

Abstract
Newly designed bivalent ligands—opioid agonist/NK1-antagonists have been synthesized. The synthesis of new starting materials—carboxy-derivatives of Fentanyl (1a–1c) was developed. These products have been transformed to ‘isoimidium perchlorates’ (2a–c). The new isoimidium perchlorates have been successfully implemented in nucleophilic addition reactions, with L-tryptophan 3,5-bis(trifluoromethyl)benzyl ester to give the target compounds—amides (3a–c). Perchlorates (2a–c) successfully undergo reactions with other nucleophiles such as alcohols, amines or hydrazines. The obtained compound 3b exhibited μ-opioid agonist activity and NK1-antagonist activity and may serve as a useful lead compound for the further design of a new series of opioid agonist/NK1-antagonist compounds.
Keywords: Analgesic, Bivalent ligands, μ-Opioids, NK1 antagonist, Fentanyl
1. Introduction
Opioid analgesics are the mainstay for treatment of moderate to severe pain. Although opioids are still the drugs of choice for the treatment of severe pain, they are less effective in the chronic pain states and generally are accompanied by undesired secondary effects. On the other hand the tachykinin peptide family members, such as Substance P, are known to play an important role as a transmitter of pain signals and the actions of tachykinins are mediated by the neurokinin-1 (NK1) receptor. Impressive results of attempts to combine action of two pharmacophores—opioid agonist and NK1 antagonist have been successfully demonstrated in our laboratory.1–9
In these earlier studies novel bivalent peptides with opioid agonist and NK1 antagonist activities were designed and synthesized. The synthesized peptides (Fig. 1) have excellent agonist activity for both μ- and δ-opioid receptors and exhibit high antagonist activity for NK1 receptors both in vitro and in vivo. These results indicate that the rational design of bifunctional ligands with opioid agonist and NK1 antagonist activities could be accomplished and might provide a tool for treatment of chronic pain states. Bivalent ligands are likely to yield new drugs with improved properties. The use of novel single molecules possessing multiple analgesic targets, particularly to bind to opioid and NK1 receptors, and to produce antinociception is becoming increasingly attractive.10
Figure 1.
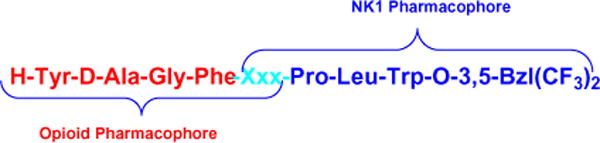
Protoptype peptide dimers.
Compounds that contain two pharmacophores or a single pharmacophore and a non-pharmacophore recognition unit linked through a connecting spacer or without it have been termed ‘bivalent ligands’.11,12 Over the past few decades, bivalent ligands have been developed for a variety of G protein-coupled receptor targets, including opioids.13 With the aim to improve the pharmacological profile of potential analgesics while minimizing common opioid-induced side-effects, dimers of monovalent ‘parent’ opioids have been prepared and include: peptide dimers, mixed peptide-non-peptide bivalent ligands and dual non-peptide dimers. Structures, using an opioid pharmacophore in combination with a non-opioid pharmacophore, have also been prepared.14
μ-Opioid receptors (MORs) are G-protein-coupled receptors (GPCRs) that mediate the physiological effects of endogenous opioid neuropeptides and synthetic opioid drugs. MORs are coexpressed with NK1 receptors in several regions of the central nervous system (CNS) that control opioid dependence and reward.15 Therefore, the principal aim of this article is to design and synthesize bivalent non-peptide compounds which combine μ-opioid receptor agonist and NK1 receptor antagonist pharmacophores in one molecule.
Novel small molecules represent variations of combinations of structures of the opioid agonist Fentanyl and NK1 antagonist pharmacophore L732138 and are presented in Figure 2. Ionic pair compounds in which both cation and anion constituents theoretically could play equal roles and thus could represent a new generation of drugs.
Figure 2.
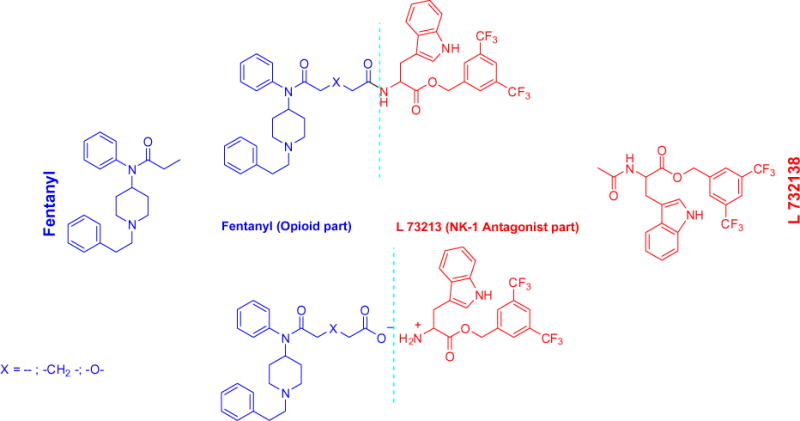
Synthetic approach.
The analogue approach was demonstrated in a patent16 for opioid-NSAID (non-steroid anti-inflammatroy drug) ion pairs wherein the opioid represents a cation and a NSAID represents an anion. Under the conditions prescribed for their use, that ion pair compounds exhibit poor or complete insolubility, but excellent chemical stability in low pH environments, such as those found in the stomach.
2. Results and discussion
2.1. Chemistry
The synthesis of one of the starting materials α-N-Boc protected 3,5-bis(trifluoromethyl)benzyl ester of L-tryptophan, has been described.17,18 The large scale synthesis of functionalized Fentanyls—carboxyfentanyl (1a), carboxymethylfentanyl (1b) and fentanyl derivative (1c) starting from 1-phenethyl-N-phenylpiperidin-4-amine19 and appropriate acid anhydride was carefully worked out by us (Scheme 1). The three compounds differ essentially only in the construction of the side acid chain.
Scheme 1.
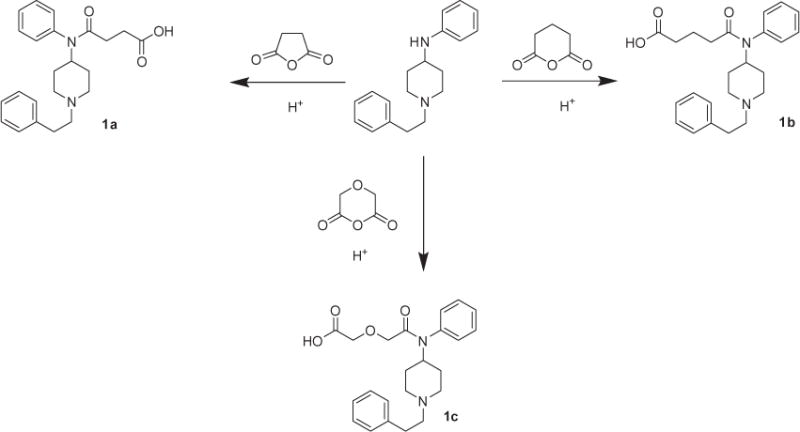
Synthesis of functionalized Fentanyls.
Crystal structures of all three compounds were obtained.20 Two of the crystal structures (1b,c) share several structural similarities, including the length of the chain, while the third (1a), with a shorter chain, is quite different. In particular the structures of 1b and 1c are solvated zwitterions featuring hydrogen bonding between adjacent ions. Proton transfer occurs via these hydrogen bonds, and in most cases is complete. Compound 1a does not form the same dimer motif and has not formally formed a zwitterion. NMR data confirm the obtained X-ray results. 1H NMR for the carboxylic acids suggests that a zwitterion may exist on (1c), but not (1a) or (1b). The protons of the piperidine ring H(2,6-ax) appear as 12 Hz triplets at δ 2.80 (1a), 2.79 (1b), 3.01 (1c). H(2,6-eq) appear as 12 Hz doublets at δ 3.42 (1a), 3.41 (1b), 3.56 (1c). 2-Phenethyl methylene protons are enantiotopic and appear at δ 3.00 and 2.90 (1a), 2.99 and 2.89 (1b), 3.15 and 2.95 (1c). Note should be taken of the downfield shift observed for (1c) and not for (1a) and (1b). H(4-ax) appears as a triple-triplet at δ 4.70 (1a), 4.72 (1b), 4.74 (1c) and does not show a relative downfield shift. The H(2,6-ax) signals appear as triplets and not quartets, which probably is due to rapid exchange on the HN with the methanol-d4 solvent.
The acids obtained could not be converted into acid chlorides by standard methods. A convenient method for creation of substituted fentanyls was developed by us based on the idea of sporadically and rarely reported ‘succinisoimidium perchlorates chemistry’ which was ‘born’ over 100 years ago21 and was minimally developed by the late 1970s.22–25 Now it has become revitalized, developed, simplified and extended, including new isoimidium compounds—glutarisoimidium and 3-oxaglutarisoimidium perchlorates (Scheme 2, 2b,c). All three isoimidium perchlorates (2a–c) were treated with L-tryptophan 3,5-bis(trifluoromethyl)benzyl ester for creation of the main target of this paper—amides (3a–c). The isoimidium perchlorate products obtained (2a–c) have been also checked for their ability to participate in different nucleophilic addition reactions. This method as developed could be implemented for any other nucleopiles as alcohols (4a), amines (5b) or hydrazines (6c), as well as to other amino acids or peptides. These results show that the isoimidium perchlorates (2a–c) could be successfully implemented in reactions with other nucleophiles (Scheme 2).
Scheme 2.
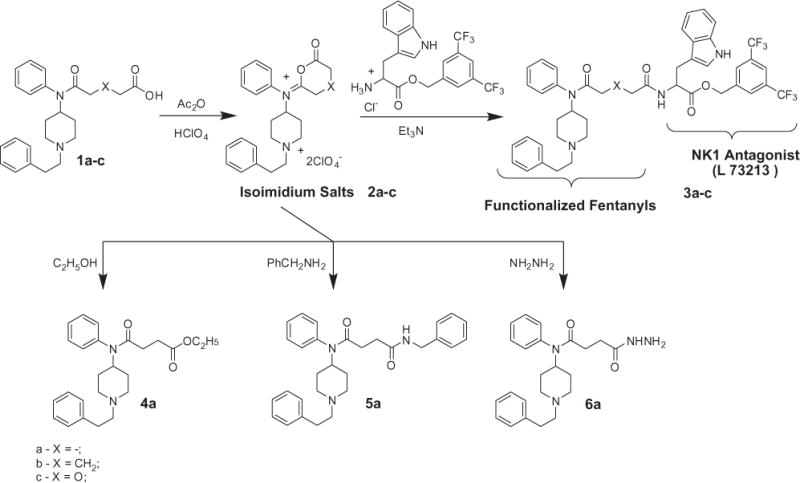
Synthesis of succinisoimidium salts and their use in nucleophilic addition reactions.
A ‘peptide chemistry’ methods with the carbodiimide activation of carboxyl function with BOP/HOBt [benzotriazole-1-yl-oxy-tris(dimethylamino)-phosphonium hexafluorophosphate]/[1-hydroxybenzotriazole] or with EDAC/HOBt [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide]/[1-hydroxybenzotriazole] were also successfully implemented for creation of covalently bonded Fentanyl carboxylic acids with tryptophan 3,5-bis(trifluoromethyl) benzyl ester with high yields.
The structures of isoimidium perchlorates (2a–c) produced were confirmed by NMR methods using 1H–13C HSQC and DQF-COSY. Existing earlier data on confirming the structures of isoimidium perchlorates was based only on IR data.22–25 Compound 2a exists in two conformations on the NMR scale based on the HSQC data (Table S1). The major conformation is listed first; the minor conformation is listed second. The two forms were distinguished by HSQC using relative 1H integration (1.0 major: 0.8 minor) and 13C peak heights. The largest differences in chemical shifts between the two conformations occur around the C=N group. The difference in chemical shift decreases proportionally with the distance from the C=N group. Compounds 2b and 2c, which are both six-membered rings, exist in one conformation on the NMR scale based on the HSQC data (Tables S2 and S3). Isoimidium perchlorates 2a–c are all protonated at the N(1) position due to a large 13 Hz quartet splitting of the H(2,6-ax) signal and a broad singlet at 6.8 ppm corresponding to the H(1N).
The next part of our investigation was devoted to creation of ionic pair compounds (7a–c) Scheme 3 based on the newly synthesized acids (1a–c) and L-tryptophan 3,5-bis(trifluoromethyl) benzyl ester. The method we developed for synthesis is very simple. The potassium salts of acids (1a–c) obtained from the corresponding acids and potassium hydroxide are simply mixed with L-tryptophan 3,5-bis (trifluoromethyl)benzyl ester hydrochloride followed by removal of the resulting KCl (Scheme 3).
Scheme 3.
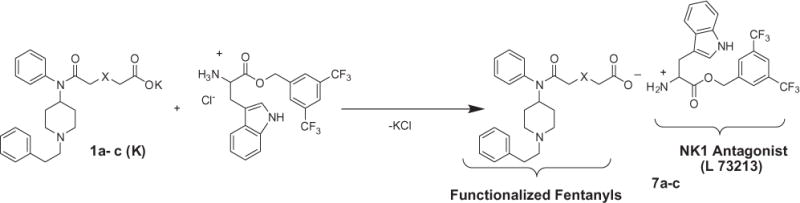
Synthesis of ionic bifunctional opioid/NK1 antagonist compounds.
1H–13C HSQC and DQF-COSY were used to confirm the six compounds (3a–c) and (7a–c) with covalent and ionic links. NMR for covalent compounds showed two different conformers or diastereomers present in solution—1a (1.0:0.7), 1b (1.0:0.9), 1c (1.0:0.7). The variation in chemical shift (1H and 13C) happens mostly in the Trp-O-3,5-Bzl(CF3)2 moiety, with Fentanyl chemical shifts appearing as broadened averages. This may be caused by the chiral center on the Trp-O-3,5-Bzl(CF3)2 moiety or slow rotation of the aromatic rings. It is worth noting that H(4-ax) in Fentanyl appears at two different chemical shifts, unlike other Fentanyl protons. Ionic compounds only show a difference in chemical shifts in the benzyl moiety, appearing in a 1:1 ratio. The covalent link can be identified in 1H NMR spectra by the amide doublet at 6–7 ppm. The ionic link can be easily seen in (7c) with a prominent ammonium peak of the tryptophan at 5.4 ppm, which is weakly observed in (7a) and (7c).
Most of the compounds showed enantiotopic (AA′BB′) methylene groups in the phenethyl moiety, confirmed by 1H–13C HSQC and spin simulation using MestreNova. Instead of two triplets, there often appear as two multiplets (m*) that correspond to the trans conformation of the phenethyl group observed, which is consistent with X-ray structures. There may be hindered rotation of the phenethyl group that cause those multiplets, which would appear as two triplets due to motional averaging (3JHH 6–8 Hz). Simulation parameters (Fig. 3): 600 MHz, 1.5 Hz linewidth, δA,A′ = 3.75, δB,B′ = 2.63, 2JAA′ = 2JBB′ = 12 Hz (geminal), 3JAB = 3JA′B′ = 11 Hz (anti), 3JAB′ = 3JA′B = 5 Hz (gauche).
Figure 3.
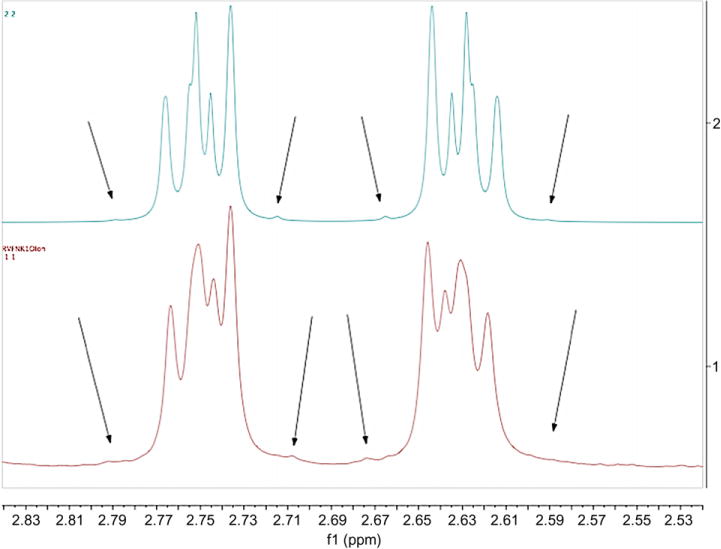
Simulated spectrum (top) and actual phenethyl methylenene multiplets in 7b (bottom) as an example. Arrows point to small outer peaks of the multiplets to show consistency between simulation and experimental.
2.2. Pharmacology
The affinities of the bivalent compounds 3a–c and 7a–c to opioid and NK1 receptors have been measured using competitive radioligand binding assays in recombinant cell lines expressing human μ or human NK1 receptors, respectively, as described previously.1–9 The functional characterization of the same compounds 3a–c and 7a–c, and the starting compounds 1a–c at δ- and μ-opioid receptors, has been carried out using guinea pig isolated ileum/longitudinal muscle myenteric plexus (GPI/LMMP) as a source of μ opioid and NK1 receptors and mouse vas deferens (MVD) as a source of δ opioid receptors as is described in the part 4.2 ‘Biological assays’. The results are presented in Tables 1–3.
Table 1.
Ki values of the bivalent ligands and the parent compounds
| Compd | Ki [3H]DAMGO (nM) | Ki [3H]Substance P (nM) |
|---|---|---|
| 3a | 130 | 13 ± 1.2 |
| 3b | 120 | 6.8 ± 0.5 |
| 3c | 400 | 31 ± 1.2 |
| 7a | >10,000 | 21 ± 4.7 |
| 7b | 3900 | 44 ± 2.3 |
| 7c | 1300 | 23 ± 9.8 |
Table 3.
Results for the MVD and GPI/LMMP testing of starting compounds 1a–c
| Compd | MVD | GPI/LMMP | DPDPE antagonism at 1 μM in the MVD | PL-017 antagonism at 1 μM in the GPI/LMMP |
|---|---|---|---|---|
|
| ||||
| (agonist activity at 1 μM or nM IC50 ± SEM) | ||||
| 1a | 10 μM–6.9% | 10 μM–2% | None at 10 μM | None at 10 μM |
| 1b | 10 μM–19.3% | 10 μM–10% | None at 10 μM | None at 10 μM |
| 1c | 10 μM–10.4% | 10 μM–0% | None at 10 μM | None at 10 μM |
| Fentanyl citrate | (n = 1) 7.2 nM | 1.9 ± 0.70 | – | – |
2.3. Discussion
The biological assays indicate that the synthesized starting functionalized Fentanyl derivatives—(1a–c) themselves dramatically lose analgesic activity in comparison with Fentanyl (Table 3), which probably can be explained with the appearance of the ‘bulky’ carboxyl group with negative charge in the Fentanyl structure, and which is more or less restored on distancing of the carboxyl group from the ethyl part of the propionyl group, (compare (1a–c) as well as on transformation of a charged group back to covalently bonded derivatives.
At the same time the behavior of the tryptophan 3,5-bis(trifluoromethyl) benzyl ester does not change so sharply on variations of substituents on the nitrogen atom, even on its transformation to a charged form in the ionic series. The obtained data suggests that both ionic and covalently linked bivalent ligands compete with high affinity for [3H] Substance P binding sites in recombinant CHO cells expressing the human NK1 receptor. Bivalent ligands with covalent linkage between the two pharmacophors exhibit reduced affinities, relative to the parent opioid (Fentanyl) to the rat MOR and for [3H]DAMGO binding sites with very low affinities, confirming that introduction of a bulky carboxyl group with negative charge into the Fentanyl pharmacophore may interfere with μ opioid receptor recognition.
In functional assays, all compounds, where opioid and NK1 antagonist are bound covalently or by ionic bonds showed NK1 antagonism in vitro with no NK1 agonist activity up to 1 μM. Compound 3b was the most potent NK1 antagonist by more than 10-fold in comparison with the others. The same compound 3b exhibited the best agonist potency at the μ-opioid receptor, indicating that this compound may serve as a useful lead compound for further design of new, improved bivalent opioid-agonist/NK1-antagonist compounds. The other compounds in the series exhibited low affinities for the hMOR in radioligand binding assays and accordingly were weak agonists at the μ- and δ-opioid receptors in in vitro functional assays.
3. Conclusions
The overall conclusion is that the results obtained in these studies confirm that properties obtained for peptide molecules could be successfully transferred for creation of novel small molecules and particularly bivalent ligands by maintaining appropriate distance between the two active pharmacophores. Probably we can take into account data published concerning bivalent ligands containing μ-opioid agonist and CCK2 receptor antagonist pharmacophores linked through spacers containing 16–22 atoms, which indicate that for the pair of pharmacophores (μ-opioid agonist and CCK2 receptor antagonist) spacers cannot be shorter than nine atoms.26
In the case of close fusion of two pharmacophores it is more probable to get a molecule with new pharmacological properties, unless these pharmacophores can overlap (share common structural features).27 The particular conclusion of this work is that the place of possible conjugation for Fentanyl and any other known pharmacophore molecule has been chosen correctly and the series of novel hybrid covalently bonded Fentanyl/NK1 antagonist molecules will be continued: a. by variations of the length and the nature of the linker between opioid and NK1 antagonist parts; and b. what is conceptually the same, by elongation of the peptide chain to synthesize Fent-Leu-Trp-O-3,5-Bzl(CF3)2, Fent-Pro-Leu-Trp-O-3,5-Bzl(CF3)2 or Fent-Xxx-Pro-Leu-Trp-O-3,5-Bzl(CF3)2 chimeric compounds according to previously obtained data.1–9
4. Experimental
4.1. Chemistry
4.1.1. General method for the synthesis of the 4-oxo-4-((1-phenethylpiperidin-4-yl)(phenyl)amino) alcanoic acids (1a–c)
A mixture of (0.01) mol of [1-(2-phenyl)-ethyl-piperidin-4-yl]-phenyl-amine, 0.011 mol of appropriate acid anhydride (succinic, glutaric or diglycolic) and 2–3 drops of acetic acid in 30 mL of dichloromethane was heated in a closed pressure proof flask on a boiling water bath for 5 h and left for the night at room temperature. The solvent was evaporated; ether was added and on cooling (ice) 0.022 mol of NaHCO3 as a 5% solution was added. The ether layer was separated, and the water solution was added 50 mL of chloroform. The mixture was neutralized on cooling and stirring by 0.022 mol of acetic acid. The chloroform solution was dried on MgSO4 and after evaporation of solvent the product was dissolved in the minimum quantity of boiling ethyl acetate from which on cooling crystals of the appropriate acid was separated with 75–90% yield.
4.1.1.1. 4-Oxo-4-((1-phenethylpiperidin-4-yl)(phenyl)amino) bu tanoic acid 1a
Crystalline solid (89.7%), mp 119–120 °C, 1H NMR (600 MHz, methanol-d4) δ 7.50 (t, J = 7.3 Hz, 2H), 7.48–7.43 (m, 1H), 7.32–7.25 (m, 4H), 7.25–7.18 (m, 3H), 4.70 (tt, J = 12.0, 3.5 Hz, 1H), 3.42 (d, J = 12.2 Hz, 2H), 3.00 (m, 2H), 2.90 (dd, J = 10.5, 6.1 Hz, 2H), 2.80 (t, J = 12.0 Hz, 2H), 2.42 (t, J = 6.8 Hz, 2H), 2.18 (t, J = 6.8 Hz, 2H), 2.00 (d, J = 12.2 Hz, 2H), 1.62 (dq, J = 3.3, 13.1 Hz, 2H). 13C NMR (150 MHz, methanol-d4) δ 172.77, 138.08, 137.48, 130.16, 129.40, 128.65, 128.31, 126.44, 58.09, 51.92, 50.76, 30.85, 30.47, 30.25, 28.14. EI-MS: m/z 381; HRMS calcd for C23H29N2O3: 381.2178; found (ESI, [M+H]+): 381.2183.
4.1.1.2. 5-Oxo-5-((1-phenethylpiperidin-4-yl)(phenyl)amino) pentanoic acid 1b
Crystalline solid (84.9%), mp 104–105 °C, 1H NMR (600 MHz, methanol-d4) δ 7.49 (t, J = 7.3 Hz, 2H), 7.45 (d, J = 7.2 Hz, 1H), 7.29 (t, J = 7.2 Hz, 2H), 7.24– 7.19 (m, 5H), 4.72 (tt, J = 12.0, 3.6 Hz, 1H), 3.42 (d, J = 12.3 Hz, 2H), 2.99 (m, 2H), 2.89 (m, 2H), 2.79 (t, J = 11.9 Hz, 2H), 2.10 (t, J = 7.3 Hz, 2H), 2.05–1.95 (m, 4H), 1.79 (dd, J = 14.7, 7.3 Hz, 2H), 1.61 (dd, J = 12.8, 3.2 Hz, 2H). 13C NMR (150 MHz, methanol-d4) δ 173.27, 138.07, 137.56, 130.05, 129.44, 128.66, 128.32, 126.43, 58.11, 51.87, 50.78, 34.52, 33.99, 30.89, 28.19, 21.05. EI-MS: m/z 395; HRMS calcd for C24H31N2O3: 395.2325; found (ESI, [M+H]+): 395.2323.
4.1.1.3. 2-(2-Oxo-2-((1-phenethylpiperidin-4-yl)(phenyl)amino) ethoxy) acetic acid 1c
Crystalline solid (75.7%), mp 143–144 °C, 1H NMR (600 MHz, methanol-d4) δ 7.55–7.45 (m, 3H), 7.35–7.26 (m, 4H), 7.25–7.22 (m, 3H), 4.74 (tt, J = 12.1, 3.6 Hz, 1H), 3.86 (d, J = 6.0 Hz, 4H), 3.56 (d, J = 12.3 Hz, 2H), 3.15 (dd, J = 10.3, 6.6 Hz, 2H), 3.01 (t, J = 12.2 Hz, 2H), 2.95 (dd, J = 10.3, 6.6 Hz, 2H), 2.09 (d, J = 12.7 Hz, 2H), 1.72 (dd, J = 12.8, 2.8 Hz, 2H). 13C NMR (150 MHz, methanol-d4) δ 174.52, 170.05, 136.78, 136.17, 129.90, 129.70, 129.28, 128.46, 128.33, 126.67, 69.40, 68.52, 57.60, 51.69, 50.81, 30.36, 27.41, 19.81. EI-MS: m/z 397; HRMS calcd for C23H29N2O4: 397.2127; found (ESI, [M+H]+): 397.2123.
4.1.2. General method for the synthesis of the isoimidium perchlorates (2a–c)
The stirred mixture of 0.5 mmol of one of the amino acids (1a–c) in 1.5 mL of acetic anhydride was heated slightly until the beginning of dissolution of the starting amino acids and the heater was turned off (but remains under the flask). The stirring was continued for 1 h. Then the mixture was cooled (ice bath) and 0.15 mL of 70% HClO4 was added dropwise. The solution obtained was allowed to come to room temperature overnight and 5 mL of dry ether was added with stirring. After 15–20 min the liquid was decanted from the precipitated perchlorate, and the residue was washed with three 5 mL portions of ether. For analytical purposes the residue was dried overnight in a vacuum-desiccator over phosphorous pentoxide. For synthetic purposes it was dissolved in 5 mL CHCl3/1 mL CH3CN mixture and added to a solution of the nucleophile. 1H–13C HSQC and DQF-COSY investigations of the obtained compounds are summarized in the Supplementary data.
4.1.3. Methods for the synthesis of the amides composed of L-tryptophane 3,5-bis(trifluoromethyl) benzyl esters and carboxy Fentanyls (3a–c)
A. General method for the addition of isoimidium perchlorates to nucleophiles
To the stirred solution of 0.5 mmol of the 3,5-bis(trifluoromethyl)benzyl ester of tryptophan hydrochloride dissolved in 5 mL CHCl3 at 0 °C was added dropwise triethylamine (2.2 mmol (4.4 equiv) dissolved in 5 mL CHCl3. After 15 min. solution of 0.5 mmol of one of isoimidium perchlorates (2a–c) dissolved in 5 mL CHCl3/1 mL CH3CN mixture was added dropwise to a solution of nucleophile. The mixture was left for a night, cooled (ice bath) and was washed consecutively with 1.0 mmol NaHCO3 5% solution of, 5% acetic acid and water. The organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residual oil were finally purified by flash chromatography on SiO2 column using 4:1 CHCl3/MeOH solvent system, which gave compounds (3a–c).
B. General coupling method
To the stirred mixture of one of the amino acids (1a–c), HOBt (1.1 equiv), BOP (1.1 equiv) and 3,5-bis(trifluoromethyl)benzyl ester tryptophan hydrochloride (1.1 equiv) dissolved in CHCl3 (1.5 mL/mmol) at 0 °C was added dropwise triethylamine (4.4 equiv). After stirring for 30 min at 0 °C, the mixture warmed up to room temperature and stirred for an additional 3–5 h until the disappearance of starting components (monitored by TLC). The reaction mixture was cooled (ice bath) and was washed consecutively with 5% NaHCO3 solution (three times), 5% acetic acid (two times) and water. The organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residual oil represents pure compounds (3a–c) solidified at room temperature. Yields of compounds obtained by both methods A and B are comparable and are around 80%. 1H–13C HSQC and DQF-COSY investigations of obtained compounds are summarized in the Supplementary data.
4.1.3.1. 3,5-Bis(trifluoromethyl)benzyl 3-(1H-indol-3-yl)-2-(4-oxo-4-((1-phenethylpiperidin-4-yl)(phenyl)amino)butanamido) propanoate 3a
EI-MS: m/z 793; HRMS calcd for C43H43F6N4O4: 793.3183; found (ESI, [M+H]+): 793.3175.
4.1.3.2. 3,5-Bis(trifluoromethyl)benzyl 3-(1H-indol-3-yl)-2-(5-oxo-5-((1-phenethylpiperidin-4-yl)(phenyl)amino)pentanamido) propanoate 3b
EI-MS: m/z 807; HRMS calcd for C44H45F6N4O4: 807.3340; found (ESI, [M+H]+): 807.3333.
4.1.3.3. 3,5-Bis(trifluoromethyl)benzyl 3-(1H-indol-3-yl)-2-(2-(2-oxo-2-((1-phenethylpiperidin-4-yl)(phenyl)amino)ethoxy) acetamido)propanoate 3c
EI-MS: m/z 809; HRMS calcd for C43H43F6N4O5: 809.3132; found (ESI, [M+H]+): 809.3128.
4.1.4. General method for the addition of nucleophiles to isoimidium perchlorates 4a, 5a, 6a
To the stirred solution of 0.5 mmol nucleophile [C2H5OH, PhCH2NH2, H2NNH2 (10-fold excess)] dissolved in 5 mL CHCl3 at 0 °C was added dropwise triethylamine (2.2 mmol, 4.4 equiv) dissolved in 5 mL CHCl3. After 15 min. solution of 0.5 mmol of isoimidium perchlorate (2a) dissolved in 5 mL CHCl3/1 mL CH3CN mixture was added dropwise to a solution of nucleophile. The mixture was left for a night, cooled (ice bath) and was washed consecutively with 1.0 mmol NaHCO3 5% solution of, 5% acetic acid and water. The organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residual oil were finally purified by flash chromatography on SiO2 column using 4:1 CHCl3/MeOH solvent system, which gave compounds (4a, 5a, 6a).
4.1.4.1. Ethyl 4-oxo-4-((1-phenethylpiperidin-4-yl)(phenyl)amino) butanoate 4a
Crystalline solid (75.7%), mp 145–146 °C, 1H NMR (600 MHz, CDCl3) δ 7.57 (d, J = 7.5 Hz, 2H), 7.48–7.33 (m, 6H), 7.13 (d, J = 7.4 Hz, 2H), 4.68 (t, J = 11.5 Hz, 1H), 4.09 (q, J = 7.1 Hz, 2H), 3.36 (br s, 2H), 2.76 (br s, 2H), 2.52 (t, J = 6.4 Hz, 2H), 2.20 (t, J = 6.3 Hz, 2H), 2.14–2.07 (m, 2H), 1.93 (d, J = 13.1 Hz, 2H), 1.23 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 172.98, 171.85, 131.48, 130.18, 130.14, 130.03, 129.89, 129.77, 129.36, 129.29, 60.62, 51.60, 45.93, 30.00, 29.37, 27.59, 14.29. EI-MS: m/z 409; HRMS calcd for C25H33N2O3: 409.2486; found (ESI, [M+H]+): 409.2478.
4.1.4.2. N1-Benzyl-N4-(1-phenethylpiperidin-4-yl)-N4-phenyl-succinamide 5a
Crystalline solid (75.7%), mp 134–135 °C, 1H NMR (600 MHz, CDCl3) δ 7.42–7.34 (m, 3H), 7.33–7.27 (m, 2H), 7.27–7.19 (m, 8H), 7.06 (dd, J = 7.9, 1.3 Hz, 2H), 6.52 (s, 1H), 4.57 (tt, J = 12.1, 3.8 Hz, 1H), 4.40 (d, J = 5.8 Hz, 2H), 3.43 (s, 2H), 2.86 (d, J = 11.6 Hz, 2H), 2.50–2.37 (t, J = 6.4 Hz, 2H), 2.24 (t, J = 6.4 Hz, 2H), 2.06 (t, J = 11.6 Hz, 2H), 1.69 (d, J = 12.0 Hz, 2H), 1.38 (qd, J = 12.2, 3.3 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 172.44, 171.80, 138.70, 138.42, 138.21, 130.40, 129.58, 129.22, 128.67, 128.63, 128.27, 127.74, 127.37, 127.13, 63.04, 53.06, 52.91, 43.54, 31.66, 31.21, 30.52. EI-MS: m/z 407; HRMS calcd for C30H36N3O2: 470.2802; found (ESI, [M+H]+): 470.2797.
4.1.4.3. 4-Hydrazinyl-4-oxo-N-(1-phenethylpiperidin-4-yl)-N-phenylbutanamide 6a
Crystalline solid (75.7%), mp 215–216 °C, 1H NMR (600 MHz, CDCl3) δ 7.85 (s, 1H), 7.44–7.33 (m, 3H), 7.24 (t, J = 7.5 Hz, 2H), 7.18–7.09 (m, 5H), 4.61 (tt, J = 12.1, 3.9 Hz, 1H), 3.59 (br s, 2H), 2.99 (d, J = 11.7 Hz, 2H), 2.75–2.67 (m*, 2H), 2.59–2.48 (m*, 2H), 2.37 (t, J = 6.5 Hz, 2H), 2.26 (t, J = 6.5 Hz, 2H), 2.13 (td, J = 11.9, 1.7 Hz, 2H), 1.79 (d, J = 12.0 Hz, 2H), 1.44 (qd, J = 12.3, 3.8 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 173.24, 171.56, 140.19, 138.22, 130.33, 129.51, 128.61, 128.60, 128.38, 126.04, 60.37, 53.02, 52.72, 33.75, 30.70, 30.44, 29.42. EI-MS: m/z 395; HRMS calcd for C23H31N4O2: 395.5173; found (ESI, [M+H]+): 395.5167.m*—The ethylene group multiplets are enantiotopic (AA′BB′). Spin simulation was used to model the multiplets with good accuracy. Simulation parameters (Fig. 3): 600 MHz, 1.5 Hz linewidth, δA,A′ = 3.0, δB,B′ = 3.3, 2JAA′ = 2JBB′ = 12 Hz (geminal), 3JAB = 3JA′B′ = 11 Hz (anti), 3JAB′ = 3JA′B = 5 Hz (gauche). Trans-conformation was taken from the X-ray structures.
4.1.5. General method for the synthesis of the ion pairs composed of L-tryptophane 3,5-bis(trifluoromethyl)benzyl esters and carboxyFentanyls (7a–c)
To the stirred solution of 0.5 mmol KOH in 2 mL of CH3OH on stirring was added 0.5 mmol of one of amino acids (1a–c) and the mixture was left for a night. Solution of 0.5 mmol of 3,5-bis(trifluoromethyl)benzyl ester tryptophan hydrochloride dissolved in 1.5 mL of CH3OH was added carefully on cooling (ice bath) and stirring to obtained mixture. Ice bath was removed and mixture was allowed to come to room temperature. After one hour of stirring CH3OH was evaporated, CHCl3 was added and KCl was washed out with 3 × 5 mL of water. The CHCl3 was evaporated and traces of water was azeotropically removed by benzene 3 × 10 mL. Remaining residual oil represents pure compounds (7a–c) which solidified at room temperature, with 70–80% yield. Data from 1H–13C HSQC and DQF-COSY investigations of obtained compounds are summarized in the Supplementary data.
Salt of 5-oxo-5-((1-phenethylpiperidin-4-yl)(phenyl)amino) pentanoic acid (1b) and 3,5-bis(trifluoromethyl)benzyl ester tryptophan (7a).
Salt of 5-oxo-5-((1-phenethylpiperidin-4-yl)(phenyl)amino) pentanoic acid (1b) and 3,5-bis(trifluoromethyl)benzyl ester tryptophan (7b).
Salt of 2-(2-oxo-2-((1-phenethylpiperidin-4-yl)(phenyl) amino) ethoxy)acetic acid (1c) and 3,5-bis(trifluoromethyl) benzyl ester tryptophan (7c)
4.2. Biological assays
4.2.1. Tissue bioassays
4.2.1.1. Isolated guinea pig ileum/longitudinal muscle with myenteric plexus
Male Hartley guinea pigs under CO2 anesthesia were sacrificed by decapitation and a non-terminal portion of the ileum removed. The longitudinal muscle with myenteric plexus (LMMP) was carefully separated from the circular muscle and cut into strips as described previously.8 These tissues were tied to gold chains with suture silk and mounted between platinum wire electrodes in 20 mL organ baths at a tension of 1 g and bathed in oxygenated (95:5 O2/CO2) Kreb’s bicarbonate buffer at 37 °C, then stimulated electrically (0.1 Hz, 0.4 ms duration) at supramaximal voltage. Following an equilibration period, compounds were added cumulatively to the bath in volumes of 14–60 μL until maximum inhibition was reached. A PL-017 dose-response curve was constructed to determine tissue integrity before analog testing.
4.2.1.2. Mouse isolated vas deferens preparation
Male ICR mice under CO2 anesthesia were sacrificed by cervical dislocation and the vasa deferentia removed. Tissues were tied to gold chains with suture silk and mounted between platinum wire electrodes in 20 ml organ baths at a tension of 0.5 g and bathed in oxygenated (95:5 O2/CO2) magnesium free Kreb’s buffer at 37 °C, then stimulated electrically (0.1 Hz, single pulses, 2.0 ms duration) at supramaximal voltage as previously described.8 Following an equilibration period, compounds were added to the bath cumulatively in volumes of 14–60 μL until maximum inhibition was reached. A DPDPE dose– response curve was constructed to determine tissue integrity before analog testing.
4.2.1.3. Agonist and antagonist testing
Compounds were tested as agonists by adding cumulatively to the bath until a full dose–response curve was constructed or to a concentration of 1 μM. Compounds were tested as antagonists by adding to the bath 2 min before beginning the cumulative agonist dose-response curves of the delta (DPDPE) or μ (PL-017) opioid or NK1 (Substance P) agonists.
4.2.1.4. Analysis
Percentage inhibition was calculated using the average tissue contraction height for 1 min preceding the addition of the agonist divided by the contraction height 3 min after exposure to the dose of agonist. IC50 values represent the mean of not less than four tissues. IC50 and Emax estimates were determined by computerized non-linear least-squares analysis (FlashCalc).
4.2.2. Radioligand binding analysis
CHO cells, stably transfected with the human neurokinin receptor-1, were obtained from Dr. James Krause (University of Washington Medical School, St. Louis, MI) and cultured as described elsewhere.1–9 Upon 80–100% confluency, a crude membrane preparation was prepared from the cells for [3H]-SP inhibition binding studies. The protein concentration was determined by the method of Bradford and the membranes were stored at −80 °C. On the day of the experiments, membranes were thawed and diluted with working buffer (50 mM Tris-Mg buffer, pH: 7.4, 50 μg/mL bacitracin, 30 μM bestatin, 10 μM captopril, 100 μM PMSF, 1 mg/mL BSA, 10–15 μg/tube) and were co-incubated with 0.5 nM [3H]-SP (final concentration, 0.1 mCi/mL, 135 Ci/mmol, Perkin Elmer) and various concentrations (10−5 to 10−10 M) of unlabeled ligands, in a final volume of 1 mL. Nonspecific binding was measured by 10 μM unlabeled SP and subtracted from total binding. The reaction mixtures were incubated in a shaking water bath for 20 min at 25 °C and were filtered onto 0.3% PEI pre-soaked Whatman GF/B glass fiber filter (Gaithersburg, MD). Each vial was washed three times with cold saline. Filters were loaded into scintillation vials and EcoLite scintillation cocktail (MP Inc.) was added then the filter-bound radioactivities were measured by liquid scintillation counter (Beckman LS 6000SC). The IC50 values for each compound was determined from nonlinear regression analysis of data points, which were then converted to inhibitory constants (Ki, nM) using the Cheng–Prusoff equation (GraphPad Prism 4 software, San Diego, CA). All experiments were carried out in duplicate and are means of at least three independent experiments.
4.3. Nuclear magnetic resonance spectroscopy
NMR samples were prepared by dissolving approx. 20 mg of each compound in 0.6 mL CDCl3 or CD3OD. All NMR spectra were acquired on a Bruker DRX-600 spectrometer equipped with 5-mm Narolac triple-resonance single z-axis gradient probe at 25 °C with XWinNMR. Spectra were processed with MestreNova software.
Supplementary Material
Table 2.
Binding data values of compounds 3a–c and 7a–c at δ- and μ-opioid receptors
| Compd | Delta opioid activity
|
Mu opioid activity
|
NK1 activity
|
|||
|---|---|---|---|---|---|---|
| MVD inhibition contraction height at 1 μM (%) | MVD antagonism at 1 μM | GPI inhibition of contraction height at 1 μM (or IC50 (nM)) | GPI Antagonism at 1 μM | Concentration tested as NK1 antagonism | NK1 antagonist nM Ke ± SEM | |
| 3a | 14 | None | 410 ± 42 | – | 1 μM | 240 ± 39 |
| 3b | 13 | None | 55 ± 12 | – | 100 nM | 21 ± 4.3 |
| 3c | 30 | None | 30% | None | 1 μM | 480 ± 12 |
| 7a | 4.3 | None | 1.8% | None | 1 μM | 210 ± 42 |
| 7b | 19.8 | None | 19.5% | None | 1 μM | 500 ± 130 |
| 7c | 17.1 | None | 11% | None | 1 μM | 490 ± 68 |
Acknowledgments
This work was supported from the U.S. Public National Institute of Health DA 13449, DA 06284 and DA 06789.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2011.08.027.
References and notes
- 1.Yamamoto T, Nair P, Ma S, Davis P, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. Bioorg Med Chem. 2009;17:7337. doi: 10.1016/j.bmc.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, Davis P, Ma S, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Med Chem. 2009;52:5164. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto T, Nair P, Largent-Milnes T, Jacobsen NE, Davis P, Ma S, Navratilova E, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. Pept Sci. 2008;45:137. [Google Scholar]
- 4.Yamamoto T, Nair P, Jacobsen NE, Davis, Ma S, Navratilova E, Moye S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. J Med Chem. 2008;51:6334. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto T, Nair P, Largent-Milnes T, Jacobsen NE, Davis P, Ma S, Navratilova E, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby V. J Pept Sci. 2007;44:53. [Google Scholar]
- 6.Yamamoto T, Nair P, Vagner J, Largent-Milnes T, Davis P, Ma S, Navratilova E, Moye S, Tumati S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby V. J Med Chem. 2008;51:1369. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hruby V, Nair P, Yamamoto T. U.S. Pat. 2,008,039,404. Chem Abstr. 2008;148:262898.
- 8.Yamamoto T, Nair P, Davis P, Ma S, Navratilova E, Moye S, Tumati S, Lai J, Vanderah TW, Yamamura HI, Porreca F, Hruby VJ. J Med Chem. 2007;50:2779. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langent-Milnes TM, Yamamoto T, Nair P, Moulton JW, Hruby VJ, Lai J, Porreca F, Vanderah TW. J Pharm. 2010;161:986. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonney IM, Foran SE, Marchand JE, Lipkowski AW, Carr DB. Eur J Pharm. 2004;488:91. doi: 10.1016/j.ejphar.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 11.Peng X, Neumeyer JL. Curr Top Med Chem. 2007;7:363. doi: 10.2174/156802607779941251. [DOI] [PubMed] [Google Scholar]
- 12.Mammen M, Chio S, Whitesides GM. Angew Chem, Int Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Hruby VJ, Porreca F, Yamamura HI, Tollin G, Agnes RS, Lee YS, Cai M, Alves I, Cowell S, Varga E, Davis P, Salamon Z, Roeske W, Vanderah T, Lai J. Am Assoc Pharm Sci J. 2006;8:E450. doi: 10.1208/aapsj080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballet S, Pietsch M, Abell AD. Protein Pept Lett. 2008;15:668. doi: 10.2174/092986608785133672. [DOI] [PubMed] [Google Scholar]
- 15.Yu YJ, Arttamangkul S, Evans ChJ, Williams JT, von Zastrow M. J Neurosci. 2008;29:22. doi: 10.1523/JNEUROSCI.4315-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancilio FD, Stowell GW, Whittall LB, White D, Whittle RR. U.S. Pat. 2,005,203,115. Chem Abstr. 2005;143:311957.
- 17.MacLeod AM, Merchant KJ, Cascieri MA, Sadowski S, Ber E, Swain ChJ, Baker R. J Med Chem. 1993;36:2044. doi: 10.1021/jm00066a015. [DOI] [PubMed] [Google Scholar]
- 18.Millet R, Goossens J-F, Bertrand-Caumont K, Chavatte P, Houssin R, Henichart J-P. Lett Pept Sci. 1999;6:221. doi: 10.1002/psc.326. [DOI] [PubMed] [Google Scholar]
- 19.Vardanyan R, Vijay G, Nichol GS, Liu L, Kumarasinghe I, Davis P, Vanderah T, Porreca F, Lai J, Hruby VJ. Bioorg Med Chem. 2009;17:5044. doi: 10.1016/j.bmc.2009.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nichol GS, Kumirov VK, Vardanyan R, Hruby VJ. Cryst Eng Commun. 2010;12:3651. doi: 10.1039/B923698H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoogewerff S, van Dorp WA. Recl Trav Chim. 1892;11:84. [Google Scholar]
- 22.Baydar AE, Boyd GV. J Chem Soc Perkin Trans 1. 1978;11:1360. [Google Scholar]
- 23.Boyd GV, Monteil RL. J Chem Soc Perkin Trans 1. 1978;11:1338. [Google Scholar]
- 24.Balaban AR, Balaban TS, Boyd GV. Synthesis. 1987;6:577. [Google Scholar]
- 25.Mazurkiewicz R. Wiadomosci Chemiczne. 1987;41:755. [Google Scholar]; Chem Abstr. 1989;110:134324. [Google Scholar]
- 26.Zheng Y, Akgun E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. J Med Chem. 2009;52:247. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YS, Agnes RS, Cain JP, Kulkarni V, Cai M, Salibay C, Ciano K, Petrov R, Mayorov A, Vagner J, Trivedi D, Davis P, Ma S, Lai J, Porreca F, Vardanyan R, Hruby VJ. Pept Sci. 2008;90:433. doi: 10.1002/bip.20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.