

Abstract
Platelet-activating factor (PAF) promotes tumour metastasis via activation of the transcription factor nuclear factor-κB (NF-κB). We here investigated the role of the protein kinase CK2 (formerly Casein Kinase 2 or II) in PAF-induced NF-κB activation and tumour metastasis, given that PAF has been reported to increase CK2 activity, and that CK2 plays a key role in NF-κB activation. PAF increased CK2 activity, phosphorylation and protein expression in vivo as well as in vitro. CK2 inhibitors inhibited the PAF-mediated NF-κB activation and expression of NF-κB-dependent pro-inflammatory cytokines and anti-apoptotic factors. Pre-treatment with the antioxidant N-Acetyl-L-Cysteine (NAC) resulted in a significant inhibition in PAF-induced enhancement of CK2 activity, phosphorylation and protein expression in vivo as well as in vitro. H2O2 and known reactive oxygen species inducers, lipopolysaccharide (LPS) and tumour necrosis factor-α (TNF-α) enhanced CK2 activity, phosphorylation and protein expression, which was again inhibited by antioxidant. PAF, LPS and TNF-α induced increased CK2 activity, phosphorylationand protein expression, which were inhibited by p38 inhibitor. PAF, LPS or TNF-α increased pulmonary metastasis of B16F10, which was inhibited by antioxidants, CK2 inhibitor and p38 inhibitor. Our data suggest that (i) reactive oxygen species activate CK2 via p38, which, in turn, induces NF-κB activation, and (ii) PAF, LPS and TNF-α increase pulmonary tumour metastasis via the induction of the reactive oxygen species (ROS)/p38/CK2/NF-κB pathway.
Keywords: casein kinase 2, nuclear factor-κB, platelet-activating factor, reactive oxygen species, tumour metastasis
Introduction
Inflammation has been increasingly recognized as an important component of tumorigenesis, and the causal relationship between inflammation and cancer has been broadly accepted.1–3 Previous reviews have focused on various aspects of the relationship between cancer and inflammation, including the roles of a variety of inflammatory cells,3–5 mediators,3,6,7 and signalling pathways8 in cancer.
Platelet-activating factor (PAF), which is produced by a variety of inflammatory cells, is a potent lipid mediator that is involved in cellular activation and intracellular signalling, as well as a myriad of inflammatory reactions.9–11 We have previously demonstrated that PAF has the ability to enhance tumour growth and metastasis.12 Our subsequent studies have also shown that PAF exerts such effects via activation of the transcription factor, nuclear factor-κB (NF-κB).13,14 Additionally, PAF has been reported to induce NF-κB activation in intestinal epithelial cells by enhancing IκBα phosphorylation.15,16 The expression of NF-κB-dependent molecules, including various angiogenic factors, such as tumor necrosis factor-α (TNF-α), vascular endothelial growth factor,17,18 matrix metalloproteinase-9,19 and anti-apoptotic factors (Bcl-2 and Bcl-xL),20 have roles in tumour growth and metastasis. These findings have provided us with fairly detailed insights into the role of inflammation in tumour promotion. However, the mechanism underlying PAF-mediated activation of NF-κB remains to be elucidated.
Protein kinase CK2 (formerly Casein Kinase 2 or II) is a highly conserved and ubiquitous protein serine/threonine kinase which is localized in the cell nucleus and cytoplasm. It consists of catalytic subunits [molecular weights (MW) 42 000 α, 38 000 α′] and a regulatory subunit (28 000 MW β), producing holoenzyme structures, linking the catalytic subunits through the β subunit into α2β2, αα′β2, or α′2β2 tetramers.21 CK2 impacts over 300 potential substrates in the cell, as well as a wide range of pathways that pertain to cell growth, proliferation and apoptosis.21,22
CK2 is implicated in tumorigenesis and transformation. CK2 inhibits the activity and stability of tumour suppressor proteins [p53 and phosphatase and tensin homolog deleted on chromosome 10 (PTEN)], promotes cell survival signalling pathways through the up-regulation of proto-oncogenic products (c-Myc, c-Myb, and c-Jun) and transcriptional activators (β-catenin and Max).23 CK2 also regulates inflammation by phosphorylating, and thereby modulating the actions of several transcription factors, including NF-κB, a signal transducer and activator of transcription, cAMP response element (CRE) binding protein, CRE modulator protein, PU.1, specificity protein-1 and steroid hormone receptors.24 Among them, NF-κB has received considerable attention pertaining to its association with CK2 signalling. CK2 has been found to enhance the DNA transactivation potential of NF-κB through phosphorylating multiple sites of IκBα25–28 as well as NF-κB.29
Regarding the molecular mechanism underlying the regulation of CK2, we have recently reported that PAF increases the enzymatic activity of CK2 in vivo.30 This suggests the possibility that CK2 is involved in biological roles of PAF. In this study, we investigated whether PAF could induce NF-κB activation and tumour metastasis via enhancing CK2 activity. We found that PAF induced NF-κB activation via the pathway involving reactive oxygen species (ROS)/p38/CK2.
Materials and methods
Animals
Specific pathogen-free C57BL/6 mice were obtained from OrientBio (Seongnam, Gyounggi, Korea) and housed in clean, pathogen-free rooms in an environment with controlled temperature (23°), humidity (55%), and a 12 hr light/dark cycle. All mice were used at 6–7 weeks of age. All experiments were conducted in accordance with the guidelines of the Chonnam National University Institutional Animal Care and Use Committee.
Cell culture
The MH-S mouse alveolar macrophage cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The B16F10 mouse melanoma cell line metastatic to the lungs of C57BL/6 mice was supplied originally by the Tumor Repository of the National Cancer Institute (Bethesda, MD). These cells were maintained in RPMI-1640 (Gibco Invitrogen, Carlsbad, CA) supplemented with 10% (volume/volume) fetal bovine serum (Gibco) at 37° in a humidified atmosphere of 5% CO2 and 95% air.
Antibodies and reagents
The following antibodies were used for Western blotting: anti-phospho-CK2α (Thr306, Ser362) (Sigma-Aldrich, St Louis, MO); anti-CK2α, anti-p65, anti-phospho-p65 (Ser539), and anti-phospho-p38 (Thr180, Tyr182) (Cell Signaling Technology, Beverly, MA); anti-GAPDH, anti-c-Rel, anti-p50, anti-RelB, anti-p52, anti-normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). PAF (1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphoryl-choline), lipopolysaccharide (LPS) derived from Escherichia coli (O127:B8, L3024), and antioxidants, N-Acetyl-L-Cysteine (NAC) were purchased from Sigma-Aldrich. The p38 mitogen-activated protein kinase (MAPK) inhibitor, SB202190 and CK2 inhibitors, TBB and TBCA, were obtained from Calbiochem Merck (Darmstadt, Germany). Recombinant mouse TNF-α was purchased from R&D Systems (Minneapolis, MN). The PAF receptor antagonist, WEB-2086, was obtained from Santa Cruz Biotechnology.
CK2 activity assay
Lung specimens were homogenized in cell lysis buffer (50 mm Tris–HCl, pH 7·5, 150 mm NaCl, 1% Triton X-100, 0·5% sodium deoxycholate, 0·1% SDS, 1 mm EGTA, 1 mm EDTA, 10 mm sodium fluoride, 1 mm sodium orthovanadate, 1 mm dithiothreitol and 1 mm PMSF). The phosphotransferase activity of CK2 was measured using a CK2 assay kit (Millipore, Temecula, CA) in accordance with the manufacturer's recommendations. Briefly, cell lysates, substrate peptide, protein kinase A inhibitor cocktail, and γ-[32P]ATP in the assay dilution buffer were incubated for 10 min at 30°. The phosphorylated substrate was then separated on P81 phosphocellulose paper and quantified with a scintillation counter.
Western blot analysis
Whole cell extracts were prepared from the lung and MH-S cells using PhosphoSafe Extraction Reagent (Novagen Merck, Darmstadt, Germany) with PMSF protease inhibitor (Sigma, St Louis, MO). Nuclear extracts were prepared from the lung and MH-S cells using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific Inc., Waltham, MA) with 10 mm sodium fluoride, 1 mm sodium orthovanadate, in accordance with the manufacturer's instructions. Equal amounts of cell lysates were separated on 10% SDS–polyacrylamide gel under reducing conditions, and were then transferred onto Protran Nitrocellulose Transfer Membranes (Whatman, Dassel, Germany). The membranes were blocked by 30 min of incubation at room temperature in 5% BSA in Tris-buffered saline (TBS)–0·1% Tween-20, followed by a 2-hr incubation at 4° with primary antibodies in 5% BSA in TBS–0·1% Tween-20. The blots were washed for 30 min with three changes of TBS–0·1% Tween-20 solution, followed by incubation for 1 hr at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG antibody (Santa Cruz Biotechnology). The blots were again washed three times for 30 min each, and finally developed in LumiGLO reagent (Cell Signaling Technology). GAPDH was used as a loading control.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from the lung and MH-S cells using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific Inc.), in accordance with the manufacturer's instructions. An oligonucleotide harbouring the Igκ-chain-binding (κB, 5′-CCGGTTAAC-AGAGGGGGCTTTCCGAG-3′) was synthesized as a probe for use in the gel retardation assay. The two complementary strands were then annealed and labelled with (α-32P)dCTP. Labelled oligonucleotides (10 000 counts/min), 10 μg of nuclear extracts, and binding buffer [10 mm Tris–HCl, pH 7·6, 500 mm KCl, 10 mm EDTA, 50% glycerol, 100 ng of poly(dIdC) and 1 mm dithiothreitol] were incubated for 30 min at room temperature in a final volume of 20 μl. The reaction mixture was analysed via electrophoresis on a 5% polyacrylamide gel in 0·5 × Tris–borate buffer. Specific binding was controlled by competition with 50-fold excess of α-32P-unlabelled (cold) NF-κB oligonucleotides as a competitor, or CRE oligonucleotides. For band specificity, 1 μg of specific supershifting antibodies against c-Rel, p50, p52, p65, RelB components of NF-κB, or normal IgG were incubated with the nuclear extract for 1 hr at 4° before the addition of labelled oligonucleotides to the binding reaction.
Immunohistochemistry
The lungs were fixed in 4% neutral buffered formalin (Sigma Chemical, St Louis, MO) overnight at room temperature, paraffin-embedded, and sectioned at 4 μm. Immunoperoxidase staining was performed at room temperature using a Histostain-Plus kit (ZYMED Lab. Inc., San Francisco, CA), in accordance with the manufacturer's instructions. In brief, the deparaffinized sections were treated with 3% H2O2 in methanol for 5 min to reduce endogenous peroxidase activity. The sections were incubated with serum block solution for 30 min, followed by incubation with one of the diluted CK2α antibodies overnight at 4°, and washing in PBS. The specimens were subsequently incubated with the corresponding biotinylated secondary antibodies for 10 min, and then horseradish peroxidase–streptoavidin complex for an additional 10 min. Colour was developed with horseradish peroxidase substrate for 3 min. The sections were counterstained with haematoxylin.
Real-time RT-PCR
Total RNA was extracted from lung and MH-S cell line using TRIzol reagent (Invitrogen), in accordance with the manufacturer's instructions. Real-time RT-PCR was performed as described previously.20 The primers were as follows: mouse TNF-α; 5′-AAA ATT CGA GTG ACA AGC CTG TAG-3′ and 5′-CCC TTG AAG AGA ACC TGG GAG TAG-3′,: mouse macrophage inflammatory protein 2 (MIP2); 5′-GTG AAC TGC GCT GTC AAT GC-3′ and 5′-CGC CCT TGA GAG TGG CTA TG-3′,: mouse Bcl-2; 5′-TTC GCA GAG ATG TCC AGT CAG CT -3′ and 5′-TGA AGA GTT CCT CCA CCA CCG T-3′,: mouse Bcl-xL; 5′-AAT GAA CTC TTT CGG GAT GGA G-3′ and 5′-CCA ACT TGC AAT CCG ACT CA-3′, and β-actin; 5′-GAT CTG GCA CCA CAC CTT CT-3′ and 5′-GGG GTG TTG AAG GTC TCA AA-3′. The relative levels of mRNA were calculated using the standard curve generated from the cDNA dilutions. The mean cycle threshold (Ct) values from quadruplicate measurements were employed in the calculation of gene expression, with normalization to β-actin employed as an internal control. Calculations of the relative level of gene expression were conducted using complementary computer software (Corbett Research, Mortlake, Australia) with a standard curve.
Lung colonization assay
B16F10 tumour cells were washed in PBS, and 1.5 × 105 tumour cells (> 95% viability by trypan blue exclusion assay) in 0·1 ml of PBS were injected intravenously into the lateral tail veins of C57BL/6 mice (n = 5 for each group). The mice were subjected to necropsy 14 days later. The lungs were removed and fixed in Bouin's solution (Sigma), and the number of surface lung colonies was counted under a dissecting microscope.
Statistical analysis
Data are expressed as means ± SE Statistical significance was determined via one-way analysis of variance (StatView; Abacus Concepts Inc., Berkeley, CA). A value of P < 0·05 was regarded as statistically significant. All experiments were conducted at least twice. Reproducible results were obtained and representative data are, therefore, provided in the figures.
Results
PAF increases the activity, phosphorylation and protein expression of CK2
We first assessed the effect of PAF on CK2 activity in vivo. CK2 activity was increased in the lungs upon PAF injection, and reached a peak at 20 min post-challenge injection, and declined thereafter (Fig. 1a). The kinetics of PAF-mediated CK2 phosphorylation was increased by 10–20 min and declined thereafter (Fig. 1b). We also examined the effect of PAF on CK2 protein expression by Western blotting and immunohistochemistry. Western blotting revealed that PAF increased CK2 expression in the lungs, which was maximal at 2–4 hr (Fig. 1c). Immunohistochemistry also demonstrated that PAF increased CK2 expression with similar kinetics observed to those of Western blotting (Fig. 1d). CK2 was strongly expressed in airway epithelial cells, and also in pneumocytes and endothelial cells. CK2 was stained in cytoplasm and nucleus at the relatively early time-points (0–2 hr), but was stained mainly within the nucleus at 2–4 hr. All the increases in activity, phosphorylation, and protein expression of CK2 were blocked by pre-treatment with the PAF antagonist, WEB or the CK2 inhibitors, TBB and TBCA (Fig. 1a–d).
Figure 1.

Platelet-activating factor (PAF) enhances CK2 activation. Mice were injected intraperitoneally with PAF (0·1 mg/kg) and the lungs were obtained at the indicated time for measurement of CK2 activity (a), phosphorylation (b), protein expression by Western blotting (c), and immunohistochemistry (d). PAF antagonist, WEB2086 (50 mg/kg), CK2 inhibitors, TBB (25 or 50 mg/kg) and TBCA (12·5 or 25 mg/kg) were administered intraperitoneally 1 hr before PAF injection. (e–g) MH-S cells (2 × 106) were treated with 2 μm of PAF for the indicated times. WEB2086 (50 μm), TBB (50 μm), or TBCA (25 μm) was added 30 min before PAF treatment. Data in (a)–(c) represent the mean ± SE of three independent experiments with three to five mice/time-point/experiment. Data in (d) are from representatives of three independent experiments with three to five mice/time-point/experiment. Original magnification 400×. Scale bar = 50 μm. Data in (e)–(g) represent the mean ± SE of three independent experiments with three to five dishes/time-point/experiment. *P < 0·05; **P < 0·01 versus control group. #P < 0·05; ##P < 0·01 versus PAF group (a–c, e–g).
We examined the in vitro effect of PAF using the murine alveolar macrophage cell line MH-S. Treatment of the cells with PAF resulted in increases in activity (Fig. 1e), phosphorylation (Fig. 1f), and protein expression (Fig. 1g) of CK2, all of which were inhibited by PAF antagonist or CK2 inhibitors.
CK2 is involved in the PAF-induced activation of NF-κB and the mRNA expression of NF-κB-dependent molecules
We next examined the role of CK2 in the PAF-induced activation of NF-κB. Injection of PAF resulted in NF-κB activation in the lung (Fig. 2a), which was inhibited by pre-treatment of animals with PAF antagonists. Complete blocking of NF-κB mobilization by addition of the cold competitor, but not of an irrelevant motif, CRE, indicated the specificity of NF-κB binding. Additionally, PAF increased the translocation of p50 and p65 subunits into the nucleus, and phosphorylation of p65 at Ser536 (Fig. 2b). In addition to NF-κB activation, NF-κB-dependent pro-inflammatory molecules such as TNF-α and MIP2 as well as anti-apoptotic factors such as Bcl-2 and Bcl-xL were induced in response to PAF (Fig. 2c). We examined the extent to which the PAF-mediated phenomena are CK2-dependent. The CK2 inhibitors significantly inhibited not only the PAF-mediated NF-κB activation (Fig. 2a,b), but also the expression of mRNA of TNF-α and MIP2 and anti-apoptotic factors (Fig. 2c). These data indicate that CK2 plays a key role in NF-κB activation.
Figure 2.

CK2 inhibitor inhibits platelet-activating factor (PAF) -induced nuclear factor-κB (NF-κB) activation. (a, b) Mice were injected intraperitoneally with PAF (0·1 mg/kg) and the nuclear extract of lungs was obtained 30 min thereafter for the measurement of NF-κB activation. WEB2086 (50 mg/kg), TBB (50 mg/kg) and TBCA (25 mg/kg) were administered intraperitoneally 1 hr before PAF injection. (c) The lungs were obtained 4 hr after PAF for the measurement of mRNA expression. Data in (a) are representative of three independent experiments with three mice per group. Data in (b) and (c) represent the mean ± SE of three independent experiments, with three mice per group. (d, e) MH-S cells (2 × 106) were treated with 2 μm of PAF for the 30 min. Nuclear extract was prepared for the measurement of NF-κB activation. WEB2086 (50 μm), TBB (50 μm), or TBCA (25 μm) was added 30 min before PAF treatment. (f) MH-S cells were harvested 4 hr after PAF treatment for the measurement of mRNA expression. Data in (d) are representative of three independent experiments with three to five dishes/experiment. Data in (e) and (f) represent the mean ± SE of three independent experiments, with three to five dishes/experiment. *P < 0·01 versus control group. #P < 0·05; ##P < 0·01 versus PAF group (b–c, e–f).
Similar findings were observed in MH-S cells. PAF-induced NF-κB activation (Fig. 2d), increased the translocation of p50 and p65 subunits into the nucleus, and phosphorylation of p65 at Ser536 (Fig. 2e) and increases in mRNA expression of TNF-α, MIP2 and anti-apoptotic factors (Fig. 2f) were nearly completely inhibited by PAF antagonist or CK2 inhibitors.
ROS is involved in PAF-induced CK2 activation
We have previously shown that the ability of PAF to activate NF-κB is a ROS-dependent phenomenon.31 Additionally, PAF can directly stimulate the production of ROS by various inflammatory cells such as neutrophils, eosinophils and macrophages.32–34 This suggests the possibility that PAF could enhance CK2 activity and protein expression via ROS generation. Pre-treatment with the antioxidant, NAC, resulted in significant inhibition in PAF-induced enhancement of CK2 activity (Fig. 3a), phosphorylation (Fig. 3b) and protein expression, as assessed by Western blotting (Fig. 3c) and immunohistochemistry (Fig. 3d) in the lungs.
Figure 3.

Reactive oxygen species (ROS) are involved in platelet-activating factor (PAF) -induced CK2 activation. Mice were injected intraperitoneally with PAF (0·1 mg/kg) and the lungs were obtained 20 min later for measurement of CK2 activity (a), phosphorylation (b). Mice were injected intraperitoneally with PAF and the lungs were obtained after 2 hr for the assessment of protein expression by Western blotting (c) and immunohistochemistry (d). NAC (5 or 20 mg/kg) was intraperitoneally injected 1 hr before PAF. Data in (a) represent the mean ± SE of two independent experiments with five mice per group. Data in (b) and (c) represent the mean ± SE of three independent experiments with three mice per group. Data in (d) are from a representative of three independent experiments with three mice per group. Original magnification 400×. Scale bar = 50 μm. MH-S cells (2 × 106) were treated with H2O2 (0·3 μm) or PAF (2 μm) for 20 min (e, f) or 2 h (g). NAC (30 μm) was added 30 min before H2O2 or PAF. Data in (e) represent the mean ± SE of two independent experiments with three dishes/experiment. Data in (f) and (g) represent the mean ± SE of three independent experiments with three to five dishes/experiment. *§P < 0·01 versus control group. #P < 0·01 versus H2O2 group. ‡P < 0·01 versus PAF group (a–c, e–g). MH-S cells (2 × 106) were treated with of H2O2 for 30 min and nuclear extract was prepared for the super shift assay (h). Super shift assay was conducted with 1 μg of anti-c-Rel, p50, p65, RelB, p52, or control rabbit IgG antibody. Data in (h) are representative of three independent experiments with three dishes/experiment.
We examined the direct role of ROS in CK2 activation and protein expression in vitro. The addition of H2O2 to MH-S cells resulted in increases in CK2 activity (Fig. 3e), phosphorylation (Fig. 3f) and protein expression (Fig. 3g). Pre-treatment of NAC blocked the H2O2 effects (Fig. 3e–g). H2O2 or PAF-induced increase in CK2 activity, phosphorylation and protein expression were also blocked by pre-treatment of NAC (Fig. 3e–g). H2O2 or PAF-induced enhancement of CK2 activity was also blocked by another antioxidant, pyrrolidinedithiocarbamate (PDTC) (data not shown), further ensuring that the effects are due to the antioxidant properties of NAC. We determined which pathway of NF-κB activation (canonical or non-canonical) is modulated by ROS-dependent mechanisms. Supershift analysis (Fig. 3h) demonstrated that addition of H2O2 to the super shift assay of antibody specific for the p65 and p50 subunits almost completely and significantly reduced the intensity of the DNA-binding activity, with a visible shift in the mobility of the complex, respectively, in MH-S cells. The NF-κB complexes were not supershifted by antibodies against p52 and RelB. These results suggest that activation of NF-κB species in response to ROS result in the occurrence of two heterodimer types, p50–p65, i.e. canonical NF-κB.
LPS and TNF-α increase CK2 activity and protein expression in a ROS-dependent manner
The findings described above (Fig. 3) suggest that, if ROS is the effector molecule modulating CK2 activity, any exogenous stimulus capable of producing ROS is able to enhance CK2 activity. We attempted to verify this hypothesis using other stimuli, LPS and TNF-α, because these reagents are ROS inducers.35 Both reagents enhanced CK2 activity (Fig. 4a), phosphorylation (Fig. 4b), and protein expression assessed by Western blotting (Fig. 4c) and immunohistochemistry (Fig. 4d). Pre-treatment with NAC blocked all the effects of LPS and TNF-α on CK2 (Fig. 4a–d).
Figure 4.

Effects of lipopolysaccharide (LPS) and tumour necrosis factor-α (TNF-α) on CK2 activation. Mice were injected intraperitoneally with LPS (0·5 mg/kg), or TNF-α (40 μg/kg) and the lungs were obtained 20 min thereafter for measurement of CK2 activity (a), phosphorylation (b). Mice were injected intraperitoneally with LPS or TNF-α and the lungs were obtained after 2 hr for protein expression by Western blotting (c) and immunohistochemistry (d). NAC (5 mg/kg) was intraperitoneally injected 1 hr before LPS, or TNF-α. Data in (a)–(c) represent the mean ± SE of three independent experiments with three to five mice per group. Data in (d) are from a representative of three independent experiments with three mice per group. Original magnification 200×. Scale bar = 100 μm. MH-S cells (2 × 106) were treated with LPS (0·2 μg/ml), or TNF-α (10 ng/ml) for 20 min (e, f) or 2 hr (g). NAC (30 μm) was added 30 min before LPS, or TNF-α. Data in (e) represent the mean ± SE of two independent experiments with three dishes/experiment. Data in (f) and (g) represent the mean ± SE of three independent experiments with three to five dishes/experiment. *§P < 0·01; **§§P < 0·001 versus control group. #P < 0·01; ##P < 0·001 versus LPS group. ‡P < 0·01; ‡‡P < 0·001 versus TNF-α group (a–c, e–g).
The addition of LPS or TNF-α to MH-S cells resulted in increases in CK2 activity (Fig. 4e), phosphorylation (Fig. 4f) and protein expression (Fig. 4g). Pre-treatment of NAC blocked the effects of LPS and TNF-α (Fig. 4e–g). LPS- or TNF-α-induced enhancement of CK2 activity was also blocked by PDTC (data not shown).
ROS-dependent p38 is involved in CK2 activation
We next attempted to determine how ROS activates CK2. It has been shown that CK2 is activated in a p38-dependent manner by UV radiation and stress signalling,28,36 and it is well known that ROS mediates the activation of MAPK, including p38.37–39 Hence, we examined whether PAF activates CK2 via ROS-dependent p38 activation. PAF, LPS and TNF-α induced p38 phosphorylation, all of which were blocked by pre-treatment of NAC (Fig. 5a). Pre-treatment with the p38 inhibitor, SB202190, blocked PAF-, LPS- and TNF-α-induced increases in activity (Fig. 5b), phosphorylation (Fig. 5c), and protein expression of CK2 (Fig. 5d,e) in the lungs. These data strongly suggest that the pro-inflammatory mediators produce ROS, which, in turn, induces CK2 activation and protein expression via p38 activation.
Figure 5.
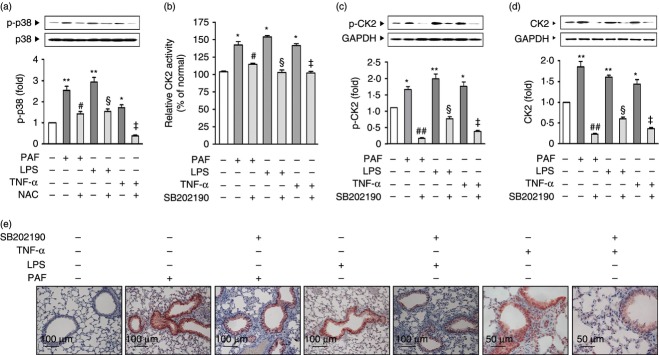
Reactive oxygen species (ROS) -dependent p38 is involved in CK2 activation. Mice were injected intraperitoneally with platelet-activating factor (PAF; 0·1 mg/kg), lipopolysaccharide (LPS; 0·5 mg/kg) or tumour necrosis factor-α (TNF-α; 40 μg/kg) and the lungs were obtained 20 min thereafter for measurement of p38 phosphorylation (a), CK2 activity (b), phosphorylation (c). Mice were injected intraperitoneally with PAF, LPS or TNF-α and the lungs were obtained 2 hr after injection for protein expression assessed by Western blotting (d) and immunohistochemistry (e). NAC (5 mg/kg) was injected intraperitoneally 1 hr before PAF, LPS or TNF-α. SB202190 (5 mg/kg) was administered intraperitoneally 2 days and 1 day before PAF, LPS or TNF-α. Data in (a)–(d) represent the mean ± SE of three independent experiments with three to five mice per group. Data in (e) are from a representative of three independent experiments, with three mice per group. *P < 0·01; **P < 0·001 versus control group. #P < 0·01; ##P < 0·001 versus PAF group. §P < 0·01 versus LPS group. ‡P < 0·01 versus TNF-α group (a–d). Original magnification 200×. Scale bar = 50 or 100 μm (e).
ROS/p38/CK2 pathway is involved in PAF-induced tumour metastasis
We finally evaluated the role of CK2 in the PAF-induced enhancement of B16F10 melanoma metastasis. Mice were treated with TBB and TBCA before PAF injection. TBB and TBCA inhibited PAF-induced metastasis of B16F10 (Fig. 6a). Consistent with the findings of linkage between ROS/p38 and CK2 described above, PAF-induced metastasis was also inhibited by pre-treatment of NAC or SB202190 (Fig. 6a). Likewise, LPS- or TNF-α-induced metastasis of B16F10 was inhibited by pre-treatment with NAC, SB202190 or CK2 inhibitors (Fig. 6b,c).
Figure 6.

Reactive oxygen species (ROS)/p38/CK2 pathway is involved in platelet-activating factor (PAF), lipopolysaccharide (LPS) and tumour necrosis factor-α (TNF-α) -induced tumour metastasis. (a) PAF (0·1 mg/kg), (b) LPS (1·0 mg/kg), and (c) TNF-α (40 μg/kg) were injected intraperitoneally for three consecutive days (days on 0–2) after the administration of B16F10 cells (1·5 × 105). WEB2086 (50 mg/kg), TBB (50 mg/kg), TBCA (25 mg/kg), NAC (5 mg/kg) were administered intraperitoneally 1 hr before PAF, LPS or TNF-α injection. SB202190 (5 mg/kg) was administered intraperitoneally 2 days and 1 day before the injection of B16F10 cells. The lungs were removed on day 14, and the numbers of surface colonies were counted. *P < 0·01 versus control group. #P < 0·05; ##P < 0·01 versus PAF, LPS or TNF-α group. Data represent the mean ± SE of three independent experiments with five mice per group.
Discussion
We13,14,40 and others41,42 have reported that PAF is invo-lved in various inflammatory reactions via the activation of NF-κB. However, the mechanism underlying PAF-mediated activation of NF-κB remains to be elucidated. In this study, we showed that PAF increased CK2 activity, phosphorylation and protein expression, and that CK2 plays a key role in the PAF-induced activation of NF-κB, by demonstrating that injection of PAF resulted in NF-κB activation, which was demonstrated by gel shift assay, Western blotting and phosphorylation of the p65 subunit at Ser536, as well as induction of NF-κB-dependent pro-inflammatory molecules and anti-apoptotic factors.
Previous studies have shown that CK2 is involved in mediating the activation of NF-κB by phosphorylating a number of sites of IκBα, including serines 283, 289, 293 and threonine 291.25–28 In addition to IκB, CK2 has also been shown to phosphorylate the NF-κB family member RelA/p65 following TNF-α stimulation or microbial infection, which results in the phosphorylation of serine 529,29 and of both serine 536 and serine 276,43 thereby enhancing the DNA transactivation potential of NF-κB. In this study, we provided evidence that CK2 is also involved in PAF-induced NF-κB activation. Borthakur et al15,16 have reported that the adaptor protein Bcl10 is required for IκBα phosphorylation for PAF-induced NF-κB activation. Further studies are required to address the linkage between ROS/CK2/p38 and Bcl10.
Many signals, such as LPS, TNF-α, interleukin-1, transforming growth factor-β and interferon-γ have been found to enhance the constitutive activity of CK2.23 In the present study, we confirmed the results of our previous study, showing that PAF enhances CK2 activity.30 Additionally, we further demonstrated that PAF enhanced CK2α phosphorylation and its protein expression. CK2α has been reported to be modified by C-terminal phosphorylation at four specific sites (Thr344, Thr360, Ser362, Ser370) by Cdk1/cyclin B, but the role of such phosphorylation on CK2 function is not clear.44,45 Our data indicate that PAF phosphorylated CK2α, at least in part, at the site of Thr360 and Ser362. Our studies revealed a similar timing of CK2 activity and phosphorylation (Fig. 1), suggesting that CK2 phosphorylation may affect cellular CK2 activity. CK2 is a nuclear-matrix-associated ubiquitous serine/threonine kinase and has been regarded as a constitutive, non-regulated protein kinase. However, abnormally high levels of CK2 have been observed in a number of cancers, including those of the mammary gland, prostate, lung, head and neck, and kidney.46 Moreover, it has been reported that CK2 levels correspond to cell proliferation rates, as cells with higher proliferation rates generally exhibit higher levels of CK2.21 Therefore, together with those findings, we provided evidence that CK2 protein levels are increased in some states, such as under conditions of stress.
Importantly, we here demonstrated that ROS plays a key role in CK2 activation and protein expression. PAF-induced enhancement of CK2 activity, phosphorylation and protein expression were inhibited by pre-treatment with antioxidants in vivo as well as in vitro (Fig. 3), suggesting that, given that PAF has a capability to produce ROS,32–34 PAF might have enhanced CK2 activity and protein expression via ROS generation. We strengthened this notion by demonstrating that H2O2 increased CK2 activity as well as protein expression in MH-S cells. Therefore, if ROS is critically involved in CK2 activation, it is likely that any exogenous stimulus that is capable of producing ROS might enhance CK2 activity. Hence, we investigated the effects of LPS and TNF-α on CK2 activation and protein expression, as these mediators are known to generate ROS via stimulating NADPH oxidase activity.36 As expected, LPS and TNF-α were capable of enhancing CK2 activity, which was again inhibited by antioxidants, suggesting that these pro-inflammatory reagents increase CK2 activity via generation of ROS. Taken together, it is likely that any exogenous stimulus capable of producing ROS is able to enhance CK2 activity.
What is the possible mechanism of ROS-mediated CK2 activation? Recent studies revealed that CK2 is activated by UV radiation and osmotic stress in a p38-dependent manner, and that it regulates the stress response.29,47,48 Hence, we investigated the role of p38 in ROS-mediated CK2 modulation. Oxidative stress-induced MAPK phosphorylation, including that of p38 MAPK, was first reported over 20 years ago.49 Consistent with these findings, we demonstrated that p38 phosphorylation occurred in response to PAF, LPS and TNF-α, which was blocked by pre-treatment with NAC, indicating that ROS generated by these mediators are responsible for p38 phosphorylation. Indeed, the p38 inhibitor, SB202190, was shown to inhibit CK2 activation and protein expression (Fig. 5), indicating that p38 kinase activity is involved in ROS-mediated CK2 activation and protein expression. In particular, the fact that SB202190 also abrogated CK2 phosphorylation suggests that, in addition to being an allosteric regulator of CK2,47 p38 MAPK directly phosphorylates CK2α, in turn, enhancing its activity. Another possibility regarding CK2 activation is that ROS acts as a modulator of protein functions and cellular signalling pathways through the reversible oxidation of cysteine residues in proteins,48 and there is a report that cysteine residues in CK2 play a role in its catalytic activity.49 Therefore, the possibility that ROS might modulate CK2 activity by oxidation of cysteine residues in CK2 cannot be excluded.
We showed that the ROS/p38/CK2 pathway leading to NF-κB activation described above was the key upstream pathway in PAF-induced metastasis of B16F10, and the pathway also played a key role in LPS-induced and TNF-α-induced tumour metastasis (Fig. 6). Therefore, our data suggest that any exogenous stimulus capable of producing ROS is likely to enhance tumour metastasis via activation of the CK2/NF-κB pathway. We have shown that PAF is implicated in tumour growth and metastasis via the expression of NF-κB-dependent molecules, including various angiogenic factors, matrix metalloproteinase-9 and anti-apoptotic factors.17–20 However, besides NF-κB, CK2 plays a critical role in tumour biology by phosphorylating a variety of critical molecules which are implicated in tumorigenesis and transformation.23 Therefore, further studies are required to determine whether the critical molecules are also ROS dependent.
In summary, we here demonstrated ROS as the key regulator for CK2 activation, so providing an important clue explaining (i) action mechanisms of pro-inflammatory mediators, and (ii) the reason why both ROS and CK2 are involved in the pathogenesis of a variety of human diseases, such as cancer, and many inflammatory and metabolic disorders. Given that targeting CK2 is emerging as a novel and potentially key strategy for targeted cancer therapy, our findings suggest that the down-regulation of CK2 activity and protein expression using antioxidants appears to be more beneficial and safer than other strategies targeting CK2 directly.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2010-0022555) and Chonnam National University, 2009.
Disclosures
The authors disclose no potential conflicts of interest.
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx J. Cancer research. Inflammation and cancer: the link grows stronger. Science. 2004;306:966–8. doi: 10.1126/science.306.5698.966. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Littlepage LE, Werb Z. The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol. 2005;70:383–8. doi: 10.1101/sqb.2005.70.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Zlotnik A. Chemokines and cancer. Int J Cancer. 2006;119:2026–9. doi: 10.1002/ijc.22024. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Gay JC. Mechanism and regulation of neutrophil priming by platelet-activating factor. J Cell Physiol. 1993;156:189–97. doi: 10.1002/jcp.1041560125. [DOI] [PubMed] [Google Scholar]
- Shukla SD. Platelet-activating factor receptor and signal transduction mechanisms. FASEB J. 1992;6:2296–301. doi: 10.1096/fasebj.6.6.1312046. [DOI] [PubMed] [Google Scholar]
- Braquet P, Touqui L, Shen TY, Vargaftig BB. Perspectives in platelet-activating factor research. Pharmacol Rev. 1987;39:97–145. [PubMed] [Google Scholar]
- Im SY, Ko HM, Kim JW, et al. Augmentation of tumor metastasis by platelet-activating factor. Cancer Res. 1996;56:2662–5. [PubMed] [Google Scholar]
- Im SY, Han SJ, Ko HM, Choi JH, Chun SB, Lee DG, Ha TY, Lee HK. Involvement of nuclear factor-κB in platelet-activating factor-mediated tumor necrosis factor-α expression. Eur J Immunol. 1997;27:2800–4. doi: 10.1002/eji.1830271109. [DOI] [PubMed] [Google Scholar]
- Choi JH, Ko HM, Kim JW, Lee HK, Han SS, Chun SB, Im SY. Platelet-activating factor-induced early activation of NF-κB plays a crucial role for organ clearance of Candida albicans. J Immunol. 2001;166:5139–44. doi: 10.4049/jimmunol.166.8.5139. [DOI] [PubMed] [Google Scholar]
- Borthakur A, Bhattacharyya S, Alrefai WA, Tobacman JK, Ramaswamy K, Dudeja PK. Platelet-activating factor-induced NF-κB activation and IL-8 production in intestinal epithelial cells are Bcl10-dependent. Inflamm Bowel Dis. 2010;16:593–603. doi: 10.1002/ibd.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthakur A, Bhattacharyya S, Kumar A, Anbazhagan AN, Tobacman JK, Dudeja PK. Lactobacillus acidophilus alleviates platelet-activating factor-induced inflammatory responses in human intestinal epithelial cells. PLoS One. 2013;8:e75664. doi: 10.1371/journal.pone.0075664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HM, Seo KH, Han SJ, et al. Nuclear factor κB dependency of platelet-activating factor-induced angiogenesis. Cancer Res. 2002;62:1809–14. [PubMed] [Google Scholar]
- Seo KH, Ko HM, Choi JH, Jung HH, Chun YH, Choi IW, Lee HK, Im SY. Essential role for platelet-activating factor-induced NF-κB activation in macrophage-derived angiogenesis. Eur J Immunol. 2004;34:2129–37. doi: 10.1002/eji.200424957. [DOI] [PubMed] [Google Scholar]
- Ko HM, Park YM, Jung B, Kim HA, Choi JH, Park SJ, Lee HK, Im SY. Involvement of matrix metalloproteinase-9 in platelet-activating factor-induced angiogenesis. FEBS Lett. 2005;579:2369–75. doi: 10.1016/j.febslet.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Heon Seo K, Ko HM, Kim HA, Choi JH, Jun Park S, Kim KJ, Lee HK, Im SY. Platelet-activating factor induces up-regulation of antiapoptotic factors in a melanoma cell line through nuclear factor-κB activation. Cancer Res. 2006;66:4681–6. doi: 10.1158/0008-5472.CAN-05-3186. [DOI] [PubMed] [Google Scholar]
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta. 2008;1784:33–47. doi: 10.1016/j.bbapap.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Singh NN, Ramji DP. Protein kinase CK2, an important regulator of the inflammatory response? J Mol Med (Berl) 2008;86:887–97. doi: 10.1007/s00109-008-0352-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Wada T, Suzuki F, Takagi T, Hasegawa J, Handa H. Casein kinase II interacts with the bZIP domains of several transcription factors. Nucleic Acids Res. 1998;26:3854–61. doi: 10.1093/nar/26.16.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny JA, Trushin SA, Bren GD, Chester N, Paya CV. Casein kinase II phosphorylates IκBα at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol Cell Biol. 1996;16:899–906. doi: 10.1128/mcb.16.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EM, Van Antwerp D, Verma IM. Constitutive phosphorylation of IκBα by casein kinase II occurs preferentially at serine 293: requirement for degradation of free IκBα. Mol Cell Biol. 1996;16:3554–9. doi: 10.1128/mcb.16.7.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Channavajhala P, Seldin DC, Sonenshein GE. Phosphorylation by the protein kinase CK2 promotes calpain-mediated degradation of IκBα. J Immunol. 2001;167:4919–25. doi: 10.4049/jimmunol.167.9.4919. [DOI] [PubMed] [Google Scholar]
- Kato T, Jr, Delhase M, Hoffmann A, Karin M. CK2 Is a C-Terminal IκB Kinase Responsible for NF-κB Activation during the UV Response. Mol Cell. 2003;12:829–39. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592–7. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- Kang NI, Yoon HY, Kim HA, et al. Protein kinase CK2/PTEN pathway plays a key role in platelet-activating factor-mediated murine anaphylactic shock. J Immunol. 2011;186:6625–32. doi: 10.4049/jimmunol.1100007. [DOI] [PubMed] [Google Scholar]
- Choi JH, Chung WJ, Han SJ, et al. Selective involvement of reactive oxygen intermediates in platelet-activating factor-mediated activation of NF-κB. Inflammation. 2000;24:385–98. doi: 10.1023/a:1007068010645. [DOI] [PubMed] [Google Scholar]
- Chanez P, Dent G, Yukawa T, Barnes PJ, Chung KF. Generation of oxygen free radicals from blood eosinophils from asthma patients after stimulation with PAF or phorbol ester. Eur Respir J. 1990;3:1002–7. [PubMed] [Google Scholar]
- Hartung HP, Parnham MJ, Winkelmann J, Englberger W, Hadding U. Platelet activating factor (PAF) induces the oxidative burst in macrophages. Int J Immunopharmacol. 1983;5:115–21. doi: 10.1016/0192-0561(83)90002-4. [DOI] [PubMed] [Google Scholar]
- Kato M, Tokuyama K, Morikawa A, Kuroume T, Barnes PJ. Platelet-activating factor-induced enhancement of superoxide anion generation in guinea-pigs. Eur J Pharmacol. 1993;232:7–12. doi: 10.1016/0014-2999(93)90721-s. [DOI] [PubMed] [Google Scholar]
- Chiarugi P, Fiaschi T. Redox signalling in anchorage-dependent cell growth. Cell Signal. 2007;19:672–82. doi: 10.1016/j.cellsig.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Sayed M, Kim SO, Salh BS, Issinger OG, Pelech SL. Stress-induced activation of protein kinase CK2 by direct interaction with p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:16569–73. doi: 10.1074/jbc.M000312200. [DOI] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–3. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin. II Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–9. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- Han SJ, Choi JH, Ko HM, Yang HW, Choi IW, Lee HK, Lee OH, Im SY. Glucocorticoids prevent NF-κB activation by inhibiting the early release of platelet-activating factor in response to lipopolysaccharide. Eur J Immunol. 1999;29:1334–41. doi: 10.1002/(SICI)1521-4141(199904)29:04<1334::AID-IMMU1334>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Pan Z, Han J, Herbert JM, Ulevitch RJ, Ye RD. Platelet-activating factor induces NF-κB activation through a G protein-coupled pathway. J Biol Chem. 1995;270:14928–34. doi: 10.1074/jbc.270.25.14928. [DOI] [PubMed] [Google Scholar]
- Roth M, Nauck M, Yousefi S, Tamm M, Blaser K, Perruchoud AP, Simon HU. Platelet-activating factor exerts mitogenic activity and stimulates expression of interleukin 6 and interleukin 8 in human lung fibroblasts via binding to its functional receptor. J Exp Med. 1996;184:191–201. doi: 10.1084/jem.184.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon SM, Wang B, Rixter D, et al. Synergistic activation of NF-κB by nontypeable H. influenzae and S. pneumoniae is mediated by CK2, IKKβ-IκBα, and p38 MAPK. Biochem Biophys Res Commun. 2006;351:368–75. doi: 10.1016/j.bbrc.2006.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield DW, Luscher B, Lozeman FJ, Eisenman RN, Krebs EG. Phosphorylation of casein kinase II by p34cdc2 in vitro and at mitosis. J Biol Chem. 1992;267:13943–51. [PubMed] [Google Scholar]
- Bosc DG, Slominski E, Sichler C, Litchfield DW. Phosphorylation of casein kinase II by p34cdc2. Identification of phosphorylation sites using phosphorylation site mutants in vitro. J Biol Chem. 1995;270:25872–8. doi: 10.1074/jbc.270.43.25872. [DOI] [PubMed] [Google Scholar]
- Munstermann U, Fritz G, Seitz G, Lu YP, Schneider HR, Issinger OG. Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur J Biochem. 1990;189:251–7. doi: 10.1111/j.1432-1033.1990.tb15484.x. [DOI] [PubMed] [Google Scholar]
- Scaglioni PP, Yung TM, Cai LF, et al. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell. 2006;126:269–83. doi: 10.1016/j.cell.2006.05.041. [DOI] [PubMed] [Google Scholar]
- Zhang P, Davis AT, Ahmed K. Mechanism of protein kinase CK2 association with nuclear matrix: role of disulfide bond formation. J Cell Biochem. 1998;69:211–20. [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]