

Abstract
Cryopyrin-associated periodic syndrome (CAPS) is characterized by dysregulated inflammation with excessive interleukin (IL)-1β activation and secretion. Neonatal-onset multi-system inflammatory disease (NOMID) is the most severe form. We explored cytokine responses in 32 CAPS patients before and after IL-1β blocking therapy. We measured cytokines produced by activated peripheral blood monuclear cells (PBMCs) from treated and untreated CAPS patients after stimulation for 48 h with phytohaemagglutinin (PHA), PHA plus IL-12, lipopolysaccharide (LPS) or LPS plus interferon (IFN)-γ. We measured IL-1β, IL-6, IL-10, tumour necrosis factor (TNF), IL-12p70 and IFN-γ in the supernatants. PBMCs from three untreated CAPS patients were cultured in the presence of the IL-1β blocker Anakinra. Fifty healthy individuals served as controls. CAPS patients had high spontaneous production of IL-1β, IL-6, TNF and IFN-γ by unstimulated cells. However, stimulation indexes (SIs, ratio of stimulated to unstimulated production) of these cytokines to PHA and LPS were low in NOMID patients compared to controls. Unstimulated IL-10 and IL-12p70 production was normal, but up-regulation after PHA and LPS was also low. LPS plus IFN-γ inadequately up-regulated the production of IL-1β, IL-6, TNF and IL-10 in CAPS patients. In-vitro but not in-vivo treatment with Anakinra improved SIs by lowering spontaneous cytokine production. However, in-vitro treatment did not improve the low stimulated cytokine levels. Activating mutations in NLRP3 in CAPS are correlated with poor SIs to PHA, LPS and IFN-γ. The impairment in stimulated cytokine responses in spite of IL-1β blocking therapy suggests a broader intrinsic defect in CAPS patients, which is not corrected by targeting IL-1β.
Keywords: Anakinra, cytokine, CAPS, NOMID
Introduction
Mutations in NLRP3 [the gene nucleotide-binding oligomerization domain (NOD)-like receptor family, pyrin domain containing 3, also known as cold-induced autoinflammatory syndrome 1, CIAS1] and its product NALP3 (Nacht leucine rich repeat and pyrin domain containing protein 3, or cryopyrin) are associated with a group of disorders with overlapping symptoms, called cryopyrin-associated periodic syndrome (CAPS) [1]. These diseases are autoinflammatory because of excess production of interleukin (IL)-1β. This innate immune cytokine is a potent endogenous pyrogen that causes a cytokine amplification loop [2,3]. All patients with cryopyrinopathies exhibit fever, urticarial rash, arthralgia/arthritis, neutrophil-mediated inflammation and elevated acute-phase reactants. Familial cold autoinflammatory syndrome (FCAS, OMIM #120100), the least severe form of CAPS, presents with self-limited cold-triggered episodes of fever, rash and arthralgia, combined with conjunctivitis and headaches [1,4]. In Muckle–Wells syndrome (MWS, OMIM #191900), the intermediate form of CAPS, fever, rash and arthralgia are often continuous with exacerbations, and patients develop hearing loss over time. A third of untreated MWS patients develop amyloidosis which progresses to kidney failure [1]. The most severe form of CAPS, neonatal-onset multi-system inflammatory disease (NOMID or CINCA, OMIM #607115), exhibits inflammatory symptoms similar to FCAS and MWS but also presents with an increased intracranial pressure and progressive hearing and vision loss early in life due to cochlear and ocular inflammation. Approximately 50% of NOMID patients, whether treated or not, develop exophytic growth of the patella and epiphyses of the long bones. Childhood mortality in untreated NOMID is high (20%) [5].
CAPS is caused by autosomal-dominant missense mutations in exon 3 that lead to gain-of-function [6,7]. These mutations can occur de novo and somatic mosaicism in NLRP3 causes disease in a third of clinical NOMID patients [8]. Some missense mutations that do not occur in superficial binding sites of the molecule in NLRP3 are asymptomatic [5,7]. NALP3 is the first intracellular pattern recognition receptor identified in humans [9]. When activated, NALP3 oligomerizes in a multi-molecular complex, the NALP3 inflammasome (Supporting information, Fig. S1). This leads to the activation of caspase-1, a protease that cleaves inactive pro-IL-1β and pro-IL-18 precursors to their active forms [3]. In turn, active IL-1β stimulates its own mRNA transcription via nuclear factor (NF)-κB [10].
Optimized IL-1β blocking therapies lead to complete remission of the inflammatory manifestations of CAPS [1]. In 2006, we showed high spontaneous and stimulated IL-1β production in 18 NOMID patients [11]. In the present report, we expand the cytokine panel and include evaluation of unstimulated and stimulated production of IL-1β, IL-6, IL-10, interferon (IFN)-γ, tumour necrosis factor (TNF) and IL-12p70 by peripheral blood mononuclear cells (PBMCs) from patients with FCAS, MWS and NOMID before and during Anakinra therapy.
Methods
Subjects and IL-1β blocking therapy
Thirty-two patients with mutations in NLRP3 and/or CAPS disease were recruited from the outpatient clinic of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), NIH, Bethesda (Table 1). All patients were followed under approved protocols, and either they or their guardians gave informed consent. The patients were categorized according to their CAPS phenotype: three individuals with a R488K mutation in NLRP3 without phenotype (asymptomatic, patients 1–3), three FCAS (patients 4–6), five MWS (patients 7–11) and 21 NOMID patients (patients 12–32) (Table 1). Part of this group was reported previously (patients 13, 14, 16, 19–21, 23–30, 32) [11]. Mutation detection was performed for all patients. Fifty healthy controls were obtained from the NIH Blood Bank under appropriate protocols.
Table 1.
Cryopyrin-associated periodic syndrome (CAPS) phenotype, NLRP3 mutation and treatment of patients
| Patient | Phenotype | NLRP3 mutation | Age (years)* | CRP, mg/dl* | Treatment |
|---|---|---|---|---|---|
| 1 | No phenotype | R488K | 34 | < 0·4 | None |
| 2 | No phenotype | R488K | 59 | 0·71 | None |
| 3 | No phenotype | R488K | 7 | < 0·4 | None |
| 4 | FCAS | L353P | 11 | 0·2 | Anakinra |
| 5 | FCAS | L353P | 41 | 1·7 | Rilonacept |
| 6 | FCAS | L353P | 58 | 3·4 | Rilonacept |
| 7 | MWS | T348M | 34 | 5·8 | Anakinra |
| 8 | MWS | F523C | 7 | 1·2 | Anakinra |
| 9 | MWS | E627G | 42 | 6·2 | Rilonacept |
| 10 | MWS | M659K | 20 | 3·1 | Rilonacept |
| 11 | MWS | E627G | 64 | 8·6 | Rilonacept |
| 12 | NOMID, no MR | L264F | 1 | 4·8 | Anakinra |
| 13 | NOMID, no MR | D303N | 15 | 3·5 | Anakinra |
| 14 | NOMID, no MR | D303N | 8 | 4·1 | Anakinra |
| 15 | NOMID, no MR | G755A | 1 | 6·6 | None |
| 16 | NOMID, no MR | G326E | 4 | 5·8 | Anakinra |
| 17 | NOMID, no MR | T348M | 12 | 3·6 | Anakinra |
| 18 | NOMID, no MR | D303N | 12 | 8·3 | Anakinra |
| 19 | NOMID, mild MR | V262A | 11 | 2·5 | Anakinra |
| 20 | NOMID, mild MR | L632F | 8 | 4·9 | Anakinra |
| 21 | NOMID, mild MR | D303N | 28 | 5·0 | Anakinra |
| 22 | NOMID, mild MR | V351L | 13 | 6·6 | Anakinra |
| 23 | NOMID, mild MR | G569R | 9 | 11·1 | Anakinra |
| 24 | NOMID, mild MR | Somatic K568N (9·4%) | 6 | 3·2 | Anakinra |
| 25 | NOMID, mild MR | Negative | 4 | 15·3 | Anakinra |
| 26 | NOMID, severe MR | F443L | 16 | 4·0 | Anakinra |
| 27 | NOMID, severe MR | A374N | 9 | 5·6 | Anakinra |
| 28 | NOMID, severe MR | Somatic F566L (14·6%) | 8 | 10·5 | Anakinra |
| 29 | NOMID, severe MR | Somatic K355N (20·1%) | 18 | 7·3 | Anakinra |
| 30 | NOMID, severe MR | L264F | 7 | 7·3 | Anakinra |
| 31 | NOMID, severe MR | Q600P | 19 | 4·3 | Anakinra |
| 32 | NOMID, severe MR | Somatic E567K (16·2%) | 11 | 12·3 | Anakinra |
At enrolment. CAPS = cryopyrin-associated periodic sydromes; FCAS = familial cold autoinflammatory syndrome; MR = mental retardation; MWS = Muckle–Wells syndrome; NOMID = neonatal-onset multi-system inflammatory disease; CRP = C-reactive protein.
PBMCs of all individuals were assayed for cytokine production and response. Seventeen CAPS patients (patients 4–10, 12–15, 19–22, 26 and 31) were assayed, while the patients were untreated. During the study, all but one patient with NOMID (patient 15) were started on Anakinra (1–2 mg/kg/day by subcutaneous injection). Any dose of steroids, non-steroidal anti-inflamatory drugs (NSAIDs) or disease-modifying anti-rheumatic drugs (DMARDs) had to be stable for 4 weeks prior to enrolment. PBMCs of NOMID patients were tested for cytokine production before treatment (10 patients: 12–15, 19–22, 26, 31), at 3 months (eight patients: 12, 14, 19–21, 26, 27, 32), 6 months (14 patients: 14, 16, 19–21, 23–30, 32), 12 months (13 patients: 14, 16–18, 20, 21, 23, 25–30) and 18 months (12 patients: 14, 16–18, 21, 23–25, 29–32) after initiation of Anakinra therapy.
Two FCAS (patients 5 and 6) and three MWS patients (patients 9–11) were treated with Rilonacept (100 mg/week preceded by a loading dose of 300 mg in 3 days' time by subcutaneous injection). All but one FCAS patient (patient 5) underwent a dose-escalation to 160 mg/week after 3 months of therapy because remission criteria [no symptoms of inflammation, low inflammatory markers such as C-reactive protein (CRP)] were not met. The Rilonacept patients were tested for cytokine production before treatment (patients 5, 6, 10 and 11), at 3 months of therapy with 100 mg/week (patients 5, 9, 10 and 11) and after 1 (patients 9–11) and 3 months (patients 6, 9, 10) of the dose-escalation to 160 mg/week. Because patient availability for testing varied, the size and composition of the patient groups under both therapies (Anakinra, Rilonacept) varied at the different time-points.
PBMC stimulation and cytokine determination
PBMCs were isolated by density gradient centrifugation of heparinized whole blood through lymphocyte separation medium as described elsewhere [12]. Fresh patient and control PBMCs were stimulated in RPMI-1640, supplemented with 20 mM Hepes, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal calf serum (FCS) at 106 cells per condition. Cells were cultured for 48 h at 37°C in 5% CO2, either unstimulated or with phytohaemagglutinin (PHA) (1%), PHA plus IL-12p70 (1 ng/ml), lipopolysaccharide (LPS) (200 ng/ml) or LPS plus IFN-γ (1000 U/ml). The production of IL-1β (detection limits 3·2–3261 pg/ml), IL-6 (2·3–18 880 pg/ml), IL-10 (2·2–8840 pg/ml), TNF (5·8–95 484 pg/ml), IL-12p70 (3·3–13 099 pg/ml) and IFN-γ (92·6–52 719 pg/ml) was measured from frozen supernatants with a Bioplex cytokine assay (Bio-Rad, Hercules, CA, USA). This assay is specific for active IL-1β.
In another experiment, PBMCs from 15 of the 50 healthy controls were similarly stimulated with 10 ng IL-1β for 48 h, after which IL-6, TNF, IFN-γ, IL-12p70 and IL-10 were measured. Furthermore, 1 μg Anakinra was added to PBMCs of two MWS patients and one NOMID patient (patients 8, 10 and 13) who were untreated at the time and all cytokines, including IL-1β, were measured in the same experimental design.
Calculation of stimulation indexes
Cytokine stimulation indices were determined for PHA, IL-12p70, LPS and IFN-γ. In the case of PHA and LPS, these indices were calculated as the ratio of stimulated to unstimulated cytokine production. Indices for IL-12p70 and IFN-γ were calculated as the ratio of cytokines produced after stimulation with PHA plus IL-12p70 or LPS plus IFN-γ to production after stimulation with PHA or LPS alone. To compute the ratios, all cytokine values between 0 and 0·1 pg/ml were set to 0·1 pg/ml. The raw values from the stimulation indices in Figs 2–5 are listed in the Supporting information.
Figure 2.

Low cytokine stimulation indices to phytohaemagglutinin (PHA), high interferon (IFN)-γ stimulation index to interleukin (IL)-12p70 in neonatal-onset multi-system inflammatory disease (NOMID) patients. Peripheral blood mononuclear cells (PBMCs) (106 cells per condition) were cultured for 48 h with and without 1% phytohaemagglutinin (PHA) and cytokine production was measured in the supernatants using a Bioplex cytokine assay. PBMCs from 50 healthy controls (○), three individuals with the R488K NLRP3 variation (▪), three familial cold autoinflammatory syndrome (FCAS) patients (▲), four Muckle–Wells syndrome (MWS) (MWS) patients (▼) and 10 NOMID patients (◆) were assayed. Stimulation indices of IL-1β (A), IL-6 (B), IL-10 (C), tumour necrosis factor (TNF) (d), IL-12p70 (e) and IFN-γ (f) in response to PHA were calculated as the ratio of stimulated to unstimulated cytokine production. To obtain the IFN-γ stimulation index to IL-12p70 (g), 1 ng/ml IL-12p70 was added to the cultures in addition to PHA. The stimulation index was calculated as the ratio of IFN-γ production stimulated with PHA and IL-12p70 to IFN-γ production stimulated with PHA only. To calculate ratios, cytokine values 0 to 0·1 pg/ml were set to 0·1 pg/ml. The Mann–Whitney U-test was used for the comparison of cytokine production by PBMCs from the different groups of cryopyrin-associated periodic syndrome (CAPS) patients (asymptomatic subjects, FCAS, MWS, NOMID) with the cytokine production by healthy controls. Only significant differences are depicted. Bar indicates the mean cytokine production by healthy controls and NOMID patients.
Figure 5.

Improved cytokine stimulation indices to phytohaemagglutinin (PHA) and LPS in neonatal-onset multi-system inflammatory disease (NOMID) patients by the addition of Anakinra in vitro. Peripheral blood mononuclear cells (PBMCs) (106 cells per condition) were cultured for 48 h with and without stimulus, and with and without the addition of 1 μg Anakinra, after which cytokines were measured in the supernatants using a Bioplex cytokine assay. Interleukin (IL)-1β, IL-6, tumour necrosis factor (TNF) and interferon (IFN)-γ stimulation indices are shown in response to 1% PHA or 200 ng/ml LPS in 50 healthy controls and of three untreated cryopyrin-associated periodic syndrome (CAPS) patients [two Muckle–Wells syndrome (MWS) patients and one NOMID patient]. Healthy controls: ○; CAPS patients: •. Cytokine stimulation indices to PHA and LPS were calculated as the ratio of stimulated to unstimulated cytokine production. To calculate ratios, cytokine values 0 to 0·1 pg/ml were set to 0·1 pg/ml.
Statistical analysis
The Mann–Whitney U-test was used for non-parametric comparison of cytokine production and cytokine stimulation indices of unpaired groups. When comparing more than two groups, the Kruskal–Wallis test was used. Results were considered to be statistically significant when P < 0·05 (two-sided).
Results
Patients
The patients and their mutations are listed in Table 1. Included are three referred individuals with the NLRP3 variant R488K [13], but atypical inflammatory symptoms. R488K is thought to be a non-CAPS causing variant. Four of 21 NOMID patients (19%) had somatic mutations, with genetic mosaicism varying from 9 to 20%. We could not find a mutation in NLRP3 in one patient.
Unstimulated cytokine production is high in untreated CAPS patients
CAPS patients are known to have constitutively high levels of inflammatory cytokines in their blood, notably of IL-1β [2,3,11,14–17], but also of IL-6 and TNF [17–19]. We previously reported high levels of IL-1β in cultures of unstimulated PBMCs from 16 CAPS patients [11]. We show here that production of IL-6 (P < 0·001), TNF (P < 0·0001) and IFN-γ (P < 0·003) is also excessive in the supernatants of these unstimulated PBMCs (Fig. 1a–c). The unstimulated levels of IL-12p70 (Fig. 1d) and IL-10 (data not shown) were normal. FCAS patients produced smaller amounts of cytokines than MWS and NOMID patients: when the three FCAS patients were excluded from the analysis, differences in IL-6 and IFN-γ production by untreated MWS/NOMID patients (14 patients) and healthy controls were more pronounced (data not shown; IL-6: P < 0·0001 and IFN-γ: P < 0·002).
Figure 1.
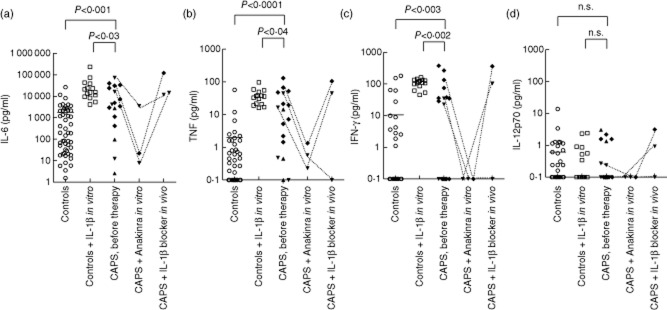
High unstimulated cytokine production in cryopyrin-associated periodic syndrome (CAPS) patients and marked effect of co-culture with Anakinra in vitro. Concentrations of interleukin (IL)-6, tumour necrosis factor (TNF), interferon (IFN)-γ and IL-12p70 in the supernatants of peripheral blood mononuclear cells (PBMCs) (106 cells per condition), cultured for 48 h and measured using a Bioplex cytokine assay. PBMCs from healthy controls were assayed with and without the addition of 10 ng IL-1β in vitro [50 controls without IL-1β (○), 15 of these 50 controls with IL-1β (□)] and from 17 CAPS patients [three with familial cold autoinflammatory syndrome (FCAS) (▲); four with Muckle–Wells syndrome (MWS) (▼) and 10 with neonatal-onset multi-system inflammatory disease (NOMID) (◆)] before treatment with Anakinra or Rilonacept was started. The Mann–Whitney U-test was used for the comparison of cytokine production by CAPS patients and controls, and for the comparison of cytokine production by CAPS patients and controls co-cultured with IL-1β. One μg Anakinra was added to cultures of three CAPS patients (two MWS, one NOMID) who were not treated at the time. Also, the cytokine production of these patients is depicted while under IL-1β blocking therapy: one MWS was treated with Anakinra for 1 month, one MWS patient was treated with Rilonacept for 3 months and the NOMID patient was treated with Anakinra for 1 month. n.s. = non-significant. Bar indicates the mean cytokine production by healthy controls.
To examine whether cytokine levels could be elevated because of excessive IL-1β production in CAPS patients, we added IL-1β to PBMCs of healthy controls. Indeed, we could mimic the CAPS cytokine profile; IL-6, TNF, IFN-γ and IL-10 were up-regulated to levels even higher than those in CAPS patients (P < 0·03, P < 0·04, P < 0·002 and P < 0·003, respectively) (Fig. 1a–c, IL-10 data not shown). In addition, we imitated IL-1β blockade in CAPS patients by adding Anakinra to cultured PBMCs from two untreated MWS patients and one untreated NOMID patient. The addition of Anakinra in vitro reduced all cytokine levels (Fig. 1a–d), including IL-1β (data not shown), to lower levels than those reached in samples from Anakinra-treated patients (Fig. 1a–d: two MWS patients treated with 1 month of Anakinra and with 3 months of Rilonacept, respectively; one NOMID patient treated with 3 months of Anakinra). Therefore, acute cytokine secretion can be manipulated by IL-1β levels in CAPS.
Defective up-regulation of cytokines in NOMID patients after stimulation with PHA, LPS and IFN-γ
While the unstimulated production of proinflammatory cytokines was high in CAPS patients, stimulation indices to PHA (Fig. 2a–f) and LPS (Fig. 3a–d) of all six measured cytokines (IL-1β, IL-6, IL-10, TNF, IL-12p70 and IFN-γ) were significantly lower in NOMID patients than in controls (LPS-stimulated IL-10 and IFN-γ data not shown). NOMID patients are unable to up-regulate the production of these cytokines to normal levels. In absolute numbers, the PHA-stimulated production of IL-6, IL-10, TNF and IFN-γ by PBMCs of NOMID patients was also significantly lower than production in healthy controls (Supporting information, Tables S2–4, S6). PHA-stimulated IL-1β production was higher in NOMID (Supporting information, Table S1), while the production of IL-12p70 did not differ (Supporting information, Table S5). Similarly, absolute amounts of IL-6, IL-10 and IL-12p70 after stimulation with LPS were lower in supernatants of PBMCs from NOMID patients than from healthy controls. IL-1β, TNF and IFN-γ were not significantly different (Supporting information, Tables S8–11, data for IL-10 and IFN-γ not shown).
Figure 3.

Low cytokine stimulation indices to lipopolysaccharide (LPS) and interferon (IFN)-γ, normal interleukin (IL)-12p70 stimulation index to IFN-γ in neonatal-onset multi-system inflammatory disease (NOMID) patients. Peripheral blood mononuclear cells (PBMCs) (106 cells per condition) were cultured for 48 h with and without 200 ng/ml LPS, and with and without the addition of 1000 U/ml IFN-γ to LPS. Cytokines were measured in the supernatants using a Bioplex cytokine assay. PBMCs from 50 healthy controls (○), three asymptomatic individuals with NLRP3 mutations (▪), three familial cold autoinflammatory syndrome (FCAS) patients (▲), four Muckle–Wells syndrome (MWS) patients (▼) and 10 NOMID patients (◆) were assayed. Stimulation indices of IL-1β, IL-6, tumour necrosis factor (TNF) and IL-12p70 in response to LPS were calculated as the ratio of stimulated to unstimulated cytokine production. Stimulation indices to IFN-γ were calculated as the ratio of cytokine production stimulated with LPS in combination with IFN-γ to cytokine production stimulated with LPS alone. To calculate ratios, cytokine values 0 to 0·1 pg/ml were set to 0·1 pg/ml. We used the Mann–Whitney U-test for the comparison of cytokine production by PBMCs from the different groups of cryopyrin-associated periodic syndrome (CAPS) patients (asymptomatics, FCAS, MWS, NOMID) with the cytokine production by healthy controls. Only significant differences are depicted. Bar indicates the mean cytokine production by healthy controls and NOMID patients.
Stimulation indices of MWS patients to LPS for IL-1β, IL-6, TNF (Fig. 3a–c) and IL-10 (data not shown) were low, but their indices for IL-12p70 (Fig. 3d) and IFN-γ (data not shown) were normal. Stimulation indices to PHA were normal, with the exception of TNF (Fig. 2a–f). FCAS patients and healthy controls up-regulated cytokines to the same extent in response to PHA and LPS (Figs 2a–f, 3a–d), although the patients' IL-10 index to LPS was low (data not shown). Indices from asymptomatic individuals with the R488K variation could even be high (Figs 2a–f, 3a–d).
The response of CAPS patients to IL-12p70 was normal to high for all cytokines (Fig. 2g, data shown for IFN-γ production only). Conversely, the stimulation index to IFN-γ was low in MWS and NOMID patients for all cytokines, with the exception of IL-12p70 (Fig. 3e–h). The IL-10 stimulation index to IFN-γ was normal in MWS patients only (data not shown). In absolute numbers, NOMID PBMCs stimulated with a combination of LPS plus IFN-γ produced significantly less IL-6, TNF, IL-10 and IL-12p70 than controls (Supporting information, Tables S13–S15, IL-10 data not shown). The IL-1β stimulation index to IFN-γ was the same across NOMID patients and controls (Supporting information, Table S12). In FCAS patients, only the IL-1β and TNF indices to IFN-γ were low, while asymptomatic individuals up-regulated all cytokines normally in response to IFN-γ (Fig. 3e–h, IL-10 data not shown).
No correlation of cytokine stimulation indices to clinical severity of CAPS phenotype
FCAS, MWS and NOMID all are caused by mutations in NLRP3. To investigate the relationship between cytokine stimulation index and clinical phenotype of CAPS, we separated the NOMID patients into three groups with increasing clinical severity, as judged by their neurological disease (none, mild or severe mental retardation). Indeed, CRP values of CAPS patients with more serious phenotypes at enrolment in the study were higher than those with less severe disease (Table 1). CRP values in asymptomatic individuals, FCAS and MWS patients before treatment with an IL-1β antagonist were, on average, 0·4, 1·8, and 4·1 mg/dl, respectively. These values were 4·8, 3·8 and 4·2 mg/dl in the three groups of NOMID patients with increasingly severe mental retardation. Next, we compared cytokine stimulation indices to LPS, PHA or IFN-γ in CAPS patients ranging from asymptomatic individuals to a NOMID patient with severe mental retardation. Poor cytokine stimulation indices to LPS, PHA or IFN-γ (data not shown for all cytokines) did not correlate with increasing severity in clinical phenotype, except for the IL-12p70 stimulation index to LPS, which was only slightly lower in the more severe CAPS phenotypes (P < 0·05, data not shown).
No improvement of low cytokine stimulation indices in patients treated with IL-1β blocking therapy
We assessed all but one NOMID patient after Anakinra was started and measured cytokine levels at 3, 6, 12 and 18 months after the start of therapy. It enabled us to analyse the effect of the IL-1R antagonist in vivo on cytokine production in NOMID patients. We noticed a slight improvement in stimulation indices to PHA (Fig. 4a–d) up to 12 months of therapy, which was lost at 18 months [data on IL-10 (P < 0·0007) and IL-12p70 (not significant: n.s.) not shown]. Poor stimulation indices to LPS [Fig. 4e–h, data on IL-10 (n.s.) and IL-12p70 (n.s.) not shown] and IFN-γ [data on all cytokines, n.s., except for IL-12p70 (P < 0·02); data not shown] did not improve with Anakinra treatment. In absolute numbers, the unstimulated cytokine production by patients treated with 3–18 months of Anakinra did not change significantly (data on IL-1β, IL-6, TNF and IFN-γ in Supporting information, Tables S16–S19). Furthermore, PHA and LPS stimulated levels remained low over time except for PHA-stimulated TNF production at 3 and 12 months (P < 0·0005 and P < 0·04, respectively) (data on IL-1β, IL-6, TNF and IFN-γ in Supporting information, Tables S16–S23). After 18 months, all absolute cytokine values reverted to pretreatment levels. Therefore, low stimulation indices in NOMID patients treated with Anakinra are indicative of both consistently high unstimulated cytokine secretion (denominator), and low stimulated production (numerator) over time.
Figure 4.

No improvement of poor cytokine stimulation indices with lengthy Anakinra treatment in neonatal-onset multi-system inflammatory disease (NOMID) patients. Peripheral blood mononuclear cells (PBMCs) (106 cells per condition) were cultured for 48 h with and without stimulus after which cytokines measured in the supernatants using a Bioplex cytokine assay. Interleukin (IL)-1β, IL-6, tumour necrosis factor (TNF) and interferon (IFN)-γ stimulation indices are shown in response to 1% phytohaemagglutinin (PHA) or 200 ng/ml lipopolysaccharide (LPS) in 50 healthy controls, 10 NOMID patients at baseline (not treated), eight NOMID patients at 3 months, 14 NOMID patients at 6 months, 13 NOMID patients at 12 months and 12 patients at 18 months of treatment with a maximum of 2 mg/kg/day of Anakinra. Healthy controls: ○; NOMID patients: •. Cytokine stimulation indices to PHA and LPS were calculated as the ratio of stimulated to unstimulated cytokine production. To calculate ratios, cytokine values 0 to 0·1 pg/ml were set to 0·1 pg/ml. The Kruskal–Wallis test was used for analysing the difference in cytokine stimulation index for NOMID patients treated with Anakinra for an increasing time-period. n.s. = non-significant. Bar indicates the mean cytokine production by healthy controls.
Similarly, treatment with Rilonacept did not influence cytokine stimulation indices in two FCAS and three MWS patients. IL-1β, IL-6, TNF and IFN-γ (data not shown) stimulation indices for PHA and LPS did not improve, regardless of Rilonacept dose and the duration of therapy. None of the absolute cytokine values under Rilonacept treatment, whether stimulated or not with IL-1β or IL-6, differed significantly from pretreatment levels (data not shown).
Improvement of low cytokine stimulation indices with IL-1β blocker added to the in-vitro cultures
Stimulation indices improved when we added 1 μg Anakinra to PBMC cultures from one NOMID and two MWS patients (Fig. 5a–h). After 48 h of stimulation, indices to PHA and LPS were much higher than those from the same patients before in-vitro treatment with an anti-IL-1β antagonist. The cytokine levels show that this improvement is due mainly to lower spontaneous cytokine production – the index denominator (Fig. 1a–c, Supporting information, Tables S24–S31). Stimulated cytokine production did not improve, even with Anakinra added to the cultures in vitro.
Discussion
CAPS patients differ from healthy individuals in the dynamics of cytokine release. Their inflammasomes are constitutively activated and their cells pre-activated. Indeed, we observed consistently high spontaneous levels of IL-1β and other proinflammatory cytokines (IL-6, TNF and IFN-γ) in non-stimulated supernatants of CAPS patients, in particular of NOMID cases. However, NOMID patients are unable to up-regulate their production of IL-1β, IL-6, IL-10, TNF, IL-12p70 and IFN-γ in response to major stimuli including PHA, LPS and IFN-γ to the same extent as do controls. In the case of IL-1β which is, on average, higher in CAPS patients, these levels dropped to control average after stimulation with LPS and LPS plus IFN-γ. Treatment for this condition with IL-1β blocking therapy, although improving clinical manifestations, does not lead to increased stimulated cytokine production in vivo in PBMCs from treated patients.
The combination of high spontaneous cytokine production with impaired stimulated production resulted in overall low cytokine stimulation indices. Stimulated cytokine production may be low because pre-activated NOMID cells are functionally exhausted and cannot sustain cytokine transcription and/or translation. Carta et al. confirm that Toll-like receptor (TLR)-2- and TLR-4-stimulated CAPS monocytes produce less IL-6 and IL-1Ra than control cells. They attribute this difference to increased redox stress, which involves relatively high levels of reactive oxygen species and anti-oxidants [20]. In addition, they conclude that the redox defect is unrelated to IL-1β levels because cytokine production was low in patients who were both untreated and treated with Anakinra. Similarly, others found lower expression by Western blot of pro-IL-1β in LPS-stimulated NOMID monocytes than in non-stimulated cells and LPS-stimulated control cells [21]. Although we did not examine the redox status in our patients' cells, our data would be consistent with observations that the underlying redox-activation status may hamper stimulated cytokine production. However, in our experiments, cytokine production of NOMID patients responded normally to IL-12p70. Thus, perhaps the redox status of a cell may only affect certain cytokine pathways, namely those stimulated with PHA, LPS and IFN-γ, but not with IL-12p70.
Low values of the stimulation indices may also be due to stimulation-induced necrosis-like death of monocytes in CAPS, which depends on cathepsin B [22]. Saito et al. found that LPS induces NLRP3 mRNA expression in CAPS patients, but kills monocytes. The death toll was independent of caspase-1, disease severity or treatment with Anakinra [23]. Others used fluorescein isothiocyanate (FACS) analysis showing dramatic loss of monocytes after the stimulation of NOMID patient cells with LPS. No such loss was seen in cell cultures from controls or from a patient deficient in an IL-1 receptor antagonist (DIRA) [17]. As monocytes die early in NOMID, control cells may release relatively greater amounts of cytokines such as IL-1β over time. Indeed, previous research shows a clearly blunted production of IL-1β in NOMID patients compared to controls after LPS stimulation [16], or no influence at all of LPS on levels of IL-1β, IL-6, IL-10 and TNF [17]. In contrast, the production of these cytokines was markedly increased in LPS-stimulated PBMCs from a control and a DIRA patient [17]. So far, only TLR ligands such as LPS have proved to induce necrosis-like cell death [17,23], but we show that other stimuli such as PHA and IFN-γ also affect monocyte-derived cytokine production.
In conclusion, functional exhaustion and stimulation-induced death of monocytes by NOMID patients could have a detrimental effect on cytokine production in our stimulation assays, and hence on cytokine stimulation indices. However, this does not translate into an increased susceptibility to infections.
It is possible that stimulated cytokine production and release in NOMID patients is, in fact, higher than in controls if measured earlier. High IL-1β and TNF production, measured only few hours after the onset of stimulation in some, but not all, cases have been reported upon TLR-stimulation in NOMID/CAPS patients [2,14,15,20,24,25]. Indeed, over the 48-h window of the experiments, CAPS cells may be releasing the bulk of IL-1β earlier than controls because their caspase-1 is activated more rapidly. LPS induces IL-1β secretion in CAPS monocytes without adenosine triphosphate (ATP) as a second stimulus, while control monocytes require stimulation by both LPS and ATP [2]. The lack of ATP needed to stimulate IL-1β release in patient cells may be because of autocrine ATP production [26]. LPS-induced IL-1β was produced in markedly greater quantities in monocytes from CAPS patients after 3 h of stimulation, but was about the same after 24 h [2]. LPS-stimulated IL-6 production in CAPS patients is also time-dependent. While IL-6 production was much lower in CAPS patients than in healthy donors at 18 h of stimulation, it was similar up to 6 h post-LPS stimulation [20].
Low responses to LPS and IFN-γ in NOMID patients recall the cytokine profile of certain patients with Mendelian susceptibility to mycobacterial disease (MSMD, OMIM #209950). These patients have genetic defects in the T helper type 1 (Th1) pathway, affecting the production of and responses to IFN-γ and IL-12 [27]. Like NOMID patients, MSMD patients with dominant-negative IFN-γ receptor-1 (IFNGR1) or partial IFN-γ receptor-2 (IFNGR2) mutations respond in a diminished but definite way to stimulation with LPS and to IFN-γ combined with LPS. This is illustrated by reduced TNF production following these stimuli in IFN-γR1-deficient patients [28], and by a low TNF stimulation index to IFN-γ in patients with autosomal-dominant IFN-γR1 or partial IFN-γR2 deficiency [29–31]. In addition, both NOMID and IFN-γR-deficient patients produce normal levels of IFN-γ in response to IL-12p70. However, there is a difference in IFN-γ-stimulated IL-12p70 production. MSMD patients with completely defective IFN-γR produce no IL-12 in response to IFN-γ, while patients with only partial defects produce a little [32,33]. In contrast, NOMID patients have normal IL-12p70 stimulation indices in response to IFN-γ. Hence, in NOMID patients, IL-12p70-stimulated IFN-γ production and IFN-γ-stimulated IL-12p70 production are unaffected. This suggests that their IFN-γ/IL-12 cytokine loop is intact, explaining in turn the absence of mycobacterial infections.
Treatment with anti-IL-1β antagonists (Anakinra, Rilonacept) achieved sustained clinical remission in all CAPS patients studied [11,34]. Surprisingly, we saw no improvement in stimulation indices or in abnormal cytokine levels in PBMCs from treated NOMID patients. In contrast, other studies highlight a positive effect of anti-IL-1β treatment in vivo on cytokine production in CAPS. In one study, Anakinra therapy led to decreased LPS-stimulated IL-1β secretion in three NOMID patients [2]. Others reported inhibited cytokine production on Anakinra treatment with a rapid decrease in serum IL-6 levels after 1 month and a slower decrease in TNF after 3 months of treatment [11,18]. Although we have shown previously that 6 months of Anakinra treatment reduced IL-1β production in NOMID patients [11], we now suggest more variation in IL-1β production with therapy. The current study uses a larger patient sample and suggests the influence of additional factors on IL-1β production such as the pre-activation state. Furthermore, responsiveness of monocytes obtained at the respective blood draw may vary on a daily basis depending on previous in-vivo exposures.
Although stimulation indices from NOMID patients did not improve with in-vivo Anakinra therapy, they increased when Anakinra was added to PBMCs of untreated patients in vitro. The improvement was due largely to a decrease in spontaneous cytokine release. One μg of Anakinra in the culture plate was estimated to equal an in-vitro concentration of about 3 ng/ml, which is close to serum concentrations of in-vivo-treated NOMID patients [35]. The positive effect of Anakinra in vitro on stimulation indices supports the idea that IL-1β up-regulates a vicious IL-1β-producing cycle in an autocrine manner that can be counteracted by the presence in the culture of IL-1β blocking drugs. Excessive spontaneous cytokine production in Anakinra-treated patients probably improves because of supraphysiological concentrations of the IL-1 receptor antagonist in the serum, but we were unable to detect this effect in patients treated with Anakinra when the blood was drawn. Indeed, when cells from treated patients are isolated and washed for the assays, it is likely that any IL-1β blocking agent is removed.
In contrast to the improvement in spontaneous cytokine release, stimulated cytokine production remains low despite the addition of Anakinra to the culture. This indicates that the defect is cell-intrinsic and unrelated to IL-1β levels. Reduced cytokine activation and secretion in PHA, LPS and IFN-γ stimulated cells, but not with IL-12p70 stimulation, needs to be confirmed by mRNA levels and protein expression studies of the cytokines and further evaluated, for example with inflammasome inhibitors.
Acknowledgments
M. H. H. was supported by grants from the Fulbright/Netherlands–America Foundation; the intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases; the Netherlands Organization for Scientific Research (NWO); the Stichting Fonds Doctor Catharine van Tussenbroek; the Prins Bernhard Cultuurfonds; the Leids Universiteits Fonds; the Stichting Dr Hendrik Muller's Vaderlandsch Fonds; the Stichting Fundatie Vrijvrouwe van Renswoude; and the Stichting Algemeen Studiefonds.
Disclosure
The authors do not state any conflicts of interest and have not received any financial support or other benefits from commercial sources.
Author contributions
M. H. H., R. G.-M. and S. M. H. designed the study, M. H. H. performed the experiments, M. H. H., R. G.-M., E. vd V. and S. M. H. wrote the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. The Nacht leucine rich repeat and pyrin domain containing protein 3 (NALP3) inflammasome activates interleukin (IL)-1β and IL-18. NALP3 has three domains: an N-terminal protein–protein interaction domain, called PYD; a central nucleotide-binding and oligomerization domain, called NACHT; and a C-terminal ligand-binding and regulatory leucin-rich repeat domain, called LRR. NACHT is encoded by exon 3. PYD interacts with a protein called ASC, which stands for ‘apoptosis-associated speck-like protein containing a caspase recruitment domain’ (CARD). This interaction then binds pro-caspase-1 to ASC via homodimerization of the respective CARD domains. This is called the NALP3 inflammasome. Upon activation by ligand binding [e.g. adenosine triphosphate (ATP)] to the LRR of NALP3 caspase-1 dimerizes, resulting in proximity-induced autoactivation. Active, p10/20-tetrameric caspase-1 is released from the complex to cleave IL-1β and IL-18 precursors into their active forms.
Fig. S2. Subdivision of patients. Flowchart of the individuals studied per experiment. Patients per group: •; patients per group with duration of treatment in months:  ; patient numbers from Table 1: #; patients with Anakinra added to their peripheral blood mononuclear cells (PBMCs) in vitro or treated with Anakinra in vivo: V. Patients per group with duration of treatment in months per dose of Rilonacept
; patient numbers from Table 1: #; patients with Anakinra added to their peripheral blood mononuclear cells (PBMCs) in vitro or treated with Anakinra in vivo: V. Patients per group with duration of treatment in months per dose of Rilonacept  .
.
Fig. S3. Diagram explaining low stimulation indices (SIs) in neonatal-onset multi-system inflammatory disease (NOMID) patients with improvement of SIs by Anakinra therapy in vitro but not in vivo.
Table S1. Raw data for Fig. 2a. Interleukin (IL)-1β production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S2. Raw data for Fig. 2b. Interleukin (IL)-6 (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S3. Raw data for Fig. 2c. Interleukin (IL)-10 (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S4. Raw data for Fig. 2d. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S5. Raw data for Fig. 2e. Interleukin (IL)-12p70 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S6. Raw data for Fig. 2f. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S7. Raw data for Fig. 2g. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA)+interleukin (IL)-12.
Table S8. Raw data for Fig. 3a. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S9. Raw data for Fig. 3b. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S10. Raw data for Fig. 3c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S11. Raw data for Fig. 3d. Interleukin (IL)-12p70 production (pg/ml) after stimulation with LPS.
Table S12. Raw data for Fig. 3e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S13. Raw data for Fig. 3f. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S14. Raw data for Fig. 3g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S15. Raw data for Fig. 3h. Interleukin (IL)-12p70 production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S16. Raw data for Fig. 4a. Interleukin (IL)-1β (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S17. Raw data for Fig. 4b. Interleukin (IL)-6 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S18. Raw data for Fig. 4c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S19. Raw data for Fig. 4d. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S20. Raw data for Fig. 4e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S21. Raw data for Fig. 4Ff. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S22. Raw data for Fig. 4g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S23. Raw data for Fig. 4h. Interferon (IFN)-γ production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S24. Raw data for Fig. 5a. Interleukin (IL)-1β production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S25. Raw data for Fig. 5b. Interleukin (IL)-6 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S26. Raw data for Fig. 5c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S27. Raw data for Fig. 5d. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S28. Raw data for Fig. 5e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S29. Raw data for Fig. 5f. IL-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S30. Raw data for Fig. 5g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S31. Raw data for Fig. 5h. Interferon (IFN)-γ production (pg/ml) after stimulation with lipopolysaccharide (LPS).
References
- 1.Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID-CINCA. Curr Rheumatol Rep. 2011;13:123–131. doi: 10.1007/s11926-011-0165-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gattorno M, Tassi S, Carta S, et al. Pattern of interleukin-1β secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 2007;56:3138–3148. doi: 10.1002/art.22842. [DOI] [PubMed] [Google Scholar]
- 3.Henderson C, Goldbach-Mansky R. Monogenic IL-1 mediated autoinflammatory and immuynodeficiency symdromes: finding the right balance in response to danger signals. Clin Immunol. 2010;135:210–222. doi: 10.1016/j.clim.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kusick VAMc. Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), 2011. Available at: http://omim.org/ (accessed 1 May 2012)
- 5.Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD. The clinical continuum of cryopyrinopathies. Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutations of a new gene encoding a putative purin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Touitou I. Infevers: an online database for autoinflammatory mutations. 2012. http://fmf.igh.cnrs.fr/ISSAID/infevers/index.php (accessed 1 May 2012)
- 8.Tanaka N, Izawa K, Saito MH, et al. High incidence of NLRP3 somatic mosaicism in chronic infantile neurological cutaneous and articular syndrome patients: results of an international multicenter collaborative study. Arthitis Rheum. 2011;63:3625–3632. doi: 10.1002/art.30512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw PJ, McDermott MF, Kanneganti T-D. Inflammasomes and autoimmunity. Trends Mol Med. 2011;17:57–64. doi: 10.1016/j.molmed.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holland SM, Dorman SE, Kwon A, et al. Abnormal regulation of interferon-γ, interleukin-12 and tumor necrosis factor-α in human interferon-γ receptor 1 deficiency. J Infect Dis. 1998;178:1095–1104. doi: 10.1086/515670. [DOI] [PubMed] [Google Scholar]
- 13.Aróstegui JI, Aldea A, Modesto C, et al. Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/PYPAF1/NALP3 gene. Arthritis Rheum. 2004;50:4045–4050. doi: 10.1002/art.20633. [DOI] [PubMed] [Google Scholar]
- 14.Janssen R, Verhard E, Lankester A, ten Cate R, van Dissel JT. Enhanced interleukin-1β and interleukin-18 release in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2004;50:3329–3333. doi: 10.1002/art.20494. [DOI] [PubMed] [Google Scholar]
- 15.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–323. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 16.Saito M, Fujisawa A, Nishikomori R, et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2005;52:3579–3585. doi: 10.1002/art.21404. [DOI] [PubMed] [Google Scholar]
- 17.Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the ILRN locus. N Engl J Med. 2009;360:2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsubayashi T, Sugiura H, Arai T, Oh-Ishi T, Inamo Y. Anakinra therapy for CINCA syndrome with a novel mutation in exon 4 of the CIAS1 gene. Acta Pediatr. 2006;95:246–249. doi: 10.1080/08035250500341451. [DOI] [PubMed] [Google Scholar]
- 19.Kuemmerle-Deschner JB, Lohse P, Koetter I, et al. NLRP3 E311K mutation in a large family with Muckle–Wells syndrome – description of a heterogeneous phenotype and response to treatment. Arthritis Res Ther. 2011;13:R196. doi: 10.1186/ar3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carta S, Tassi S, Delfino L, et al. Deficient production of IL-1 receptor antagonist and IL-6 coupled to oxidative stress in cryopyrin-associated periodic syndrome monocytes. Ann Rheum Dis. 2012;71:1577–1581. doi: 10.1136/annrheumdis-2012-201340. [DOI] [PubMed] [Google Scholar]
- 21.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID) Arthritis Rheum. 2002;46:3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujisawa A, Kambe N, Saito M, et al. Disease-associated mutations in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic cells. Blood. 2007;109:2903–2911. doi: 10.1182/blood-2006-07-033597. [DOI] [PubMed] [Google Scholar]
- 23.Saito M, Nishikomori R, Kambe N, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008;111:2132–2141. doi: 10.1182/blood-2007-06-094201. [DOI] [PubMed] [Google Scholar]
- 24.Hedrich CM, Bruck N, Paul D, Hahn G, Gahr M, Rösen-Wolff A. ‘Mutation negative’ familial cold autoinflammatory syndrome (FCAS) in an 8-year-old boy: clinical course and functional studies. Rheumatol Int. 2012;32:2629–2636. doi: 10.1007/s00296-011-2019-3. [DOI] [PubMed] [Google Scholar]
- 25.Lasiglie D, Traggiai E, Federici S, et al. Role of IL-1 beta in the development of human T(H)17 cells: lesson from NLPR3 mutated patients. PLOS ONE. 2011;6:e20014. doi: 10.1371/journal.pone.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Netea MG, Nold-Petry CA, Nold MF, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haverkamp MH, van Dissel JT, Holland SM. Human host genetic factors in nontuberculous mycobacterial infection: lessons from single gene disorders affecting innate and adaptive immunity and lessons from molecular defects in interferon-γ-dependent signaling. Microbes Infect. 2006;8:1157–1166. doi: 10.1016/j.micinf.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 28.Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-γ-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;355:1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 29.Dorman SE, Holland SM. Mutation in the signal transducing chain of the interferon-γ receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon γ receptor 1 deficiencies. Lancet. 2004;364:2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 31.Rosenzweig SD, Dorman SE, Uzel G, et al. A novel mutation in IFN-γ receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. 2004;173:4000–4008. doi: 10.4049/jimmunol.173.6.4000. [DOI] [PubMed] [Google Scholar]
- 32.Noordzij JG, Hartwig NG, Verreck FAW, et al. Two patients with complete defects in interferon gamma receptor-dependent signaling. J Clin Immunol. 2007;27:490–496. doi: 10.1007/s10875-007-9097-8. [DOI] [PubMed] [Google Scholar]
- 33.Sologuren I, Boisson-Dupuis S, Pestano J, et al. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20:1509–1523. doi: 10.1093/hmg/ddr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sibley CH, Plass N, Snow J, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease (NOMID) treated with Anakinra. Arthritis Rheum. 2012;64:2375–2386. doi: 10.1002/art.34409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fox E, Jayaprakash N, Pham T-H, Goldbach-Mansky R. The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non-human primates. J Neuroimmunol. 2010;223:138–140. doi: 10.1016/j.jneuroim.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The Nacht leucine rich repeat and pyrin domain containing protein 3 (NALP3) inflammasome activates interleukin (IL)-1β and IL-18. NALP3 has three domains: an N-terminal protein–protein interaction domain, called PYD; a central nucleotide-binding and oligomerization domain, called NACHT; and a C-terminal ligand-binding and regulatory leucin-rich repeat domain, called LRR. NACHT is encoded by exon 3. PYD interacts with a protein called ASC, which stands for ‘apoptosis-associated speck-like protein containing a caspase recruitment domain’ (CARD). This interaction then binds pro-caspase-1 to ASC via homodimerization of the respective CARD domains. This is called the NALP3 inflammasome. Upon activation by ligand binding [e.g. adenosine triphosphate (ATP)] to the LRR of NALP3 caspase-1 dimerizes, resulting in proximity-induced autoactivation. Active, p10/20-tetrameric caspase-1 is released from the complex to cleave IL-1β and IL-18 precursors into their active forms.
Fig. S2. Subdivision of patients. Flowchart of the individuals studied per experiment. Patients per group: •; patients per group with duration of treatment in months: ; patient numbers from Table 1: #; patients with Anakinra added to their peripheral blood mononuclear cells (PBMCs) in vitro or treated with Anakinra in vivo: V. Patients per group with duration of treatment in months per dose of Rilonacept .
Fig. S3. Diagram explaining low stimulation indices (SIs) in neonatal-onset multi-system inflammatory disease (NOMID) patients with improvement of SIs by Anakinra therapy in vitro but not in vivo.
Table S1. Raw data for Fig. 2a. Interleukin (IL)-1β production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S2. Raw data for Fig. 2b. Interleukin (IL)-6 (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S3. Raw data for Fig. 2c. Interleukin (IL)-10 (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S4. Raw data for Fig. 2d. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S5. Raw data for Fig. 2e. Interleukin (IL)-12p70 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S6. Raw data for Fig. 2f. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S7. Raw data for Fig. 2g. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA)+interleukin (IL)-12.
Table S8. Raw data for Fig. 3a. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S9. Raw data for Fig. 3b. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S10. Raw data for Fig. 3c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S11. Raw data for Fig. 3d. Interleukin (IL)-12p70 production (pg/ml) after stimulation with LPS.
Table S12. Raw data for Fig. 3e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S13. Raw data for Fig. 3f. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S14. Raw data for Fig. 3g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S15. Raw data for Fig. 3h. Interleukin (IL)-12p70 production (pg/ml) after stimulation with lipopolysaccharide (LPS)+interferon (IFN)-γ.
Table S16. Raw data for Fig. 4a. Interleukin (IL)-1β (pg/ml) production after stimulation with phytohaemagglutinin (PHA).
Table S17. Raw data for Fig. 4b. Interleukin (IL)-6 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S18. Raw data for Fig. 4c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S19. Raw data for Fig. 4d. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S20. Raw data for Fig. 4e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S21. Raw data for Fig. 4Ff. Interleukin (IL)-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S22. Raw data for Fig. 4g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S23. Raw data for Fig. 4h. Interferon (IFN)-γ production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S24. Raw data for Fig. 5a. Interleukin (IL)-1β production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S25. Raw data for Fig. 5b. Interleukin (IL)-6 production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S26. Raw data for Fig. 5c. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S27. Raw data for Fig. 5d. Interferon (IFN)-γ production (pg/ml) after stimulation with phytohaemagglutinin (PHA).
Table S28. Raw data for Fig. 5e. Interleukin (IL)-1β (pg/ml) production after stimulation with lipopolysaccharide (LPS).
Table S29. Raw data for Fig. 5f. IL-6 production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S30. Raw data for Fig. 5g. Tumour necrosis factor (TNF) production (pg/ml) after stimulation with lipopolysaccharide (LPS).
Table S31. Raw data for Fig. 5h. Interferon (IFN)-γ production (pg/ml) after stimulation with lipopolysaccharide (LPS).