

Abstract
Crohn's disease (CD) has been correlated with altered macrophage response to microorganisms. Considering the efficacy of infliximab treatment on CD remission, we investigated infliximab effects on circulating monocyte subsets and on macrophage cytokine response to bacteria. Human peripheral blood monocyte-derived macrophages were obtained from CD patients, treated or not with infliximab. Macrophages were infected with Escherichia coli, Enterococcus faecalis, Mycobacterium avium subsp. paratuberculosis (MAP) or M. avium subsp avium, and cytokine levels [tumour necrosis factor (TNF) and interleukin (IL)-10] were evaluated at different time-points. To evaluate infliximab-dependent effects on monocyte subsets, we studied CD14 and CD16 expression by peripheral blood monocytes before and after different infliximab administrations. We also investigated TNF secretion by macrophages obtained from CD16+ and CD16− monocytes and the frequency of TNF+ cells among CD16+ and CD16− monocyte-derived macrophages from CD patients. Infliximab treatment resulted in elevated TNF and IL-10 macrophage response to bacteria. An infliximab-dependent increase in the frequency of circulating CD16+ monocytes (particularly the CD14++CD16+ subset) was also observed (before infliximab: 4·65 ± 0·58%; after three administrations: 10·68 ± 2·23%). In response to MAP infection, macrophages obtained from CD16+ monocytes were higher TNF producers and CD16+ macrophages from infliximab-treated CD patients showed increased frequency of TNF+ cells. In conclusion, infliximab treatment increased the TNF production of CD macrophages in response to bacteria, which seemed to depend upon enrichment of CD16+ circulating monocytes, particularly of the CD14++CD16+ subset. Infliximab treatment of CD patients also resulted in increased macrophage IL-10 production in response to bacteria, suggesting an infliximab-induced shift to M2 macrophages.
Keywords: cytokine secretion, inflammatory bowel diseases, infliximab therapy, macrophages, monocyte subsets
Introduction
Crohn's disease (CD) is an immunologically mediated disorder resulting in chronic inflammation of the intestine. CD and ulcerative colitis (UC) are the two major forms of inflammatory bowel disease (IBD). CD and UC are clinically, immunologically and morphologically distinct from each other [1,2].
Tumour necrosis factor (TNF)-α is one of the most potent proinflammatory mediators in the pathophysiology of IBD, and anti-TNF treatment has been particularly effective in inducing and maintaining remission [3]. Infliximab is a chimeric monoclonal immunoglobulin (Ig)G1 antibody (75% human, 25% mouse protein), and is highly effective in neutralizing TNF biological activity [4,5]. The efficacy of infliximab is well established in CD patients with severe, steroid-refractory or fistulizing disease [6,7], as well as in patients with moderate-to-severe UC [8]. Although infliximab has been proved to be highly effective in the clinical management of IBD, its mechanism of action is not completely understood. Evidence points to (i) induction of apoptosis of immune cells by reverse signalling through transmembrane TNF [9]; (ii) neutralization of soluble and transmembrane TNF [9]; (iii) alteration of cytokine secretion, as detected in serum and in cultures of lamina propria biopsies, with decreased interferon (IFN)-γ production [10,11]; and, more recently (iv) induction of immunoregulatory macrophages [12].
Macrophages play an important role in the intestinal innate immune defences contributing to the integrity of the intestinal mucosa [13], and have been implicated in CD pathogenesis. Macrophages act both by elimination of microbial agents and by induction of adaptive immunity through antigen presentation and cytokine production [14]. Autophagy-related genes (ATG16L1, IRGM) and genes encoding pattern-recognition receptors (e.g. NOD2), were identified as CD susceptibility loci/genes by genome-wide association studies [15–17], suggesting an important contribution of innate immunity in CD immunopathology. Although it has been known that normal intestinal macrophages show low proinflammatory activity, it has been shown that in intestinal diseases, particularly in CD, their profile differs considerably [13]. Indeed, these cells showed increased expression of co-stimulatory receptors, Toll-like receptors and inflammatory cytokines [18,19].
Peripheral blood monocytes have been divided into three distinct subsets according to CD14 and CD16 marker expression [18,20,21]: one subset with high expression of CD14 and no expression of CD16 (CD14++CD16−, also designated classical subset), one with high expression of CD14 and variable expression of CD16 (CD14++CD16+, intermediate subset) and another with lower expression of CD14 and high expression of CD16 (CD14+CD16++, non-classical subset). CD16+ monocyte subsets, particularly the intermediate subset, have been associated with inflammatory functions [20] and are known to be the major producers of inflammatory cytokines such as TNF and interleukin (IL)-1β [22,23]. It was reported that, in CD patients with active disease, CD16+ monocytes were increased in peripheral blood and intestinal mucosa [24,25], possibly contributing to inflammation. So far, effects of infliximab treatment on the distribution of monocyte subsets in CD patients are largely unknown.
In a previous work, we reported deficient TNF and IL-10 secretion by macrophages isolated from CD patients in response to in-vitro bacterial infection, namely commensal strains of Escherichia coli (EC) and Enterococcus faecalis (EF), as well as Mycobacterium avium subsp. paratuberculosis (MAP) and M. avium subsp. avium (MA) human isolates [26]. Herein we present evidence that infliximab treatment is a modifying factor of the macrophage cytokine response to bacteria and that changes in TNF response are related to infliximab-induced CD16+ monocytes, particularly of the intermediate subset.
Materials and methods
Patients and samples
In these studies, 105 individuals were included: 69 CD patients under infliximab therapy (CD-IFX), 22 CD patients not treated with infliximab (CD) and 14 healthy controls (HC). Among CD-IFX patients, 47 were included in cytokine bacterial-stimulation index studies: these patients were treated with infliximab for a minimum of 14 weeks, corresponding to at least three infliximab administrations; that is, after induction treatment. The remaining 22 CD-IFX patients were included in monocyte and macrophage subpopulation studies: these patients were tested before and at 2, 6 and 14 weeks after the first administration of infliximab; that is, in three slots of the infliximab induction schedule. The age and gender of the subjects enrolled in this study are shown in Table 1 and the therapeutic regimen is included in Table 2.
Table 1.
Age and gender of Crohn's disease (CD patients enrolled into the studies
| HC | CD | CD-IFX | P-value† | |
|---|---|---|---|---|
| n | 18 | 22 | 69 | – |
| Male/female | 6/12 | 11/11 | 37/32 | n.s. |
| Mean age | 31 | 38 | 38 | n.s. |
| Age interval | 19–66 | 21–73 | 18–64 | – |
P-value for gender was calculated using χ2 test; P-value for age was calculated using Student's t-test. IFX = infliximab; n.s. = not significant; IFX = infliximab; HC = healthy controls.
Table 2.
Therapeutic regimen of Crohn's disease (CD) and CD-infliximab (IFX) patients enrolled into the studies
| CD | CD-IFX | |
|---|---|---|
| 5-ASA | 5 | 0 |
| AZA | 0 | 0 |
| CORT | 0 | 0 |
| 5-ASA + AZA | 17 | 0 |
| IFX | 0 | 38 |
| IFX + 5-ASA | 0 | 4 |
| IFX + AZA | 0 | 21 |
| IFX + AZA + CORT | 0 | 5 |
ASA = aminosalicylic acid; CORT = corticotrophin; AZA = azathioprine.
Informed consent was obtained in accordance with the institutional review board regulations at IBMC, University Fernando Pessoa and Hospital de São João.
Patients were recruited from the Gastroenterology Department at Hospital de São João. Diagnosis was based on standard clinical, endoscopic, histological and radiographic criteria [27].
Whole blood samples were obtained from every individual, and peripheral blood mononuclear cell isolation was performed as described below.
Cell culture
The blood collected from each individual (13·5 ml) was diluted 1:2 in phosphate-buffered saline (PBS) and placed on Histopaque®-1077 (Sigma, St Louis, MO, USA) in 15-ml sterile tubes. After centrifugation, the mononuclear cells present in the monolayer were collected, washed three times in Hanks's balanced salt solution (HBSS; Sigma) and resuspended in RPMI-1640 with GlutaMAX™ (Invitrogen, Carlsbad, CA, USA), supplemented with antibiotic/anti-mycotic solution (Invitrogen) [RPMI without fetal bovine serum (FBS)]. Cells were cultured for 2 h in 10-cm-diameter tissue culture plates and non-adherent cells were washed off to achieve monocyte enrichment. At this point, medium was replaced and a 10% FBS supplement (complete RPMI) was added. Cultures proceeded until day 5, when adherent cells (the monocyte-enriched population) were detached using trypsin–ethylenediamine tetraacetic acid (EDTA) solution. The cell density was adjusted to 2 × 105/ml; cells were plated in 24-well plates (1 ml/well), and cultured for an additional period of 2 days to complete monocyte differentiation into macrophages. At day 7, the cell monolayers obtained showed typical macrophage morphology. Medium was replaced by RPMI-1640 with GlutaMAX™ and 10% FBS (infection RPMI) and cultures were infected.
In-vitro infection of monocyte-derived macrophages
Macrophage cultures were either left uninfected or were infected with M. avium subsp. paratuberculosis (MAP) (ATCC 43015, an isolate from a Crohn's patient), M. avium subsp. avium (MA) (strain 101, an isolate from an AIDS patient), E. coli (EC) (ATCC 25922) or E. faecalis (EC) (ATCC 29212) at a multiplicity of infection (MOI) of 10 bacteria : 1 cell. Cultures were incubated for 3 h to allow phagocytosis of the bacteria. Monolayers infected with EC or EF were assessed at 3 h after infection only (T3h). Cells infected with MAP or MA were assessed at T3h, 3 days (T3d) and 7 days after infection (T7d). After T3h, culture supernatants were collected from some wells and used for determination of TNF, IL-12, IL-23 and IL-10 by ELISA. Cells in other wells were allowed to incubate for T3d or T7d. At those time-points, culture supernatants were also collected for cytokine determination.
TNF and IL-10 detection
After collection, culture supernatants were centrifuged at 10 000 g for 5 min to sediment remaining bacteria. The pellet was rejected and the supernatants were aliquoted and stored frozen at −70°C until assay performance. TNF and IL-10 concentration were measured, respectively, with human TNF and IL-10 ELISA Ready-Set-Go kits purchased from eBioscience (San Diego, CA, USA), according to instructions from the manufacturer. Results in pg/ml were normalized to 2 × 105 cells and expressed as pg/2 × 105 cells.
Characterization of peripheral blood monocytes and evaluation of macrophage TNF production by flow cytometry
Peripheral blood mononuclear cells were isolated as described above. After isolation, cells were resuspended in 2 ml of RPMI without FBS. One ml of the cell suspension was used for monocyte subset studies and the remaining 1 ml was used for macrophage subpopulation studies by diluting with 9 ml of RPMI without FBS and plating in a 10-cm-diameter tissue culture plate for further differentiation into macrophages, as described above.
Mononuclear cells used for monocyte subset studies were centrifuged, resuspended in 200 μl PBS and divided into four microtubes (50 μl/tube). Blocking of Fc receptors was accomplished by the addition of 15 μl of human serum to each microtube and incubation for 15 min at 4°C. Cells were centrifuged again and resuspended in 50 μl PBS. One microtube was left unstained and the others were stained with either fluorescein isothiocyanate (FITC)-conjugated anti human-CD14 (anti-CD14; ImmunoTools GmbH, Friesoythe, Germany), allophycocyanin (APC)-conjugated anti-human-CD16 (anti-CD16; ImmunoTools) or both antibodies. Cells were incubated for 30 min at 4°C, centrifuged, resuspended in 500 μl of 1% paraformaldehyde in PBS and stored at 4°C for a maximum of 1 week for flow cytometry analysis [fluorescence activated cell sorter (FACS)Calibur; BD Biosciences, San Jose, CA, USA).
For the evaluation of TNF production by macrophages by flow cytometry, macrophage cultures were either left uninfected or were infected with MAP at a MOI of 10:1, as described above. MAP was used as stimulus, because of its previously reported association with CD [28,29]. At T3h extracellular bacteria were washed off, cells were detached using trypsin–EDTA solution and stained with anti-CD14 and anti-CD16, as described for monocytes. Cells were then permeabilized by incubation with 0·1% saponin and 0·1% bovine serum albumin (BSA) in PBS (permeabilization buffer) for 5 min, and stained with phycoerythrin (PE)-conjugated anti-human TNF antibody (Biolegend, San Diego, CA, USA) in permeabilization buffer for 1 h at 4°C. After staining, cells were centrifuged and resuspended in 1% paraformaldehyde for subsequent flow cytometry analysis.
Isolation and culture of CD16+ and CD16− monocytes
Peripheral blood mononuclear cells were isolated from 50 ml of blood collected from healthy donors, as described above. After isolation, cells were treated with ammonium–chloride–potassium (ACK) lysing solution to eliminate erythrocytes. The cell suspension was centrifuged, resuspended in magnetic cell sorting (MACS) buffer (0·5% BSA and 2 mM EDTA in PBS) and monocytes were isolated using CD14 human MicroBeads (Miltenyi Biotec, Germany). In brief, CD14 microbeads were added to the cell suspension as recommended by the manufacturer, cells were incubated 20 min on ice to allow microbead binding, washed, resuspended in MACS buffer and added to a previously prepared LS column on a MidiMACS magnetic separator (Miltenyi Biotec). After cell suspension addition, the column was washed with MACS buffer to discard unbound cells, then removed from the separator and washed again with MACS buffer for collection of CD14+ cells (monocytes). Monocytes were adjusted to a density of 10 × 106 cells/70 μl PBS; a small aliquot (7 μl) was transferred to another microtube and diluted in basic sorting buffer (5 mM EDTA, 25 mM HEPES and 1–2% FBS in Ca2+/Mg2+-free PBS) (unstained cells). The remaining cells were incubated with 10 μl human serum for Fc receptor blocking, washed and stained with anti-CD16 antibody as described above. After staining, the monocytes were resuspended in basic sorting buffer at a density of 10 × 106 cells/ml; CD16+ and CD16− monocytes were separated by cell sorting (FACSAria I; BD Biosciences, Heidelberg, Germany). CD16+ and CD16− monocyte suspensions were then adjusted to a density of 2 × 105 cells/ml and cultured for 7 days in a 24-well plate (1 ml/well). At culture day 7, cells were left uninfected or were infected with MAP at a MOI of 10:1. At T3h the supernatants were collected for detection of TNF by ELISA, as described above.
Statistical analysis
Bacteria-induced cytokine secretion by macrophages (‘bacterial-stimulation index’) was expressed as the difference between cytokine levels of infected versus uninfected macrophage cultures obtained from the same individual.
The unpaired Student's t-test was used to compare differences obtained between two groups of results in each plot. In Table 1, gender of subjects in different groups was compared by χ2 test and mean age was compared using the unpaired Student's t-test. P-values less than 0·050 were considered significant. Results are presented as arithmetic mean ± standard error of the mean (s.e.m.).
Results
Macrophages from CD-IFX patients are higher TNF and IL-10 responders to bacterial infection
Macrophages obtained from CD-IFX patients showed an enhanced TNF bacterial-stimulation index in response to infection by EC (374·6 ± 67·1), MAP (169·6 ± 60·8) and MA (79·9 ± 26·5) at T3h (Fig. 1), compared to macrophages from CD patients not under infliximab treatment (respectively, 196·9 ± 60·7; 12·5 ± 12·6; 19·4 ± 10·8; all results expressed in pg/2 × 105 cells). This enhancement was maintained at T3d and T7d for MAP and MA infection.
Figure 1.
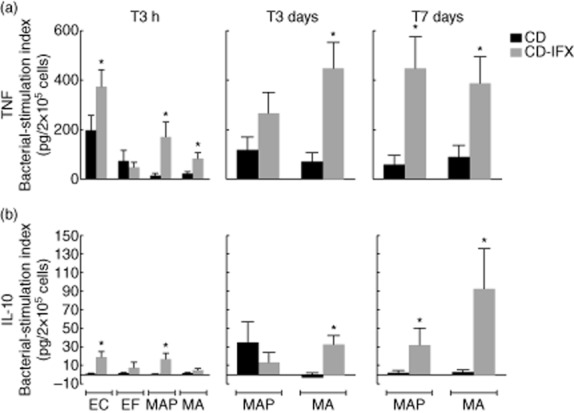
Bacterial-stimulation index (bacteria-induced cytokine secretion) by macrophages from Crohn's disease (CD) and CD- infliximab (IFX) patients after infection with Escherichia coli (EC), E. faecalis (EC), Mycobacterium avium subsp. paratuberculosis (MAP) or M. avium subsp. avium (MA) at 3 h (T3h), 3 days (T3d) and 7 days (T7d) after infection. Bacterial-stimulation index was calculated as the difference between cytokine levels measured in infected versus uninfected macrophage cultures obtained from the same subject. Significant differences between CD-IFX and CD macrophages were calculated using Student's t-test (*P < 0·005).
Macrophages from CD-IFX patients showed enhanced IL-10 secretion (bacterial-stimulation index, expressed in pg/2 × 105 cells) in response to infection by EC (18·7 ± 6·08 versus 0·451 ± 0·863 for macrophages from untreated CD patients) and MAP (16·2 ± 6·91 versus −0·192 ± 0·635) at T3h (Fig. 1). At T7d, increased IL-10 production was also observed in response to MAP (31·7 ± 17·9) and MA (92·5 ± 43·5), compared to macrophages from CD patients not treated with infliximab (respectively, 0·874 ± 3·38 and 2·40 ± 2·67).
In addition to infliximab, azathioprine (a known immunosuppressor) was also given to a group of CD-IFX patients. To test possible effects of azathioprine treatment on our results, we compared azathioprine-treated and -untreated CD-IFX macrophage cultures in their ability to respond to bacterial infection (at T3h) through TNF and IL-10 production (Table 3). We found that azathioprine treatment resulted in a statistically significant increase in IL-10 secretion in response to MAP (azathioprine-treated: 43·68 ± 21·07 pg/2 × 105 cells; azathioprine-untreated: 7·26 ± 4·64 pg/2 × 105 cells). TNF production was not affected by treatment with azathioprine.
Table 3.
Effects of azathioprine treatment of Crohn's disease-infliximab (CD-IFX) patients on the cytokine bacterial-stimulation index at T3h
| Bacterial-stimulation index (pg/2 × 105 cells) | ||||
|---|---|---|---|---|
| no AZA | AZA | P-value† | ||
| TNF-α | EC | 362·7 ± 17·4 | 175·2 ± 22·6 | n.s. |
| MAP | 98·9 ± 9·2 | 87·0 ± 32·0 | n.s. | |
| MA | 59·5 ± 6·4 | 106·2 ± 35·4 | n.s. | |
| IL-10 | EC | 11·04 ± 5·02 | 38·73 ± 17·96 | n.s. |
| MAP | 7·26 ± 4·64 | 43·68 ± 21·07 | 0·022 | |
| MA | 3·49 ± 3·15 | 7·16 ± 3·64 | n.s. | |
P-value was calculated using Student's t-test. EC = Escherichia coli; MAP = Mycobacterium avium subsp paratuberculosis; MA = M. avium subsp avium. n.s. = not significant; TNF = tumour necrosis factor; IL = interleukin; AZA = azathioprine.
Infliximab treatment increased the frequency of CD16+ peripheral blood monocytes
To investigate whether the increment in the TNF response to bacteria was associated with an infliximab-induced change in the phenotype of circulating monocytes, we studied the expression of CD14 and CD16 by peripheral blood monocytes before and after one, two or three infliximab administrations. The monocyte gate was defined according to forward- (FSC) and side-scatter (SSC) parameters (Supporting information, Fig. S1). As described in the literature [20,21], we considered three monocyte subpopulations, one with high expression of CD14 and low or no expression of CD16 (CD14++CD16−, also designated classical subset), one with high expression of CD14 and variable expression of CD16 (CD14++CD16+, intermediate subset) and another with lower expression of CD14 and high expression of CD16 (CD14+CD16++, non-classical subset) (Fig. 2). CD16+ monocytes in general were more frequent in CD patients (before infliximab treatment: 6·10% ± 0·71%) than in HC (4·01% ± 0·98%) (Fig. 3a, right panel). Infliximab treatment further increased the frequency of CD16+ monocytes (after three administrations: 11·22% ± 1·47%), with a resulting decrease in the CD16− subpopulation (Fig. 3a). The infliximab-dependent increment in the frequency of CD16+ monocytes was associated mainly with the CD14++CD16+ monocyte subset (before infliximab treatment: 4·65% ± 0·58%; after three administrations: 10·68% ± 2·23%) (Fig. 3b and Supporting information, Fig. S2). A small increase in the frequency of CD14+CD16++ monocytes was also observed, although only significant compared to HC.
Figure 2.
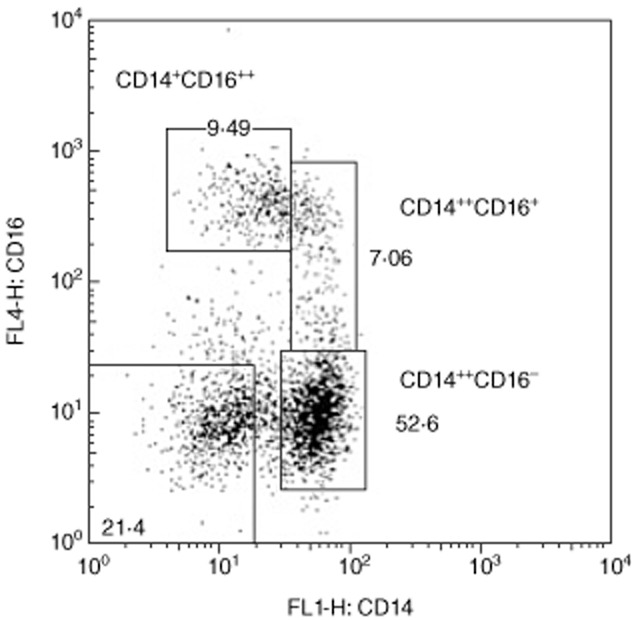
Dot-plot of peripheral blood monocytes from a representative CD patient, according to CD14 and CD16 staining. Monocytes were divided into three subsets.
Figure 3.
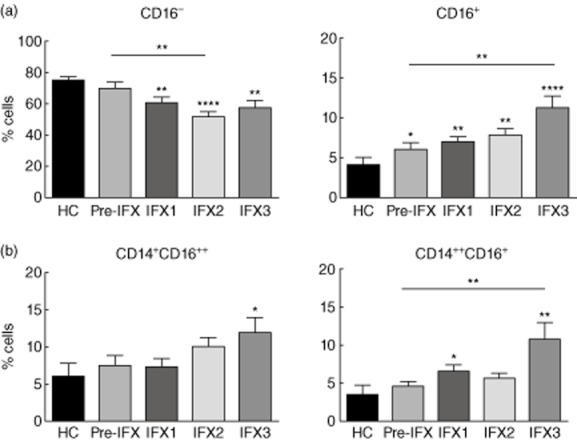
(a) Percentage of CD16− (CD14++CD16−) (left panel) and CD16+ (CD14+CD16++ and CD14++CD16+) (right panel) monocytes from healthy controls and CD patients before and after treatment with different doses of infliximab (IFX). (b) Distribution of the CD16+ monocytes by the two subpopulations. Asterisks not associated with a line indicate a significant difference with control (*P < 0·050; **P < 0·010; ***P < 0·001; ****P < 0·0001).
Macrophages obtained from CD16+ monocytes showed increased TNF secretion in response to MAP
As infliximab treatment resulted in increased frequency of CD16+ blood monocytes, we next investigated whether macrophage cultures obtained from CD16− or CD16+ monocytes differed in the TNF response to MAP. CD16+ and CD16− monocytes isolated from the blood of healthy donors by cell sorting were cultured for 7 days to allow differentiation into macrophages. At culture day 7, the resulting macrophages were infected with MAP. MAP challenge (T3h) induced high TNF secretion by macrophages obtained from CD16+ monocytes only (MAP-infected: 34·23 ± 0·16; uninfected: 10·96 ± 0·28, expressed by pg/2 × 105 cells) (Fig. 4, right panel). On the contrary, MAP-infected macrophages obtained from CD16− monocytes produced lower amounts of TNF compared to uninfected macrophages (MAP-infected: 13·28 ± 1·30; uninfected: 20·92 ± 2·85, expressed by pg/2 × 105 cells) (Fig. 4, left panel).
Figure 4.
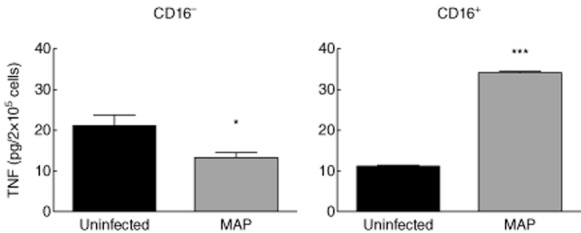
Tumour necrosis factor (TNF) production by macrophages obtained from CD14+CD16− and CD14+CD16+ peripheral blood monocytes from healthy donors, in response to challenge with Mycobacterium avium subsp. paratuberculosis (MAP) for 3 h, at a multiplicity of infection (MOI0 of 10:1. Monocyte differentiation into macrophages was accomplished by culturing the isolated monocyte populations for 7 days. *P < 0·050; ***P < 0·001.
CD16+ macrophages from CD patients showed higher frequency of TNF-producing cells
We next investigated whether CD16+ macrophages contributed to the augmented bacterial-stimulation index observed after infliximab treatment of CD patients. In fact, after three infliximab administrations, MAP infection induced an increased frequency of TNF+ cells among CD16+ macrophages only (MAP-induced increment in the frequency of TNF+ cells: before infliximab treatment, 1·05% ± 0·12%; after three infliximab administrations, 2·02% ± 0·47%;) (Fig. 5).
Figure 5.
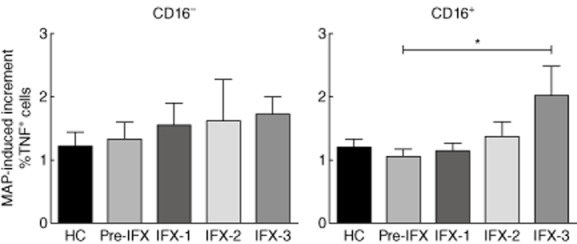
Mycobacterium avium subsp. paratuberculosis (MAP)-induced increment in the frequency of tumour necrosis factor (TNF)+ cells, as calculated by the ratio of the percentage of TNF+ cells in MAP-infected versus uninfected CD16− (left) or CD16+ (right) macrophages. *P < 0·050.
Discussion
In a previous work from our group we investigated IBD macrophage cytokine secretion in response to infection in vitro with EC, EF, MAP and MA [26]. In this study we examined the effects of infliximab treatment on bacterial-induced TNF and IL-10 secretion by macrophages from CD patients, and investigated possible infliximab-induced changes in peripheral blood monocyte subsets that could account for the alterations observed.
Macrophages from CD patients showed defective TNF secretion in response to bacterial infection [26]. Our results confirmed those by Smith and collaborators [30], showing that macrophages from CD patients expressed deficient TNF secretion in response to heat-killed EC. Infliximab treatment of CD patients resulted in increased TNF production by macrophages in response to bacterial infection, suggesting that infliximab therapy corrects the macrophage deficiency observed in CD patients. Infliximab treatment also induced an increase in CD16+ monocytes, mainly of the CD14++CD16+ phenotype, also designated intermediate monocytes. The intermediate monocytes were associated previously with a proinflammatory role, being high producers of IL-1β and TNF in response to lipopolysaccharide (LPS), but low producers of reactive oxygen species (ROS), although showing high phagocytic ability [23]. It was suggested that intermediate monocytes are probably involved in antigen presentation and T cell activation [20]. An increase in the CD14++CD16+ monocyte subset in patients with active CD compared to HC was reported previously and associated with the elevated inflammatory state of the CD patients [24]. Surprisingly, infliximab treatment resulted in a further increase of the intermediate monocyte subset, which might possibly be explained by a compensatory response of the host to TNF neutralization, as CD14++CD16+ monocytes are high TNF producers [23]. Alternatively, induction of intermediate monocytes may result from a direct effect of infliximab on monocytes. This could be achieved by signalling through Fc receptors, shown previously to be involved in induction of regulatory macrophages [12], or from reverse signalling through transmembrane TNF. Reverse signalling (cell signalling cascade resulting from binding of infliximab to transmembrane TNF) was found to induce LPS resistance in peripheral blood mononuclear cells, altering cytokine secretion and apoptotic activity [9].
The infliximab-dependent increment in CD16+ monocytes observed by us suggested the possibility that the augmented TNF bacterial-stimulation index was due to TNF production by macrophages differentiated from CD16+ monocytes. Indeed, the higher TNF response to MAP observed with HC macrophages obtained from CD14+CD16+ monocytes, compared to CD14+CD16− cells, suggested that the increment in TNF bacterial-stimulation index due to infliximab treatment was associated with an enrichment in circulating CD16+ monocytes. In addition, we found that MAP infection resulted in enhanced frequency of TNF+ cells among CD16+ macrophages from CD patients, reinforcing the role of these cells in the infliximab-dependent increment in TNF production in response to MAP.
Anti-TNF antibodies were shown to induce the differentiation of a regulatory subset of macrophages in vitro, in an Fc-dependent manner, after incubation of cells in mixed lymphocyte reactions (MLR) with infliximab, adalimumab or certolizumab-IgG [12]. These macrophages (Mφind) expressed higher CD206 (a marker of M2 macrophages) than macrophages obtained by IFN-γ treatment of cells in an MLR (Mφ1). Mφind produced elevated IL-10, compared to dendritic cells or Mφ1 macrophages, but comparable TNF and IL-1β [12], after LPS stimulation. Regulatory macrophages, expressing the markers CD206 and CD68, were also observed in infliximab-treated IBD patients with signs of mucosal healing [31]. These regulatory macrophages (M2 macrophages) suppressed T cell proliferation and promoted wound healing [31]. We observed that, in spite of increased TNF secretion, macrophages from CD-IFX patients also showed increased IL-10 production in response to bacteria, thus confirming the results obtained by Vos and collaborators [12]. Azathioprine administered concomitantly with infliximab further increased IL-10 secretion in response to MAP, which is in agreement with azathioprine's immunosuppressive role. The results we obtained concerning IL-10 are in favour of an infliximab-dependent induction of regulatory macrophages, as demonstrated by Vos and collaborators [12,31]. As CD-IFX macrophages produced both high TNF and IL-10 in response to bacteria, it is possible that they belong to M2b macrophages, a subgroup of the M2 phenotype [32,33] induced by immune complexes and known to secrete both TNF and IL-10. However, M1 and M2 subsets really describe extreme polarization states, and macrophages can also express intermediate functional features.
Reverse signalling through membrane TNF superfamily members seems to involve activation of extracellular-regulated kinase (ERK) [a mitogen-activated protein kinase (MAPK)], nuclear factor (NF)-κB and also phosphatidylinositol-3-kinase (PI3K) [34,35], although it is still not clear how these signalling molecules interact with each other in generating cell responses. Nevertheless, NF-κB activation has been connected to proinflammatory cytokine production (e.g. TNF) [36,37], while PI3K activation [through phosphorylation of protein kinase B (Akt), a downstream kinase] is associated with anti-inflammatory responses (e.g. with enhancement of IL-10 synthesis) [38]. As such, it is possible that reverse signalling through infliximab binding to transmembrane TNF generates monocytes that differentiate in vitro into macrophages with both pro- and anti-inflammatory markers.
In conclusion, infliximab treatment of CD patients modified the macrophage cytokine response to bacterial infection. Infliximab corrected the defective TNF production of CD macrophages, which seemed to depend upon the enrichment of CD16+ circulating monocytes (particularly of the CD14++CD16+ subset), as macrophages differentiated from CD16+ monocytes showed a higher TNF response to MAP. Infliximab treatment of CD patients also resulted in increased macrophage IL-10 production in response to bacteria, suggesting an infliximab-induced shift to M2 macrophages. Future studies will be important to further characterize the macrophage subsets obtained from CD16+ and CD16− monocytes of infliximab-treated CD patients, in terms of M1 and M2 markers.
Acknowledgments
This work was funded by Project PIC/IC/82802/2007 co-funded by the COMPETE Program subsidized by FEDER and by FCT (Fundação para a Ciência e Tecnologia), and Project ‘NORTE-07-0124-FEDER-000002-Host-Pathogen Interactions’ co-funded by Programa Operacional Regional do Norte (ON.2 – O Novo Norte), under the Quadro de Referência Estratégico Nacional (QREN), through the Fundo Europeu de Desenvolvimento Regional (FEDER) and by FCT. The authors wish to thank all patients and controls for their participation in this study.
Disclosure
The authors declare that they have no competing interests.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Representative forward- (FSC) and side-scatter (SSC) dot-plot of peripheral blood mononuclear cells, showing the monocyte gate.
{kind=link}
Fig. S2. Dot-plots of CD14 and CD16 expression by monocytes of two representative Crohn's disease (CD) patients, showing enrichment of the CD14++CD16+ population after infliximab (IFX) treatment. IFX1: after one administration; IFX2: after two administrations; IFX3: after three administrations.
{kind=link}
References
- 1.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 3.Magro F, Portela F. Management of inflammatory bowel disease with infliximab and other anti-tumor necrosis factor alpha therapies. BioDrugs. 2010;24(Suppl. 1):3–14. doi: 10.2165/11586290-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Caviglia R, Ribolsi M, Rizzi M, Emerenziani S, Annunziata ML, Cicala M. Maintenance of remission with infliximab in inflammatory bowel disease: efficacy and safety long-term follow-up. World J Gastroenterol. 2007;13:5238–5244. doi: 10.3748/wjg.v13.i39.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osterman MT, Lichtenstein GR. Current and future anti-TNF therapy for inflammatory bowel disease. Curr Treat Options Gastroenterol. 2007;10:195–207. doi: 10.1007/s11938-007-0013-3. [DOI] [PubMed] [Google Scholar]
- 6.Armuzzi A, De Pascalis B, Fedeli P, De Vincentis F, Gasbarrini A. Infliximab in Crohn's disease: early and long-term treatment. Dig Liver Dis. 2008;40(Suppl. 2):S271–279. doi: 10.1016/S1590-8658(08)60537-X. [DOI] [PubMed] [Google Scholar]
- 7.D'Haens G, Baert F, van Assche G, et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn's disease: an open randomised trial. Lancet. 2008;371:660–667. doi: 10.1016/S0140-6736(08)60304-9. [DOI] [PubMed] [Google Scholar]
- 8.Allen PB, Lindsay H, Tham TCK. How do patients with inflammatory bowel disease want their biological therapy administered? BMC Gastroenterol. 2010;10:1. doi: 10.1186/1471-230X-10-1. doi: 10.1186/1471-230X-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eissner G, Kirchner S, Lindner H, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol. 2000;164:6193–6198. doi: 10.4049/jimmunol.164.12.6193. [DOI] [PubMed] [Google Scholar]
- 10.Agnholt J, Dahlerup JF, Kaltoft K. The effect of etanercept and infliximab on the production of tumour necrosis factor alpha, interferon-gamma and GM-CSF in in vivo activated intestinal T lymphocyte cultures. Cytokine. 2003;23:76–85. doi: 10.1016/s1043-4666(03)00201-1. [DOI] [PubMed] [Google Scholar]
- 11.Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 12.Vos ACW, Wildenberg ME, Duijvestein M, Verhaar AP, van den Brink GR, Hommes DW. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. 2011;140:221–230. doi: 10.1053/j.gastro.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Sheikh SZ, Plevy SE. The role of the macrophage in sentinel responses in intestinal immunity. Curr Opin Gastroenterol. 2010;26:578–582. doi: 10.1097/MOG.0b013e32833d4b71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol. 2008;181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- 15.Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn's disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. doi: 10.1146/annurev-genom-082908-150013. [DOI] [PubMed] [Google Scholar]
- 16.Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
- 17.Durães C, Machado JC, Portela F, et al. Phenotype–genotype profile in Crohn's disease predicted by genetic markers in autophagy-related genes (GOIA study II) Inflamm Bowel Dis. 2012;19:230–239. doi: 10.1002/ibd.23007. [DOI] [PubMed] [Google Scholar]
- 18.Hausmann M, Obermeier F, Schreiter K, et al. Cathepsin D is up-regulated in inflammatory bowel disease macrophages. Clin Exp Immunol. 2004;136:157–167. doi: 10.1111/j.1365-2249.2004.02420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Zhang L, Yu C, Yang X-F, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker Res. 2014;2:1. doi: 10.1186/2050-7771-2-1. doi: 10.1186/2050-7771-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong KL, Yeap WH, Tai JJY, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. 2012;53:41–57. doi: 10.1007/s12026-012-8297-3. [DOI] [PubMed] [Google Scholar]
- 22.Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum. 2012;64:671–677. doi: 10.1002/art.33418. [DOI] [PubMed] [Google Scholar]
- 23.Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grip O, Bredberg A, Lindgren S, Henriksson G. Increased subpopulations of CD16(+) and CD56(+) blood monocytes in patients with active Crohn's disease. Inflamm Bowel Dis. 2007;13:566–572. doi: 10.1002/ibd.20025. [DOI] [PubMed] [Google Scholar]
- 25.Koch S, Kucharzik T, Heidemann J, Nusrat A, Luegering A. Investigating the role of proinflammatory CD16+ monocytes in the pathogenesis of inflammatory bowel disease. Clin Exp Immunol. 2010;161:332–341. doi: 10.1111/j.1365-2249.2010.04177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campos N, Magro F, Castro AR, et al. Macrophages from IBD patients exhibit defective tumour necrosis factor-α secretion but otherwise normal or augmented pro-inflammatory responses to infection. Immunobiology. 2011;216:961–970. doi: 10.1016/j.imbio.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Dignass A, Van Assche G, Lindsay JO, et al. The second European Evidence-based Consensus on the diagnosis and management of Crohn's disease: current management. J Crohns Colitis. 2010;4:28–62. doi: 10.1016/j.crohns.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Abubakar I, Myhill D, Aliyu SH, Hunter PR. Detection of Mycobacterium avium subspecies paratuberculosis from patients with Crohn's disease using nucleic acid-based techniques: a systematic review and meta-analysis. Inflamm Bowel Dis. 2008;14:401–410. doi: 10.1002/ibd.20276. [DOI] [PubMed] [Google Scholar]
- 29.Feller M, Huwiler K, Stephan R, et al. Mycobacterium avium subspecies paratuberculosis and Crohn's disease: a systematic review and meta-analysis. Lancet Infect Dis. 2007;7:607–613. doi: 10.1016/S1473-3099(07)70211-6. [DOI] [PubMed] [Google Scholar]
- 30.Smith AM, Rahman FZ, Hayee BH, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vos ACW, Wildenberg ME, Arijs I, et al. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm Bowel Dis. 2012;18:401–408. doi: 10.1002/ibd.21818. [DOI] [PubMed] [Google Scholar]
- 32.Benoit M, Desnues B, Mege J-L. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 33.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Söllner L, Shaqireen DO, Kwajah MM, Wu JT, Schwarz H. Signal transduction mechanisms of CD137 ligand in human monocytes. Cell Signal. 2007;19:1899–1908. doi: 10.1016/j.cellsig.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 35.Lim S-G, Suk K, Lee W-H. Reverse signaling from LIGHT promotes pro-inflammatory responses in the human monocytic leukemia cell line, THP-1. Cell Immunol. 2013;285:10–17. doi: 10.1016/j.cellimm.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Bode JG, Ehlting C, Häussinger D. The macrophage response towards LPS and its control through the p38MAPK–STAT3 axis. Cell Signal. 2012;24:1185–1194. doi: 10.1016/j.cellsig.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Deng W, Xie J. NOD2 signaling and role in pathogenic mycobacterium recognition, infection and immunity. Cell Physiol Biochem. 2012;30:953–963. doi: 10.1159/000341472. [DOI] [PubMed] [Google Scholar]
- 38.Lutay N, Håkansson G, Alaridah N, Hallgren O, Westergren-Thorsson G, Godaly G. Mycobacteria bypass mucosal NF-kB signalling to induce an epithelial anti-inflammatory IL-22 and IL-10 response. PLOS ONE. 2014;9:e86466. doi: 10.1371/journal.pone.0086466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative forward- (FSC) and side-scatter (SSC) dot-plot of peripheral blood mononuclear cells, showing the monocyte gate.
Fig. S2. Dot-plots of CD14 and CD16 expression by monocytes of two representative Crohn's disease (CD) patients, showing enrichment of the CD14++CD16+ population after infliximab (IFX) treatment. IFX1: after one administration; IFX2: after two administrations; IFX3: after three administrations.