

Abstract
The use of biological agents combined with methotrexate (MTX) in rheumatoid arthritis (RA) patients has strongly improved disease outcome. In this study, the effects of abatacept on the size and function of circulating B and T cells in RA patients not responding to anti-tumour necrosis factor (TNF)-α have been analysed, with the aim of identifying immunological parameters helpful to choosing suitable tailored therapies. We analysed the frequency of peripheral B and T cell subsets, B cell function and T regulatory cell (Treg) inhibitory function in 20 moderate/severe RA patients, according to the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) criteria, primary non-responders to one TNF-α blocking agent, who received abatacept + MTX. Patients were studied before and 6 months after therapy. We found that abatacept therapy significantly reduced disease activity score on 44 joints (DAS)/erythrocyte sedimentation rate (ESR) values without causing severe side effects. The size of the circulating B and T cell compartments in RA patients was not significantly different from healthy donors, but B cell proliferation and plasma cell differentiation was impaired before therapy and restored by abatacept. While Treg cell frequency was normal, its inhibitory function was absent before therapy and was partially recovered 6 months after abatacept. B and Treg cell function is impaired in RA patients not responding to the first anti-TNF-α agent. Abatacept therapy was able to rescue immune function and led to an effective and safe clinical outcome, suggesting that RA patients, in whom anti-TNF-α failed, are immunologically prone to benefit from an agent targeting a different pathway.
Keywords: abatacept, B cell subsets, CTLA-4-Ig, regulatory T cells, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is an inflammatory autoimmune disease of unknown aetiology, potentially leading to progressive joint destruction, functional disability and extra-articular manifestations [1,2].
Although not fully understood, the general consensus is that CD4 T cell activation by an unidentified agent represents one of the first inducers of RA. T cell activation subsequently leads to macrophage and B cell activation with cytokine production and chemokine release, resulting in inflammation, joint damage and autoantibody production. As co-stimulation is a prerequisite for T cell activation, blockade of the co-stimulatory signalling pathway may represent a potential therapeutic target. The most studied T cell co-stimulator is the CD28 molecule, widely expressed by all T cells in the mouse and in normal non-activated human T cells [3]. CD28 binds to CD80 and CD86, which are constitutively present on antigen-presenting cells (APCs) [4,5]. CD80/CD86 molecules are also present on activated B cells, such as in tonsils, but they are poorly expressed on resting B cells [6]. In addition to the co-stimulatory role for T cell activation, the expression of these molecules on B cells interferes with the B cell responses to interleukin 4 (IL-4) + CD40L stimulation and to the regulation of immunoglobulin (Ig)E synthesis [7]. Upon activation, T cells express a molecule called cytotoxic T lymphocyte antigen 4 (CTLA-4, also known as CD152) that competes with CD28 for the binding to CD80 and CD86, thus playing a role as a T cell co-stimulus inhibitor [8,9]. Because the affinity of the CD80/CD86 receptors is higher for CTLA-4 than for CD28, impairment of T cell activation can be achieved by using a soluble CTLA-4 engineered molecule. Abatacept is a dimeric fusion protein composed of the human CTLA-4 extracellular domain and a human FcIgG1, creating a soluble receptor able to bind with high-affinity CD80/CD86 molecules [10]. Abatacept was developed to block T cell activation by interfering with the co-stimulation signals delivered through APCs, but this molecule may also modulate the APC function. Bonelli et al. have shown recently that monocytes isolated from RA patients treated with abatacept showed a reduced capacity of adherence to endothelial cells and of transendothelial migration, thus rendering monocytes unable to penetrate the synovial tissue [11]. CTLA-4-Ig is also able to promote immunosuppressive activity of dendritic cells (DCs) by modulating cytokine production and by keeping the oxidant/anti-oxidant balance crucial for DC function [12]. Besides APCs, T cell activation is controlled by a special T cell subset, the regulatory T cells (Tregs) that play a pivotal role in the modulation of the immune responses and in the maintenance of peripheral tolerance. First identified in the mouse in the middle of the 1990s, Tregs [13] fast became the target of a large number of studies involving immunological diseases in humans [14,15]. Alterations in Treg numbers and/or function have been reported in patients affected by autoimmune diseases, including RA. Treg cells constitutively express CTLA-4; CTLA-4 is a target gene of forkhead box protein 3 (FoxP3) [16], a transcription factor necessary to generate Tregs from naive T cells. The absence of FoxP3 leads to immunodysregulation, polyendocrinopathy, enteropathy and X-linked (IPEX) syndrome, whereas the loss of CTLA-4 on Tregs results in the failure of cell-mediated suppressive function [17].
Because CTLA-4-Ig impairs T cell activation and indirectly alters the ability of T cells to provide help to B cells during the immune response, it becomes crucial to analyse B cell subsets and their function in the course of abatacept therapy. In the adult, B cells are generated in the bone marrow and migrate to the periphery at the transitional B cell stage, when they are still short-lived and functionally immature. Transitional B cells are transported by the bloodstream to the spleen, where they develop into mature B cells. Mature B cells generate switched memory B cells and antibody-producing cells after they have been stimulated, expanded and selected in the germinal centres in the presence of T cell help [18].
Tumour necrosis factor (TNF)-α blocking agents represent the first biological therapy approved for RA, and are currently the most used molecules. Although TNF-α blocking agents are generally well tolerated and show good efficacy in patients with RA, up to 40% of the patients may not respond to therapy for lack of efficacy, development of resistance or treatment-related adverse events [19], thus several other biological agents have been introduced. However, this growing availability of therapeutic weapons for RA patients is not supported by head-to-head trials or validated clinical/immunological predictive markers that can help the clinicians in the adoption of a ‘targeted therapy’, in particular in patients who respond inadequately to the first biological agent.
To evaluate if RA patients would benefit from changing the biological drug targeting a different activation pathway, we analysed the effects of abatacept therapy on the frequency and function of circulating B cell subsets in RA patients who did not respond to anti-TNF-α therapy. We also investigated the T cell compartment and the Treg suppressive capacity.
Material and methods
Patients and healthy donors
Twenty-five moderate to severe RA patients, according to the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria [20], of whom 20 primary non-responders to one TNF-α blocking agent and five naive for this therapy were recruited between 2010 and 2012 at the Division of Immunology and Rheumatology, S. Andrea Hospital, Sapienza University of Rome. All the patients agreed to participate according to the ethical guidelines of the 1975 Declaration of Helsinki. The study was approved by the local ethical committee. Twenty-five age- and sex-matched healthy blood donors (HDs) (Blood Transfusion Unit, Ospedale Pediatrico Bambino Gesù, Rome) were used as controls.
All patients received 10 mg/kg/body weight (bw) of abatacept (ORENCIA®; Bristol-Meyers Squibb, Princeton, NJ, USA) intravenously (i.v.), in addition to methotrexate (MTX) 10 mg/week i.v./subcutaneously (s.c). Disease activity score on 44 joints (DAS), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), RF (rheumatoid factor) and adverse events were assessed before initiating abatacept therapy (pre) and 6 months after (post). Patients' clinical information is described in Table 1.
Table 1.
Clinical, serological and demographic data of the 20 rheumatoid arthritis (RA) patients before (pre) and 6 months after abatacept therapy (post)
| ESRa (mm/h) | CRP (mg/l) | DASb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Patients | Age (years) | Disease duration (years) | Previous therapy | Pre | Post | Pre | Post | Pre | Post |
| 1 | 30 | 2 | IFX | 11 | 9 | 0·5 | 1 | 3·8 | 2·4 |
| 2 | 42 | 14 | ADA | 21 | 9 | 17 | 3 | 3·9 | 2 |
| 3 | 54 | 7 | ETN | 13 | 25 | 5·2 | 1·1 | 3·8 | 1·2 |
| 4 | 70 | 20·5 | ETN | 17 | 13 | 6 | 5 | 3·6 | 3 |
| 5 | 71 | 5 | ADA | 35 | 16 | 1·5 | 5 | 4·6 | 2·8 |
| 6 | 68 | 7 | IFX | 26 | 18 | 5 | 3 | 4·6 | 2·8 |
| 7 | 56 | 10 | GOL | 8 | 6 | 3 | 3 | 4 | 3·2 |
| 8 | 68 | 11 | ETN | 46 | 27 | 12 | 7 | 5·4 | 3·7 |
| 9 | 65 | 20 | ADA | 40 | 58 | 20 | 22 | 5 | 3·3 |
| 10 | 55 | 8 | ETN | 81 | 6 | 1·2 | 3 | 3·3 | 3·2 |
| 11 | 62 | 16 | IFX | 52 | 7 | 40 | 5 | 4 | 3·6 |
| 12 | 72 | 8 | ETN | 6 | 24 | 0 | 0 | 3·7 | 2·4 |
| 13 | 61 | 13 | ETN | 57 | 52 | 11 | 6 | 4·5 | 4·6 |
| 14 | 60 | 6 | IFX | 28 | 20 | 3 | 3 | 4·9 | 4·2 |
| 15 | 57 | 4 | ADA | 22 | 12 | 19 | 6 | 3·1 | 3 |
| 16 | 61 | 20 | IFX | 15 | 30 | 2 | 17 | 3·9 | 3·3 |
| 17 | 47 | 8·5 | ADA | 40 | 15 | 6 | 0·4 | 4·4 | 4·3 |
| 18 | 49 | 2 | GOL | 54 | 24 | 22 | 2 | 3·7 | 3·1 |
| 19 | 60 | 8 | ETN | 40 | – | 12 | – | 4 | – |
| 20 | 52 | 6 | ADA | 28 | – | 8 | – | 3·7 | – |
| Mean | 58 | 9·8 | 32 | 20·6 | 9·7 | 5·1 | 4·1 | 3 | |
| s.d. | 10·1 | 5·4 | 18·5 | 13·8 | 9·4 | 5·3 | 0·5 | 0·8 | |
P = 0·05 [mean erythrocyte sedimentation rate (ESR) pre versus post].
P < 0·001[mean disease activity score on 44 joints (DAS) pre versus post].
ADA = adalimumab; CRP = C-reactive protein; Etn = etanercept; GOL = golimumab; IFX = infliximab; s.d. = standard deviation.
Cell isolation and flow cytometry analysis
Heparinized peripheral blood mononuclear cells (PBMCs) were isolated by FicollPaque™ Plus (Amersham Pharmacia Biotech, Uppsala, Sweden) density-gradient centrifugation, counted and used for cell culture (see below) or stained with the appropriate combination of labelled antibodies and analysed by flow cytometry, as described previously [21]. Dead cells were excluded from analysis by side-/forward-scatter gating. All analyses were performed on a fluorescence-activated cell sorter (FACS)Canto (BD Biosciences, San Diego, CA, USA) interfaced to PC FACSDiva software. One hundred thousand events per sample were analysed.
B cell proliferation and plasma cell differentiation
Mononuclear cells were labelled with 5-chloromethylfluorescein diacetate at the final concentration of 0·1 μg/ml (CellTracker CMFDA; Molecular Probes, Eugene, OR, USA) and cultured at 2–3 × 105 cells per well in 96-well plates with RPMI-1640 (Gibco BRL, Life Technologies, Carlsbad, CA, USA), 10% heat inactivated fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT, USA), 2% l-glutamine (Gibco BRL), 5 × 10−5M 2-β-mercaptoethanol (Sigma, St Louis, MO, USA) and 20 mg/ml gentamycin (Gibco BRL), supplemented or not with 2·5 μg/ml cytosine–phosphate–guanosine (CpG)-oligodeoxynucleotide (ODN) (Hycult Biotechnology, Uden, the Netherlands) and CTLA-4-Ig (125 mg/ml, abatacept, Orencia®; Bristol-Meyers Squibb) diluted 1:1000. Cell proliferation and phenotypical analysis were performed by flow cytometry using a FACSCalibur Flow Cytometer (BD Biosciences) on day 7 [22].
Flow cytometry analysis
After 7-day culture, cells were collected and stained with the appropriate combination of labelled antibodies: monoclonal clone HIB19 (anti-CD19), clone M-T271 (anti-CD27), clone HIT2 (anti-CD38), clone UCHT1 (anti-CD3), clone B1·49·9 (anti-CD25), clone HIT8a (anti-CD8), clone RPA-T4 (anti-CD4), clone HIL-7R-M21 (anti-CD127) and clone FN50 (anti-CD69) were obtained from BD Biosciences and anti-IgM Fc5μ fragment-specific (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). All analyses were performed on a FACSCanto (BD Biosciences) interfaced to PC FACSDiva software. One hundred thousand live cells per sample were analysed.
Cell sorting
PBMCs from RA patients and HDs were isolated from heparinized peripheral blood by Ficoll-Paque™ Plus (Amersham Biosciences, Little Chalfont, UK) by density-gradient centrifugation and stained with the following antibodies: clone UCHT1 (anti-CD3), clone B1·49·9 (anti-CD25), clone HIT8a (anti-CD8) and clone RPA-T4 (anti-CD4), all purchased from BD Biosciences. Cells were sorted as follows: CD4+ T cells and CD4+ T cells without CD4+CD25+ Treg cells using a FACSvantage SE (BD Biosciences). Cell purity was > 98%.
T cell proliferation
Two hundred thousand purified CD4+ T cells and sorted CD4+ T cells without CD4+CD25+ Treg cells were stimulated with anti-CD3/CD28 beads (Invitrogen, Life Technologies, Carlsbad, CA, USA) at the ratio of 50 cells : one bead in RPMI-1640 medium (Gibco BRL) supplemented with 10% fetal calf serum (FCS; Gibco BRL) for 5 days. Cell proliferation was analysed using the Delfia® cell proliferation kit (Perkin Elmer Life Sciences, Turku, Finland), according to the manufacturer's instructions. Briefly, on the fourth day of culture cells were incubated with bromodeoxyuridine (BrdU) for 20 h, then fixed and denatured. After removal of the fixing solution, cells were incubated for 2 h with Europium (Eu)-labelled anti-BrdU antibody under agitation. Delfia inducer was added to the wells and Eu-fluorescence was measured in a time-resolved fluorometer2100 EnVision™ Multilabel Reader (PerkinElmer Life Sciences).
Statistical analysis
Data were analysed using StatView statistical program for MacIntosh (StatView Software, San Diego, CA, USA) and P-values were determined with the paired Student's -test. P-values < 0·05 were considered to be statistically significant.
Results
Clinical improvement after Aabatacept (CTLA-4-Ig) therapy
Twenty patients with moderate to severe RA, despite previous anti-TNF-α therapy, were enrolled and abatacept 10 mg/kg/bw plus MTX 10 mg/week introduced as therapeutic regimen. Two patients were lost at follow-up for reasons unrelated to therapy. Five patients with moderate to severe RA naive for anti-TNF-α therapy were also enrolled. Before starting abatacept therapy (pre), 16 of 20 (80%) of the patients presented severe disease with DAS > 3·7 and four of 20 (20%) showed moderate disease (2·4 < DAS ≤ 3·7). Six months after abatacept therapy (post) patients showed a significant reduction in disease activity (mean DAS 4·1 versus 3; P < 0·001) (Table 1); in particular, 10 of 18 patients showed a good or moderate clinical response, with four patients reaching clinical remission/low disease activity, according to the EULAR criteria (Fig. 1). A significant reduction in ESR from pre to post was also observed. Regarding safety, patients reported eight minor infections (two cystitis, two herpes simplex and four upper respiratory infections that did not compromise the course of the therapeutic programme with CTLA-4-Ig, resolved with a conventional antibiotic therapy); no major adverse events were reported.
Figure 1.
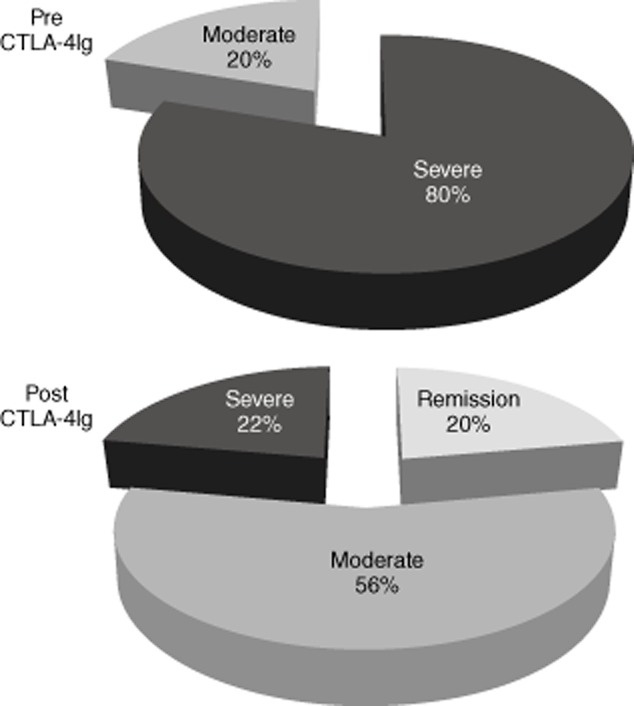
Pie diagrams indicate the distribution of disease activity according to European League Against Rheumatism (EULAR) criteria in rheumatoid arthritis (RA) patients before and 6 months after therapy with abatacept [cytotoxic T lymphocyte antigen 4-immunoglobulin (Ig) [CTLA-4-Ig]. Severe disease is represented in black, moderate disease in grey and remission or low disease in light grey.
Effects of abatacept (CTLA-4-Ig) on the frequency of B cell subsets and B cell function
Using flow cytometry, we first analysed peripheral blood mononuclear cells (PBMCs) isolated from RA patients before (pre) and 6 months after (post) CTLA-4-Ig therapy and stained for CD19, CD24, CD38, CD27, IgM and IgD. Frequency of B cells (CD19pos), as well as transitional (CD24brightCD38brightCD27negIgMposIgDpos), mature (CD24posCD38posCD27negIgMposIgDpos) and memory (CD24brightCD38negCD27pos) B cell subsets [18], was measured in patients and HDs. The size of the B cell compartments in RA patients before and after therapy was not significantly different (Fig. 2a and Supporting information, Table S1). Although a reduction in the frequency of RA patient B cells (mainly memory B cells) at both time-points, compared to HDs, was found, the difference was not statistically significant.
Figure 2.
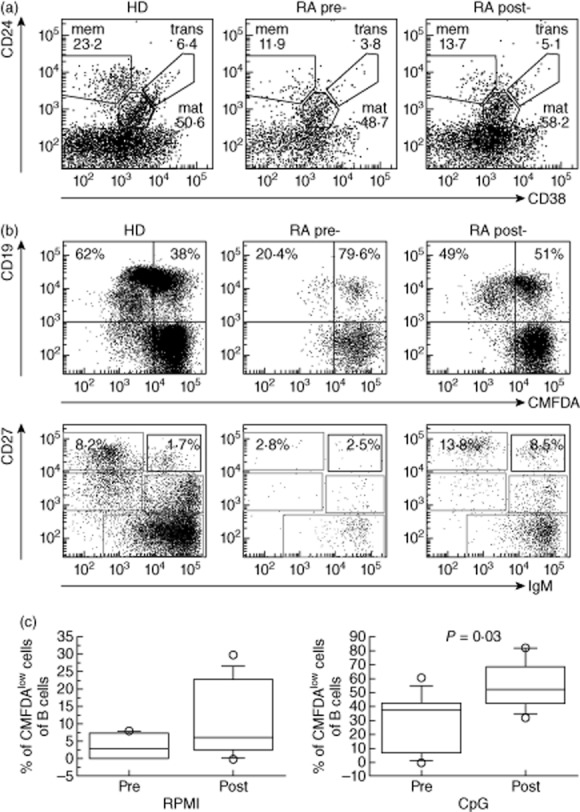
B cell subset analysis and B cell response to cytosine–phosphate–guanosine (CpG) in rheumatoid arthritis (RA) patients before and 6 months after treatment with abatacept. (a) Dot-plot shows a representative example of CD24 versus CD38 staining of peripheral blood lymphocytes (PBLs) from a healthy control (HD) and an RA patient before (pre) and after therapy (post), regions indicate: memory (mem), mature (mat) and transitional (trans) B cells and values correspond to the frequency of each B cell subset inside CD19pos cells. (b) PBLs pre-labelled with 5-chloromethylfluorescein diacetate (CMFDA) were stimulated with CpG for 7 days and analysed for B cell proliferation (upper panels) and plasma cell differentiation (lower panels). Values in the upper panel quadrants correspond to the frequency of proliferating and non-proliferating CD19pos cells. In lower panels the frequency of switched and immunoglobulin (Ig)M antibody secreting cells are indicated. (c) Box-plot graphs represent the median (solid line), interquartile ranges (boxes) and minimum and maximum non-outlier frequency values (whiskers) of proliferating CD19pos cells after culture without (RPMI) or with CpG. P-values are indicated when statistically differences were found between the two groups.
We have shown previously that B cells express Toll-like receptor (TLR)-9 and that CpG induces TLR-9-dependent B cell proliferation and antibody secretion [22]; thus, CpG stimulation is a good tool to measure, in vitro, the capacity of B cells to proliferate, differentiate and generate antibody-secreting cells. In healthy adults, stimulation of peripheral blood lymphocytes with CpG induces cell division in 48·5–70% of CD19pos cells; furthermore, upon TLR-9 stimulation memory B cells differentiate into plasma cells by up-regulating CD27 and CD38 molecules and by down-modulating CD19.
In order to study the effects of CTLA-4-Ig therapy on the function of B cells, we stimulated PBMCs isolated from RA patients before and after therapy with CpG and measured B cell proliferation, plasma cell generation and Ig production. Cells were cultured for 7 days in the presence or absence of CpG, after which they were collected and analysed by flow cytometry for the expression of CMFDA, CD19, CD27 and IgM. Concentrations of IgA, IgG and IgM were measured in culture supernatants by enzyme-linked immunosorbent assay (ELISA). We found that, before therapy, B cells from RA patients, in the absence of CpG stimulus, were lost after 7 days in culture compared to HDs and RA patients after therapy (Fig. 2c, left panel and data not shown). Moreover, the response to CpG in terms of B cell proliferation and plasma cell generation was impaired in RA patients before initiating CTLA-4-Ig therapy (Fig. 2b). In fact, the frequency of CD19pos B cells showing low expression of CMFDA was, on average, 20·4% (Fig. 2b) and the Ig concentrations in the culture supernatants were very low (Supporting information, Fig. S1). After 6 months of CTLA-4-Ig therapy B cell proliferation increased significantly and reached similar HD levels (Fig. 2b,c, P = 0·03), thus suggesting that abatacept helped to restore B cell function in the group of RA patients.
We then asked the question of whether CTLA-4-Ig could interfere with the in-vitro B cell response to CpG stimulation. Therefore, we isolated PBMCs from HDs and stimulated them for 7 days with CpG in the presence or absence of abatacept. We found that CTLA-4-Ig alone did not induce proliferation; instead it promoted B cell survival in the culture conditions deprived of CpG (Fig. 3a). In CpG + CTLA-4-Ig-stimulated cultures, B cells proliferated, generated plasma cells and produced all Ig classes, as in the cultures stimulated with only CpG (Fig. 3b).
Figure 3.
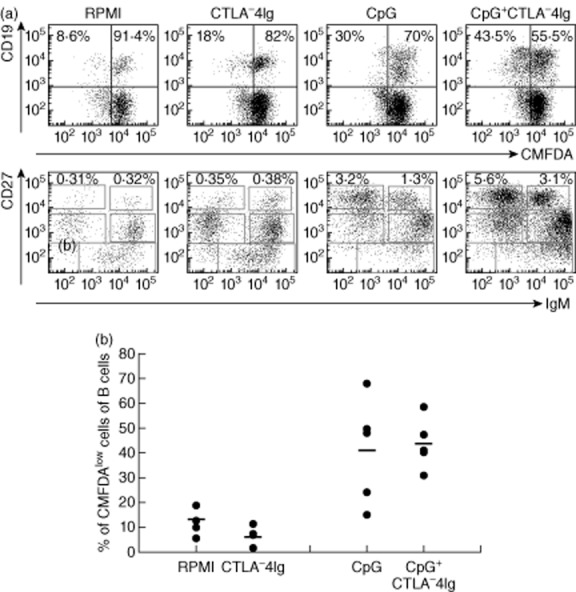
B cell response to cytosine–phosphate–guanosine (CpG) in the presence of cytotoxic T lymphocyte antigen 4-immunoglobulin (CTLA-4-Ig) of peripheral blood lymphocytes (PBLs) isolated from healthy donors (HD). Peripheral blood mononuclear cells (PBMCs) were stimulated for 7 days either with medium (RPMI), CTLA-4-Ig (1:1000), CpG or CpG + CTLA-4-Ig. (a) Dot-plot shows a representative example of PBLs pre-labelled with 5-chloromethylfluorescein diacetate (CMFDA) and stained for CD19, CD27 and IgM. Upper panels show the frequency of proliferating and non-proliferating CD19pos cells. Lower panels represent PC differentiation with the frequency of switched and IgM antibody secreting cells of total B cells. (b) Graph represents the frequency of proliferating CD19pos cells in five independent experiments (single dots) and the bar indicates the mean.
During the course of the study, the use of abatacept has been extended to patients with moderate/severe RA disease not yet treated with anti-TNF-α agents. Therefore, in five RA patients fulfilling these requirements we studied B cell proliferation before and after starting abatacept therapy. The patients' total PBMCs were stimulated with CpG and analysed as described previously. For both time-points and for all RA patients we found that 7 days after CpG stimulation the frequency of CD19posCMFDAlow was equivalent to the frequency of proliferating B cells observed in HDs (frequency of CD19posCMFDAlow before abatacept was 49·12 ± 16·6% and was 50·4 ± 14·46% 6 months after, HDs = 54·1 ± 11·3%; Supporting information, Fig. S2). This result suggests strongly that, in a situation in which the B cell function is not compromised, abatacept therapy does not interfere with it.
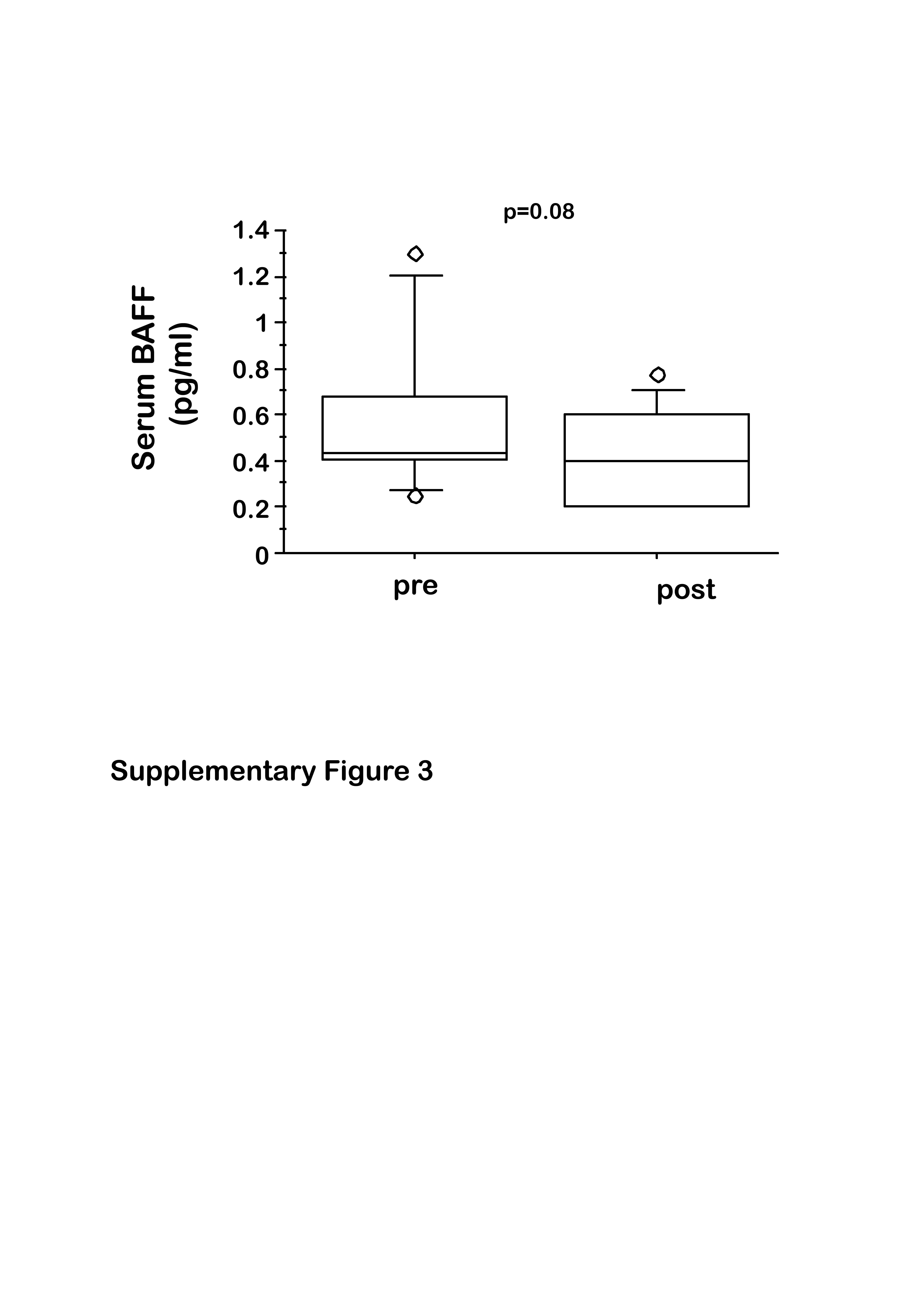
B cell activating factor (BAFF) is one of the B cell survival factors [23] often found increased in the serum of RA patients [24,25]. We also measured serum concentration of BAFF before and 6 months after abatacept in our cohort of RA patients. We found no significant difference between the serum concentration of BAFF from RA and HDs. Moreover, for each RA patient we observed that therapy with CTLA-4-Ig induced a reduction in the serum concentration of BAFF; differences were insufficient to reach significance (Supporting information, Fig. S3, P = 0·08).
Effects of CTLA-4-Ig on the frequency of T cell subsets and regulatory T cell inhibitory function
Although the main aim for the use of CLTA-4-Ig in RA is to limit APC-induced T cell activation [26,27], few studies have addressed the impact of such therapy on the number and function of T cells in humans [28–31]. We analysed the peripheral blood T cell compartments in the group of RA patients before and after CTLA-4-Ig therapy. PBMCs were stained for CD3, CD4, CD25, CD45RO, CD69 and CD127 and analysed by flow cytometry. The frequency of T (CD3pos), naive/memory (CD3posCD45ROpos) (Fig. 4a), CD4 and CD8 T cells and Treg cells (CD4pos CD25posCD69neg CD127low) (Fig. 4b) was calculated and the results are shown in Supporting information, Table S2. We found that the frequency of both CD4 and CD8 T cells were significantly lower in RA patients compared to HD, but the size of the T cell subset before and 6 months after abatacept therapy does not change significantly in RA patients (Supporting information, Table S2 [29]). Previous reports have shown that not all RA patients have a reduced Treg cell compartment [32,33]; often the number of Treg cells is normal, but cells have lost their suppressive capacity, meaning that they are unable to inhibit T cell proliferation. One of the therapeutic strategies able to rescue Treg function correlated with clinical improvement in RA patients is the block of the TNF-α signalling pathway by anti-TNF-α blocking agents [34,35]. In our study group, anti-TNF-α therapy failed, and even if the size of the Treg compartment was normal we could not establish if Treg cells were functional. Thus, we asked the following questions: first, whether Treg cells isolated from RA peripheral blood before CTLA-4-Ig therapy were able to block T cell proliferation; and secondly, whether abatacept would be able to modulate Treg function. In order to study the suppressive function of Tregs in small samples of peripheral blood, namely from patients, we decided to set up the following experimental strategy, taking advantage of the fact that in physiological conditions a low number of Treg cells are able to modulate T cell proliferation. We purified CD4pos T cells and CD4 depleted of CD25posCD4pos T cells (Fig. 5a) and compared CD4 proliferation in response to anti-CD3/anti-CD28 stimulation. As expected, the presence of Tregs limited CD4 proliferation upon stimulation with anti-CD3/anti-CD28; conversely, in the absence of CD25posCD4pos T cells, CD4 proliferation was significantly higher than in the cultures of total CD4 cells (Fig. 5b).
Figure 4.
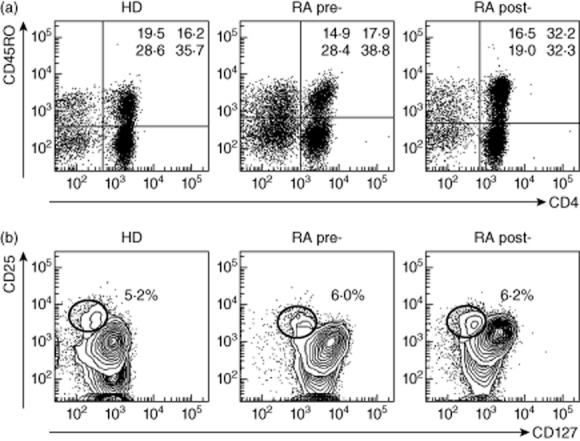
Analysis of the peripheral blood T cell subset composition in rheumatoid arthritis (RA) patients. Peripheral blood mononuclear cells (PBMCs) from healthy donors (HD) and RA patients before (pre) and after therapy (post) were stained for CD3, CD4, CD45RO, CD127 and CD25 and analysed by flow cytometry. (a) Dot-plots show a representative example of peripheral blood lymphocytes (PBLs) gated on CD3pos cells and analysed for the expression of CD4 and CD45RO, values indicate the frequency of cells for each quadrant (lower left = CD8posCD45ROneg; upper left = CD8posCD45ROpos; lower right = CD4posCD45ROneg; upper right CD8posCD45ROpos). (b) Representative dot-plot showing CD4pos cells expressing CD25 and CD127; regulatory T cells are indicated (CD25posCD127low) with correspondent frequencies.
Figure 5.
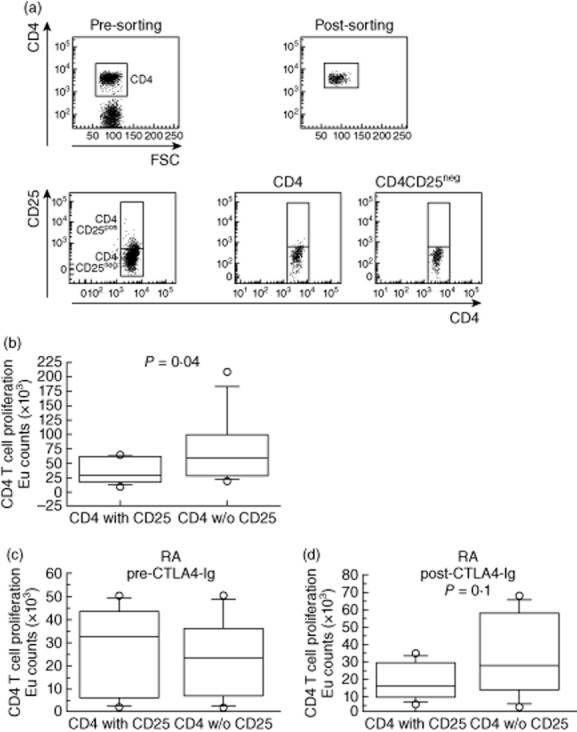
Inhibitory function of regulatory T cells (Treg) cells. (a) Peripheral blood mononuclear cells (PBMCs) were stained for CD3, CD4 and CD25; total CD4pos cells or CD4pos depleted of CD4posCD25pos cells were purified by cell sorting. (b) CD4 T cell proliferation was detected using Delfia® cell proliferation kit; europium counts are shown as box-plots of five independent experiments using healthy donor (HD) PBMCs. (c,d) T cell proliferation in the presence or absence of regulatory T cells in RA patients (n = 5) before (pre) and after (post) therapy. CD4 T cell proliferation was detected as described above with box-plot graphs. Statistical significance was determined by the t-test and P-value is indicated.
We then purified CD4pos T cells and CD4pos without CD25posCD4pos T cells from PBMCs of RA patients before and after abatacept therapy and analysed T cell proliferation upon stimulation with anti-CD3/anti-CD28 beads. We found that before therapy CD4pos T cells proliferated similarly in the presence or absence of Treg cells, suggesting that Tregs from RA patients were unable to inhibit CD4 helper cell proliferation (Fig. 5c). Although not statistically different, 6 months after abatacept therapy stimulation with anti-CD3/anti-CD28 beads induced a higher proliferation in the cultures depleted of CD4posCD25pos T cells than in cultures of total CD4pos T cells (Fig. 5d, P = 0·1). In conclusion, RA patients, not responding to anti-TNF-α agents, reacquired Treg inhibitory capacity after abatacept.
Discussion
Significant progress in understanding the molecular pathways involved in the pathogenesis and progression of RA disease allowed the identification of potential targets for therapeutic intervention using biological disease-modifying anti-rheumatic drugs (DMARDs). Historically, the first approved biological drug to be used in RA patients was anti-TNF-α, which has been demonstrated to have an effective and safety profile [36–38]. However, 40% of RA patients presented either primary/secondary therapeutic failure or developed adverse events to the drug, raising the need for the introduction of other therapeutic tools [19]. Among the biological drugs used in RA anti-CD20 chimeric monoclonal antibody (mAb) (rituximab) [39] that specifically targets B cells, thus inducing their death, humanized anti-human IL-6 receptor mAb (tocilizumab) [40], which hinders IL-6 from exerting its proinflammatory effects and CTLA-4-Ig (abatacept) that functions as an inhibitor of T cell activation [10] are available. Abatacept was first approved in 2005 by the Food and Drug Administration (FDA) [41] for its use in moderate–severe RA patients with active disease who had inadequate response to TNF-α antagonists. More recently, due to its good safety profile, abatacept therapy has also been extended to RA patients naive to anti-TNF-α agents. The efficacy of abatacept in reducing clinical signs of disease and decreasing disease symptoms has been demonstrated in at least six independent clinical trials and, by their extension phases, these studies enrolled more than 4000 patients [26]. Currently, RA management aims at ‘true remission’ by controlling disease activity and by blocking the progressive joint destruction through the use of combined therapies of biological agents plus methotrexate, possibly at early disease stages [42]. However, as a consequence of the lack of head-to-head trials and of good predictive markers, there is no general consensus on the best strategy to be adopted in patients who respond inadequately to the first biological agent. Thus, the therapeutic decision often appears to be more empirical than individually patient-driven.
In the present work we looked for the biological rationale for the use of abatacept in RA patients unresponsive to anti-TNF-α therapy, by performing a pilot study in which the effects of CTLA-4-Ig therapy on the size and function of B cells and Treg cells of RA patients naive or not responding to anti-TNF-α therapy were analysed.
From a clinical viewpoint, our results confirm previous reports by the Abatacept Trial in Treatment of Antitumour necrosis factor IN adequate responders (ATTAIN), which demonstrated a good safety and efficacy profile of abatacept therapy in these patients [26]. In fact, 6 months after abatacept treatment a significant reduction in disease activity was observed, with four patients reaching remission/low activity; no severe adverse events were reported.
The overall disease activity improvement observed in our RA patients can reflect a generalized reduction of the systemic inflammation (ESR from 32 to 20, P = 0·05). It is known that activation of DCs can be dampened by CTLA-4; both CTLA-4 expressed by Tregs and CTLA-4-Ig induce indoleamine 2,3-dioxygenase (IDO) expression on DCs, giving them regulatory and tolerogenic functions [43,44]. Moreover, CTLA-4-Ig alters cytokine secretion by T helper cells, contributing to reduce inflammation [29].
Before abatacept therapy RA patients presented a normal-sized B compartment, but B cell proliferation in response to CpG stimulation was impaired. In addition, we found that Tregs were unable to inhibit T cell proliferation after CD3/CD28 stimulation.
Considering that these patients had previously been treated with anti-TNF-α agents and that TNF-α is known to be an autocrine/paracrine factor for B cell proliferation, we investigated whether the defects on B cell proliferation could be attributed to the anti-TNF-α therapy. We measured the B cell proliferation response to CpG in five RA patients naive to TNF-α agents and found no difference on B cell function compared to HDs.
We found that in anti-TNF-α-naive patients, before starting abatacept, both B cell proliferation and differentiation to plasma cells were significantly higher than in the patients treated previously with anti-TNF-α. Because B cell function in RA patients naive to TNF-α inhibitors was not impaired, it is important to note that no further increase in B cell proliferation and differentiation was observed after abatacept therapy.
It has been reported previously that B cell activation with anti-Ig + anti-CD40 and IL-4 induces an increase in TNF-α expression on B cells and subsequent proliferation [45]. Proliferation can be blocked, in vitro, by adding monoclonal antibodies against TNF-α and can be increased by adding exogenous TNF-α. Duddy et al. showed that TNF-α secretion by B cells depends upon the type and strength of stimulatory signals [46]. TNF-α is essential for lymphoid microarchitecture; in fact, TNF-α knock-out mice completely lack primary follicles in the spleen and cannot form germinal centres upon T cell-dependent immunization [47]. The in-vivo effect of blocking the TNF-α signalling pathway in humans has been demonstrated recently by Anolik et al., who showed impairment in B cell function (via effects on FDCs) in RA patients treated with anti TNF-α agents [48].
As mentioned previously, the management of systemic autoimmune diseases such as RA still represents a challenge, considering that it is difficult to achieve and maintain the appropriate immunosuppression effective in the control of autoimmune reactions/inflammation by preserving, in the meantime, the level of immune response necessary to fight infection. Although B cells clearly play an important role in RA pathogenesis, the rescue of normal B cell function in a context of low inflammation and T cell activation by the use of CTLA-4-Ig therapy may offer a good safety profile without compromising immune reactions against pathogens [49].
In agreement with Gonzales et al., we observed a reduction in Treg function and its partial rescue after abatacept [50]. Tregs play a pivotal role in the modulation of immune responses, in particular in the maintenance of peripheral tolerance. Alterations in the Treg frequency and/or function have been reported in patients affected by autoimmune diseases such as multiple sclerosis and systemic lupus erythematosus [14,15]; in RA patients a functional defect of Tregs has been described. The loss of Treg function in RA patients can be rescued by anti-TNF-α therapy in those responding clinically to infliximab [35]. This apparent contradiction with our results can be explained by the fact that we analysed only patients not responding to anti-TNF-α agents.
In conclusion, our results indicate that RA patients not responding to the first anti-TNF-α agent presents an impairment of B cell and Treg function that can be restored by abatacept. If confirmed by larger studies, these data may help in the selection of a tailored therapy, suggesting that RA patients responding inadequately to anti-TNF-α inhibitors are immunologically prone to benefit from a biological agent with a different mechanism of action.
Acknowledgments
This study has received financial support from Bristol Myers Squibb (BMS). M. M. R. was supported by a Mérieux Research Starting Grant, Institut Merieux, Lyon, France.
Disclosures
Authors declare any potential conflicts of interest.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Immunoglobulin (Ig)A/G/M concentration (μg/ml) in culture supernatants. Peripheral blood mononuclear cells (PBMCs) from rheumatoid arthritis (RA) patients before (pre) and 6 months after cytotoxic T lymphocyte antigen 4 (CTLA-4)-Ig therapy were stimulated with cytosine–phosphate–guanosine (CpG) for 7 days and Igs tested by enzyme-linled immunosorbent assay (ELISA). Dots correspond to single RA patients and the bar to the mean concentration of IgA, IgG and IgM detected in the supernatants of CpG-stimulated PBMCs from healthy donors (HD) (n = 20).
{kind=link}
Fig. S2. B cell response to CpG of peripheral blood lymphocytes (PBLs) isolated from anti-tumour necrosis factor (TNF)-α-naive rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy. Peripheral blood mononuclear cells (PBMCs) were pre-labelled with 5-chloromethylfluorescein diacetate (CMFDA), stimulated for 7 days with CpG and analysed by flow cytometry for the expression of CD19, CD27 and IgM. Graph represents the frequency of proliferating CD19pos cells (CMFDAlow B cells) in five patients (patients1–5, single symbols) and the bar indicates the mean.
{kind=link}
Fig. S3. Serum concentration of B cell activating factor (BAFF) (pg/ml) in rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy. Box-plots indicate the median (solid line), interquartile ranges (boxes) and minimum and maximum non-outlier values (whiskers). Statistical significance was determined by the t-test and P-value is indicated.
{kind=link}
Table S1. Frequency of peripheral blood B cell subsets of patients with rheumatoid arthritis (RA) before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy and in healthy controls (HD). B cell subsets were analysed by flow cytometry, values represent the mean ± standard deviation (n = 20).
Table S2. Frequency of peripheral blood T cell subsets present in rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy and in healthy controls (HD, n = 20). T cell subsets were analysed by flow cytometry; values represent the mean ± standard deviation.
References
- 1.Genovese MC, Bathon JM, Martin RW, et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: two-year radiographic and clinical outcomes. Arthritis Rheum. 2002;46:1443–1450. doi: 10.1002/art.10308. [DOI] [PubMed] [Google Scholar]
- 2.Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–911. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- 3.Pieper J, Johansson S, Snir O, et al. Peripheral and site-specific CD4(+) CD28(null) T cells from rheumatoid arthritis patients show distinct characteristics. Scand J Immunol. 2014;79:149–155. doi: 10.1111/sji.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 5.Caux C, Vanbervliet B, Massacrier C, et al. B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med. 1994;180:1841–1847. doi: 10.1084/jem.180.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boussiotis VA, Freeman GJ, Gribben JG, Daley J, Gray G, Nadler LM. Activated human B lymphocytes express three CTLA-4 counterreceptors that costimulate T-cell activation. Proc Natl Acad Sci USA. 1993;90:11059–11063. doi: 10.1073/pnas.90.23.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeannin P, Delneste Y, Lecoanet-Henchoz S, Gauchat JF, Ellis J, Bonnefoy JY. CD86 (B7-2) on human B cells. A functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells. J Biol Chem. 1997;272:15613–15619. doi: 10.1074/jbc.272.25.15613. [DOI] [PubMed] [Google Scholar]
- 8.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. [PubMed] [Google Scholar]
- 9.Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852–863. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 10.Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discov. 2006;5:185–186. doi: 10.1038/nrd1989. [DOI] [PubMed] [Google Scholar]
- 11.Bonelli M, Ferner E, Goschl L, et al. Abatacept (CTLA-4IG) treatment reduces the migratory capacity of monocytes in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65:599–607. doi: 10.1002/art.37787. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Pillars article: immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995. J Immunol. 2011;186:3808–3821. [PubMed] [Google Scholar]
- 14.Lee JH, Wang LC, Lin YT, Yang YH, Lin DT, Chiang BL. Inverse correlation between CD4+ regulatory T-cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology. 2006;117:280–286. doi: 10.1111/j.1365-2567.2005.02306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavin MA, Rasmussen JP, Fontenot JD, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi T, Kishi A, Osaki M, et al. Construction of self-recognizing regulatory T cells from conventional T cells by controlling CTLA-4 and IL-2 expression. Proc Natl Acad Sci USA. 2013;110:E2116–2125. doi: 10.1073/pnas.1307185110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 19.Day R. Adverse reactions to TNF-alpha inhibitors in rheumatoid arthritis. Lancet. 2002;359:540–541. doi: 10.1016/S0140-6736(02)07718-8. [DOI] [PubMed] [Google Scholar]
- 20.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 21.Kruetzmann S, Rosado MM, Weber H, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003;197:939–945. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Capolunghi F, Cascioli S, Giorda E, et al. CpG drives human transitional B cells to terminal differentiation and production of natural antibodies. J Immunol. 2008;180:800–808. doi: 10.4049/jimmunol.180.2.800. [DOI] [PubMed] [Google Scholar]
- 23.Avery DT, Kalled SL, Ellyard JI, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–1319. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 25.Pers JO, Daridon C, Devauchelle V, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann NY Acad Sci. 2005;1050:34–39. doi: 10.1196/annals.1313.004. [DOI] [PubMed] [Google Scholar]
- 26.Genovese MC, Schiff M, Luggen M, et al. Longterm safety and efficacy of abatacept through 5 years of treatment in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor inhibitor therapy. J Rheumatol. 2012;39:1546–1554. doi: 10.3899/jrheum.111531. [DOI] [PubMed] [Google Scholar]
- 27.Sibilia J, Westhovens R. Safety of T-cell co-stimulation modulation with abatacept in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(5 Suppl. 46):S46–56. [PubMed] [Google Scholar]
- 28.Buch MH, Boyle DL, Rosengren S, et al. Mode of action of abatacept in rheumatoid arthritis patients having failed tumour necrosis factor blockade: a histological, gene expression and dynamic magnetic resonance imaging pilot study. Ann Rheum Dis. 2009;68:1220–1227. doi: 10.1136/ard.2008.091876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pieper J, Herrath J, Raghavan S, Muhammad K, Vollenhoven R, Malmstrom V. CTLA4-Ig (abatacept) therapy modulates T cell effector functions in autoantibody-positive rheumatoid arthritis patients. BMC Immunol. 2013;14:34. doi: 10.1186/1471-2172-14-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scarsi M, Zanotti C, Chiarini M, et al. Reduction of peripheral blood T cells producing IFN-gamma and IL-17 after therapy with abatacept for rheumatoid arthritis. Clin Exp Rheumatol. 2014;32:204–210. [PubMed] [Google Scholar]
- 31.Scarsi M, Ziglioli T, Airo P. Baseline numbers of circulating CD28-negative T cells may predict clinical response to abatacept in patients with rheumatoid arthritis. J Rheumatol. 2011;38:2105–2111. doi: 10.3899/jrheum.110386. [DOI] [PubMed] [Google Scholar]
- 32.Blache C, Lequerre T, Roucheux A, et al. Number and phenotype of rheumatoid arthritis patients' CD4+CD25hi regulatory T cells are not affected by adalimumab or etanercept. Rheumatology (Oxf) 2011;50:1814–1822. doi: 10.1093/rheumatology/ker183. [DOI] [PubMed] [Google Scholar]
- 33.Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol. 2011;31:596–605. doi: 10.1007/s10875-011-9542-6. [DOI] [PubMed] [Google Scholar]
- 34.Huang Z, Yang B, Shi Y, et al. Anti-TNF-alpha therapy improves Treg and suppresses Teff in patients with rheumatoid arthritis. Cell Immunol. 2012;279:25–29. doi: 10.1016/j.cellimm.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- 37.Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–259. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 38.Maini RN, Breedveld FC, Kalden JR, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1552–1563. doi: 10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.de la Torre I, Leandro MJ, Edwards JC, Cambridge G. Baseline serum immunoglobulin levels in patients with rheumatoid arthritis: relationships with clinical parameters and with B-cell dynamics following rituximab. Clin Exp Rheumatol. 2012;30:554–560. [PubMed] [Google Scholar]
- 40.Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x-ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. 2007;66:1162–1167. doi: 10.1136/ard.2006.068064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bristol-Myers Squibb Company. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125118s0138lbl.pdf (accessed December 2011)
- 42.Smolen JS, Landewe R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69:964–975. doi: 10.1136/ard.2009.126532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellor AL, Chandler P, Baban B, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–1401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 44.Grohmann U, Orabona C, Fallarino F, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 45.Boussiotis VA, Nadler LM, Strominger JL, Goldfeld AE. Tumor necrosis factor alpha is an autocrine growth factor for normal human B cells. Proc Natl Acad Sci USA. 1994;91:7007–7011. doi: 10.1073/pnas.91.15.7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172:3422–3427. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 47.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anolik JH, Ravikumar R, Barnard J, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. 2008;180:688–692. doi: 10.4049/jimmunol.180.2.688. [DOI] [PubMed] [Google Scholar]
- 49.Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;(2) doi: 10.1002/14651858.CD008794.pub2. CD008794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez-Quiroga C, Abud-Mendoza C, Doniz-Padilla L, et al. CTLA-4-Ig therapy diminishes the frequency but enhances the function of Treg cells in patients with rheumatoid arthritis. J Clin Immunol. 2011;31:588–595. doi: 10.1007/s10875-011-9527-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunoglobulin (Ig)A/G/M concentration (μg/ml) in culture supernatants. Peripheral blood mononuclear cells (PBMCs) from rheumatoid arthritis (RA) patients before (pre) and 6 months after cytotoxic T lymphocyte antigen 4 (CTLA-4)-Ig therapy were stimulated with cytosine–phosphate–guanosine (CpG) for 7 days and Igs tested by enzyme-linled immunosorbent assay (ELISA). Dots correspond to single RA patients and the bar to the mean concentration of IgA, IgG and IgM detected in the supernatants of CpG-stimulated PBMCs from healthy donors (HD) (n = 20).
Fig. S2. B cell response to CpG of peripheral blood lymphocytes (PBLs) isolated from anti-tumour necrosis factor (TNF)-α-naive rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy. Peripheral blood mononuclear cells (PBMCs) were pre-labelled with 5-chloromethylfluorescein diacetate (CMFDA), stimulated for 7 days with CpG and analysed by flow cytometry for the expression of CD19, CD27 and IgM. Graph represents the frequency of proliferating CD19pos cells (CMFDAlow B cells) in five patients (patients1–5, single symbols) and the bar indicates the mean.
Fig. S3. Serum concentration of B cell activating factor (BAFF) (pg/ml) in rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy. Box-plots indicate the median (solid line), interquartile ranges (boxes) and minimum and maximum non-outlier values (whiskers). Statistical significance was determined by the t-test and P-value is indicated.
Table S1. Frequency of peripheral blood B cell subsets of patients with rheumatoid arthritis (RA) before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy and in healthy controls (HD). B cell subsets were analysed by flow cytometry, values represent the mean ± standard deviation (n = 20).
Table S2. Frequency of peripheral blood T cell subsets present in rheumatoid arthritis (RA) patients before (pre) and 6 months after (post) cytotoxic T lymphocyte antigen 4 (CTLA-4)-immunoglobulin (Ig) therapy and in healthy controls (HD, n = 20). T cell subsets were analysed by flow cytometry; values represent the mean ± standard deviation.