir. Cell. Mol. Biol. 2006;34:192–203. doi: 10.1165/rcmb.2004-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste M.C., Jacoby D.B., Richards S.M., Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J. Clin. Invest. 1995;96:549–557. doi: 10.1172/JCI118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroth M.K., et al. Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am. J. Respir. Cell. Mol. Biol. 1999;20:1220–1228. doi: 10.1165/ajrcmb.20.6.3261. [DOI] [PubMed] [Google Scholar]
- Donninger H, Glashoff R, Haitchi H.-M., Syce J.A., Ghildyal R, Bardin P.G. Rhinovirus induction of the CXC chemokine epithelial-neutrophil activating peptide-78 in bronchial epithelium. J. Infect. Dis. 2003;187:1809–1817. doi: 10.1086/375246. [DOI] [PubMed] [Google Scholar]
- Spurrell J.C.L., Wiehler S, Zaheer R.S., Sanders S.P., Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;289:L85–L95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- Sanders S.P., Siekierski E.S., Porter J.D., Richards S.M., Proud D. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 1998;72:934–942. doi: 10.1128/jvi.72.2.934-942.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee L.A., Lyon N, Seo K, Huibregtse J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Hsiang T.-Y., Kuo R.-L., Krug R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl Acad. Sci. USA. 2010;107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cunha J, Knight E.J., Haas A.L., Truitt R.L., Borden E.C. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl Acad. Sci. USA. 1996;93:211–215. doi: 10.1073/pnas.93.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cunha J, Ramanujam S, Wagner R.J., Witt P.L., Knight E.J., Borden E.C. In vitro and in vivo secretion of human ISG15, an interferon-induced immunomodulatory cytokine. J. Immunol. 1996;157:4100–4108. [PubMed] [Google Scholar]
- Zaheer R.S., Proud D. Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN regulatory factor-1. Am. J. Respir. Cell. Mol. Biol. 2010;43:413–421. doi: 10.1165/rcmb.2009-0203OC. [DOI] [PubMed] [Google Scholar]
- Koetzler R, Zaheer R.S., Newton R, Proud D. Nitric oxide inhibits IFN regulatory factor 1 and nuclear factor-κB pathways in rhinovirus-infected epithelial cells. J. Allergy Clin. Immunol. 2009;124:551–557. doi: 10.1016/j.jaci.2009.04.041. [DOI] [PubMed] [Google Scholar]
- Harty R.N., Pitha P.M., Okumura A. Antiviral activity of innate immune protein ISG15. J. Innate Immun. 2009;1:397–404. doi: 10.1159/000226245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Denison C, Huibregtse J.M., Gygi S, Krug R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed protein functioning in diverse cellular pathways. Proc. Natl Acad. Sci. USA. 2005;102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater L., et al. Co-ordinated role of TLR-3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6:e1001178. doi: 10.1371/journal.ppat.1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jamaluddin M, Li K, Garofalo R.P., Casola A, Brasier A.R. Retinoic acid-inducible gene I mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007;81:1401–1411. doi: 10.1128/JVI.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark P.A.B., et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkov Y.A., et al. Budesonide and formoterol effects on rhinovirus replication and epithelial cell cytokine responses. Respir. Res. 2013;14:98. doi: 10.1186/1465-9921-14-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes A., et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma Thoraxdoi: 10.1136/thoraxjnl-2012-2029092013 [DOI] [PubMed]
- Contoli M., et al. Role of deficient type-III interferon-λ production in asthma exacerbations. Nat. Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- Ritchie K.J., Zhang D.-E. ISG15: the immunological kin of ubiquitin. Semin. Cell Dev. Biol. 2004;15:237–246. doi: 10.1016/j.semcdb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Skaug B, Chen Z.J. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudy M.H., Traves S.L., Wiehler S, Proud D. Cigarette smoke modulates rhinovirus-induced airway epithelial chemokine production. Eur. Respir. J. 2010;35:1256–1263. doi: 10.1183/09031936.00128809. [DOI] [PubMed] [Google Scholar]
- Zaheer R.S., Koetzler R, Holden N.S., Wiehler S, Proud D. Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J. Immunol. 2009;182:4854–4864. doi: 10.4049/jimmunol.0802401. [DOI] [PubMed] [Google Scholar]
- Bogunovic D, Boisson-Dupuis S, Casanova J.-L. ISG15: leading a double life as a secreted protein. Exp. Mol. Med. 2013;45:e18. doi: 10.1038/emm.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owhashi M, Taoka Y, Ishii K, Nakazawa S, Uemura H, Kambara H. Identification of a ubiquitin family protein as a novel neutrophil chemotactic factor. Biochem. Biophys. Res. Commun. 2003;309:533–539. doi: 10.1016/j.bbrc.2003.08.038. [DOI] [PubMed] [Google Scholar]
- Reich N, Evans B, Levy D, Fahey D, Knight E.J., Darnell J.E., Jr. Interferon-induced transcription of a gene encoding a 15-kDa protein depends upon an upstream enhancer element. Proc. Natl Acad. Sci. USA. 1987;84:6394–6398. doi: 10.1073/pnas.84.18.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl Acad. Sci. USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D., et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011;7:e1002070. doi: 10.1371/journal.ppat.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.-J., Hwang S.-Y., Imaizumi T, Yoo J.-Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008;82:1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour J.H., Dziejman M, Liu M.T., Leung J.H., Lane T.E., Luster A.D. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T-cell generation and trafficking. J. Immunol. 2002;168:3915–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- Trifilo M.J., et al. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 2004;78:585–594. doi: 10.1128/JVI.78.2.585-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehler S, Proud D. Interleukin-17A modulates human airway epithelial responses to human rhinovirus infection. Am. J. Physiol. Cell. Mol. Physiol. 2007;293:L505–L515. doi: 10.1152/ajplung.00066.2007. [DOI] [PubMed] [Google Scholar]
- Newcomb D.C., et al. Phosphatidylinositol 3-kinase is required for rhinovirus-induced airway epithelial cell interleukin-8 expression. J. Biol. Chem. 2005;280:36952–36961. doi: 10.1074/jbc.M502449200. [DOI] [PubMed] [Google Scholar]
- Wang X., et al. Syk is downstream of intercellular adhesion molecule-1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. J. Immunol. 2006;177:6859–6870. doi: 10.4049/jimmunol.177.10.6859. [DOI] [PubMed] [Google Scholar]
- Schmid S, Mordstein M, Kochs G, García-Sastre A, tenOever B.R. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 2010;285:42013–42022. doi: 10.1074/jbc.M110.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koetzler R, Zaheer R.S., Wiehler S, Holden N.S., Giembycz M.A., Proud D. Nitric oxide inhibits human rhinovirus-induced transcriptional activation of CXCL10 in airway epithelial cells. J. Allergy Clin. Immunol. 2009;123:201–208. doi: 10.1016/j.jaci.2008.09.041. [DOI] [PubMed] [Google Scholar]
- Gern J.E., et al. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J. Immunol. 1996;156:621–627. [PubMed] [Google Scholar]
- Churchill L, Chilton F.H., Resau J.H., Bascom R, Hubbard W.C., Proud D. Cyclooxygenase metabolism of endogenous arachidonic acid by cultured human tracheal epithelial cells. Am. Rev. Respir. Dis. 1989;140:449–459. doi: 10.1164/ajrccm/140.2.449. [DOI] [PubMed] [Google Scholar]
- Mosser A.G., et al. Similar frequency of rhinovirus-infectable cells in upper and lower airway epithelium. J. Infect. Dis. 2002;185:734–743. doi: 10.1086/339339. [DOI] [PubMed] [Google Scholar]
- Leigh R., et al. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J. Allergy Clin. Immunol. 2008;121:1238–1245. doi: 10.1016/j.jaci.2008.01.067. [DOI] [PubMed] [Google Scholar]
- Proud D, Gwaltney J.M., Jr., Hendley J.O., Dinarello C.A., Gillis S, Schleimer R.P. Increased levels of interleukin-1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J. Infect. Dis. 1994;169:1007–1013. doi: 10.1093/infdis/169.5.1007. [DOI] [PubMed] [Google Scholar]