

Abstract
Interleukin (IL)-17 plays a critical role in inflammation. Most studies to date have elucidated the inflammatory role of IL-17A, often referred to as IL-17. IL-17F is a member of the IL-17 family bearing 50% homology to IL-17A and can also be present as heterodimer IL-17AF. This study elucidates the distribution and contribution of IL-17A, F and AF in inflammatory arthritis. Neutralizing antibody to IL-17A alone or IL-17F alone or in combination was utilized in the mouse collagen-induced arthritis (CIA) model to elucidate the contribution of each subtype in mediating inflammation. IL-17A, F and AF were all increased during inflammatory arthritis. Neutralization of IL-17A reduced the severity of arthritis, neutralization of IL-17A+IL-17F had the same effect as neutralizing IL-17A, while neutralization of IL-17F had no effect. Moreover, significantly higher levels of IL-17A and IL-17F were detected in peripheral blood mononuclear cells (PBMC) from patients with rheumatoid arthritis (RA) in comparison to patients with osteoarthritis (OA). IL-17A and AF were detected in synovial fluid mononuclear cells (SFMC) in RA and OA, with IL-17A being significantly higher in RA patients. Enriched CD3+ T cells from RA PBMCs produced singnificantly high levels of IL-17A and IL-17AF in comparison to OA peripheral blood CD3+ T cells. IL-17A, F and AF were undetectable in T cells from SFMCs from RA and OA. While IL-17A, F, and AF were all induced during CIA, IL-17A played a dominant role. Furthermore, production of IL-17A, and not IL-17F or IL-17AF, was elevated in PBMCs, SFMCs and enriched peripheral blood CD3+ T in RA.
Keywords: arthritis, cytokine, inflammation
Introduction
Interleukin (IL)-17 plays an undisputed role in mediating inflammation. The IL-17 family consists of six subtypes, IL-17A, IL-17B, IL-17C, IL-17D, IL-17E and IL-17F, with a 16–50% homology of B–F to IL-17A. IL-17F has about 50% homology to IL-17A. IL-17A and F can be secreted as homodimers as well as the heterodimer IL-17AF [1,2].
The pathogenic role of IL-17A in inflammatory arthritis has been reported in studies involving neutralization of IL-17A and in IL-17A-deficient mice [3,4]. The pathogenic potential of other IL-17 subtypes has been reported in studies where T cells transduced with IL-17A, IL-17F, IL-17B or IL-17C all induced joint inflammation in the mouse model of collagen-induced arthritis (CIA) and neutralization of IL-17B reduced the severity of CIA [5]. The vast majority of studies on rheumatoid arthritis (RA) reporting increased levels of IL-17 are focused on IL-17A, frequently referred to as IL-17 [6–10]. There is limited information regarding the expression of other subtypes. IL-15-mediated induction of IL-17C, E and F has been reported in mononuclear cells from RA [11]. IL-17A, IL-17F and IL-17C were increased significantly in psoriatic skin [12]. Moreover, IL-17A and, to a lesser extent, IL-17F was shown to induce a similar set of inflammatory genes in RA synovial fibroblasts [13,14]. These reports suggest that inflammatory potential is not restricted to IL-17A alone, but also extends to other subtypes of IL-17 in preclinical models as well as in human diseases.
The studies in this report are focused on elucidating the contribution of IL-17A, F and AF responses in CIA and RA. We have also performed head-to-head studies of in-vivo neutralization of IL-17A, IL-17F, and IL-17A+IL-17F in CIA to establish the contribution each of these cytokines in mediating joint inflammation.
Materials and methods
Collagen-induced arthritis
Experiments involving animals were carried out in accordance with institutional guidelines under protocol approved by the Animal Care and Use Committee of the University of Arizona. All efforts were made to minimize suffering. Eight to 10-week-old male DBA1 mice were purchased from Taconic Farms (Hudson, NY, USA). Arthritis was induced following immunization with collagen and complete Freund's adjuvant (CFA). CFA was prepared by mixing heat-inactivated mycobacterial strain H37Ra in incomplete Freund's adjuvant (4 mg/ml) and was mixed with lympholized chicken collagen (Chondrex, Redmond, WA, USA) dissolved overnight in acetic acid (4 mg/ml) at 1:1 to form an emulsion which was injected intradermally at the base of the tail. Clinical scoring of arthritis, assay of anti-collagen antibodies and tissue histology of paws were undertaken as described previously [15].
Generation of mouse IL-17F antibody mAb (I17M502)
Solution-based panning of the Centocor pIX de-novo Fab libraries was performed based on previously used Centocor de-novo library panning techniques [16]. Specifically, Fab library phage libraries were mixed and separated by heavy chain gene, then blocked for 1 h with 3% milk in 1 × phosphate-buffered saline Tween 20 (PBST). Microcentrifuge tubes were blocked with 3% milk in 1 × PBST for 1 h; 50 μl of Dynabeads (Dynal/Life Technologies, Paisley, Scotland, UK) per library were washed with 1 × PBS and blocked with 3% milk in PBST for 1 h at room temperature (RT) rotating. Bt-mIL-17F at 100 nM was also blocked for 1 h at RT rotating with 0·3% milk in PBST, then mixed with the blocked library phage for 1 h. Blocked streptavidin beads were incubated with the phage/antigen solution for 30 min. Beads were captured magnetically and washed 10 times with PBST. Cells were plated out evenly on 3–150 mm Luria-Bertani LB broth with carbenicillin (LB/Carb)/1% glucose plates (Teknova, Hollister, CA, USA) and incubated overnight at 37°C. Cells were scraped from the agar plates into 2 × yeast extract and tryptone (YT) media and 50 μl was used to inoculate 20 ml of 2 × YT/carb and grown at 37°C shaking for 2 h. Cultures were then infected with 1 ml of VCSM13 helper phage and incubated for 30 min at 37°C without shaking before adding kanamycin (35 μg/ml) and isopropyl β-D-1-thiogalactopyranoside (IPTG) (1 mM) and incubating overnight at 30°C [17]. The panning process was repeated for a total of three rounds. Following the third round of panning, cells were harvested for plasmid DNA miniprep. DNA was cut with NheI and SpeI enzymes to remove the pIX gene and the vector DNA was gel isolated, ligated and transformed to obtain single colonies for screening. Single colonies were picked and expressed in deep 96-well plates by induction with IPTG. Cultures were centrifuged and supernatants were used for soluble Fab screening against mouse IL-17F coated on a Maxisorp plate (1 μg/ml) and for Fab expression by binding to sheep anti-human FD (The Binding Site, Birmingham, UK) coated on a Maxisorp plate. Fabs were detected in both enzyme-linked immunosorbent assays (ELISAs) using anti-kappa horseradish peroxidase (HRP) (Southern Biotech, Birmingham, AL, USA) and detection with electrochemical luminescence substrate (Roche, Basel, Switzerland). Clones that demonstrated inhibition of IL-17F binding to mouse IL-17RC were subjected to affinity maturation. Polymerase chain reaction (PCR)-amplified heavy chains were cloned into LC libraries as described [16]. Panning was executed as described above, but with lower concentrations of bt-mIL-17F, 1 nM for all three rounds. Clones were screened as detailed above. Unique clones were converted to immunoglobulin (Ig)G and further characterized for affinity and inhibition of IL-17F activity. Neutralization activity of I17M502 was demonstrated in vitro via inhibition of IL-17F-induced epithelial cell-derived chemokine production from CMT93 cells (data not shown).
Administration of neutralizing antibodies
Mouse anti-IL-17A and isotype control antibodies were a kind gift from Dr Merle Elloso (Janssen Research and Development, L.L.C., Spring House, PA, USA). Mouse anti-IL-17F was generated as above. For experiments involving administration of neutralizing antibodies, each mouse received 100 μg of specific antibody+100 μg of respective isotype control antibody or 100 μg each of both isotype control antibodies, daily via the intraperitoneal (i.p.) route.
Mouse tissue harvest and culture set-up
Splenocytes or draining inguinal lymph nodes were harvested from mice 2 weeks following immunization with collagen (initiation phase) and from arthritic mice. Single-cell suspensions (5 × 106/ml) of splenocytes were stimulated with collagen (100 μg/ml; Chondrex, Redmond, WA, USA), in the presence or absence of IL-23 (100 ng/ml; Peprotech, Rocky Hill, NJ, USA) for 6 days. Culture supernatants were collected and frozen in −80°C for further analyses.
Human subjects
The study was approved by the Human Research Ethics Committee of the University of Arizona and the participant's written consent was obtained in accordance with the terms of the ethics committee of the University of Arizona. Forty patients who fulfilled the 1987 American College of Rheumatology (ACR) criteria for RA and 19 patients with osteoarthritis (OA) were recruited for this study. Blood samples were collected from all subjects, although in some cases the yield was insufficient for all assays. Synovial fluid mononuclear cells (SFMCs) were collected from eight patients with RA and nine patients with OA at the University of Michigan, under a protocol approved by the Human Research Ethics Committee of the University of Michigan.
Collection of peripheral blood and processing
Peripheral blood was collected by venipuncture into heparinized tubes or DNA tubes. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood using Ficoll-Paque density gradient (GE Healthcare, Little Chalfont, UK). PBMCs or synovial fluid mononuclear cells (SFMCs) were cultured at 5 × 106 cells/ml with phorbol myristate acetate (PMA) (5 ng/ml)/ionomycin (500 ng/ml) for 6 h or 24 h and culture supernatants were collected and stored at −80°C until further analyses. DNA was prepared from blood, and shared epitope alleles were analysed at the University of Arizona Genetics Core genotyping core, University of Arizona, using the human leucocyte antigen (HLA)-DRB1 genotyping kit from Qiagen (Venlo, the Netherlands).
CD3+ T cell isolation from PBMCs and SFMCs
CD3+ T cells were enriched from PBMCs or SFMCs from some RA and OA patients using a human T cell enrichment kit (Miltenyi Biotech, San Diego, CA, USA), according to manufacturer's protocol. Purified CD3+ T cells were then cultured at 5 × 106 cells/ml with PMA (5 ng/ml)/ ionomycin (500 ng/ml) for 24 h and culture supernatants were collected and stored at −80°C until further analyses.
Flow cytometry
For intracellular flow cytometry, cells were stimulated briefly with PMA/ionomycin and brefeldin A, followed by surface staining of CD4 (anti-CD4 cloneGK1·5; Biolegend, San Diego, CA, USA) and intracellular staining with anti-IL-17A (clone TC11-18H10·1; Biolegend) and anti-IL-17F (clone eBio18F10; eBioscience, San Diego, CA, USA). Fluorescently labelled isotype-matched antibodies were used as controls. Flow cytometry data was acquired with BD LSR and analysed with FlowJo software (TreeStar, Inc., Ashland, OR, USA).
Cytokine analysis
Mouse and human IL-17A, IL-17F and IL-17AF in culture supernatants were analysed utilizing kits from eBioscience. We validated the manufacturer's data and found no cross-reactivity between ELISA for IL-17A, IL-17F and IL-17AF (data not shown). IL-6, IFN-γ, IL–1β, TNF-α, granulocyte colony-stimulating factor (G-CSF) and granulocyte–macrophage colony-stimulating factor (GM-CSF) were measured in mouse sera by Luminex assay using kits from R&D Systems (Minneapolis, MN, USA).
Statistical analysis
All ELISA and real-time PCR assays were performed in triplicate. Differences among groups were analysed by Student's t-test or Mann–Whitney U-test. Correlation was analysed by Pearson's correlation analysis. P-value of <0·05 was considered significant.
Results
IL-17A, F and AF are induced during CIA
IL-17A, IL-17F and IL-17AF were analysed in draining inguinal lymph nodes or splenocytes in naive mice, mice from the initiation phase of CIA (2 weeks following immunization with collagen and CFA) and from arthritic mice. Data in Fig. 1a show that IL-17A and F were not present in naive mice. IL-17A is induced first, during the initiation phase, with 0·488% CD4 cells being positive for IL-17A, which increases to 1·20% of total CD4 cells with the onset of arthritis. IL-17F is only induced upon the onset of arthritis with 1·23% of CD4 cells being positive for IL-17F. Interestingly, only a very small percentage of CD4 cells stained positive for both IL-17A and IL-17F. Similar results were obtained with draining inguinal lymph nodes (data not shown).
Figure 1.
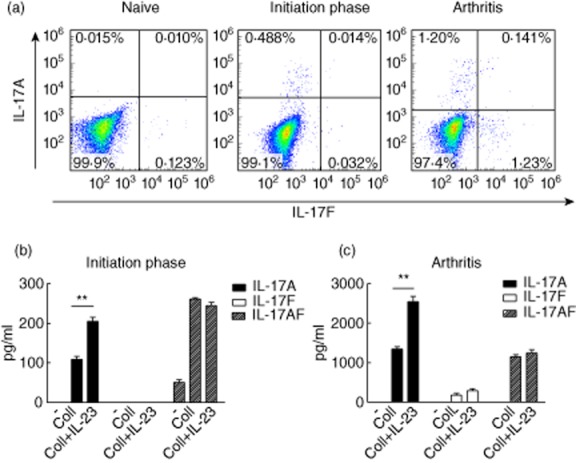
Interleukin (IL)-17A, F and AF responses are induced in collagen-induced arthritis (CIA). (a) Splenocytes from naive mice, mice from initiation phase (2 weeks following immunization with collagen and complete Freund's adjuvant (CFA) and arthritic mice were stimulated for 6 h with phorbol myristate acetate (PMA)/ionomycin and brefeldin A and stained for anti-CD4, IL-17A and IL-17F. Data shown are gated on CD4. Data are representative of three separate experiments. (b,c) Splenocytes of mice from initiation phase and arthritic mice were stimulated with collagen in the presence or absence of IL-23 for 7 days, followed by analyses of IL-17A, F and AF responses in culture supernatants by enzyme-linked immunosorbent assay (ELISA). Data are representative of two separate experiments. **P < 0·005.
It is well accepted that IL-17A is induced during CIA as a consequence of anti-CII responses [15,18]. Figure 1b shows that during the initiation phase both IL-17A and IL-17AF were induced in a collagen-specific manner. In keeping with the undetectable levels of IL-17F by flow cytometry during the initiation phase (Fig. 1a), IL-17F was not detectable in culture supernatants. IL-23 induced a significant increase in IL-17A only. There was no increase in IL-17AF levels in the cultures when IL-23 was added. Figure 1c shows that IL-17A, IL-17F and AF were detectable in cultures of splenocytes from arthritic mice stimulated with collagen, with IL-17F levels lower than IL-17A or IL-17AF. IL-23 selectively induced IL-17A without any significant change in IL-17F or IL-17AF.
To summarize, IL-17A and IL-17F were induced during arthritis and were produced by discrete cell populations. Furthermore, IL-17AF is also produced during CIA.
IL-17A plays a dominant pathogenic role in CIA
So far our studies showed that IL-17A, IL-17AF and, to a lesser extent, IL-17F were induced during CIA. In order to elucidate the pathogenic potential of IL-17A, F and AF during CIA, mice were immunized with collagen and CFA followed by administration of neutralizing antibodies or matched isotype controls daily starting from day 20. The experimental groups included (i) anti-IL-17A+isotype control for anti-IL-17F; (ii) anti-IL-17F+isotype control for anti-IL-17A; (iii) anti-IL-17A+anti-IL-17F; and (iv) isotype control for anti-IL-17A+isotype control for anti-IL-17F. Data in Fig. 2a show that mice receiving anti-IL-17A or anti-IL-17A+anti-IL-17F antibodies had significantly reduced clinical severity of arthritis compared to mice that received anti-IL-17F alone or isotype control antibodies. Figure 2b depicts representative micrographs from each group. Figure 2c shows that joint inflammation, synovitis, cartilage damage and bone erosion scores were all reduced significantly in mice that received anti-IL-17A alone or anti-IL-17A+anti-IL-17F in comparison to mice that received anti-IL-17F alone or isotype control antibody.
Figure 2.

Interleukin (IL)-17A plays a dominant pathogenic role in collagen-induced arthritis (CIA). Thirty-two DBA1 mice were immunized to induce arthritis. Mice were divided into four groups of eight mice each (1) anti-IL-17A+isotype of anti-IL-17F; (2) anti-IL-17F+isotype of anti-IL-17A; (3) anti-IL-17A+anti-IL-17F; (4) isotype of IL-17A+isotype of IL-17F. All mice received 100 μg of antibodies daily starting at day 21 post-immunization with collagen and complete Freund's adjuvant (CFA) (shown as arrow). (a) Data shown as cumulative arthritis score per group. *P = 0·01. (b) Data show the histology of haematoxylin and eosin staining of representative paws from the four groups of mice, magnification ×10. (c) Data show the histological scoring of paws from various groups. *P < 0·05; ** P < 0·005; ***P < 0·0005.
Anti–IL-17A antibody treatment suppresses serum IgG2b and proinflammatory cytokines IL-6 and G-CSF
Anti-collagen antibodies play a critical pathogenic role in CIA, hence it is plausible that the differential effects of IL-17A and IL-17F on clinical arthritis were associated with differences in anti-collagen antibody responses. IgG1, IgG2a and IgG2b anti-collagen antibody responses were measured in sera from mice receiving various interventions. Data in Fig. 3a show that there were no differences in IgG1 anti-collagen antibody responses between the groups. Administration of anti-IL-17A alone or anti-IL-17F alone did not alter IgG2a responses, whereas a combination of IL-17A+IL-17F reduced IgG2a anti-collagen antibodies. In contrast, administration of anti-IL-17A alone, anti-IL-17F alone or anti-IL-17A+anti-IL-17F significantly reduced IgG2b responses. These findings show that both IL-17A and IL-17F have similar effects on anti-collagen antibody responses; however, neutralization of IL-17A significantly reduced arthritis severity.
Figure 3.

Anti-interleukin (IL)-17A antibody treatment suppresses serum immunoglobulin (Ig)G2b and proinflammatory cytokines IL-6 and granulocyte colony-stimulating factor (G-CSF). (a) Experimental design was the same as for Fig. 2 above. Serum drawn at the time of harvest was analysed for anti-collagen antibody using enzyme-linked immunosorbent assay (ELISA), as outlined in Materials and methods. **P < 0·005; ***P < 0·0005; n.s. = not significant. Data are from five to eight mice in each group. (b) Serum from various groups of mice was analysed for cytokine IL-6, interferon (IFN)-γ, IL–1β, G-CSF, GM-CSF and tumour necrosis factor (TNF)-α by Luminex assay. *P < 0·05; **P < 0·005; ***P < 0·0005; n.s. = not significant. Data are from five to eight mice in each group.
In addition to intact anti-collagen antibody responses, joint inflammation in CIA is dependent upon robust cytokine responses, including IL-6, IL-1β, IFN–γ, TNFα, G-CSF and GM-CSF. Of these cytokines, IL-17A has been reported to directly induce the secretion of G-CSF, IL-6, IL-1β, GM-CSF and TNF-α [18–20]. In order to elucidate if the differential effect of anti-IL-17A and anti-IL-17F on arthritis was due to differences in the regulation of the above cytokines in vivo, IL-6, IFN-γ, IL-1β, TNF-α, G-CSF and GM-CSF were measured in sera from mice receiving anti-IL-17A, anti-IL-17F, anti-IL-17A+anti-IL-17F or isotype control antibodies. IL-6 and G-CSF were suppressed significantly in mice treated with anti-IL-17A or anti-IL-17A+anti-IL-17F antibody in comparison to mice receiving isotype control antibody (Fig. 3b). As noted above, mice receiving anti-IL-17A or anti-IL-17A+anti-IL-17F antibody had significantly reduced severity of arthritis. There was no suppression of IL-6 or G-CSF in mice receiving anti-IL-17F antibody and mice in this group did not have any reduction in their arthritis severity. Surprisingly, IL-6 levels were increased in mice receiving anti-IL-17F antibody in comparison to mice receiving isotype control. IFN-γ, IL-1β, TNF-α and GM-CSF were not altered among the various groups of mice (Fig. 3b).
While we did not find suppression of IL-1β, TNF-α, IFN-γ or GM-CSF in any group of mice, the data depicting the suppression of IL-6 and G-CSF by anti-IL-17A are in line with previous reports supporting the augmentation of IL-6 and G-CSF by IL-17A during inflammation [3,21–23]. Although previous studies have reported the induction of TNF-α and IL-1β by IL-17A in human macrophages, we did not find any change in systemic levels of TNF-α or IL-1β upon administration of anti-IL-17A during CIA [21]. Furthermore, the unaltered levels of TNF-α do not rule out the pathogenic role of TNF-α, either alone or in synergy with IL-17A. Our data, showing the unaltered levels of IFN-γ with the administration of anti-IL-17A, are in line with a previous study elucidating unchanged levels of T helper type 1 (Th1) responses following administration of anti-IL-17A antibody during inflammatory arthritis [24]. Studies elucidating the effector function of IL-17A or IL-17F on RA synovial cells have reported that while IL-17A and IL-17F may induce similar sets of genes, IL-17A was potent in inducing most inflammatory genes, including IL-6 and G-CSF, in comparison to IL-17F [13,25]. Thus, our data on IL-6 and G-CSF are in keeping with previous data, supporting the possibility of a more dominant role of IL-17A over IL-17F during arthritis. These findings suggest that the protective effect of anti-IL-17A or anti-IL-17A+anti-IL-17F on arthritis may be due to the selective suppression of IL-6 and G-CSF.
Collectively, our data demonstrate that IL-17A plays a dominant role over IL-17F in the regulation of inflammatory arthritis. Additionally, while IL-17A and IL-17F may have similar effects on the regulation of B cell production of anti-collagen antibodies, IL-17A has a dominant effect on IL-6 and G-CSF in-vivo.
IL-17A, F, and AF in peripheral blood mononuclear cells of patients with RA
In order to corroborate our data pertaining to IL-17A, F and AF obtained in the preclinical model of CIA with the human disease RA, IL-17A, F and AF were analysed in PBMCs of patients with RA. Clinical and demographic characteristics of the patients with RA are outlined in Table 1. PBMCs from patients with RA or OA were stimulated with PMA/ionomycin for 6 or 24 h followed by analysis of IL-17A, IL-17F and IL-17AF by ELISA. Upon stimulation of PBMCs with PMA/ionomycin, IL-17A, F and AF were not detectable at 6 h (data not shown). Figure 4 shows the levels of IL-17A, F and AF in PBMCs of patients with RA or OA stimulated for 24 h. Levels of IL-17A and F in culture supernatants of PBMCs from RA were higher than in OA, whereas the levels of IL-17AF were similar in the two groups. In our preliminary studies, only very small numbers of cells were positive for IL-17A or IL-17F by intracellular flow cytometry and hence this technique was not utilized for further assays involving human samples. In our studies the serum levels of IL-17A, F and AF were too low (below 5 pg/ml) to be able to detect reliably without interference of heterophilic antibodies [26]. Hence, serum levels of IL-17A, F and AF were not utilized for this study.
Table 1.
Demographic and clinical features of subjects with rheumatoid arthritis (RA) [pertaining to Fig. 4: RA peripheral blood mononuclear cells (PBMC)]
| RA cohort (n = 40) | |
|---|---|
| n (%) | |
| Female subjects [n (%)] | 32 (80) |
| Anti-CCP + [n (%)] (in n = 38 patients)† | 23 (60) |
| SE 1 copy (in n = 32 patients)‡ | 16 (50) |
| SE 2 copies (in n = 32 patients)‡ | 3 (9·3) |
| On prednisone | 10 (25) |
| On DMARDS (methotrexate, azathioprine, leflunomide) | 35 (87·5) |
| On NSAIDS and narcotic pain medications | 5 (12·5) |
| On anti-TNF therapy | 3 (7·5) |
| Mean (s.d.) | |
| DAS28 | 4·0 (1·6) |
| Tender swollen joint count 44 | 12·33 (10·85) |
| ESR | 24·3 (23·8) |
| CRP (in n = 36 patients)§ | 10·28 (13·51) |
Data on anti-cyclic citrullinated peptide (CCP) status was not available in two patients.
Data on shared epitope not available for eight patients.
Data on level of C-reactive protein (CRP) not available for four patients.
ESR = erythrocyte sedimentation rate; DAS28 = disease activity score 28; TNF = tumour necrosis factor; NSAID = non-steroidal anti-inflammatory drugs; DMARD = disease-modifying anti-rheumatic drugs; SE = shared epitope; s.d. = standard deviation.
Figure 4.

Interleukin (IL)-17A, F and AF in peripheral blood mononuclear cells of patients with rheumatoid arthritis (RA). Peripheral blood monuclear cells (PBMCs) from patients with RA or osteoarthritis (OA) were stimulated with phorbol myristate acetate (PMA)/ionomycin for 24 h and IL-17A, IL-17F and IL-17AF were analysed in culture supernatant by enzyme-linked immunosorbent assay (ELISA). *P < 0·05; n.s. = not significant.
Clinically, RA is a heterogeneous disease with variable levels of serological markers and a range of clinical severity. More than 50% of patients with RA are positive for anti-cyclic citrullinated peptides (CCP) antibody and patients positive for this antibody also have increased severity of arthritis, suggesting that perhaps the IL-17 responses of patients positive for anti-CCP antibody might be distinct from patients negative for this antibody. In our studies, however, the levels of IL-17A, F or AF following stimulation with PMA/ionomycin were similar among patients with RA who were anti-CCP-positive versus anti-CCP-negative (data not shown). While the aetiology of RA remains to be unravelled, it is thought that a combination of genetic risk alleles and environmental triggers plays a key role in this disease. To date, HLA-DRB1 alleles that contain the shared epitope (SE) have been shown to have the strongest association with RA [27,28]. The SE is contained in the HLA-DRB1 alleles *0401, *0404, *0405, *0408, *0413, *0101, *0102, *1402 and *1001 [27,29]. We wanted to evaluate whether patients carrying one or two alleles of the SE had higher levels of IL-17A, F or AF following stimulation with PMA/ionomycin in comparison to patients who did not have any copies of the SE alleles. Our studies showed no differences in the levels of IL-17A, F or AF among patients carrying one or two copies of the shared epitope (data not shown). Furthermore, we did not find any significant correlations between levels of IL-17A, F and AF with duration of disease, clinical severity as assessed by disease activity score 28 (DAS28) score, 44 tender and swollen joint count, erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) (data not shown).
In summary, IL-17A and IL-17F were higher in patients with RA in comparison to patients with OA, irrespective of serological status, presence or absence of shared epitope or disease activity.
IL-17A, F and AF responses in SFMCs
It is very well-recognized that systemic responses may not be the same as joint-specific responses. In order to elucidate joint-specific IL-17A, F and AF responses, we stimulated SFMCs from patients with RA or OA with PMA/ionomycin for 6 or 24 h. IL-17A, F or AF were not detectable in SFMCs cultured with PMA/ionomycin for 6 h (data not shown). Our data in Fig. 5 show that IL-17A and AF were detectable in supernatants of SFMCs in RA and OA cultured for 24 h. RA SFMC cultures produced more IL-17A than OA cultures, but equal amounts of IL-17AF. IL-17F was not detectable in cultures of RA or OA SFMCs (data not shown). These findings suggest that IL-17A and IL-17AF are predominantly present in target tissues in comparison to IL-17F. The very small numbers of IL-17A- or IL-17F-producing cells in our preliminary assays precluded us from utilizing intracellular flow cytometry to study IL-17A and IL-17F responses in a reliable manner in joint SFMCs.
Figure 5.

Interleukin (IL)-17A, F and AF responses in synovial fluid mononuclear cells (SFMCs). Mononuclear cells from synovial fluid of patients with rheumatoid arthritis (RA)or osteoarthritis (OA) were stimulated with phorbol myristate acetate (PMA)/ionomycin for 24 h and analysed for IL-17A, F and AF by enzyme-linked immunosorbent assay (ELISA). **P < 0·005; n.s. = not significant.
IL-17A, F and AF responses in purified CD3+ T cells from PBMCs or SFMCs
It has been reported earlier that IL-17A is expressed mainly by T cells, and hence we wanted to elucidate IL-17A, F and AF responses from purified CD3+ T cells from PBMCs or SFMCs. CD3+ T cells from PBMCs or SFMCs of patients with RA or OA were cultured with PMA/ionomycin for 24 h. Our data in Fig. 6 show that IL-17A and AF were detectable in supernatants of CD3+ T cells from PBMCs from patients with RA cultured with PMA/ionomycin for 24 h and were significantly higher than T cells from patients with OA. IL-17F was not detectable (data not shown). Additionally, IL-17A, F and AF were not detectable in supernatants of CD3+ T cells isolated from SFMCs from patients with RA (n = 5) or OA (n = 5) (data not shown). These findings suggest that peripheral blood T cells from patients with RA produce increased amounts of IL-17A and IL-17AF. Furthermore, SFMC T cells are probably not the major producer of IL-17A, F or AF.
Figure 6.

Interleukin (IL)-17A, F and AF responses in purified CD3+ T cells from peripheral blood monuclear cells (PBMCs) or synovial fluid mononuclear cells (SFMCs). Purified CD3(+) T cells from patients with rheumatoid arthritis (RA) (n = 9) or osteoarthritis (OA) (n = 6) were stimulated with phorbol myristate acetate (PMA)/ionomycin for 24 h and IL-17A, IL-17F and IL-17AF were analysed in culture supernatant by enzyme-linked immunosorbent assay (ELISA). **P < 0·005; ***P < 0·0005.
Discussion
IL-17A and IL-17F can form homodimers or a heterodimer IL-17AF [1]. In this report we show that IL-17A, F and AF were all induced during CIA. IL-17A is induced first, following immunization with collagen, and increases upon onset of arthritis, while IL-17F is induced upon arthritis onset. Moreover, IL-17A and IL-17F are produced by discrete sets of CD4 T cells, and there is differential regulation of IL-17A, IL-17F and IL-17AF, with IL-23 preferentially inducing IL-17A over IL-17F and IL-17AF. These findings are in keeping with recent reports supporting the differential regulation of IL-17A and IL-17F in primary cell cultures, lymphoma and lupus [30–34].
Even though IL-17A, F and AF were increased during CIA, administration of anti-IL-17A reduced the severity of arthritis, suggesting a dominant role of IL-17A over IL-17F. Furthermore, while heterodimer IL-17AF was detectable during CIA, neutralization with anti-IL-17A was sufficient to reduce arthritis, further supporting the dominant role of IL-17A. Our findings confirm previous studies reporting the amelioration of CIA upon administration of anti-IL-17A polyclonal antibody and reduced severity of CIA in IL-17A-deficient mice, and provide direct evidence that IL-17F plays a lesser role. Blocking of IL-17A may be sufficient to reduce the effects of the heterodimer IL-17AF [3,4].
IL-17A and IL-17F both bind to the same receptor complex comprised of IL-17RA and IL-17RC, and induce expression of similar inflammatory cytokines and chemokines [13,14,25,35] In keeping with this similarity, anti-IL-17A or anti-IL-17F had similar effects on anti-collagen antibody responses and neutralization of both (anti-IL-17A+anti-IL-17F) had a more profound effect than neutralization of either IL-17A or IL-17F. Interestingly, while neutralization of IL-17A was associated with reduced anti-collagen IgG2b responses and a reduced severity of arthritis, the reduced pathogenic anti-collagen IgG2b response seen with neutralization of IL-17F was not associated with a reduction in the severity of arthritis. This result suggests that other pathways, including IL-17A, may be critical for mediating arthritis, specifically in the absence of IL-17F.
The cytokines IL-6, IL-1β, TNFα, IFN-γ, G-CSF and GM-CSF have all been reported to be associated with inflammatory arthritis. While IL-17A and IL-17F may potentially induce similar sets of inflammatory genes, IL-17A is a more potent inducer of most inflammatory cytokines, including IL-6, IL-1β, G-CSF and GM-CSF, than is IL-17F [13,18–20,25]. In our study we found that mice which received anti-IL-17A or anti-IL-17A+anti-IL-17F had significantly reduced arthritis, along with significant and robust reduction of the levels of serum IL-6 and G-CSF. Administration of anti-IL-17F was not associated with reduced levels of IL-6 or G-CSF or reduced severity of arthritis. These findings are in keeping with previous studies in RA synovial fibroblasts reporting a differential effect of IL-17A and IL-17F [13,25]. It is possible that the leading role of anti-IL-17A over IL-17F may be mediated via its preferential effects on IL-6 and/or G-CSF in vivo.
IL-17A and IL-17F levels were higher in cultured PBMCs from RA in comparison to OA. There were no differences in the levels of IL-17AF produced by PBMCs from RA and OA. IL-17A, F and AF responses in RA were similar in subjects with RA who were anti-CCP+ versus subjects who were anti-CCP–, or among patients positive or negative for the SE allele. Furthermore, there was no correlation with duration or severity of RA, suggesting that the activation of the IL-17 pathway is not restricted to certain patients within the RA population but is elevated with inflammation in a large number of patients with RA. Stimulation of SFMCs of subjects with RA and OA showed increased levels of IL-17A in RA. There were no significant differences in the levels of IL-17AF. IL-17F was undetectable in these cultures. In keeping with the increased levels of IL-17A in PBMCs from patients with RA in comparison to patients with OA, IL-17A was also significantly higher in peripheral blood CD3+ T cells in patients with RA. Interestingly, while IL-17F levels were elevated in RA PBMCs versus OA PBMCs, peripheral blood CD3+ T cells did not produce IL-17F, suggesting that in RA, IL-17F is probably produced by non-T cells in the peripheral blood. Peripheral blood CD3+ T cells from patients with RA produced significantly increased levels of IL-17AF in comparison to peripheral blood CD3+ T cells of patients with OA. This is in contrast to similar levels of IL-17AF from PBMCs among patients with RA or OA. This is due probably to varying percentages of T cells in peripheral blood in the donor population. IL-17A was elevated significantly in SFMCs from patients with RA; however, neither IL-17A, F nor AF were detectable in CD3+ T cells from SFMCs of patients with RA or OA. This is in keeping with a previous study reporting that mast cells and not Th17 cells are the major source of IL-17A in the RA joint [36]. It is possible that the IL-17 subtypes (A, F and AF) are produced by discrete cells and regulated differentially in peripheral blood and joints of patients with RA and OA.
Other subtypes of IL-17, including B, C, D and E, may also mediate inflammation. It has been reported that IL-17B and IL-17C can induce joint inflammation in CIA [5]. Furthermore, IL-17C, E and F are induced by IL-15 in joint mononuclear cells from patients with RA, and IL-17C has been shown to be increased in psoriatic skin [11,12]. Further studies are needed to elucidate comprehensively the role of each subtype in inflammation.
Overall, our data in an animal preclinical model establish that while IL-17A, IL-17F and IL-17AF are all present in inflammatory arthritis, IL-17A plays a dominant role over IL-17F. Moreover, blockade of IL-17A was sufficient to neutralize the effect of IL-17AF. Furthermore, production of IL-17A, and not IL-17F or IL-17AF, was elevated in PBMCs and SFMCs in RA. Our studies support the concept that IL-17A is the major contributor to IL-17-related inflammatory effects in RA. Clinical trials with anti-IL-17A antibodies are ongoing (Secukinumab NCT 01640938 and Ixekizumab NCT 00966875), the results of which will shed additional light on the importance of IL-17A in mediating inflammation in RA.
Acknowledgments
This study was supported in part by grants from NIH AR054323, AR38477, University of Arizona Faculty Seed Grant and SCARI grant. We acknowledge the assistance of Kayla Iwahashi and Jessica Sonder for assistance with collection of peripheral blood from patients.
Author contributions
S. S. conceived and designed the study, and was involved in conduction of experiments, data analysis and wrote the manuscript; S. J. made substantial contribution to conduction of experiments and wrote the manuscript; M. B. conducted experiments and carried out data analysis; J. E. and D. A. F. collected and provided some patient samples; F. A. conducted experiments and performed data analyses; B. W., J. W. and B. H. J. generated the anti-IL-17F antibody; X. Z. and S. R. B. conducted some experiments. All authors read and approved the final manuscript.
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 2.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 3.Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 4.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi Y, Fujio K, Shoda H, et al. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- 6.Shahrara S, Huang Q, Mandelin AM, 2nd, Pope RM. TH-17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziolkowska M, Koc A, Luszczykiewicz G, et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 8.Metawi SA, Abbas D, Kamal MM, Ibrahim MK. Serum and synovial fluid levels of interleukin-17 in correlation with disease activity in patients with RA. Clin Rheumatol. 2011;30:1201–1207. doi: 10.1007/s10067-011-1737-y. [DOI] [PubMed] [Google Scholar]
- 9.Rosu A, Margaritescu C, Stepan A, Musetescu A, Ene M. IL-17 patterns in synovium, serum and synovial fluid from treatment-naive, early rheumatoid arthritis patients. Rom J Morphol Embryol. 2012;53:73–80. [PubMed] [Google Scholar]
- 10.Moran EM, Mullan R, McCormick J, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Res Ther. 2009;11:R113. doi: 10.1186/ar2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang SY, Kim HY. Expression of IL-17 homologs and their receptors in the synovial cells of rheumatoid arthritis patients. Mol Cells. 2005;19:180–184. [PubMed] [Google Scholar]
- 12.Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. 2009;160:319–324. doi: 10.1111/j.1365-2133.2008.08902.x. [DOI] [PubMed] [Google Scholar]
- 13.Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112–3120. doi: 10.4049/jimmunol.0801967. [DOI] [PubMed] [Google Scholar]
- 14.Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis. 2011;70:727–732. doi: 10.1136/ard.2010.143768. [DOI] [PubMed] [Google Scholar]
- 15.Sarkar S, Cooney LA, White P, et al. Regulation of pathogenic IL-17 responses in collagen-induced arthritis: roles of endogenous interferon-gamma and IL-4. Arthritis Res Ther. 2009;11:R158. doi: 10.1186/ar2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi L, Wheeler JC, Sweet RW, et al. De novo selection of high-affinity antibodies from synthetic fab libraries displayed on phage as pIX fusion proteins. J Mol Biol. 2010;397:385–396. doi: 10.1016/j.jmb.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 17.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar S, Fox DA. Targeting IL-17 and Th17 cells in rheumatoid arthritis. Rheum Dis Clin North Am. 2010;36:345–366. doi: 10.1016/j.rdc.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 20.Kawaguchi M, Adachi M, Oda N, Kokubu F, Huang SK. IL-17 cytokine family. J Allergy Clin Immunol. 2004;114:1265–1273. doi: 10.1016/j.jaci.2004.10.019. quiz 1274. [DOI] [PubMed] [Google Scholar]
- 21.Jovanovic DV, Di Battista JA, Martel-Pelletier J, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 22.Cai XY, Gommoll CP, Jr, Justice L, Narula SK, Fine JS. Regulation of granulocyte colony-stimulating factor gene expression by interleukin-17. Immunol Lett. 1998;62:51–58. doi: 10.1016/s0165-2478(98)00027-3. [DOI] [PubMed] [Google Scholar]
- 23.Schwarzenberger P, Huang W, Ye P, et al. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 24.Zwerina K, Koenders M, Hueber A, et al. Anti IL-17A therapy inhibits bone loss in TNF-alpha-mediated murine arthritis by modulation of the T-cell balance. Eur J Immunol. 2012;42:413–423. doi: 10.1002/eji.201141871. [DOI] [PubMed] [Google Scholar]
- 25.Hot A, Zrioual S, Toh ML, Lenief V, Miossec P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis. 2011;70:341–348. doi: 10.1136/ard.2010.132233. [DOI] [PubMed] [Google Scholar]
- 26.DeForge LE, Loyet KM, Delarosa D, et al. Evaluation of heterophilic antibody blocking agents in reducing false positive interference in immunoassays for IL-17AA, IL-17FF, and IL-17AF. J Immunol Methods. 2010;362:70–81. doi: 10.1016/j.jim.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 28.Bax M, van Heemst J, Huizinga TW, Toes RE. Genetics of rheumatoid arthritis: what have we learned? Immunogenetics. 2011;63:459–466. doi: 10.1007/s00251-011-0528-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weyand CM, Hicok KC, Conn DL, Goronzy JJ. The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis. Ann Intern Med. 1992;117:801–806. doi: 10.7326/0003-4819-117-10-801. [DOI] [PubMed] [Google Scholar]
- 30.Adamik J, Henkel M, Ray A, Auron PE, Duerr R, Barrie A. The IL17A and IL17F loci have divergent histone modifications and are differentially regulated by prostaglandin E2 in Th17 cells. Cytokine. 2013;64:404–412. doi: 10.1016/j.cyto.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melton AC, Melrose J, Alajoki L, et al. Regulation of IL-17A production is distinct from IL-17F in a primary human cell co-culture model of T cell-mediated B cell activation. PLOS ONE. 2013;8:e58966. doi: 10.1371/journal.pone.0058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krejsgaard T, Litvinov IV, Wang Y, et al. Elucidating the role of interleukin-17F in cutaneous T-cell lymphoma. Blood. 2013;122:943–950. doi: 10.1182/blood-2013-01-480889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauen T, Hedrich CM, Juang YT, Tenbrock K, Tsokos GC. cAMP-responsive element modulator (CREM)alpha protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem. 2011;286:43437–43446. doi: 10.1074/jbc.M111.299313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedrich CM, Rauen T, Kis-Toth K, Kyttaris VC, Tsokos GC. cAMP-responsive element modulator alpha (CREMalpha) suppresses IL-17F protein expression in T lymphocytes from patients with systemic lupus erythematosus (SLE) J Biol Chem. 2012;287:4715–4725. doi: 10.1074/jbc.M111.323261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toy D, Kugler D, Wolfson M, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 36.Hueber AJ, Asquith DL, Miller AM, et al. Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol. 2010;184:3336–3340. doi: 10.4049/jimmunol.0903566. [DOI] [PubMed] [Google Scholar]