

Abstract
We report magic angle spinning, dynamic nuclear polarization (DNP) experiments at magnetic fields of 9.4 T, 14.1 T, and 18.8 T using the narrow line polarizing agents 1,3-bisdiphenylene-2-phenylallyl (BDPA) dispersed in polystyrene, and sulfonated-BDPA (SA-BDPA) and trityl OX063 in glassy glycerol/water matrices. The 1H DNP enhancement field profiles of the BDPA radicals exhibit a significant DNP Overhauser effect (OE) as well as a solid effect (SE) despite the fact that these samples are insulating solids. In contrast, trityl exhibits only a SE enhancement. Data suggest that the appearance of the OE is due to rather strong electron-nuclear hyperfine couplings present in BDPA and SA-BDPA, which are absent in trityl and perdeuterated BDPA (d21-BDPA). In addition, and in contrast to other DNP mechanisms such as the solid effect or cross effect, the experimental data suggest that the OE in non-conducting solids scales favorably with magnetic field, increasing in magnitude in going from 5 T, to 9.4 T, to 14.1 T, and to 18.8 T. Simulations using a model two spin system consisting of an electron hyperfine coupled to a 1H reproduce the essential features of the field profiles and indicate that the OE in these samples originates from the zero and double quantum cross relaxation induced by fluctuating hyperfine interactions between the intramolecular delocalized unpaired electrons and their neighboring nuclei, and that the size of these hyperfine couplings is crucial to the magnitude of the enhancements. Microwave power dependent studies show that the OE saturates at considerably lower power levels than the solid effect in the same samples. Our results provide new insights into the mechanism of the Overhauser effect, and also provide a new approach to perform DNP experiments in chemical, biophysical, and physical systems at high magnetic fields.
INTRODUCTION
The last decade has witnessed a renaissance in the use of high frequency dynamic nuclear polarization (DNP) to enhance sensitivity in nuclear magnetic resonance (NMR) experiments. In particular, the development of gyrotron and other high frequency microwave sources permits DNP to be performed at magnetic fields used in contemporary NMR experiments (5—20 T).1–8 To date these experiments, which have focused mostly on insulating solids formed from glassy, frozen solutions of proteins and other nonconducting materials, have relied primarily on narrow line monoradicals and the solid effect (SE)1,9–11 or nitroxide biradicals and the cross effect (CE)12–18 to mediate the polarization process. These approaches have resulted in large signal enhancements and have enabled many experiments that would otherwise be impossible.19–23 Nevertheless, with the exception of an early example on a 1D conductor,24 the Overhauser effect (OE), which was the initial DNP mechanism proposed by Overhauser25 and confirmed by Carver and Slichter,26 has not been identified or utilized during the course of this renaissance. Although the possibility of an OE in insulator was discussed by Abragam,27 the conventional wisdom is that Overhauser DNP is important only in systems with mobile electrons such as conductors (metals and low dimensional conductors) or in liquid solution.28,29 Accordingly, it was not expected to be a significant polarization mechanism in non-conducting solids. In contrast to these expectations, we report here the observation of significant Overhauser enhancements using the narrow line radicals SA-BDPA in glycerol/water and 1,3-bisdiphenylene-2-phenylallyl (BDPA) in polystyrene glassy matrices, both insulating solids.
Recently we described the development of a class of narrow line radicals, sulfonated BDPA (SA-BDPA), with the intent of improving SE enhancements in aqueous media.30 In experiments at 5 T (211 MHz for 1H) these radicals showed the expected SE enhancement at ωSE = ω0S ± ω0I and in addition a relatively weak, positive enhancement when microwaves were applied at ω0S. We have continued investigations of SA-BDPA and at higher fields, 9.4 and 14.1 T (400 and 600 MHz for 1H), and observed larger enhancements around ω0S. Furthermore, BDPA itself dispersed in polymer matrices also exhibits enhancements at ω0S that are one order of magnitude larger than the SE at 18.8 T. We attributed this central peak to an Overhauser effect even though the samples are nonconducting solids. In particular, the lineshape is characteristic of an OE in which the sign of the DNP enhancement does not depend upon the offset of the microwave frequency. Interestingly, trityl OX063 radical,31 which was designed to optimize Overhauser effects in solution by eliminating hyperfine couplings,32,33 shows only SE enhancements at ω0S ± ω0I. Thus, the OE effect appears to require the presence of multiple hyperfine or dipolar couplings that permit zero quantum (ZQ) or double quantum (DQ) relaxation, respectively, and Overhauser enhancements. Furthermore, the field dependent studies indicate that the size of the OE enhancements scales at least weakly with ω0I, in contrast to the or dependence observed for the CE or SE.13,18,34,35 Finally, simulations suggest that a large hyperfine coupling and the dominance of the ZQ or DQ relaxation rate in the polarization process lead to the observed positive or negative Overhauser enhancements, respectively.
BACKGROUND
Part of our results (vide infra) share a characteristic feature with the Overhauser effect in solution where an enhancement is observed around the electron resonance frequencies. In 1H OE-DNP in solutions this enhancement is usually negative and positive enhancements are generally not observed except in the case of scalar relaxation.36 It is, therefore, convenient to briefly review the concepts leading to the enhancements in liquid-DNP and explain how they can be applied to DNP in rotating solids. In an electron–nuclear system the source of the OE-DNP enhancement is the difference between the ZQ and the DQ cross-relaxation rates. The rates, Γ1,ZQ and Γ1,DQ, are governed by fluctuating electron–nuclear couplings and are active between the electron-nuclear ZQ and DQ transitions, respectively. The imbalance of the two rates generates a population redistribution during on-resonance microwave (μw) irradiation, resulting in enhanced positive or negative nuclear polarization depending on whether Γ1,ZQ > Γ1,DQ or Γ1,ZQ < Γ1,DQ. In general, for DNP in liquids Γ1,DQ dominates and the OE enhancements are negative for 1H. In addition to observations in liquids, the Overhauser effect was observed earlier in conducting solids28,29 and in heavily doped semiconductors,37 where it was treated as a three spin effect. It has been neglected in insulating solids.
We note that the OE mechanism differs from the SE in that the μw radiation is applied to the single quantum (SQ) electron transitions and the enhancement is generated by ZQ and DQ cross relaxation. In contrast, in the SE the μw irradiation is applied at the “forbidden” transitions (either a ZQ or DQ transition) and the enhancement is due to the single quantum relaxation of the electron.
Our experimental results discussed below show that both ZQ and DQ relaxation can dominate the OE process, and data with positive and negative enhancements are observed. When the fluctuating interactions are the hyperfine couplings, the scalar part of the hyperfine coupling leads to the ZQ cross relaxation rate Γ1,ZQ, while the dipolar part leads to the DQ cross relaxation rate Γ1,DQ. All these effects characterizing OE-DNP are relevant in solution as well as in rotating samples, although the origin of the fluctuations of the hyperfine coupling is different.
EXPERIMENTAL
Samples
Polystyrene doped with 2% BDPA or d21-BDPA was prepared by a film casting method.1,38 Briefly, 2.4 mg of BDPA in complex with benzene or 2 mg of d21-BDPA and 95 mg of PS-d8 together with 5 mg of PS-d5 were dissolved in 2 ml of chloroform. The solution was then spread on a glass surface. A thin film of PS doped with 2% BDPA or d21-BDPA was collected and ground thoroughly upon evaporation of the solvent. Residual solvent was then removed under vacuum for at least 12 h.
The preparation of DNP samples containing SA-BDPA and trityl OX063 were described in Refs. 30 and 34. For each sample, the corresponding radical was dissolved in a mixture of glycerol-d8/D2O/H2O (60/30/10 volume ratio) supplemented with 100 mM 13C, 15N Proline. The final concentrations of SA-BDPA and trityl OX063 were 40 mM.
DNP experiments
DNP experiments at 9.4 T and 14.1 T were performed using two NMR/DNP spectrometers (Bruker BioSpin (Billerica, MA)) operating at 400 MHz/263 GHz and 600 MHz/395 GHz of 1H/electron Larmor frequencies.6 Experiments at 18.8 T were conducted on an NMR/DNP spectrometer at Utrecht University operating at 800 MHz/527 GHz of 1H/electron Larmor frequencies. The temperature of the sample was ∼105 K with μw's on and ∼100 K without microwaves as calibrated from the T1 of 79Br on a KBr sample under the same conditions.39,40 The spinning frequency, which does not affect the DNP efficiency in our study, was chosen to be ωr/2π = 8 kHz as a compromise between sample heating and signal intensity. The sweep coil supports up to ±20 A corresponding to ±75 mT at 400 MHz, ±128 mT at 600 MHz and at 800 MHz the corresponding numbers are ±10 A and ±45 mT. The room temperature shim set provides fine adjustments within the range of 240 ppm at 9.4 T, 150 ppm at 14.1 T, and 100 ppm at 18.8 T.
The DNP enhancement field profiles were obtained by comparing the intensity of NMR signals with and without microwaves at different magnetic fields of the NMR magnet. All experiments started with a series of saturation pulses followed by a recovery period. The magnetically dilute 1H NMR signals present in “DNP Juice” were narrowed by magic angle spinning (MAS) and detected using a rotor-synchronized Hahn echo pulse sequence. The spin-lattice relaxation T1 and the DNP buildup time constant TB were measured by varying the recovery time. We found that the T1 and TB for each sample at each magnetic field were essentially identical. For the BDPA/PS sample, the T1 and TB were 4.6, 6.0, and 7.2 s at 9.4, 14.1, and 18.8 T, respectively. The T1 and TB for SA-BDPA in glycerol/water sample were 37 and 45 s at 9.4 and 14.1 T, respectively.
RESULTS
Figures 1(a)–1(c) show 1H DNP-MAS enhancement field profiles obtained from samples containing the polarizing agents trityl OX063, SA-BDPA, BDPA, and perdeuterated BDPA (d21-BDPA) at 9.4 T using 9 W of microwave power (see Figures 2 and 3 for the molecular structures and EPR spectra, respectively). The data were recorded by observing the intensity of the dilute 1H signals with and without μw irradiation. The enhancement was then calculated according to ɛ = (I/I0) − 1. The field profiles in Figure 1 clearly show the expected SE enhancements at ω0S ± ω0I for all the radicals. In addition, in the case of SA-BDPA and BDPA there is a strong positive enhancement at the center of the field profile due to the Overhauser effect with ZQ relaxation dominating and yielding a positive enhancement. We also have observed a weak negative OE (ɛ = −0.6) when using d21-BDPA (Figure 1(c)), a result that is consistent with DQ relaxation dominating the enhancement.
FIG. 1.

1H DNP enhancement field profiles of (a) trityl OX063, (b) BDPA (red, solid circle) and SA-BDPA (blue, open circle), and (c) d21-BDPA at 9.4 T. All the field profiles were obtained with about 9 W of microwave power. A well-resolved solid effect is observed for all three radicals. The dashed line indicates the position of the isotropic g-value and the BDPA's are upfield from trityl. In addition, in (b) a significant Overhauser DNP enhancement is also present when irradiating at the EPR transition in BDPA and SA-BDPA. The positive enhancement indicates the DNP process is dominated by a zero-quantum (ZQ) process; (c) The field profile for perdeuterated BDPA shows a negative OE indicating it is dominated by a double quantum (DQ) process.
FIG. 2.

Molecular structures of (a) trityl (OX063), (b) sulfonated BDPA (SA-BDPA) and (c) BDPA. Note that the EPR spectrum of trityl exhibits no hyperfine structure due to 1H couplings. BDPA has a total of 21 1H's coupled to the electron and SA-BDPA has similar couplings but 5 fewer due to the addition of –SO3 groups to the rings.
FIG. 3.

Liquid solution EPR spectra of the BDPA's and trityl shown in Figure 2. BDPA and d21-BDPA were dissolved in toluene whereas SA-BDPA and trityl were dissolved in water. The radical concentration was 50 μM for BDPA's samples and 1 mM for the trityl sample. Note that trityl exhibits no hyperfine structure due to 1H couplings. In contrast BDPA has a total of 21 1H's coupled to the electron.41,42 SA-BDPA has 5 fewer 1H's as suggested by Haze et al.30 due to the addition of –SO3 groups to the rings. The simulation for BPDA uses isotropic hyperfine coupling constants of −0.15 MHz (2), −0.50 MHz (3), 1.09 MHz (4), 1.38 MHz (4), −5.29 MHz (4), and −5.54 MHz (4), where the number in parenthesis indicates the number of 1H’s. These are slightly different from the literature values of with coupling constants of −0.29, −0.50, 0.97, 1.37, −5.29, and −5.55 MHz. The simulation for SA-BDPA includes 16 1H's with isotropic couplings of −0.28 MHz (2), −0.5 MHz (2), 0.98 MHz (4), −5.04 MHz (4), and −5.26 MHz (4). The line broadening in SA-BDPA is most likely due to the slow tumbling of the radical in water.
Similarly, the field profiles of the same samples at 14.1 T were recorded with 13 W of microwave power and are illustrated in Figure 4, showing the same features as in Figure 1. All the field profiles exhibited well-resolved DNP solid effect enhancements with the negative and the positive SE corresponding to the zero quantum and double quantum SE transitions, respectively. Note that at 14.1 T the SE enhancement profiles begin to display the presence of a small g-anisotropy from the BDPA’s. Furthermore, in contrast to previous SE data where we observed a 10%–20% asymmetry in the ZQ and DQ enhancements,12 we find that the maximum positive and negative enhancements are essentially equal. It is important to note that a comparison of Figures 1 and 4 shows that at 14.1 T the OE becomes the dominant DNP mechanism in the BDPA/PS sample and increases to ∼40% of the intensity of the SE transition in SA-BDPA. Thus, the OE exhibits a ∼ω0I dependence as opposed to the inverse dependences predicted for the SE1,9–11 and CE.12–18
FIG. 4.

1H DNP enhancement field profile of BDPA (The circles are filled with black.), SA-BDPA (blue, open circle) at 14.1 T. All the field profiles were obtained with 13 W of microwave power. The Overhauser effect in BDPA and SA-BDPA became more efficient at this magnetic field. Note that at this magnetic field the Overhauser effect dominates the SE in the BDPA/PS sample.
We further investigated the field dependence of the OE and SE in the BDPA/PS sample. Shown in Figure 5 is the comparison of the DNP field profiles of this sample at three different magnetic fields. The OE exhibits a ∼ω0I dependence increasing from 13 at 9.4 T to 17 at 14.1 T and to 20 at 18.8 T. The SE, on the other hand, shows a dependence decreasing from 12 at 9.4 T to 6 at 14.1 T and to 2 at 18.8 T.
FIG. 5.
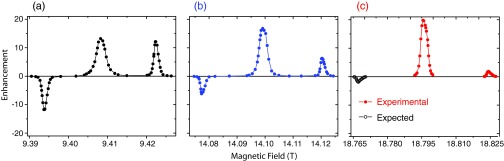
1H DNP enhancement field profile of BDPA at 9.4 T (a), 14.1 T (b), and 18.8 T (c). At 18.8 T, due to the limited experimental time, we only measured the Overhauser effect and the positive solid effect peaks (red, solid circle) using 14 W of microwave power. The peak of the negative solid effect (black, open circle) was then deduced from that of the positive solid effect. Note that the enhancement of the Overhauser effect appears to scale with B0 whereas that of the solid effect scales very close to B0−2. At 18.8 T, the maximum enhancement of the Overhauser effect is one order of magnitude larger than that of the solid effect.
We have also examined the μw power dependence for a BDPA sample as shown in Figure 6(a). Note that the DNP enhancement via the OE displays a sharp rise and saturates at very low microwave powers (<2 W). This indicates that the Overhauser peak relies on irradiation of the allowed EPR transitions at lower microwave power rather than the nominally forbidden SE transitions. In contrast, the enhancement of the DNP solid effect increases monotonically and does not saturate at even at the highest microwave powers.
FIG. 6.

(a) Experimental 1H DNP enhancements of BDPA as a function of μw power at 9.4 T. The Overhauser enhancement (red, solid circles) showed a sharp rise at low power, whereas the enhancement via the solid effect (blue, open circles) gradually increased with μw power. (b) Simulations of the power dependence of the OE and SE using the parameters for BDPA included in the caption of Figure 8.
DISCUSSION
Similar to the published data at 5 T,30 the field profile at 9.4 T of SA-BDPA showed a DNP enhancement symmetrically disposed about ω0S corresponding to μw irradiation on resonance with the EPR transition. We attribute this central peak to an Overhauser effect even though the samples are nonconducting solids. In particular, the lineshape of the center peak is characteristic of the OE in which the sign of the DNP enhancement does not depend upon the offset of the microwave frequency.
As mentioned above,27 Abragam predicted that the Overhauser effect would dominate in insulating materials given that the isotropic hyperfine interaction is larger than the nuclear Zeeman interaction. The solid effect would otherwise be the dominating DNP mechanism. The prediction is valid for low magnetic fields where the nuclear Zeeman interaction is small and the two DNP mechanisms can overlap. In our study at high fields (>9 T), the hyperfine coupling is two orders of magnitude smaller than the nuclear Zeeman interaction and the two DNP mechanisms are well separated by the nuclear Larmor frequency. Therefore, the central peaks in the field profiles of SA-BDPA and BDPA are purely due to the Overhauser effect.
Abragam and co-workers also briefly reported observations of OE enhancements in charcoal and graphite, although the state of the samples was not well characterized.43 In a more recent study by Dementyev et al.,37 a negative Overhauser effect was observed at 2.35 T and 1.1 K in a single crystal of doped semiconductors Si:P. Even though the doped semiconductor was in an insulating state, the dopant concentration was sufficiently high to form clusters of electrons. The spin system leading to DNP therefore consists of two electrons, two 31P nuclei and a single 29Si, but the Hamiltonian was simplified to one containing three spins. In this system the strong exchange coupling of up to 100 GHz between the electrons is comparable to the electron Zeeman interaction (66 GHz), and modulates the electron-nuclear coupling, leading to a negative Overhauser effect. Thus, this system is very different from the systems and experiments studied and reported here. In particular, B0 ≥ 9.4 T and the electron concentration is ∼40 mM, yielding an exchange coupling that is at least 2–3 orders of magnitude smaller than the electron Zeeman energy. In addition, if this mechanism was present in our experiments, the Overhauser effect would have been observed for all our samples, in particular with the same sign and intensity of BDPA and d21-BDPA. Thus, the fluctuations leading to the OE enhancements cannot be explained by the exchange coupling.
Figure 2 shows the molecular structures and Figure 3 shows the solution X-band EPR spectra of all the paramagnetic polarizing agents used in our study, together with simulations of the EPR spectra of SA-BDPA and BDPA performed with EasySpin.44 As is clear from Figure 3 both BDPA and SA-BDPA exhibit a rich array of hyperfine splittings whereas trityl OX063 and d21-BDPA do not. Specifically, the simulations for BDPA used isotropic hyperfine couplings to 21 1H's with coupling constants of −0.15 MHz (2), −0.50 MHz (3), 1.09 MHz (4), 1.38 MHz (4), −5.29 MHz (4), and −5.54 MHz (4) similar to the published data,41,42 where the number in parenthesis indicates the number of 1H’s. The sulfonation process removes 5 1H's30 and therefore the simulation of the SA-BDPA spectrum shown in Figure 3 includes 16 1H's with isotropic couplings of −0.28 MHz (2), −0.5 MHz (2), 0.98 MHz (4), −5.04 MHz (4), and −5.26 MHz (4). In contrast, trityl has a maximum proton hyperfine coupling of 0.05 MHz.33 As we will see below this is the likely source of the Overhauser effect observed in the SA-BDPA and BDPA and absent in trityl OX063. The fact that there is a large enhancement at ω0S could possibly be explained as electron decoupling and is discussed in Ref. 45. However, the fact that trityl does show paramagnetic quenching in MAS experiments45 but does not exhibit an enhancement at ω0S excludes this possibility.
The relationship between isotropic hyperfine coupling and the Overhauser effect in SA-BDPA and BDPA is also strongly supported by the data on d21-BDPA. Deuteration effectively removes all the hyperfine couplings, which is evident in the EPR spectrum of d21-BDPA in Figure 3. Thus, any DNP processes in d21-BDPA are mediated by the anisotropic dipolar coupling between the radical and 1H's of polystyrene, which is consistent with the observation of a negative Overhauser effect (see Figure 1(c)). Moreover, such a coupling is expected to be of smaller amplitude and leads to lower DNP enhancements for both the solid effect and Overhauser effect. This result also implies that in SA-BDPA or BDPA, the main pathway for the polarization transfer starts from the 1H's on the radicals.
It is worth noting that the field profile of trityl (Figure 1(a)) exhibits a reproducible asymmetric feature near its center reminiscent of a cross effect field profile. The low field side of this feature is slightly negative and the center field corresponding to the location where OE's are observed is a crossing point of zero intensity. At high field there is a positive lobe to the profile. At the moment we do not have a satisfactory explanation for this feature.
To further explain the experimental data and other observations outlined above, we performed simulations of the SE and the OE field profiles at 8 kHz MAS following the approach described by Mentink-Vigier et al.17 with the addition of specific ZQ and DQ cross-relaxation pathways and rates as illustrated in Figure 7. The evolution superoperator for one rotor period is obtained by step integration and is then applied repetitively to obtain the time evolution of the density matrix, and from this the NMR signals. For the SE case the original calculations involved a system composed of an electron coupled to a proton via a dipolar hyperfine interaction. The same system is sufficient to represent the fundamental physics of the Overhauser DNP mechanism during sample rotation when an isotropic hyperfine interaction is added to the system and ZQ and DQ relaxation mechanisms are introduced. Because of the high static magnetic field the hyperfine interactions can be truncated and the laboratory frame spin Hamiltonian during an MAS DNP experiment on the two-spin system can be written as
where
and the microwave driving term is
Here ω0S = gisoβeB0 is the frequency of the isotropic g value and Ωt accounts for the time dependent orientation imparted by the magic angle spinning. This Hamiltonian permits computation of the energy levels via a diagonalization procedure.17 The relaxation is then introduced in the eigenframe at each angular step and the propagator is computed in Liouville space. During the SE simulations, the relaxation parameters are computed assuming fluctuations of and operators in the eigenframe of the Hamiltonian. In the case of the MAS Overhauser effect we also assumed fluctuations of the hyperfine coupling and added those to the source of relaxation. As illustrated in Figure 7, the spin operators of the hyperfine coupling support additional cross relaxation pathways with ZQ (Δm = 0) relaxation originating from the operators of its isotropic and anisotropic parts and DQ (Δm = 2) relaxation from the operators of only its anisotropic part. In the presence of strong isotropic contributions the ZQ relaxation time is expected to be shorter than the DQ relaxation time, which leads to a positive Overhauser effect.
FIG. 7.

Energy level diagram showing the ZQ and DQ, ΓZQ and ΓDQ, relaxation pathways in the two spins system. Γe and Γn are the direct electronic and nuclear relaxation rates. The dashed arrows indicate the positions in time where the μw irradiation is on resonance for the SQ transitions.
In our calculations, we defined two cross-relaxation rates and that are different and that are the coefficients of the relaxation rates derived from the ZQ and DQ operators, respectively. Realizing that in our case the hyperfine coefficients satisfy , we can expect the cross relaxation rates to satisfy ΓZQ > ΓDQ. The introduction of the two relaxation pathways appears necessary to achieve a buildup time of the order of T1n.
Based on this model we performed a series of simulations for the case of BDPA's where the external magnetic field, B0, and the μw power were swept. The simulations employed the parameters compiled in the figure captions which were chosen to approximate the experimental values.
Figure 8 illustrates the intensity of the predicted field profile for SA-BDPA and BDPA at two different fields (a) 9.4 T and (c) 14.1 T. Figure 8(b) presents the simulated field profile of d21-BDPA. At this point there are significant uncertainties in the values of experimental parameters, so the simulations cannot predict the absolute enhancement values, but only rather their relative intensities and accordingly are presented as normalized plots in Figure 8. Nevertheless, we observe the expected SE signal at ω0S ± ω0I as well as a positive enhancement at ω0S. Experimentally, the Overhauser enhancement is larger in BDPA than in SA-BDPA, presumably because of the stronger coupling to 21 1H’s, and we have increased Aiso in the BDPA simulations to account for this fact. We also note that for the results presented here the ratio εOE / εSE increases with B0. For SA-BDPA it increases from 0.13 to 0.46 and for BDPA from 1.25 to 2.9 at 9.4 and 14.1 T, respectively. These results are in good agreement with the experimental data. It is worth noting that the shapes of the simulated field profiles do not depend strongly on either or but are primarily determined by the ratio Γ1,ZQ / Γ1,DQ.
FIG. 8.

Normalized simulations of the DNP field profiles for SA-BDPA and BDPA at (a) 9.4 and (c) 14.1 T and (b) for d21-BDPA at 9.4 T at T = 100 K assuming (ω1S/2π)∼0.85 MHz. The following parameters were used in the simulations: BDPA:Aiso = 2.5 MHz, Adip = 1.5 MHz (in the principal axis frame), is the same order of magnitude as the published data.41,42 We employed an average value of the isotropic hyperfine value. , , Γ1n = (T1n)−1 = 0.16 s−1 @ 9 T and 14 T, SA-BDPA:Aiso = 2 MHz, Adip = 1.5 MHz (in the principal axis frame), assuming the sulfonation may affect the isotropic values by reducing the number of protons on the rings. , , Γ1n = (T1n)−1 = 0.03 s−1 @ 9 T and Γ1n = (T1n)−1 = 0.02 s−1 @ 14 T. d21-BDPA: A purely dipolar coupling of 0.1 MHz which corresponds to e−-1H distance of 0.9 nm, and cross relaxation rates , were used.
The origin of the observation that the Overhauser effect enhancement appears to scale with B0 (Figure 5) is not well established, again due to the lack of experimental parameters, especially electronic relaxations at high fields. We suggest that the cross relaxation rates increase with B0 as calculated by Pines et al.46 Note that the cross relaxation rates ΓZQ and ΓDQ are proportional to the spectral density J(ω0S ± ω0I), respectively, both of which can be approximated as J(ω0S) since ω0S ≫ ω0I. Thus, faster cross relaxation rates would be evident in shorter electron T1e at high fields in the case of SA-BDPA and BDPA. A similar result was reported for trityl OX063 in glycerol/water glassy matrix.47
In addition, as illustrated in Figure 1(c) we have observed a weak negative OE when using d21-BDPA as the polarizing agent. In this case dipolar mediated DQ relaxation rather than ZQ processes dominate the Overhauser effect enhancement profile. Simulations shown in Figure 8(b) reproduce the essential features of the negative OE.
Finally, as shown in Figure 6(b) we are able to correctly predict the DNP enhancement as a function of the μw field strength ω1S for both the SE and Overhauser case. The Overhauser enhancement increases steeply with ω1S and saturates, while the SE increases more slowly. Again the simulations reproduce the essential features of the experimental power dependence of the Overhauser and solid effects.
CONCLUSIONS
In summary we present here the initial experimental observation of microwave driven OE DNP around ω0S in insulating solids at high field using the narrow line radicals SA-BDPA and BDPA. It is present along with the expected solid effect occurring at ω0S ± ω0I. The experimental and simulated enhancement field profiles demonstrate a field dependence ∼ωOI, in contrast to the inverse dependences predicted for the SE and CE. The relative intensity of the two contributions depends strongly on the ratio ΓZQ / ΓDQ and the sizes and/or numbers of the nuclear hyperfine couplings. These observations suggest that OE's could be seen in other systems and should stimulate the development of additional narrow line radicals that possess the features. They should be useful for MAS DNP at higher magnetic fields.
ACKNOWLEDGMENTS
We thank Johan Van der Zwan and Deni Mance for their assistance with recording the 800 MHz data and Leo Tometich (Bruker BioSpin) for extensive technical assistance. This research was supported by grants to R.G.G. from the National Institutes of Biomedical Imaging and Bioengineering, Grant Nos. EB-002804 and EB002026 and to T.M.S. from the National Institutes of Health of General Medical Sciences, Grant No. GM095843. S.V. acknowledges the support of a grant from the German-Israeli Project Cooperation of the DFG through a special allotment by the Ministry of Education and Research (BMBF) of the Federal republic of Germany. B.C. was supported by the Deutsche Forschungsgemeinschaft (DFG research fellowship CO 802/1-1). M.B. is supported by an NWO Vici fellowship (Grant No. 700.10.443).
REFERENCES
- 1.Becerra L. R., Gerfen G. J., Temkin R. J., Singel D. J., and Griffin R. G., Phys. Rev. Lett. 71, 3561–3564 (1993). 10.1103/PhysRevLett.71.3561 [DOI] [PubMed] [Google Scholar]
- 2.Gerfen G. J., Becerra L. R., Hall D. A., Griffin R. G., Temkin R. J., and Singel D. J., J. Chem. Phys. 102, 9494–9497 (1995). 10.1063/1.468818 [DOI] [Google Scholar]
- 3.Becerra L. R., Gerfen G. J., Bellew B. F., Bryant J. A., Hall D. A., Inati S. J., Weber R. T., Un S., Prisner T. F., McDermott A. E., Fishbein K. W., Kreischer K. E., Temkin R. J., Singel D. J., and Griffin R. G., J. Magn. Reson., Ser. A 117, 28–40 (1995). 10.1006/jmra.1995.9975 [DOI] [Google Scholar]
- 4.Hornstein M. K., Bajaj V. S., Griffin R. G., Kreischer K. E., Mastovsky I., Shapiro M. A., Sirigiri J. R., and Temkin R., IEEE Trans. Electron Devices 52, 798–807 (2005). 10.1109/TED.2005.845818 [DOI] [Google Scholar]
- 5.Bajaj V. S., Hornstein M. K., Kreischer K. E., Sirigiri J. R., Woskov P. P., Mak-Jurkauskas M. L., Herzfeld J., Temkin R. J., and Griffin R. G., J. Magn. Reson. 189, 251–279 (2007). 10.1016/j.jmr.2007.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosay M., Tometich L., Pawsey S., Bader R., Schauwecker R., Blank M., Borchard P. M., Cauffman S. R., Felch K. L., Weber R. T., Temkin R. J., Griffin R. G., and Maas W. E., Phys. Chem. Chem. Phys. 12, 5850–5860 (2010). 10.1039/c003685b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosay M., Zeri A.-C., Astrof N. S., Opella S. J., Herzfeld J., and Griffin R. G., J. Am. Chem. Soc. 123, 1010–1011 (2001). 10.1021/ja005659j [DOI] [PubMed] [Google Scholar]
- 8.Thurber K. R., Yau W.-M., and Tycko R., J. Magn. Reson. 204, 303–313 (2010). 10.1016/j.jmr.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith A. A., Corzilius B., Barnes A. B., Maly T., and Griffin R. G., J. Chem. Phys. 136, 015101 (2012). 10.1063/1.3670019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenckebach W. T., Appl. Magn. Reson. 34, 227–235 (2008). 10.1007/s00723-008-0121-9 [DOI] [Google Scholar]
- 11.Hovav Y., Feintuch A., and Vega S., J. Magn. Reson. 207, 176–189 (2010). 10.1016/j.jmr.2010.10.016 [DOI] [PubMed] [Google Scholar]
- 12.Hu K.-N., Bajaj V. S., Rosay M., and Griffin R. G., J. Chem. Phys. 126, 044512 (2007). 10.1063/1.2429658 [DOI] [PubMed] [Google Scholar]
- 13.Hu K.-N., Debelouchina G. T., Smith A. A., and Griffin R. G., J. Chem. Phys. 134, 125105 (2011). 10.1063/1.3564920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu K.-N., Song C., Yu H.-h., Swager T. M., and Griffin R. G., J. Chem. Phys. 128, 052302 (2008). 10.1063/1.2816783 [DOI] [PubMed] [Google Scholar]
- 15.Hu K. N., Yu H. H., Swager T. M., and Griffin R. G., J. Am. Chem. Soc. 126, 10844–10845 (2004). 10.1021/ja039749a [DOI] [PubMed] [Google Scholar]
- 16.Matsuki Y., Maly T., Ouari O., Karoui H., Le Moigne F., Rizzato E., Lyubenova S., Herzfeld J., Prisner T., Tordo P., and Griffin R. G., Angew. Chem., Int. Ed. 48, 4996–5000 (2009). 10.1002/anie.200805940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mentink-Vigier F., Akbey U., Hovav Y., Vega S., Oschkinat H., and Feintuch A., J. Magn. Reson. 224, 13–21 (2012). 10.1016/j.jmr.2012.08.013 [DOI] [PubMed] [Google Scholar]
- 18.Thurber K. and Tycko R., J. Chem. Phys. 137, 084508 (2012). 10.1063/1.4747449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maly T., Debelouchina G. T., Bajaj V. S., Hu K. N., Joo C. G., Mak-Jurkauskas M. L., Sirigiri J. R., van der Wel P. C. A., Herzfeld J., Temkin R. J., and Griffin R. G., J. Chem. Phys. 128, 052211 (2008). 10.1063/1.2833582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes A. B., De Paepe G., van der Wel P. C. A., Hu K. N., Joo C. G., Bajaj V. S., Mak-Jurkauskas M. L., Sirigiri J. R., Herzfeld J., Temkin R. J., and Griffin R. G., Appl. Magn. Reson. 34, 237–263 (2008). 10.1007/s00723-008-0129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni Q. Z., Daviso E., Can T. V., Markhasin E., Jawla S. K., Temkin R. J., Herzfeld J., and Griffin R. G., Acc. Chem. Res. 46, 1933–1941 (2013). 10.1021/ar300348n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akbey U., Franks W. T., Linden A., Lange S., Griffin R. G., van Rossum B. J., and Oschkinat H., Angew. Chem., Int. Ed. 49, 7803–7806 (2010). 10.1002/anie.201002044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossini A. J., Zagdoun A., Lelli M., Canivet J., Aguado S., Ouari O., Tordo P., Rosay M., Maas W. E., Copéret C., Farrusseng D., Emsley L., and Lesage A., Angew. Chem., Int. Ed. 51, 123–127 (2012). 10.1002/anie.201106030 [DOI] [PubMed] [Google Scholar]
- 24.Un S., Prisner T., Weber R. T., Seaman M. J., Fishbein K. W., McDermott A. E., Singel D. J., and Griffin R. G., Chem. Phys. Lett. 189, 54–59 (1992). 10.1016/0009-2614(92)85152-Z [DOI] [Google Scholar]
- 25.Overhauser A. W., Phys. Rev. 92, 411–415 (1953). 10.1103/PhysRev.92.411 [DOI] [Google Scholar]
- 26.Carver T. R. and Slichter C. P., Phys. Rev. 92, 212–213 (1953). 10.1103/PhysRev.92.212.2 [DOI] [Google Scholar]
- 27.Abragam A., Phys. Rev. 98, 1729–1735 (1955). 10.1103/PhysRev.98.1729 [DOI] [Google Scholar]
- 28.Griesinger C., Bennati M., Vieth H. M., Luchinat C., Parigi G., Höfer P., Engelke F., Glaser S. J., Denysenkov V., and Prisner T. F., Prog. Nucl. Mag. Reson. Spectrosc. 64, 4–28 (2012). 10.1016/j.pnmrs.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 29.Cheng C. and Han S., Annu. Rev. Phys. Chem. 64, 507–532 (2013). 10.1146/annurev-physchem-040412-110028 [DOI] [PubMed] [Google Scholar]
- 30.Haze O., Corzilius B., Smith A. A., Griffin R. G., and Swager T. M., J. Am. Chem. Soc. 134, 14287–14290 (2012). 10.1021/ja304918g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ardenkjær-Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thaning M., and Golman K., Proc. Natl. Acad. Sci. U.S.A. 100, 10158–10163 (2003). 10.1073/pnas.1733835100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy T. J., Iwama T., Halpern H. J., and Rawal V. H., J. Org. Chem. 67, 4635–4639 (2002). 10.1021/jo011068f [DOI] [PubMed] [Google Scholar]
- 33.Trukhan S. N., Yudanov V. F., Tormyshev V. M., Rogozhnikova O. Y., Trukhin D. V., Bowman M. K., Krzyaniak M. D., Chen H., and Martyanov O. N. J., Magn. Reson. 223, 29–36 (2013). 10.1016/j.jmr.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corzilius B., Smith A. A., and Griffin R. G., J. Chem. Phys. 137, 054201 (2012). 10.1063/1.4738761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hovav Y., Feintuch A., and Vega S., J. Chem. Phys. 134, 074509 (2011). 10.1063/1.3526486 [DOI] [PubMed] [Google Scholar]
- 36.Loening N. M., Rosay M., Weis V., and Griffin R. G., J. Am. Chem. Soc. 124, 8808–8809 (2002). 10.1021/ja026660g [DOI] [PubMed] [Google Scholar]
- 37.Dementyev A. E., Cory D. G., and Ramanathan C., J. Chem. Phys. 134, 154511 (2011). 10.1063/1.3576133 [DOI] [PubMed] [Google Scholar]
- 38.Afeworki M., Mckay R. A., and Schaefer J., Mater. Sci. Eng. A162, 221–228 (1993). 10.1016/0921-5093(90)90046-6 [DOI] [Google Scholar]
- 39.Sarkar R., Concistre M., Johannessen O. G., Beckett P., Denning M., Carravetta M., al-Mosawi M., Beduz C., Yang Y., and Levitt M. H., J. Magn. Reson. 212, 460–463 (2011). 10.1016/j.jmr.2011.08.021 [DOI] [PubMed] [Google Scholar]
- 40.Thurber K. R. and Tycko R., J. Magn. Reson. 196, 84–87 (2009). 10.1016/j.jmr.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalal N. S., Kennedy D. E., and McDowell C. A., J. Chem. Phys. 61, 1689 (1974). 10.1063/1.1682163 [DOI] [Google Scholar]
- 42.Weis V., Bennati M., Rosay M., Bryant J. A., and Griffin R. G., J. Magn. Reson. 140, 293–299 (1999). 10.1006/jmre.1999.1841 [DOI] [PubMed] [Google Scholar]
- 43.Abragam A., Landesman A., and Winter J. M., C. R. Acad. Sci. 247, 1852–1853 (1958). [Google Scholar]
- 44.Stoll S. and Schweiger A., J. Magn. Reson. 178, 42–55 (2006). 10.1016/j.jmr.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 45.Corzilius B., Andreas L. B., Smith A. A., Ni Q. Z., and Griffin R. G., J. Magn. Reson. 240, 113 (2014). 10.1016/j.jmr.2013.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pines D., Bardeen J., and Slichter C. P., Phys. Rev. 106, 489–498 (1957). 10.1103/PhysRev.106.489 [DOI] [Google Scholar]
- 47.Lumata L., Kovacs Z., Sherry A. D., Malloy C., Hill S., Tol J. v., Yu L., Song L., and Merritt M. E., Phys. Chem. Chem. Phys. 15, 9800–9807 (2013). 10.1039/c3cp50186h [DOI] [PMC free article] [PubMed] [Google Scholar]