

Abstract
As part of the SAMPL4 blind challenge, filtered AutoDock Vina ligand docking predictions and large scale binding energy distribution analysis method binding free energy calculations have been applied to the virtual screening of a focused library of candidate binders to the LEDGF site of the HIV integrase protein. The computational protocol leveraged docking and high level atomistic models to improve enrichment. The enrichment factor of our blind predictions ranked best among all of the computational submissions, and second best overall. This work represents to our knowledge the first example of the application of an all-atom physics-based binding free energy model to large scale virtual screening. A total of 285 parallel Hamiltonian replica exchange molecular dynamics absolute protein-ligand binding free energy simulations were conducted starting from docked poses. The setup of the simulations was fully automated, calculations were distributed on multiple computing resources and were completed in a 6-weeks period. The accuracy of the docked poses and the inclusion of intramolecular strain and entropic losses in the binding free energy estimates were the major factors behind the success of the method. Lack of sufficient time and computing resources to investigate additional protonation states of the ligands was a major cause of mispredictions. The experiment demonstrated the applicability of binding free energy modeling to improve hit rates in challenging virtual screening of focused ligand libraries during lead optimization.
Keywords: Binding free energy, Reorganization free energy, Free energy ligand screening, BEDAM, HIV Integrase
Introduction
Molecular recognition is an essential component for virtually all biological processes. In pharmaceutical research there is a great interest in computer models capable of predicting accurately the strength of protein-small molecule association. These models can help in the identification of novel medicinal drugs and optimize their potency. Thermodynamically, the strength of the association between a ligand molecule and its target receptor is measured by the standard binding free energy or, equivalently, the corresponding dissociation/association equilibrium constants. Because it is very challenging to model these quantities from first principles, empirical models are most often employed in applied research. Docking & scoring methods are widely used in virtual screening campaigns whereby likely binders are selected from large libraries in terms of their geometrical and energetic compatibility with known or assumed structures of the protein receptor and, by so doing, narrow down the search for a lead compound.
While docking & scoring methods have reached a level of maturity [1–5], particularly with respect to structural predictions [6, 7], improvements in virtual screening accuracy are needed to make in-silico screening a more effective tool. Including a higher level of theory and physical realism is one potential route towards this goal. In addition, it is hoped that models which incorporate some aspects of dynamical behavior might better address important aspects of drug development such as lead optimization, specificity, toxicity, and resistance. High resolution molecular mechanics potentials coupled with molecular dynamics conformational sampling are the basis of physics-based binding free energy models, which originate from clear statistical mechanics formalisms [8–18]. However the general applicability of physics-based binding free energy models for the prediction of protein-ligand binding affinities remains uncertain [19–22]. Part of the reason is that it is difficult to draw general conclusions from the body of work represented in the existing literature, which is scattered over a large array of methods each focused on very small datasets.
The SAMPL4 HIV integrase blind challenge [23, 24] offers a unique opportunity to address some of these questions. Our first objective was to assess feasibility. That is to establish whether current software and hardware technologies are up to the task of performing the hundreds of binding free energy calculations required by virtual screening. The second objective, assuming that the first could be met, was to assess in an unbiased fashion whether free energy estimates actually lead to improved screening results. In addition to these methodological questions, our groups share a deep interest in contributing to the development of novel HIV anti-viral therapies [25–30]. The HIV integrase enzyme is a relatively new medicinal target with several potential inhibition sites, including the LEDGF site, which could be further exploited [31, 32]. Modeling of the SAMPL4 ligand candidates will add to our understanding of this important target.
As part of the HIV integrase virtual screening SAMPL4 challenge [33], in this work we apply the binding energy distribution analysis method (BEDAM) binding free energy model [14, 34–36] and AutoDock Vina [4] and filtering criteria to screen likely binders to the LEDGF site of integrase. As described in the sister paper in this volume [37], AutoDock Vina plus filtering and visually inspection (hereafter referred to as “AD Vina”) are used to predict the structures of the complexes and to prioritize free energy calculations. BEDAM calculations, started from these initial structures, provided binding free energy estimates to rank likely binders. BEDAM estimates the protein-ligand binding free energy by simulating an alchemical thermodynamic path along which the ligand is transferred from the water solution to the protein binding site. In addition to net protein-ligand interaction energies, the binding free energy so obtained is influenced by intramolecular strain and entropic losses of both the receptor and the ligand. These effects, collectively referred to as reorganization free energy contributions [15, 38], while affecting binding significantly, are often not captured by empirical scoring functions. Slow convergence rates caused by incomplete conformational sampling along the alchemical path is a well known problem in molecular dynamics-based binding free energy methods. To address these challenges BEDAM employs advanced conformation sampling methods based on parallel Hamiltonian replica exchange (HREM) [16], and pre-computed conformational reservoirs [34].
The SAMPL4 set is a focused library composed of similar ligands grown from variations on a few common molecular scaffolds developed for lead optimization. This kind of ligand database is more difficult to screen than the diverse databases normally processed in virtual screening applications during hit discovery. In addition, the LEDGF binding site of integrase is large and known to be capable of accommodating a variety of ligand sizes. Ligand flexibility is also likely to play a key role in the thermodynamics of binding. These features make the screening task quite chal lenging for traditional methods such as docking, and particularly suitable for testing the added value of binding free energy-based screening protocols in these challenging situations. The blind set up of the SAMPL4 experiment adds further depth to these aspects allowing for a realistic and unbiased assessment of theories, methods, and practices.
Methods
System preparation
The dimer of the CCD domain of HIV integrase was prepared from known crystal structures [37]. Bound ligands, water molecules and crystallization ions were removed and protein sidechain protonation states were assigned assuming neutral pH. HIS171, a key residue of the LEDGF binding site, was protonated at the Nδ position (see below). Preparatory molecular dynamics simulations of the dimer with explicit solvation and the OPLS force field conducted with the DESMOND [39] program were employed to probe the flexibility of the dimer and guide further setup choices for the LEDGF binding site. These studies, while confirming the flexibility of the “140s” and “190s” loops [40], showed relatively little flexibility around the LEDGF site with the notable exception of the ARG167-HIS171 loop next to the LEDGF binding site. Based on this information a truncated model of the LEDGF binding site was constructed for the purpose of BEDAM binding free energy calculations which includes residues 68–90 and 158–186 of chain A and residues 71–138 of chain B. This truncated model contains at a minimum all receptor residues with at least one atom closer than 12 Å of any atom of the ligand in the 3NF8 crystal structure. Additional residues were included to minimize the number of chain breaks. Chain terminals were capped by acetyl and N-methylacetamide groups.
The molecular models for the 311 compounds provided as part of the SAMPL4 challenge after removing duplicated entries, were prepared using the LigPrep workflow of the 2012 version of the Maestro program (Schrödinger, LLC). Protonation and tautomerization states were assigned based on a pH of 7 ± 2. Ionization penalties were computed at pH 7 using Epik [41], also part of the Schrödinger molecular modeling suite. The protonation states provided by SAMPL4 were used but some of them resulted in ionization penalties larger than 4 kcal/mol. These occurrences were considered unlikely to lead to strong binding and were not further considered. The resulting database of 451 unique protonation states of the 311 ligands was the subject of the AD Vina virtual screening campaign conducted at the Scripps Research Institute as described in the sister publication in this volume [37]. This resulted in a set of structural predictions for the poses of the ligands to the LEDGF binding site of the integrase CCD domain. The corresponding complexes were prioritized based on docking rankings and subject to BEDAM binding free energy analysis using the docked pose as the starting conformation. BEDAM binding free energies were ultimately obtained for 285 of the 451 models of the ligands (see “Discussion” section).
BEDAM binding free energy protocol
The BEDAM method [14] computes the absolute binding free energy between a receptor A and a ligand B employing a λ-dependent effective potential energy function with implicit solvation of the form
| (1) |
where r = (rA, rB) denotes the atomic coordinates of the complex, with rA and rB denoting those of the receptor and ligand, respectively,
| (2) |
is the potential energy of the complex when receptor and ligand are dissociated, and
| (3) |
is the binding energy function defined for each conformation r = (rB, rA) of the complex as the difference between the effective potential energies U(r) with implicit solvation [42] of the bound and dissociated conformations of the complex without internal conformational rearrangements.
It is easy to see from Eqs. (3), (1), and (2) that Uλ = 1 corresponds to the effective potential energy of the bound complex and Uλ= 0 corresponds to the state in which the receptor and ligand are not bound. Intermediate values of λ trace an alchemical thermodynamic path connecting these two states. The binding free energy ΔGb is by definition the difference in free energy between these two states. The standard free energy of binding is related to this by the relation [8]
| (4) |
where C° is the standard concentration of ligand molecules (C° = 1 M, or equivalently 1,668 Å–3) and Vsite is the volume of the binding site (see below).
To improve convergence of the free energy near λ = 0, in this work we employ a modified “soft-core” binding energy function of the form
| (5) |
where umax is some large positive value (set in this work as 1,000 kcal/mol). This modified binding energy function, which is used in place of the actual binding energy function [Eq. (3)] wherever it appears, caps the maximum unfavorable value of the binding energy while leaving unchanged the value of favorable binding energies [35].
The multistate Bennett acceptance ratio estimator (MBAR) [35, 43] is used here to compute the binding free energy ΔGb from a set of binding energies, u, sampled from molecular dynamics simulations at a series of λ values. For later use we introduce here the reorganization free energy for binding defined by the expression [15]
| (6) |
where ΔEb = 〈u〉1 is the average binding energy of the complex and is the standard binding free energy. The former is computed from the ensemble of conformations of the complex collected at λ = 1 and is computed by difference using Eq. (6).
Conformational sampling
Rather than simulating each λ state independently, BEDAM employs a HREM λ-hopping strategy whereby simulations (replicas) periodically attempt to exchange λ values through Monte Carlo (MC) λ-swapping moves. λ-exchanges are accepted with the Metropolis probability min[1, exp(–βΔλΔu)] [14] where Δλ is the difference in λ's being exchanged and Δu is the difference in binding energies between the replicas exchanging them. Replica exchange strategies of this kind yield superior conformational sampling and more rapid convergence rates by allowing conformational transitions to occur at the value of λ at which they are most likely to occur and to be then propagated to other states [16].
Conformational reservoirs were employed to further enhance conformational sampling of internal degrees of freedom of the ligands [34, 36]. In this technique the ensemble of conformations for the separate receptor and ligand in solution are obtained from temperature replica exchange calculations [44] and saved in repositories referred to as conformational reservoirs. The conformational reservoirs then take the place of the λ = 0 replica of conventional BEDAM calculations. k-exchanges with the repositories follow the same acceptance rule as for the other replicas. When an exchange is requested, one conformation for the ligand is selected at random from the reservoir and placed in a random position and orientation within the binding site volume of the receptor. The binding energy of the resulting complex is evaluated and inserted in the Metropolis acceptance step as above. If the exchange is accepted the conformation from the reservoir is passed on to the next replica and begins to be propagated by MD and λ-exchanges as in conventional BEDAM.
Because the reservoir represents a canonical ensemble of conformations, overall the method is canonical, while providing greater coverage of the internal degrees of freedom of the ligands. In addition, the method is computationally efficient because, due to the small system size, the cost of obtaining ligand reservoirs is negligible relative to the computational cost of the binding free energy calculations.
Computational details
In the current implementation BEDAM employs an effective potential in which the effect of the solvent is represented implicitly by means of the AGBNP2 implicit solvent model [42] together with the OPLS-AA [45, 46] force field for covalent and non-bonded interatomic interactions. Force field parameters were assigned using Schrödinger's automatic atomtyper [47].
Parallel molecular dynamics simulations were conducted with the IMPACT program [47]. The simulation temperature was set to 300 K. We employed 20 intermediate steps at λ = 0, 0.001, 0.002, 0.0033, 0.0048, 0.006, 0.008, 0.01, 0.04, 0.07, 0.1, 0.25, 0.35, 0.45, 0.55, 0.65, 0.71, 0.78, 0.85, and 1 [35]. The binding site volume was defined as any conformation in which the center of mass of the ligand was within 6 Å of the center of mass of the LEDGF binding site defined in terms of the Cα atoms of residues 168–174 and 178 of chain A and residues 95, 96, 98, 99, 102, 125, 128, 129, and 132 of chain B (residue and chain designations according to the 3NF8 crystal structure). The ligand was sequestered within this binding site volume by means of a flat-bottom harmonic potential. Based on this definition the volume of the binding site, Vsite, is calculated as 904 Å3 corresponding to –kT ln C° Vsite = 0.69 kcal/mol. The Cα atoms of the residues of the CCD domain of integrase were restrained to the input structure by means of spherical harmonic restraints with force constant of 1 kcal/mol Å2, with the exception of the residues closest to the binding site (residues 167–178 of chain A and 95–102 and 124–132 of chain B) which were left free.
The temperature replica exchange simulations to obtain conformational reservoirs for the ligand utilized eight replicas distributed between 300 and 600 K and were 10 ns in length. The collected ensembles at a temperature of 300 K, saved every 10 ps, constituted the conformational reservoirs used in the parallel HREM calculations.
BEDAM calculations were performed for 1.1 ns of MD per replica (22 ns total for each complex) starting from the docked ligand poses. Data from the last 600 ps/replica was used for free energy analysis. Binding energies were sampled with a frequency of 1 ps for a total of 20 × 600 = 12,000 binding energy samples per complex. Uncertainties in the binding free energies were estimated by the difference between the results from data from the 400–1,000 and 600–1,100 ps sections of the trajectory.
Approximately two thirds of the BEDAM free energy calculations were conducted on the Gordon cluster at the San Diego Supercomputing Center as part of the XSEDE NSF consortium. Additional calculations were conducted at the BioMaPS High Performance Computing Center at Rutgers University and on the Garibaldi cluster at the Scripps Research Institute at San Diego. Each BEDAM calculation was typically performed on 80 processing cores concurrently and took two days to complete. The computational campaign lasted approximately seven weeks from late May to July 2013 and consumed approximately 1.2 million CPU hours. Throughput was limited by the amount of available CPU resources and our XSEDE allocation (see below).
Ultimately we were able to collect binding free energy estimates for 285 of the 451 protonation models of the 311 ligands and only for the LEDGF site of integrase. Computed binding free energies were combined with ionization penalties (see above) to yield binding free energy scores. The binding free energy score for a ligand was set as the most favorable free energy score among all of its protonation states. Ligands with free energy scores of –4 kcal/mol or better were predicted as LEDGF integrase binders. This cutoff value is approximately equivalent to a milli-molar dissociation constant, which corresponds to the weakest LEDGF site binders that have been crystallized [48]. 67 of the 311 ligands fell in the category. Ligands with missing free energy score were considered as non-binders. Ligand rankings were based on the available binding free energy scores with top binders having the most favorable binding free energies; ligands with missing data were ranked last in random ranking order. The set of predicted binders together with the overall rankings thus computed were submitted to SAMPL4 on July 16 2013 and assigned prediction ID #135.
Results and performance
Binding free energy predictions
The BEDAM binding free energy estimates to the LEDGF site of integrase for 285 of the 451 protonation states of the 311 ligands are listed in Table S1 of the supplementary information, which includes uncertainties. The results for the top 10 binders are given in Table 1. The free energy score (“FE Score”), obtained by the adding the ionization penalty (“I.P.”) to the BEDAM binding free energy, has been used to rank ligands. Ligands denoted in bold face in Table 1 are confirmed LEDGF integrase binders.
Table 1.
Computed BEDAM binding free energies, , ionization penalties, “I.P.”, and resulting free energy scores, “FE Score”, for the top ten predicted binders of the LEDGF site of integrase
| Ligand ida | Ligand structure | FE scoreb | b | I.P. b,c | Δ Eb b | b |
|---|---|---|---|---|---|---|
| AVX17557_3 |
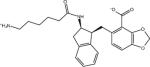
|
–8.9 | –8.9 | 0.0 | –51.6 | 42.7 |
| AVX101124_0 |
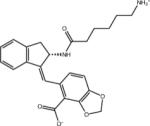
|
–8.4 | –8.4 | 0.0 | –50.1 | 41.7 |
| AVX17556_3 |
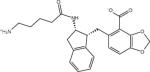
|
–8.2 | –8.2 | 0.0 | –47.1 | 38.9 |
| AVX17556_1 |
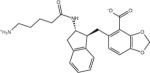
|
–7.9 | –7.9 | 0.0 | –46.0 | 38.1 |
| AVX101133_0 |
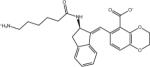
|
–7.5 | –7.5 | 0.0 | –46.8 | 39.3 |
| AVX17556_0 |
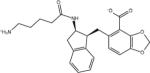
|
–7.4 | –7.4 | 0.0 | –48.8 | 41.4 |
| AVX17285_0 |
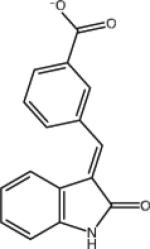
|
–7.4 | –7.4 | 0.0 | –37.9 | 30.5 |
| AVX38753_3_1 |
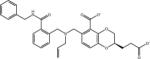
|
–7.4 | –7.9 | 0.5 | –47.3 | 39.4 |
| AVX101124_1 |
|
–7.3 | –7.3 | 0.0 | –48.0 | 40.7 |
| AVX38789_2_1 |
|
–7.3 | –7.8 | 0.5 | –45.4 | 37.6 |
Also listed are thermodynamic decomposition results of into average binding energy, ΔEb, and reorganization free energy, , components. Ligands in bold face are crystallographically confirmed LEDGF binders
See text for a description of ligand identifiers. The list in this table follows the rankings submitted blindly to SAMPL with the exception of the removal of ligand AVX17268_1, later excluded from the challenge
In kcal/mol
Ionization penalty
Ligand identifiers are those provided by the SAMPL organizers whereby a sequential index denotes a specific stereo-isomer of the compound. This is followed at times by a second sequential index added by us to denote a specific protonation state. So for example AVX17557_3 denotes the fourth stereo-isomer of compound AVX17557 and AVX38753_3_1 denotes the second protonation state of the fourth stereoisomer of compound AVX38753. A missing second index signifies the protonation state as provided by SAMPL, which for many ligands is the only relevant protonation state.
While inclusion of ionization penalties did not significantly affect the ligand rankings, they were helpful at discounting at the outset ligand forms with very unfavorable protonation states unlikely to contribute to binding. In any case, the exploration of multiple protonation states has been key in a number of cases. For example AVX38753_3 was correctly recognized as a LEDGF binder only after examining its unprotonated form (AVX38753_3_1 in Table 1) corresponding to a binding free energy score of –7.4 kcal/mol. The original protonated form resulted in a binding free energy of only –0.7 kcal/mol which alone would have resulted in a misclassification of AVX38753_3 as a non-binder.
It should be noted that in the provided ligand library [23] protonated alkyl nitrogen centers have been considered as potential chiral centers even though the resulting conformational stereoisomers are in equilibrium in solution and could not have been assayed independently. This is particularly evident when, as for the AVX38753_3_1 entry in Table 1, the deprotonated form is considered. AVX38753_3_1 is in dynamic equilibrium with AVX38753_2_1 (not shown) by nitrogen inversion. (The 2D chemical representations in Tables 1 and S1 do not distinguish between the two conformational stereoisomers.) The BEDAM calculations, which were restricted to one or the other of the two conformational stereoisomers, predicted more favorable binding for AVX38753_3_1 than for AVX38753_2_1 (–7.4 and –1.8 kcal/mol, respectively). In this case, assuming equal populations of the two conformational stereoisomers in solution, the binding would be largely dominated by the first stereoisomer, which has been indeed verified by crystallography.
In general the binding free energies listed in Tables 1 and S1 should be considered reflective of hypothetical binding affinities conditional on protonation state and conformational stereoisomer form, not of the actual binding affinities that would be measured by assaying the parent compound. We have not attempted to integrate the conditional binding affinities, and conformational populations and protonation state populations in solution to arrive at binding free energy estimates comparable to measurable binding affinities. Rather, computed conditional binding free energies were used to rank ligand forms to predict the most likely forms observed in crystallographic screening experiments.
Thermodynamic decomposition
Tables 1 and S1 also report the decomposition of the computed binding free energies into the average binding energy, ΔEb, and the reorganization free energy . The average binding energy measures the thermodynamic driving force towards binding provided by favorable receptor-ligand interactions. Conversely, the reorganization free energy measures configurational entropy loss and intramolecular energetic strain caused by complex formation, which, collectively, oppose binding [15]. As previously observed, binding energy and reorganization free energy contributions oppose each other and binding free energies, being much smaller in magnitude, result from a large compensation between these opposing effects.
In general the strongest binders tend to have some of the most favorable binding energies. However the opposite is often not the case. Ligands with very favorable binding energies are not necessarily good binders. For example among the top ten with respect to binding energy only four correspond to binding free energies better than –4 kcal/mol, the cutoff considered here to discriminate binders from non-binders. Furthermore, as further discussed below, rankings based on binding energies are significantly worse than binding free energy rankings in predicting true binders. Conversely, ligand forms with only moderate binding energies sometimes result in very favorable binding free energies. This typically happens with relatively small ligands, such as AVX17285_0 in Table 1, which incur small reorganization penalties. In this respect, the inclusion of reorganization free energies helps overcome the overly strong correlation between interaction energy-based scores with ligand size.
Prediction performance
Only a fraction of the ligands and ligand forms were subjected to BEDAM binding free energy analysis and only binding to the LEDGF site was probed. For the purpose of the SAMPL submission, ligands lacking computed binding free energy data were considered non-binders and ranked last after those with estimated but unfavorable binding free energies. As a result, screening prediction performance metrics based on prediction accuracy for each ligand in the database, such as the area under the Receiver Operating Characteristic (ROC) curve, are not fully applicable to our prediction set. Early recovery metrics such as the enrichment factor, which probe the ability to pick out top binders, are more relevant to judge performance of our prediction protocol both in terms of the efficacy of prioritizing calculations and of correctly assigning binding free energy scores to the ligands that were considered.
Of the ten ligands listed in Table 1 half are confirmed LEDGF binders and half are not. This 50 % ratio can be compared to the fraction of the 53 unique confirmed LEDGF binders in the original ligand database of 322 ligands (16.4 %) to yield an enrichment factor of 3. The enrichment factor represents the overall prediction performance of the method over random. This level of performance can also be compared to a perfect prediction in which all 10 top ligands are binders corresponding to a maximum enrichment factor of only 6 (100/16.4 %).
The enrichment factor for the top 10 % of the database (top 30 ligands approximately) has been used to compare the performance of SAMPL submissions. The value for our “in-silico” predictions is 2.21 (Fig. 1) second best among the submissions in the screening challenge behind only the effort of Voet et al. [49], which benefited from extensive prior experience with this medicinal target. The level of enrichment we obtained is very promising also considering that it has been evaluated with respect to all integrase binders, including the binders to the fragment and Y3 sites, which we have not explored. When considering only the confirmed LEDGF binders the enrichment factor for the top 10 % of our predicted binders increases to 2.45. This result should be compared to the maximum achievable 10 % enrichment factor, which is only 5.66, much lower than in typical virtual screening applications with large and diverse ligand datasets [3, 28].
Fig. 1.
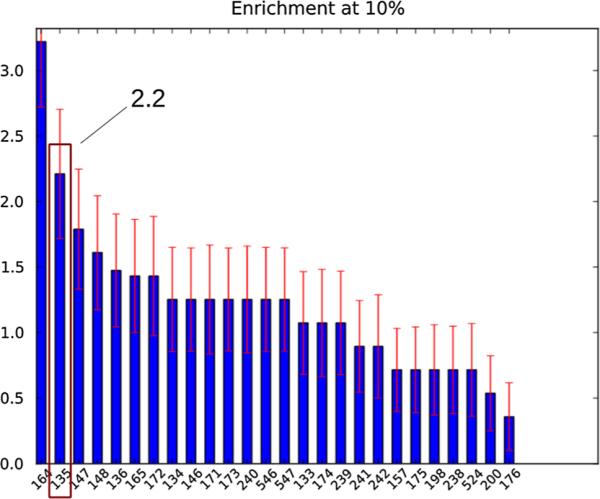
10 % enrichment factors for all of the HIV integrase SAMPL4 submission as computed by the SAMPL4 organizers. The docking and binding free energy-based submission described here corresponds to id 135, second from left
The early ROC curve for our predictions of LEDGF IN binders depicted in Fig. 2 shows that ligand rankings based on binding free energy scores are significantly better than random at picking true binders. In this analysis a true positive is defined as a confirmed LEDGF binder. The true positive rate is the fraction of true binders recovered while progressively going down the ranked list of complexes. The corresponding false positive rate is the ratio of the number of non-binders in the same set relative to the total number of non-binders in the database. At a false positive rate of 10 % (corresponding to the top 44 compounds in terms of binding free energy scores) approximately 33 % of the true binders are recovered, or 3.3 times better than random. At a false positive rate of 20 % (corresponding to the top 75 compounds) 44 % of the LEDGF binders are recovered. In comparison, as Fig. 2 shows, predictions based on the binding energy rankings alone are notably worse (see Fig. 2), achieving on average only 60 % of the prediction performance of the free energy rankings according to this measure. The ROC results also show that the additional discrimination power of the binding free energy rankings is not evident in the reorganization free energy scores, which represent physical effects neglected by the binding energy scores, as the best rankings developed from them perform worse than random (Fig. 2). These results indicate that the main source of added discrimination power of binding free energies resides in the combination of energetic and reorganization effects rather than in one of them alone.
Fig. 2.
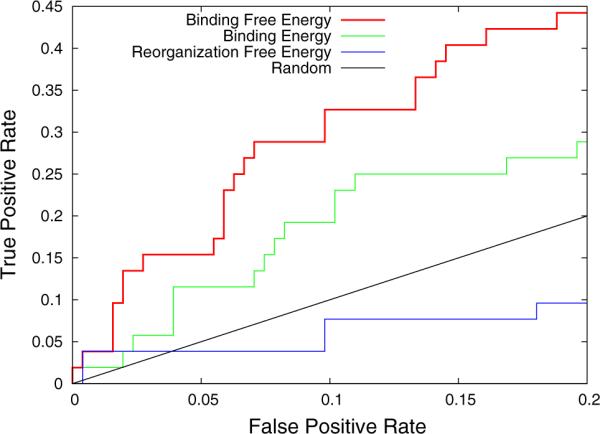
Early ROC curve for the classification of binders and non-binders to the LEDGF site of integrase. The ROC curve based on binding free energy rankings is in red (top), the ones based on binding energy rankings and reorganization free energy rankings are in green (middle) and blue (bottom) respectively. The straight black line is the 1:1 line corresponding to random picking. Results for sorting reorganization free energies from low to high are shown here, the reverse rankings yielded poorer performance
Analysis of binding free energy results for the LEDGF binders
Crystal structures of integrase inhibitors bound to the LEDGF site of integrase constitute a solid benchmark on which to judge our in-silico screening protocol. Ideally we would like the free energy screening method to score favorably all of the confirmed binders, and it is instructive to consider which factors have contributed in a favorable or unfavorable way towards this goal. The analysis of the prediction performance on the confirmed LEDGF binders is summarized in Table 2. Of the 53 confirmed LEDGF binders, 23 were correctly predicted (true positives). These had one or more protonation states with computed binding free energies of –4 kcal/mol or better. The remaining 30 binders were incorrectly classified as non-binders (false negatives). The largest fraction of false negatives (43 %) correspond to cases with missing binding free energy data, that is data not acquired due to insufficient time and computing resources. In this class of ligands none of the protonation states investigated yielded a favorable binding free energy, however some of the protonation states were not investigated and the corresponding ligands were arbitrarily classified as non-binders anyway. A smaller fraction (27 %) of false negatives correspond to ligands with intermediate binding free energy scores between –4 and –2 kcal/mol. These borderline scores, while above the cutoff we established, are not sufficiently unfavorable to clearly discount binding and are not considered useful to uncover clear shortcomings of the prediction methodology. Only 9 of the 30 false negatives (30 %) had substantially unfavorable free energy scores and, in addition, most of these can be attributed to incorrect starting structures for the BEDAM free energy simulations rather than incorrect free energy scoring (see below).
Table 2.
Summary of the analysis of results for the confirmed LEDGF binders
| 53 confirmed LEDGF binders | 23 correctly predicted (43 %) | 8 bad structures (35 %) | |
| 15 good structures (65 %) | |||
| 30 incorrectly predicted (57 %) | 13 not computed (43 %) | ||
| 8 intermediate score (27 %) | |||
| 9 bad score (30 %) | 7 bad structures (80 %) | ||
| 2 good structures (20 %) |
Moving left to right cases are classified as either correctly and incorrectly predicted (true positives and false negatives). False negatives are further classified based on the binding free energy scores (third column). Both true positives and false negatives are classified based on the quality of the initial binding mode (4th column)
In summary, these results show that the prediction methodology we employed was very successful at correctly describing confirmed LEDGF binders. A very good fraction of these (43 %) were correctly classified based on the binding free energy value. Only 9 out of 53 cases (17 %) can be traced to clear shortcomings of the docking and free energy analysis procedure, while most other misclassification correspond to either borderline or missing data not useful for drawing conclusions.
Importance of structural predictions
Initial structures for the BEDAM binding free energy calculations were provided by docking binding mode predictions as described in the accompanying report in this volume [37]. In general, molecular dynamics trajectories which constitute part of the BEDAM protocol were of insufficient length to modify these bound initial structures to a significant extent. Because of this and the fact that binding free energy predictions were limited to the LEDGF site, we decided to submit a single set of structural (phase II) predictions based only on the docking experiments as described [37].
The other important implication is that the binding free energy predictions submitted to SAMPL are, essentially, conditional on the starting conformation, and, consequently, their prediction accuracy is attributable to both the ability of the docking and filtering procedure to provide structures close to the actual binding modes and the accuracy of the binding free energy protocol to score favorably these binding modes. Indeed we observed a very strong correlation between the correct identification of true binders and the quality of the structural predictions from docking. The large majority of the clear false negative cases (seven out of nine), that is binders predicted to be non-binders with clearly unfavorable binding free energy scores (–2 kcal/mol or higher), had initial structures clearly incompatible with crystallographic observations (Table 2). In comparison, only a minority (35 %) of the true positives and none of the borderline false negatives correspond to initial structures distinct from the crystallo-graphic pose. On the other hand only 7 of the 30 false negatives are attributable to structural prediction errors.
These findings underscore both the dependency of the binding free energy protocol on the availability of accurate structural predictions, and the very good performance of the AD Vina protocol in this respect, as also confirmed by the excellent performance of this method on the structure prediction phase II SAMPL challenge [37].
Explicit solvent modeling
Finally, we also performed double decoupling calculations (DDM) [8, 50, 51] in explicit solvent to explore at a higher level of detail the thermodynamics of binding of this system in a few cases. These calculations were also performed blindly, prior to obtaining experimental data from the SAMPL organizers. The protein receptor is modeled with the Amber ff99sb-ILDN force field [52], and the ligands are described by the Amber GAFF parameters set. The partial charges of the ligands are obtained using the AM1-BCC method. A DDM calculation involves two legs of simulation, in which a restrained ligand is gradually decoupled from the receptor binding pocket or from the aqueous solution [8]. In each leg of the decoupling simulations, the Coulomb interaction is turned off first using 11 λ windows, and the Lennard-Jones interactions are then turned off in 17 λ windows. (Electrostatic decoupling: λ = 0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0; Lennard-Jones decoupling: λ = 0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.55, 0.6, 0.65, 0.7, 0.75, 0.8, 0.85, 0.9, 0.94, 0.985, 1.0). The simulations were carried out using the Gromacs4.6.1 package [53].
The results of the explicit solvent calculations are given in Table 3. While the DDM calculations overestimate binding free energies in general, a clear separation between binders and non-binders appears achievable only when all of the following conditions are satisfied: (a) starting from a good initial structure; (b) the Nδ atom of His171 is protonated; (c) the ligand is relatively small, i.e. contains less than 30 heavy atoms. In the simulated structures of the binders, the hydrogen atom on the Nδ of His171 was observed to interact with the carboxylate moiety on the ligands. It is likely that the both the doubly protonated His171 and the monoprotonated Nδ are significantly populated in the bound state: using PROPKA [54] we calculated that the pKa of His171 to be shifted from 6.3 in unbound state to 7.3 when a ligand carrying a benzoxole carboxylate moiety is bound. We obtained very similar binding free energies using either the doubly protonated or Nδ protonated forms of His171 (not shown). Compared with the Nε protonated form, the protonation at Nδ is estimated to enhance binding affinity by approximately –4 kcal/mol, due to the favorable electrostatic interaction between Nδ-H and the carboxylate group on the ligand. Significant errors in the calculated free energies using DDM are attributed to hysteresis for the decoupled ligand state in the gas phase. The strong, unscreened ligand intramolecular interaction in the gas phase leads to a highly rugged energy surface, causing conformational trapping in the nanoseconds-length simulations. Simulation of the intermediate gas phase reference state is avoided naturally in the implicit solvent alchemical scheme used in BEDAM.
Table 3.
Binding free energies to the LEDGF site of integrase computed with explicit solvation for multiple protonation states of the receptor
| Ligand | Binder/nonbinder | a | Simulation setup |
|---|---|---|---|
| AVX17375_2 | Binder | –4.5 | Bad initial pose, H171 protonated at Nε |
| AVX17375_2 | Binder | –8.7 | Good initial pose, H171 doubly protonated |
| AVX17734_1 | Nonbinder | –4.7 | Large ligand, gas phase sampling not converged |
| AVX17375_3 | Nonbinder | –7.6 | Same as above |
| AVX38743_5 | Binder | –2.1 | Good initial pose, H171 protonated at Nε |
| AVX38743_5 | Binder | –7.9 | Good initial pose, H171 doubly protonated |
| AVX40813_0 | Nonbinder | –4.9 | H171 protonated at Nε |
| AVX62525_3 | Nonbinder | –4.1 | H171 protonated at Nε |
In kcal/mol
Discussion
The SAMPL integrase virtual screening experiment offered a real world example of the use of structure-based computational methods to identify likely binders among a focused database of candidates. This task involved a set of challenges very different from those posed by early hit identification from a large database of diverse molecular scaffolds. In the latter situation, docking-based high throughput virtual screening strategies can narrow down the set of likely binders, with the primary goal of identifying some actual binders, rather than necessarily minimizing the number of false positives. Also, because only few likely binders are dispersed among possibly thousands of clear non-binders, relatively large enrichment factors can be expected only by considering, for example, steric and geometrical factors.
In contrast, in the current application participants were confronted with a focused library of candidate ligands derived from an already established molecular scaffold. This was particularly noticeable for the LEDGF binding site, to which virtually all of the ligands in the database could be successfully docked. Discrimination of binders from non-binders in this case had to therefore rely on other less coarse grained metrics such as the free energy of binding. The performance illustrated by the present blind predictions indicates that binding free energy methods can be successfully combined with accurate structural predictions from docking to tackle difficult virtual screening applications.
The usefulness of docking methods for binding mode prediction is well documented. The accuracy of AD Vina structural predictions on these difficult targets have been key to achieve a high level of screening performance. In contrast, the reliability and range of applicability of free energy methods, even when applied to known structures, remains to be fully established. Retrospective studies on small datasets, while abundant, have not been successful at giving a general and unbiased picture of the state of the field. The SAMPL challenge [34, 55] has offered us a unique opportunity, building upon recent work on host-guest systems [36], to validate our free energy protocol on a large scale and in an unbiased fashion. We believe that only by studying large datasets patterns can emerge that can be useful to establish advantages and shortcomings of the methodology.
In the past few years we have been refining our BEDAM protocol for binding free energy estimation. The method relies on a physics based effective potential with implicit solvation developed in our lab (OPLS/AGBNP2) [42, 56] and advanced Hamiltonian-hopping conformational sampling strategies. The implicit solvent description, while in principle less accurate than explicit solvation models, affords a number of simplifications particularly the ability to model directly the alchemical transformation from the unbound to the bound state without going through a gas phase intermediate. The latter requirement, typical of double decoupling methods with explicit solvation [8, 57] is a source of poor convergence especially for large and charged ligands as in the present application. The BEDAM method also relies on advanced parallel HREM conformational sampling strategies and conformational reservoirs techniques, as well as efficient statistical reweighting techniques [43, 58] to optimally merge data obtained along the binding thermodynamic path. We believe that all of these technologies have contributed to achieve the reasonably high level of prediction accuracy in the present SAMPL challenge, suggesting that free energy calculations can be used together with docking to improve enrichment over docking alone.
Binding free energies include effects such as conformational entropy loss and intramolecular strain (collectively quantified here by the binding reorganization free energy values), aspects that are neglected by more approximate models but are shown here to be significant and justify the use of free energy simulations to distinguish binders from nonbinders. This is clearly illustrated by the results in Fig. 2 which shows that rankings based on binding energies alone, which neglect reorganization free energy contributions, yield significantly lower prediction performance.
The binding free energy is the difference in free energy between the bound and unbound states and therefore it depends on the energetic and dynamical nature of both. The binding energy, that is the ligand-receptor net interaction energy, probes essentially only the bound state and is not sufficient in general to capture all of the effects contributing to the binding affinity. For example, the interplay between energetic and reorganization effects has been key to correctly identify as binders a number of small compounds with relatively weak ligand-receptor interaction energies and unusually small reorganization penalties. Some examples, illustrative of about 25 % of the correctly identified binders, are shown in Fig. 3. Most of these would have been mis-classified as non-binders using binding energy scores alone.
Fig. 3.
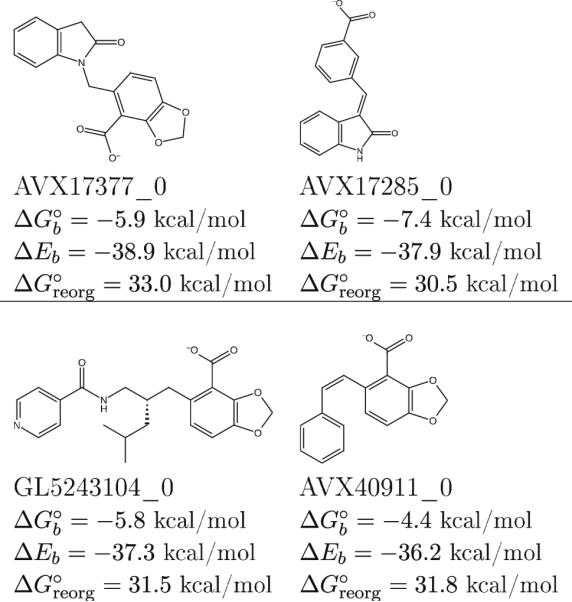
Examples of LEDGF integrase binders characterized by binding energies and reorganization free energies of small magnitude
Conversely, reorganization free energies helped to correctly identify non-binders, such as those illustrated in Fig. 9, characterized by large and favorable binding energies offset by equally large reorganization free energy losses. Such non-binders would have been incorrectly considered as actives by methods based only on ligand-receptor interaction energies. The examples in Fig. 9, are only few of the many similar occurrences of avoided false positives we observed.
Fig. 9.

Examples of nonbinders that have both correlated hydrogen bonds and occupied P1 sub-pockets. The binding of these ligands is severely hindered by the large reorganization free energy penalties, which are not clearly evident from the structures alone
These examples suggest that scoring strategies based mainly on ligand-receptor interaction energies, such as single-structure and single-trajectory versions of the MM-PB/GB/SA method [59], constitutes a less accurate model as compared with BEDAM and that significantly improved rankings can be obtained by including reorganization effects. The separate trajectory version of the MM-PB/SA method includes the portion of the reorganization free energy related to intramolecular strain and should in principle improve accuracy. However only the additional inclusion of conformational entropies would model the full binding free energies and fully capture the effects described here.
We observed that reorganization free energies correlate strongly with the size and flexibility of the ligand as for example measured by the number of rotatable bonds as in Fig. 4a. On the other hand the flexibility of the receptor also plays a major role as illustrated in the correlation plot in Fig. 4b. Inspection of BEDAM molecular dynamics trajectories, in which the interaction coupling parameter λ varies with time, reveals a progressive decrease of conformational fluctuations of both the receptor and the ligand as their interactions are turned on. Conversely, fluctuations tend to increase when interactions are turned off. This behavior is more evident for larger ligands and is observed with both binders and non-binders. Large ligands that are also binders have the ability to form numerous contacts with the receptor without causing large entropic losses.
Fig. 4.
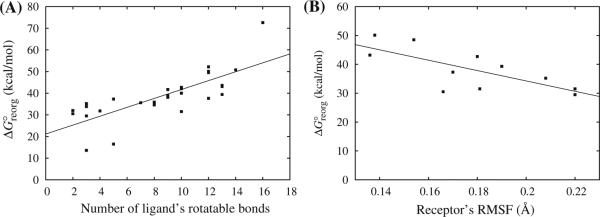
Reorganization free energies for a subset of complexes versus, a number of rotatable bonds of the ligand and, b root mean square fluctuation of the residues of the LEDGF binding site. The correlation coefficients are 0.59 and 0.53, respectively
Computational aspects
One of the goals for the participation in the SAMPL4 screening integrase challenge has been to establish the applicability of binding free energy calculations in virtual screening. The promising results obtained in the SAMPL challenge clearly indicate that binding free energy simulations can provide added value when used in conjunction with docking. This outcome was not inevitable as it is often the case that more rigorous theories do not lead to improved predictions in practice [36]. In the integrase screening challenge the homogeneity of the candidate compounds and the more central role of receptor and ligand flexibility has likely tipped the balance towards binding free energy measures relative to simpler interaction energy and empirical scoring-based methods. It is thus expected that binding free energy-based ligand screening will be useful in other similar circumstances; e.g. at a second stage of virtual screening, when a challenging focused pool of candidates is being considered.
Having established the usefulness of a binding free energy-based screening protocol, the second question to consider is whether the superior results obtained have been worth the extra effort. This question can be addressed only by looking at the practical aspects of the project. One immediate consideration is that a project such as this involving hundreds of binding free energy calculations simply could not have been considered without a high level of automation. Easy to use tools such as LigPrep and Epik have been essential in prepping the ligand models virtually without human intervention. Similarly, Schrödinger's OPLS automatic force field atomtyper available in the IMPACT MD engine has been key to streamlining system set up. The binding free energy project has also greatly benefited from automated protocols in the AD Vina structure prediction and prioritization phase.
Automation built into BEDAM binding free energy implementation has also played a key role. Binding free energy protocols are generally very difficult to streamline. We implemented a self-contained BEDAM utility written in Python which processes a list of inputs not much more extensive than those required for a docking calculation. None of these inputs had to be adjusted manually and individually for each ligand. The utility took care of producing the structural and procedural inputs for the IMPACT MD engine, automated the creation of job submission scripts, and performed the analysis of the simulation results. Without one or more of these aspects of automation the project could not have been completed, as it would have required an unsustainable commitment of human resources.
Despite the high level of automation it has not been possible to complete all of the binding free energy calculations. Part of the reason is that the CPU allocation on XSEDE resources, the source of most of the computational throughput, had to be shared among several projects and consequently were prematurely exhausted. On the other hand, meeting of SAMPL deadlines and the necessary coordination with preparatory docking studies, meant that only a few weeks were available to complete the binding free energy calculations. The imposed time schedule was not unreasonable as it is representative of scenarios in pharmaceutical applications. So in addition to the CPU allocation, the computational throughput, that is the number of ligands completed per day given the amount and capabilities of available CPU resources, has also been an important determinant. We are currently exploring the use of grid resources and distributed replica exchange algorithms to maximize throughput.
It is hard to say how much our predictions would have improved if all of the free energy calculations could have been completed and/or extended to longer simulation times. Longer simulation times would have yielded better converged estimates less dependent on initial conditions. Only 285 of the 451 ligand models were analyzed resulting in full coverage of only 59 % of the SAMPL library (127 of the 311 unique ligands had one or more protonation states not evaluated). Assuming that the observed recognition efficiency of the method on the sampled portion of the library is applicable to the portion not sampled, better coverage would have resulted in superior predictions.
Structural and thermodynamic insights
In addition to validating our in-silico virtual screening protocol, in this work we have also gained significant insights into the structural and energetics determinants governing ligand binding to the Integrase LEDGF site. As shown in Fig. 5, all 53 LEDGF site binders in SAMPL4 have the benzoic acid or benzodioxole/dioxine carboxylate core. In fact, all the known LEDGF inhibitors developed so far contain a carboxylate group that mimics the bidendate hydrogen bond interaction of LEDGF-derived peptides with the integrase amino groups of Glu170 and His171 [31]. The carboxylate moiety is clearly the main determinant for the ligand binding at the LEDGF site. However, while the absence of the correct chemical scaffold (i.e. the templates in Fig. 5) explains why many of the nonbinders do not bind, there are also many ligands that contain the required benzoic acid or benzodioxole/dioxine carboxylate groups yet they do not bind. So what are the causes for these molecules not to bind? Although obviously experimental structures for nonbinders are not available, we can use the simulated structures of the complexes and estimated binding free energies from BEDAM to gain insights on binding determinants.
Fig. 5.
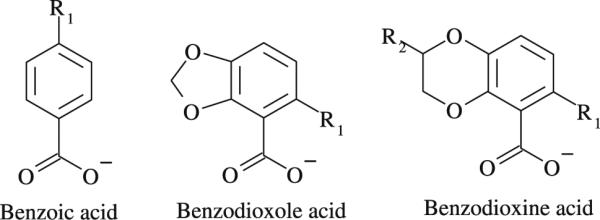
Chemical scaffolds of the integrase LEDGF binders in SAMPL4
Crystal structures show that most LEDGF binders (51 out of 53 binders in the SAMPL4 dataset) are found to adopt a dominant binding mode illustrated in Fig. 6. Three key interactions are identified: (1) the interaction of the carboxylate group of the ligand with the backbone amino groups of His171, Glu170 and Thr174 of chain A, which are well reproduced by AD Vina-predicted structures and maintained in the BEDAM HREM simulations. (2) The R1 group (see Fig. 5) occupies the hydrophobic P1 sub-pocket formed by Met178 and Leu102 on chain B, and Trp132 and Ala129 on chain A. (3) Dioxole/dioxine groups occupy the less hydrophobic P2 sub-pocket, formed by Glu95, Thr125 and Tyr99 on chain A and Lys173 on chain B. In the benzoic acid containing binders, the P2 sub-pocket is unoccupied.
Fig. 6.
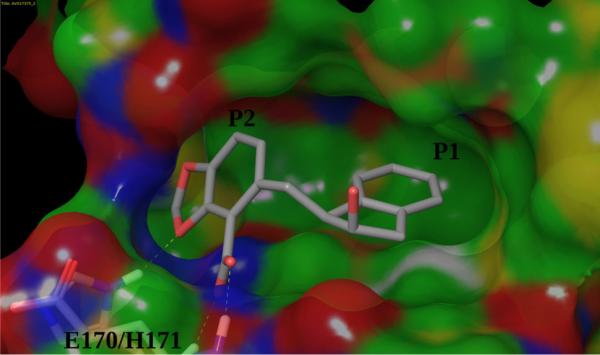
The dominant binding mode adopted by the LEDGF inteegrase binders
The non-binders in Fig. 7 illustrate the determinant role of the hydrophobic P1 subpocket and the complex interplay between the potentially exclusive requirements of satisfying the binding hotspots of the LEDGF site. In both of these cases, correctly predicted by the AD Vina/BEDAM virtual screening protocol, the formation of the key carboxylate-backbone interactions precludes the occupation of the hydrophobic P1 sub-pocket leading to poor binding.
Fig. 7.

Modeled structures of the complexes of two non-binders (AVX40910_0 and AVX62730_0) with the LEDGF site of integrase. These examples illustrate that occlusion of the P1 hydrophobic site is necessary for strong binding
On the other hand satisfying both hydrogen bonding and hydrophobic enclosure in the P1 sub-pocket was found to be a necessary but not sufficient condition for binding. One illustrative case is shown in Fig. 8. Both ligands carry a benzoic acid group connected with an indolinone group by a double bond; the only difference is in the substitution position of the carboxylate on the benzene ring. The ortho-substituted isomer is a nonbinder, while the meta-substituted isomer is a binder. Both are correctly predicted. In the modeled structures the carboxylate moieties on both ligands form the correlated hydrogen bonds with the receptor; and in both cases, the P1 sub-pocket is occupied by the indolinone groups. However, due to steric constraints involved in occupying the P1 subpocket while maintaining the carboxylate-backbone interactions, the amide group of AVX17284_0 ends up buried deeply inside the hydrophobic P1 sub-pocket. This arrangement is thermodynamically unfavorable since the desolvation cost for burying the polar amide is not compensated by the formation of ligand-receptor electrostatic interactions. In contrast, due to more favorable steric compatibility, the amide group of the AVX17285_0 binder can point into the solvent and the associated desolvation cost is much smaller.
Fig. 8.

Modeled structures of the complexes of AVX17284_0 (a non-binder) and AVX17285_0 (a binder) with the LEDGF site of integrase. The buried amide group is responsible for the poor binding of the former
Examples of non-binders that escape simple structural rationalization are illustrated in Fig. 9. These ligands satisfy all of the LEDGF site hotspots yet they are not observed to bind integrase. The BEDAM binding free energy analysis shows that this is because the large and favorable ligand-receptor interaction energies are offset by equally large reorganization free energy penalties. Unlike binders of similar size, these ligands pay a substantial free energy cost for assuming binding competent conformations and/or for forcing the receptor to do the same. Clearly the key in successful ligand design is to optimize ligand-receptor interactions while minimizing the associated costs in terms of intramolecular energetic strain and entropic losses. Proper incorporation of all these effects is necessary to make computational models as effective as possible for use in drug discovery.
Conclusions
We have employed a combined protocol including docking and binding free energy calculations to screen a focused library of ligands to the LEDGF site of HIV integrase. The exercise has been conducted as part of the SAMPL4 ligand screening blind challenge whereby experimental binders were not known to participants. Our blind predictions produced the best enrichment factor among all of the computational submissions. We found that the accuracy of the structural predictions from AD Vina were key to achieve the observed level of prediction performance. Reorganization free energy costs, included in the overall binding free energy estimates, have been the main determinant of the superior ranking performance of our method over scoring methods based primarily on ligand-receptor interaction energy strengths.
Overall, participation in the SAMPL4 HIV integrase challenge has been a very instructive experience to test the applicability of advanced physics-based modeling with a firm foundation in statistical mechanics to in-silico drug development pipelines. Our results show that substantial improvements in virtual screening performance are possible by augmenting docking protocols with binding free energy analyses, especially during the lead optimization phase.
Supplementary Material
Acknowledgments
This work has been supported in part by Research Grants from the National Institute of Health (GM30580 and P50 GM103368). The calculations reported in this work have been performed at the BioMaPS High Performance Computing Center at Rutgers University funded in part by the NIH shared instrumentation Grants Nos. 1 S10 RR022375 and 1 S10 RR027444, on XSEDE resources under National Science Foundation allocation Grant No. TG-MCB100145, and on the Garibaldi cluster at the Scripps Research Institute. We thank David Mobley and the SAMPL4 organizers.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10822-014-9711-9) contains supplementary material, which is available to authorized users.
Contributor Information
Emilio Gallicchio, Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA.
Nanjie Deng, Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA.
Peng He, Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA.
Lauren Wickstrom, Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA.
Alexander L. Perryman, Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA
Daniel N. Santiago, Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA
Stefano Forli, Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA.
Arthur J. Olson, Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA
Ronald M. Levy, Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA
References
- 1.Goodsell DS, Morris GM, Olson AJ. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Curr Opin Chem Biol. 2002;6:439–446. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Z, Felts AK, Friesner RA, Levy RM. J Chem Inf Model. 2007;47:1599–1608. doi: 10.1021/ci7000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trott O, Olson AJ. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Expert Opin Drug Discov. 2010;5:597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Repasky MP, Murphy RB, Banks JL, Greenwood JR, Tubert-Brohman I, Bhat S, Friesner RA. J Comput Aided Mol Des. 2012;26:787–799. doi: 10.1007/s10822-012-9575-9. [DOI] [PubMed] [Google Scholar]
- 8.Gilson MK, Given JA, Bush BL, McCammon JA. Biophys J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boresch S, Tettinger F, Leitgeb M, Karplus M. J Phys Chem B. 2003;107:9535–9551. [Google Scholar]
- 10.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Proc Natl Acad Sci U S A. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jorgensen WL. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 12.Zhou H-X, Gilson MK. Chem Rev. 2009;109:4092–4107. doi: 10.1021/cr800551w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mobley DL, Dill KA. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallicchio E, Lapelosa M, Levy RM. J Chem Theory Comput. 2010;6:2961–2977. doi: 10.1021/ct1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallicchio E, Levy RM. Adv Prot Chem Struct Biol. 2011;85:27–80. doi: 10.1016/B978-0-12-386485-7.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallicchio E, Levy RM. Curr Opin Struct Biol. 2011;21:161–166. doi: 10.1016/j.sbi.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Berne BJ, Friesner RA. Proc Natl Acad Sci U S A. 2012;109:1937–1942. doi: 10.1073/pnas.1114017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mobley DL, Klimovich PV. J Chem Phys. 2012;137:230901. doi: 10.1063/1.4769292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirts MR, Mobley DL, Brown SP. Free energy calculations in structure-based drug design. Cambridge University Press; Cambridge, MA: 2010. [Google Scholar]
- 20.Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS. Curr Opin Struct Biol. 2011;21:150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lapelosa M, Gallicchio E, Levy RM. J Chem Theory Comput. 2012;8:47–60. doi: 10.1021/ct200684b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mobley DL. J Comput Aided Mol Des. 2012;26:93–95. doi: 10.1007/s10822-011-9497-y. [DOI] [PubMed] [Google Scholar]
- 23.Peat TS, Dolezal O, Newman J, Mobley D, Deadman JJ. Interrogating HIV integrase for compounds that bind: a sample challenge. J Comput Aided Mol Des. 2014 doi: 10.1007/s10822-014-9721-7. doi:10.1007/s10822-014-9721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mobley DL, Liu S, Lim NM, Wymer KL, Perryman AL, Forli S, Deng N, Su J, Branson K, Olson AJ. Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aided Mol Des. 2014 doi: 10.1007/s10822-014-9723-5. doi:10.1007/s10822-014-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su Y, Gallicchio E, Das K, Arnold E, Levy RM. J Chem Theory Comput. 2007;3:256–277. doi: 10.1021/ct600258e. [DOI] [PubMed] [Google Scholar]
- 26.Lapelosa M, Gallicchio E, Arnold GF, Arnold E, Levy RM. J Mol Biol. 2009;385:675–691. doi: 10.1016/j.jmb.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frenkel YV, Gallicchio E, Das K, Levy RM, Arnold E. J Med Chem. 2009;52:5896–5905. doi: 10.1021/jm900282z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felts AK, Labarge K, Bauman JD, Patel DV, Himmel DM, Arnold E, Parniak MA, Levy RM. J Chem Inf Model. 2011;51:1986–1998. doi: 10.1021/ci200194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallicchio E. Mol Biosci. 2012;2:7–22. doi: 10.4236/cmb.2012.21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng N, Zheng W, Gallicchio E, Levy RM. J. Am. Chem. Soc. 2011;133:9387–9394. doi: 10.1021/ja2008032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelman A, Kessl JJ, Kvaratskhelia M. Curr Opin Chem Biol. 2013;17:339–345. doi: 10.1016/j.cbpa.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, et al. Proc Natl Acad Sci. 2013;110:8690–8695. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mobley DL, Liu S, Lim NM, Wymer KL, Perryman AL, Forli S, Deng N, Su J, Branson K, Olson AS. Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aided Mol Des. 2013 doi: 10.1007/s10822-014-9723-5. doi:10.1007/s10822-014-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallicchio E, Levy RM. J Comput Aided Mol Des. 2012;25:505–516. doi: 10.1007/s10822-012-9552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan Z, Gallicchio E, Lapelosa M, Levy RM. J Chem Phys. 2012;136:144102. doi: 10.1063/1.3701175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wickstrom L, He P, Gallicchio E, Levy RM. J Chem Theory Comput. 2013;9:3136–3150. doi: 10.1021/ct400003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perryman AL, Santiago DN, Forli S, Santos-Martins D, Olson AJ. Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: participation in the SAMPL4 protein-ligand binding challenge. J Comput Aided Mol Des. 2014 doi: 10.1007/s10822-014-9709-3. doi:10.1007/s10822-014-9709-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chia-en C, Chen W, Gilson MK. Proc Natl Acad Sci USA. 2007;104:1534–1539. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the ACM/IEEE conference on supercomputing (SC06) IEEE; Tampa, FL: 2006. [Google Scholar]
- 40.Perryman AL, Forli S, Morris GM, Burt C, Cheng Y, Palmer MJ, Whitby K, McCammon JA, Phillips C, Olson AJ. J Mol Biol. 2010;397:600–615. doi: 10.1016/j.jmb.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenwood JR, Calkins D, Sullivan AP, Shelley JC. J Comput Aided Mol Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 42.Gallicchio E, Paris K, Levy RM. J Chem Theory Comput. 2009;5:2544–2564. doi: 10.1021/ct900234u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirts MR, Chodera JD. J Chem Phys. 2008;129:124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okumura H, Gallicchio E, Levy RM. J Comput Chem. 2010;31:1357–1367. doi: 10.1002/jcc.21419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 46.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 47.Banks JL, Beard JS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. J Comput Chem. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peat TS, Rhodes DI, Vandegraaff N, Le G, Smith JA, Clark LJ, Jones ED, Coates JAV, Thienthong N, Janet N, et al. PloS one. 2012;7:e40147. doi: 10.1371/journal.pone.0040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voet ARD, Kumar A, Berenger F, Zhang KYJ. Combining in silico and in cerebro approaches for virtualscreening and pose prediction in SAMPL4. J Comput Aided Mol Des. 2014 doi: 10.1007/s10822-013-9702-2. doi:10.1007/s10822-013-9702-2. [DOI] [PubMed] [Google Scholar]
- 50.Boresch S, Tettinger F, Leitgeb M, Karplus M. 107:9535–9551. [Google Scholar]
- 51.Deng N, Zhang P, Cieplak P, Lai L. J Phys Chem B. 2011;115:11902–11910. doi: 10.1021/jp204047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Proteins Struct Funct Bioinforma. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Spoel D, et al. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bas DC, Rogers DM, Jensen JH. Proteins Struct Funct Bioinforma. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 55.Muddana HS, Daniel VC, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK. J Comput Aided Mol Des. 2012;6:475–487. doi: 10.1007/s10822-012-9554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gallicchio E, Levy RM. J Comput Chem. 2004;25:479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 57.Deng Y, Roux B. J Phys Chem B. 2009;113:2234–2246. doi: 10.1021/jp807701h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gallicchio E, Andrec M, Felts AK, Levy RM. J Phys Chem B. 2005;109:6722–6731. doi: 10.1021/jp045294f. [DOI] [PubMed] [Google Scholar]
- 59.Brown SP, Muchmore SW. J Chem Inf Model. 2006;46:999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.