

Abstract
Aims
Aspirin is widely used as an anti-platelet agent for cardiovascular prophylaxis. Despite aspirin treatment, many patients experience recurrent thrombotic events, and aspirin resistance may contribute to this. We examined the prevalence of aspirin resistance in a healthy population, and investigated whether the platelet proteome differed in aspirin-resistant subjects.
Methods
Ninety-three healthy subjects received aspirin 300 mg daily for 28 days. Before and at the end of treatment, urine was taken to determine 11-dehydrothromboxane B2, and blood was taken to measure arachidonic acid (AA)-induced aggregation of platelet-rich plasma and to interrogate the platelet proteome by mass spectrometric analysis with further confirmation of findings using Western blotting.
Results
In two of the 93 subjects, neither AA-induced aggregation nor urinary 11-dehydrothromboxane B2 was effectively suppressed by aspirin, despite measurable plasma salicylate concentrations, suggesting the presence of true aspirin resistance. Despite no detectable differences in the platelet proteome at baseline, following aspirin a marked increase was seen in platelet glycoprotein IIIa expression in the aspirin-resistant but not aspirin-sensitive subjects. An increase in platelet glycoprotein IIIa expression with aspirin resistance was confirmed in a separate cohort of 17 patients with stable coronary artery disease on long term aspirin treatment, four of whom exhibited aspirin resistance.
Conclusions
In a healthy population, true aspirin resistance is uncommon but exists. Resistance is associated with an increase in platelet glycoprotein IIIa expression in response to aspirin. These data shed new light on the mechanism of aspirin resistance, and provide the potential to identify aspirin-resistant subjects using a novel biomarker.
Keywords: aspirin resistance, glycoprotein IIIa, platelets, proteomics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Decreased platelet responsiveness to aspirin, so called aspirin resistance, is believed to contribute to the occurrence of recurrent thrombotic events in patients treated with aspirin. However, its true prevalence in a healthy population and its underlying mechanism are not known. We examined the prevalence of aspirin resistance in a cohort of healthy subjects, using two independent methods to verify its presence, and examined differences in the platelet proteome between subjects found to be sensitive and resistant to aspirin.
WHAT THIS STUDY ADDS
Our study demonstrates that aspirin resistance exists in healthy subjects, but is uncommon. We found an apparently higher prevalence in an older population of patients with established coronary heart disease on long term aspirin therapy. The presence of aspirin resistance is associated with an increase in platelet expression of glycoprotein IIIa, and this not only reveals an underlying previously unrecognized mechanism but may form the basis for a future diagnostic test for aspirin resistance.
Introduction
Aspirin is widely used and highly effective for cardiovascular prophylaxis [1–4]. It irreversibly inhibits platelet production of thromboxane A2 (TXA2) through acetylating the enzyme cyclo-oxygenase-1 (COX-1) at the Ser-529 residue [5,6]. However, 5–45% of patients exhibit reduced antiplatelet effectiveness of aspirin, so-called aspirin resistance [7–9]. This wide variation in reported prevalence can be explained to a large extent by there being no standard definition of, or validated methodology for defining, aspirin resistance.
Some studies originally defined aspirin resistance at a clinical level [10]. However, clinical lack of effect of aspirin can often be due to factors external to the platelet [11]. The current prevailing view, therefore, is to define aspirin resistance biochemically and/or functionally, measuring platelet function and/or TXA2 production. Platelet light transmission aggregometry is the gold standard test for measuring the effect of antiplatelet drugs on platelet function [12,13]. However, it is labour-intensive and not readily applied in the clinical situation. Newer point of care tests including PFA-100 [14,15] and VerifyNow® [16] are increasingly being used. The measurement of the TXA2 metabolites, thromboxane B2 (TXB2) in the serum or 11-dehydro TXB2 (a spontaneous breakdown product) in the urine, are also useful indicators of aspirin resistance [17]. However, the different assays often give widely divergent results as regards aspirin sensitivity/resistance within individual patients. Moreover, the mechanism(s) underlying aspirin resistance at the level of the platelet remain entirely unclear.
Studies of platelet expression of candidate proteins for aspirin resistance have proved negative or at best inconclusive in identifying differences between aspirin-sensitive and resistant individuals [18–20]. Modern technology allows analysis of the whole platelet proteome and such an approach, using two-dimensional gel electrophoresis in 19 aspirin-sensitive and 19 aspirin-resistant subjects with underlying ischaemic heart disease, revealed that aspirin-resistant subjects had increased expression of fibrinogen γ chain, haptoglobin and vitamin D binding protein [21]. However, this study employed PFA-100 testing for identifying aspirin resistance and this technique has proved highly non-specific in this regard [22].
The isobaric labelling reagent Tandem Mass Tags (TMT®) enables global quantitation of protein expression levels in entire proteomes. Available in six isobaric flavours (TMT®sixplex), TMT tags label peptides and proteins at amino groups, allowing a multiplexed comparison of up to six different samples in the same experiment [23]. In the present study, an intact protein labelling approach was adopted in order to profile the platelet proteome with the aim of identifying novel protein biomarkers of aspirin resistance. Labelling at an intact protein level reduces sample complexity and minimizes variation between different experimental groups as a result of sample processing [24,25].
The aims of the present study were threefold. Firstly, to identify the prevalence of aspirin resistance in a cohort of healthy subjects being treated with aspirin. Secondly, using a mass spectrometry (MS) approach, to identify proteins whose expression may differ between platelets from aspirin-resistant and aspirin-sensitive healthy subjects. Finally, to confirm our findings from a study of a healthy young population in a cohort of older patients being treated long term with aspirin for coronary heart disease.
Methods
An expanded Methods section is available in Appendix S1.
Ninety-three healthy subjects of either gender with no history of heart disease, stroke or any other significant medical condition were recruited by email advertisement of staff and students at King's College London. Their characteristics are shown in Tables 1 and 2. They were on no regular medication and had not taken any anti-inflammatory drugs or antiplatelet agents within 30 days of enrolment in the study. All study procedures were performed at the Department of Clinical Pharmacology, St Thomas' Hospital, London, UK.
Table 1.
Healthy subject characteristics
| Number of subjects | 93 (61 female, 32 male) |
|---|---|
| Age (years) | 29.9 ± 1.2 |
| Body mass index (kg m−2) | 22.9 ± 0.4 |
| Ethnicity | Caucasians 78, Asian 9, African/Caribbean 6 |
| Smokers | 17 |
Table 2.
Haematological and biochemical profile of healthy subjects
| Reference range | ||
|---|---|---|
| Homocysteine (μmol l−1) | 15.85 ± 0.69 | 0.0–17.0 |
| Total cholesterol (mmol l−1) | 4.47 ± 0.09 | |
| HDL cholesterol (mmol l−1) | 1.72 ± 0.04 | 0.9–2.00 |
| LDL cholesterol (mmol l−1) | 2.27 ± 0.09 | |
| Triglycerides (mmol l−1) | 1.02 ± 0.8 | <1.7 |
| Glucose (mmol l−1) | 4.58 ± 0.15 | 3.3–5.5 |
| HbA1c (mmol mol−1) | 30.9 ± 5.0 | 22–44 |
| Platelets (×109 l−1) | 251 ± 7.01 | 150–400 |
| White blood cells (×109 l−1) | 6.12 ± 0.16 | 4.0–11.0 |
| Neutrophils (×109 l−1) | 3.41 ± 0.12 | 1.5–7.0 |
| Lymphocytes (×109 l−1) | 2.04 ± 0.070 | 1.2–3.5 |
| Monocytes (×109 l−1) | 0.50 ± 0.042 | 0.2–1.0 |
| Urea (mmol l−1) | 4.48 ± 0.12 | 1.7–8.3 |
| Creatinine (μmol l−1) | 74.05 ± 1.53 | 45–84 |
HbA1C, glycated haemoglobin; HDL, high density lipoprotein; LDL, low density lipoprotein.
Subjects received aspirin 300 mg daily for 28 days. Before and at the end of the treatment period, urine samples were taken to determine 11-dehydroTXB2 concentrations, and blood samples were taken for light transmission aggregometry of platelet-rich plasma and for proteomic analysis of platelet lysates.
No subjects were lost to follow-up and none experienced complications of treatment necessitating drop-out from the study.
To confirm the proteomic association identified in platelets from healthy subjects, patients were identified from the database of patients in the Cardiology Department at St Thomas' Hospital who had stable coronary artery disease and were receiving anti-platelet monotherapy with 75 mg aspirin once daily long term. Seventeen such patients were recruited. Their characteristics are shown in Table S1. Aspirin resistance was identified in platelets from these patients by light transmission aggregometry, and Western blotting for glycoprotein IIIa was performed on platelet lysates, as described fully in Appendix S1.
Ethical approval for the study was obtained from the Riverside Research Ethics Committee, London, UK. All subjects gave written informed consent.
Statistical methods
Intra- and inter-assay coefficients of variation for platelet aggregation, urinary 11-dehydroTXB2 and serum salicylate measurements were all < 10%. All platelet aggregation and urinary 11-dehydroTXB2 data were expressed as mean ± SEM, and analyzed by one way analysis of variance (anova) with repeated measures and Bonferroni post hoc test. Data for the proteomic assays were analyzed as outlined in the individual sections. In all cases, P < 0.05 (two-tailed) was taken as significant.
Results
Aspirin resistance is rare but does occur in healthy subjects
In our cohort of 93 healthy subjects, after 28 days' treatment with aspirin (300 mg daily), platelet aggregation in response to 1.6 mmol l−1 AA decreased markedly when compared with basal levels (76.07 ± 0.99% and 75.74 ± 1.04% at visits 1 and 2 respectively vs. 8.96 ± 1.37% at visit 3, P < 0.001). Three subjects exhibited AA-induced aggregation >20% following aspirin, suggesting the presence of aspirin resistance. In each of these three subjects, AA-induced aggregation was very similar pre- and post-aspirin, and AA-induced aggregation post-aspirin was markedly higher than all other subjects (Figure 1A). Basal levels of aggregation in response to AA did not differ significantly between visits 1 and 2.
Figure 1.
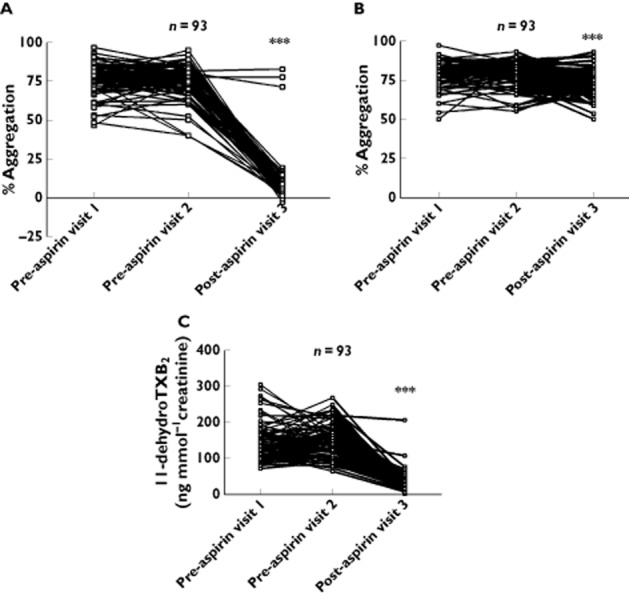
Aspirin resistance in a cohort of 93 healthy subjects. Aggregation responses are shown to (A) arachidonic acid (AA, 1.6mmol l−1) and (B) ADP 30 μmol l−1 in platelet-rich plasma. Also shown are urinary 11-dehydrothromboxane B2 (11-dehydroTXB2) (C). Visits 1 and 2 represent values 1 week apart. Visit 3 represents values after subjects were treated with 300 mg aspirin daily for 28 days. ***P < 0.001 as compared with both visit 1 and visit 2
Platelet aggregation in response to ADP 30 μmol l−1 also decreased after 1 month's treatment with aspirin, when compared with basal levels at visits 1 and 2 (79.43 ± 0.73% and 79.40 ± 0.77% at visits 1 and 2 respectively vs. 72.86 ± 0.81% at visit 3, P < 0.001) (Figure 1B). Although significant, this decrease was small in magnitude and much less than the effect seen with AA. Basal levels of aggregation in response to ADP did not differ significantly between visits 1 and 2.
Urinary levels of 11-dehydro TXB2 decreased after 1 month of 300 mg aspirin daily when compared with basal levels measured at visits 1 and 2 (Figure 1C): 139.17 ± 5.40 and 150.50 ± 4.43 ng mmol−1 creatinine at visits 1 and 2 respectively vs. 33.47 ± 2.69 ng mmol−1 creatinine at visit 3, P < 0.001. Four subjects had 11-dehydroTXB2 concentrations >68 ng mmol−1 creatinine, suggesting the presence of aspirin resistance by this criterion. Of these, the two subjects with the highest levels of 11-dehydroTXB2 were also identified as aspirin resistant as defined by AA-induced aggregation.
Serum salicylate concentrations were measured at visit 3 in all 93 subjects, in order to assess their adherence to aspirin treatment. Salicylate concentrations were only detectable in 29 of the 93 subjects (Table S2), reflecting both the relative insensitivity of the assay at these doses of aspirin and the short half-life of salicylate in the circulation (2–4 h following therapeutic doses). The 64 subjects who had undetectable levels nevertheless all had significant and marked inhibition of AA-induced aggregation following aspirin treatment, suggesting aspirin was being taken by all of these subjects, at least to some degree. Detectable salicylate concentrations were, however, present in all three subjects defined as aspirin resistant by AA-induced aggregation (subjects 59, 85 and 99). Pill counts performed at visit 3 also suggested that all subjects were fully adherent to treatment.
For the purposes of all further experiments and analyses, only the two subjects in whom aspirin failed to adequately suppress both AA-induced aggregation and urinary 11-dehydroTXB2 were considered to be truly aspirin resistant.
To determine whether responses to aspirin when applied in vitro differ between aspirin-resistant and aspirin-sensitive subjects, 31 of the subjects who took part in the original study (including the two subjects who were identified as aspirin-resistant by both AA-induced aggregation and by urinary 11-dehydro TXB2) were invited back after at least 3 months off aspirin treatment and, following incubation of PRP with different concentrations of aspirin in vitro, AA-induced aggregation responses were assessed. All concentrations of aspirin above 50 μmol l−1 caused a reduction in AA-induced aggregation, compared with basal in the absence of aspirin. A concentration-dependent inhibition of AA-induced aggregation was seen. However, there was no difference between the effects of 500 and 1000 μmol l−1 aspirin, both concentrations reducing aggregation by approximately 90% (Figure 2A). At the lower aspirin concentrations (50, 100 and 200 μmol l−1), considerable heterogeneity was seen in AA responses. This was not the case at higher concentrations (500 and 1000 μmol l−1) of aspirin (Figure 3). In the case of the two aspirin-resistant subjects, AA-induced aggregation was inhibited to a considerably lesser extent at both 50 and 100 μmol l−1 as compared with the 29 aspirin-sensitive subjects; whereas at higher aspirin concentrations, response to AA was inhibited in all subjects (Figure 2B).
Figure 2.
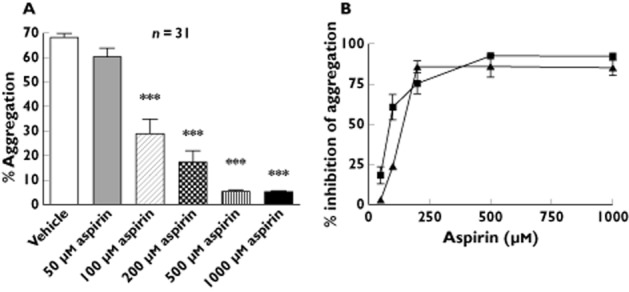
Concentration–response relationship of aspirin on arachidonic acid-mediated platelet aggregation in vitro. (A) Arachidonic acid (AA)-induced platelet aggregation at a concentration of 1.6 mmol l−1 was measured in the presence of either vehicle (ethanol) or increasing aspirin concentrations. *** P < 0.001 as compared with vehicle. (B) Percentage inhibition of AA (1.6 mmol l−1)-induced aggregation in the presence of different concentrations of aspirin, in aspirin-resistant vs. aspirin-sensitive subjects.  , sensitivity (n = 29);
, sensitivity (n = 29);  , resistant (n = 2)
, resistant (n = 2)
Figure 3.

Inter-individual heterogeneity in platelet responses to aspirin at different concentrations in vitro. Frequency distribution graphs are shown demonstrating percentage inhibition by aspirin (50 μmol l−1: A, 100 μmol l−1: B, 200 μmol l−1: C, 500 μmol l−1: D and 1000 μmol l−1: E) on arachidonic acid 1.6 mmol l−1-induced aggregation
Platelet glycoprotein IIIa expression is increased following aspirin therapy in aspirin-resistant but not aspirin-sensitive subjects
Proteomic analysis of platelet lysates revealed an increase in expression of the A isoform of glycoprotein IIIa in aspirin-resistant compared with aspirin-sensitive subjects, following (but not prior to) aspirin treatment. This finding was confirmed by Western blotting analysis. Full details are given in the expanded Results section in Appendix S1.
To confirm the validity of this finding in patients receiving long term aspirin, we studied 17 patients with stable coronary artery disease receiving ongoing treatment with 75 mg aspirin (but no other concomitant anti-platelet therapy). Apart from aspirin, seven of these subjects were also taking a proton pump inhibitor, 13 were on a β-adrenoceptor blocker, 13 were taking a calcium channel blocker, 11 were on a statin and 10 were taking an angiotensin converting enzyme inhibitor or angiotensin receptor blocker. Five of these 17 individuals were found to be aspirin-resistant by light transmission aggregometry. PRP aggregation in one of these subjects normalized following incubation with acetylsalicylic acid 100 μmol l−1, suggesting poor medication adherence or possibly pharmacokinetic factors as the principal underlying mechanism. The four patients whose platelets did not respond to acetylsalicylic acid added in vitro were assigned the status of aspirin-resistant. As compared with the other subjects, we observed an increase in platelet glycoprotein IIIa expression in those identified as being aspirin-resistant (Figure 4).
Figure 4.
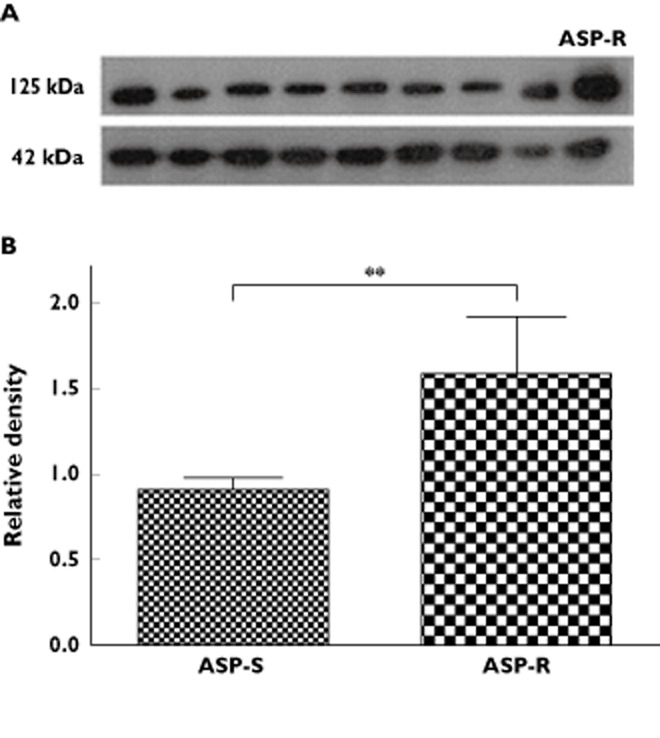
Platelet glycoprotein IIIa (GPIIIa) expression in aspirin-resistant vs. aspirin-sensitive subjects with coronary artery disease. (A) Typical Western blot of GPIIIa (125 kDa) and beta-actin (42 kDa), in platelets from an aspirin-resistant subject (ASP-R) compared to those from several aspirin-sensitive subjects. (B) Bar chart of expression of GPIIIa expressed relative to beta-actin, as assessed by Western blotting. ASP-R and ASP-S refer to aspirin-resistant and aspirin-sensitive subjects respectively. ** P < 0.01 vs. aspirin-sensitive subjects
Discussion
Aspirin remains the drug of first choice for the prophylaxis of cardiovascular events in those at risk [2–4]. However, recent studies have shown that the antiplatelet effect of aspirin is not equal in all patients and this has given rise to the term ‘aspirin resistance’ [26]. Its reported prevalence has varied greatly between studies due to the lack of a standard definition or validated methodology for identifying it. The first aim of this work was therefore to investigate the prevalence of aspirin resistance within a healthy population, on the basis of two independent criteria. In order to determine the degree of inhibition of COX-1 by aspirin, AA-induced platelet aggregation and urinary 11-dehydroTXB2 were measured. The results suggest that prevalence is low in healthy subjects, with only two out of 93 subjects being identified as truly aspirin resistant.
A number of other studies have defined aspirin resistance by investigating platelet pathways activated by agonists indirectly inhibited by aspirin, which has resulted in many more subjects being defined as aspirin resistant. In the majority of these studies, the PFA-100 has been used [27–31]. The apparent differences in prevalence between studies, based on using these different techniques, suggest that many of these techniques may give unreliable results or, at any rate, are measuring parameters other than COX-1 inhibition.
Recent studies have suggested that in clinical practice, individual patients may require different doses of aspirin in order to achieve an adequate antiplatelet effect [32]. Our data are consistent with these findings. When investigating the effects of different concentrations of aspirin in vitro on AA-induced aggregation, we found that the higher aspirin concentrations used inhibited platelet aggregation to AA in almost all subjects tested, whilst lower concentrations of aspirin inhibited platelet responses to different degrees in different subjects. When translated to the in vivo situation, these results suggest that heterogeneity may occur in responses to low doses of aspirin as used for cardiovascular prophylaxis. However this requires confirmation through further in vivo studies, and insufficient dosage is unlikely to explain the whole phenomenon of aspirin resistance.
To date, very little information is available as to whether differences in the platelet proteome between aspirin-resistant and sensitive subjects could be a contributing factor in aspirin resistance and where such studies have been done, the definition of aspirin resistance has been suboptimal, since non-specific tests such as the PFA-100 have been used for definition. Nevertheless, in recent years, major technological advances in the field of proteomics have allowed the identification and quantitation of a greater number of proteins within complex samples [33–35]. Therefore, we used relative quantitative MS to investigate differences in the platelet proteome between the aspirin-resistant and aspirin-sensitive subjects, in conjunction with strict definition of aspirin resistance based on high levels of both AA-induced aggregation and urinary 11-dehydroTXB2 coupled with measurable concentrations of serum salicylate in subjects taking aspirin.
We identified glycoprotein IIIa as increasing in expression in platelets after aspirin treatment. Although possible changes in certain other proteins were suggested by our discovery experiments, these were not confirmed by selective reaction monitoring (SRM). However, SRM of platelet lysates suggested that, whereas baseline glycoprotein IIIa expression was not different between aspirin-resistant and aspirin-sensitive subjects, its level of expression was increased after 28 days of aspirin treatment, in aspirin-resistant as compared with aspirin-sensitive subjects. This increase was confined to the A isoform. This finding was confirmed by Western blotting, which showed that, despite no difference in baseline expression, post-aspirin there was an increase in glycoprotein IIIa (isoform A) expression in aspirin-resistant but not aspirin-sensitive subjects. In our cohort of 17 patients with stable coronary artery disease, we found that four had functional aspirin resistance and these exhibited increased glycoprotein IIIa expression as compared with aspirin-sensitive patients. From a mechanistic viewpoint, administration of aspirin to aspirin-resistant patients, by giving rise to an increase in platelet expression of glycoprotein IIIa, may be expected to augment platelet aggregability by increasing binding of (and hence cross-linking of platelets by) fibrinogen, thereby overcoming the antiplatelet effect of aspirin. Platelet glycoprotein IIIa upregulation may also explain, at least in part, why major adverse cardiovascular events in aspirin-treated patients appear to be predicted especially well by COX-1-independent indices of platelet function, in particular PFA-100 collagen-ADP closure time [36].
The results presented here demonstrate the discovery and evaluation of selected candidates for aspirin resistance by monitoring changes in protein expression pre- to post-aspirin treatment. Application of TMT® technology enabled the same sample set to be used for both the discovery and evaluation stage. Samples were labelled at an intact protein level to reduce the technical variation often seen as a result of sample processing and digestion when labelling at the peptide level. To reduce sample complexity and maximize the number of proteins identified in the discovery phase, labelled samples were mixed and analyzed by GeLC-MS/MS. This approach enabled the identification of 406 platelet proteins. To our knowledge, these results represent the most comprehensive profiling of the platelet proteome to date. Furthermore, quantitative proteomics using TMTsixplex® enabled identification of novel candidate protein biomarkers of aspirin resistance, and of those identified, glycoprotein IIIa expression was most clearly different between aspirin-resistant and aspirin-sensitive subjects, post-aspirin treatment.
The most important limitation of this work was the fact that only two truly aspirin-resistant subjects were identified, out of 93. The fact that only two subjects were identified limited the statistical analyses that could be undertaken. However, the fact that our finding as regards platelet glycoprotein IIIa expression was replicated in a separate, patient cohort in a small series of subjects receiving aspirin for coronary artery disease bears out the validity of this finding. Also, it should be noted that the doses of aspirin were different in the two cohorts studied: 300 mg daily in the healthy subjects and 75 mg daily in those with coronary artery disease. We used a higher than normal dose in the healthy subject cohort simply to reduce the possibility that any observed decrease in aspirin responsiveness might be attributable to pharmacokinetic rather than pharmacodynamic factors but in the patient cohort, subjects were being treated according to standard clinical care.
In conclusion, we have found that aspirin resistance, as defined by strict criteria, exists in healthy subjects and is associated with an increase in platelet expression of glycoprotein IIIa in response to aspirin treatment. Similar results were observed in patients with stable coronary artery disease. This provides not only novel insight into the molecular basis of aspirin resistance, but in principle a novel diagnostic test to identify aspirin resistance. This may be of particular applicability to certain patient groups, such as those with diabetes, in whom aspirin resistance appears to be particularly common [37]. However, the precise mechanism by which aspirin treatment causes upregulation of platelet glycoprotein IIIa expression in aspirin-resistant subjects remains to be elucidated. Moreover, further work is required to define the specificity of platelet glycoprotein IIIa expression as a diagnostic marker of aspirin resistance.
Authors' contributions
Drs Floyd and Goodman recruited the subjects and took blood/urine samples for analysis. Drs Floyd, Goodman, Becker and Chen and Ms Mustafa performed the laboratory work. Drs Schofield, Campbell and Ward assisted with the proteomic analyses. Dr Sharma and Professor Ferro supervised the work. All authors contributed to writing the manuscript. Drs Floyd and Goodman contributed equally to this work as first authors. Dr Sharma and Professor Ferro contributed equally to this work as senior authors.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). The authors declare no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years.
Drs Schofield, Campbell and Ward are employed by Proteome Sciences plc. This work was supported by studentships from Guy's and St Thomas' Charity (to Dr Floyd), the Biotechnology and Biological Sciences Research Council (to Dr Goodman) the China Scholarship Council (to Dr Chen), and by grants from Heart Research UK and the British Heart Foundation (to Dr Sharma and Professor Ferro).
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1
TMT® workflow for characterization of the platelet proteome in aspirin-sensitive and aspirin-resistant subjects
Figure S2
Protein profiling of the platelet proteome
Figure S3
Change in platelet protein expression levels pre- to post-aspirin differs between aspirin-resistant and sensitive subjects
Figure S4
Candidate platelet proteins for aspirin resistance
Figure S5
Total ion chromatogram of selected peptides analyzed by selective reaction monitoring (SRM)
Figure S6
Identification of analysis of platelet glycoprotein IIIa as a candidate protein biomarker of aspirin resistance
Table S1
Characteristics of subjects with coronary heart disease on long term aspirin therapy
Table S2
Serum salicylate concentrations in the 29 subjects in whom levels could be detected
Table S3
TMT® labeling strategy
Table S4
Summary of the peptides selected for selective reaction monitoring (SRM) quantitation from each of the candidate proteins
Appendix S1
Expanded methods and result, supplementary tables and figures
References
- 1.ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction. Lancet. 1988;2:349–360. [PubMed] [Google Scholar]
- 2.Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy – II: maintenance of vascular graft or arterial patency by antiplatelet therapy. BMJ. 1994;308:159–168. [PMC free article] [PubMed] [Google Scholar]
- 3.Antiplatelet Trialists' Collaboration. Collaborative overview of randomised trials of antiplatelet therapy – III: reduction in venous thrombosis and pulmonary embolism by antiplatelet prophylaxis among surgical and medical patients. BMJ. 1994;308:235–246. [PMC free article] [PubMed] [Google Scholar]
- 4.Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest. 1975;56:624–632. doi: 10.1172/JCI108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci U S A. 1975;72:3073–3076. doi: 10.1073/pnas.72.8.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gum PA, Kottke-Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, Sapp SK, Topol EJ. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88:230–235. doi: 10.1016/s0002-9149(01)01631-9. [DOI] [PubMed] [Google Scholar]
- 8.McKee SA, Sane DC, Deliargyris EN. Aspirin resistance in cardiovascular disease: a review of prevalence, mechanisms, and clinical significance. Thromb Haemost. 2002;88:711–715. [PubMed] [Google Scholar]
- 9.Hovens MM, Snoep JD, Eikenboom JC, van der Bom JG, Mertens BJ, Huisman MV. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175–181. doi: 10.1016/j.ahj.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 10.Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 11.Goodman T, Sharma P, Ferro A. The genetics of aspirin resistance. Int J Clin Pract. 2007;61:826–834. doi: 10.1111/j.1742-1241.2007.01344.x. [DOI] [PubMed] [Google Scholar]
- 12.Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194:927–929. doi: 10.1038/194927b0. [DOI] [PubMed] [Google Scholar]
- 13.Michelson AD, Frelinger AL, III, Furman MI. Current options in platelet function testing. Am J Cardiol. 2006;98:S4–10. doi: 10.1016/j.amjcard.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Mammen EF, Alshameeri RS, Comp PC. Preliminary data from a field trial of the PFA-100 system. Semin Thromb Hemost. 1995;21(Suppl. 2):113–121. doi: 10.1055/s-0032-1313613. [DOI] [PubMed] [Google Scholar]
- 15.Hayward CP, Harrison P, Cattaneo M, Ortel TL, Rao AK. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J Thromb Haemost. 2006;4:312–319. doi: 10.1111/j.1538-7836.2006.01771.x. [DOI] [PubMed] [Google Scholar]
- 16.Smith JW, Steinhubl SR, Lincoff AM, Coleman JC, Lee TT, Hillman RS, Coller BS. Rapid platelet-function assay: an automated and quantitative cartridge-based method. Circulation. 1999;99:620–625. doi: 10.1161/01.cir.99.5.620. [DOI] [PubMed] [Google Scholar]
- 17.Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24:1980–1987. doi: 10.1161/01.ATV.0000145980.39477.a9. [DOI] [PubMed] [Google Scholar]
- 18.Damas JK, Waehre T, Yndestad A, Otterdal K, Hognestad A, Solum NO, Gullestad L, Froland SS, Aukrust P. Interleukin-7-mediated inflammation in unstable angina: possible role of chemokines and platelets. Circulation. 2003;107:2670–2676. doi: 10.1161/01.CIR.0000070542.18001.87. [DOI] [PubMed] [Google Scholar]
- 19.Nadar S, Blann AD, Lip GY. Effects of aspirin on intra-platelet vascular endothelial growth factor, angiopoietin-1, and p-selectin levels in hypertensive patients. Am J Hypertens. 2006;19:970–977. doi: 10.1016/j.amjhyper.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Boncler M, Luzak B, Rozalski M, Golanski J, Rychlik B, Watala C. Acetylsalicylic acid is compounding to antiplatelet effect of C-reactive protein. Thromb Res. 2007;119:209–216. doi: 10.1016/j.thromres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Farre AJ, Mateos-Caceres PJ, Sacristan D, Azcona L, Bernardo E, de Prada TP, Alonso-Orgaz S, Fernandez-Arguero M, Fernandez-Ortiz A, Macaya C. Relationship between vitamin D binding protein and aspirin resistance in coronary ischemic patients: a proteomic study. J Proteome Res. 2007;6:2481–2487. doi: 10.1021/pr060600i. [DOI] [PubMed] [Google Scholar]
- 22.Favaloro EJ. Clinical utility of the PFA-100. Semin Thromb Hemost. 2008;34:709–733. doi: 10.1055/s-0029-1145254. [DOI] [PubMed] [Google Scholar]
- 23.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 24.Sinclair J, Timms JF. Quantitative profiling of serum samples using TMT protein labelling, fractionation and LC-MS/MS. Methods. 2011;54:361–369. doi: 10.1016/j.ymeth.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Engmann O, Campbell J, Ward M, Giese KP, Thompson AJ. Comparison of a protein-level and peptide-level labeling strategy for quantitative proteomics of synaptosomes using isobaric tags. J Proteome Res. 2010;9:2725–2733. doi: 10.1021/pr900627e. [DOI] [PubMed] [Google Scholar]
- 26.Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006;367:606–617. doi: 10.1016/S0140-6736(06)68040-9. [DOI] [PubMed] [Google Scholar]
- 27.Lordkipanidze M, Pharand C, Schampaert E, Turgeon J, Palisaitis DA, Diodati JG. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J. 2007;28:1702–1708. doi: 10.1093/eurheartj/ehm226. [DOI] [PubMed] [Google Scholar]
- 28.Macchi L, Christiaens L, Brabant S, Sorel N, Ragot S, Allal J, Mauco G, Brizard A. Resistance in vitro to low-dose aspirin is associated with platelet PlA1 (GP IIIa) polymorphism but not with C807T(GP Ia/IIa) and C-5T Kozak (GP Ibalpha) polymorphisms. J Am Coll Cardiol. 2003;42:1115–1119. doi: 10.1016/s0735-1097(03)00921-5. [DOI] [PubMed] [Google Scholar]
- 29.Fontana P, Nolli S, Reber G, de Moerloose P. Biological effects of aspirin and clopidogrel in a randomized cross-over study in 96 healthy volunteers. J Thromb Haemost. 2006;4:813–819. doi: 10.1111/j.1538-7836.2006.01867.x. [DOI] [PubMed] [Google Scholar]
- 30.Bernardo E, Angiolillo DJ, Ramirez C, Cavallari U, Trabetti E, Sabate M, Hernandez R, Moreno R, Escaned J, Alfonso F, Banuelos C, Costa MA, Bass TA, Pignatti PF, Macaya C, Fernandez-Ortiz A. Lack of association between gene sequence variations of platelet membrane receptors and aspirin responsiveness detected by the PFA-100 system in patients with coronary artery disease. Platelets. 2006;17:586–590. doi: 10.1080/09537100600881412. [DOI] [PubMed] [Google Scholar]
- 31.Gurbel PA, Bliden KP, DiChiara J, Newcomer J, Weng W, Neerchal NK, Gesheff T, Chaganti SK, Etherington A, Tantry US. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115:3156–3164. doi: 10.1161/CIRCULATIONAHA.106.675587. [DOI] [PubMed] [Google Scholar]
- 32.Lee PY, Chen WH, Ng W, Cheng X, Kwok JY, Tse HF, Lau CP. Low-dose aspirin increases aspirin resistance in patients with coronary artery disease. Am J Med. 2005;118:723–727. doi: 10.1016/j.amjmed.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 33.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Choe L, D'Ascenzo M, Relkin NR, Pappin D, Ross P, Wiliamson B, Guertin S, Pribil P, Lee KH. 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics. 2007;7:3651–3660. doi: 10.1002/pmic.200700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casey T, Solomon PS, Bringans S, Tan KC, Oliver RP, Lipscombe R. Quantitative proteomic analysis of G-protein signalling in Stagonospora nodorum using Isobaric Tags for Relative and absolute quantification (iTRAQ) Proteomics. 2010;10:38–47. doi: 10.1002/pmic.200900474. [DOI] [PubMed] [Google Scholar]
- 36.Frelinger AL, 3rd, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, Furman MI, Michelson AD. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–2596. doi: 10.1161/CIRCULATIONAHA.109.900589. [DOI] [PubMed] [Google Scholar]
- 37.Simpson SH, Abdelmoneim AS, Omran D, Featherstone TR. Prevalence of high on-treatment platelet reactivity in diabetic patients treated with aspirin. Am J Med. 2014;127:95.e1–959. doi: 10.1016/j.amjmed.2013.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
TMT® workflow for characterization of the platelet proteome in aspirin-sensitive and aspirin-resistant subjects
Figure S2
Protein profiling of the platelet proteome
Figure S3
Change in platelet protein expression levels pre- to post-aspirin differs between aspirin-resistant and sensitive subjects
Figure S4
Candidate platelet proteins for aspirin resistance
Figure S5
Total ion chromatogram of selected peptides analyzed by selective reaction monitoring (SRM)
Figure S6
Identification of analysis of platelet glycoprotein IIIa as a candidate protein biomarker of aspirin resistance
Table S1
Characteristics of subjects with coronary heart disease on long term aspirin therapy
Table S2
Serum salicylate concentrations in the 29 subjects in whom levels could be detected
Table S3
TMT® labeling strategy
Table S4
Summary of the peptides selected for selective reaction monitoring (SRM) quantitation from each of the candidate proteins
Appendix S1
Expanded methods and result, supplementary tables and figures