

Abstract
Primary Sjögren syndrome (pSS) is an autoimmune disorder characterized by an epithelial injury surrounded by dense lymphocytic infiltrates. The conditions for the long-term maintenance of human salivary gland epithelial cells from pSS patients and a co-culture system with pSS lymphocytes were used to assess the effect of Rituximab (RTX) on the inflammatory condition and progression in pSS. Quantitative real-time PCR, genes and protein array analysis, Western blot, flow cytometry, small interfering RNA transfection and nuclear factor-κB (NF-κB) DNA binding assays were used as methods. Supporting the benefits of RTX, this study demonstrates that RTX decreases NF-κB activity and interrupts the NF-κB signalling pathway through the up-regulation of the Raf-1 kinase inhibitor protein (RKIP). Over-expression of RKIP down-regulates interleukins, their receptors and the expression of genes encodes proteins that attracted lymphocytes. Silencing of the RKIP gene leads to significantly increased expression and release of pro-inflammatory mediators supporting that RKIP expression could be involved in the suppression of NF-κB activation in pSS salivary gland epithelial cells.
Keywords: array, nuclear factor-κB, Raf-1 kinase inhibitor protein, Rituximab, Sjögren syndrome
Introduction
Primary Sjögren syndrome (pSS) is a systemic autoimmune disease characterized by chronic inflammation of salivary and lachrymal glands preferentially, but may frequently also involve other exocrine glands, for example those of the respiratory tract, gastrointestinal tract and skin.1 Focal infiltrates of T and B lymphocytes, dendritic cells and macrophages with subsequent impairment of the salivary and lachrymal glandular function are hallmarks of the disease and result clinically in xerostomia and keratoconjunctivitis sicca.2 Although the process that underlies the cellular and humoral autoimmune response in pSS is not known, it is well established that both T and B lymphocytes are involved and that interactions of activated salivary gland epithelial cells (SGEC) and endothelial cells with the infiltrating lymphoid and dendritic cells contribute to the perpetuation and progression of the disease as well as to systemic lymphocyte derangements.3 Characteristic features indicating B-cell disturbances in pSS include the presence of several autoantibody populations such as the rheumatoid factor and anti-SSA/SSB antibodies, circulating immune complexes, hypergammaglobulinaemia, characteristic disturbances of peripheral B-cell subsets, formation of ectopic lymphoid tissue with germinal centre-like structures, oligoclonal B-cell proliferations and, finally, the occurrence of non-Hodgkin's lymphoma, which is the most serious complication of pSS.4 About 5% of patients with SS develop a malignant B-cell non-Hodgkin's lymphoma, usually of the mucosa-associated lymphoid tissue type and frequently located in the parotid glands.5 Currently, there is a lack of evidence-based intervention therapy that may influence pSS-related chronic inflammation and lymphoproliferation. In clinical practice, B-cell down-regulation may therefore provide a target for treatment. Rituximab (RTX), an engineered chimeric monoclonal antibody against CD20 antigen, a transmembrane protein found on mature, immature and malignant B cells, is then a putative therapy for both sicca syndrome and SS-related B-cell lymphoproliferation.6 Although the mechanisms of action responsible for the effects of RTX are not fully understood, some evidence on the mechanisms of action of RTX has been published supporting the hypothesis that the anti-CD20 monoclonal antibody mediates the inhibition of the constitutive activity of the nuclear factor-κB (NF-κB) pathway. RTX seems to decrease NF-κB DNA binding activity and interrupts the NF-κB signalling pathway through the up-regulation of Raf-1 kinase inhibitor protein (RKIP) expression.7 RKIP has been shown to down-regulate IκB kinase (IKK) activity directly and by interfering with IKK activators it dampens the effects of interleukins on the activation of NF-κB. The loss of RKIP expression in cancers leads to transcriptional activation of NF-κB, resulting in a dramatic inhibition of apoptosis and the development of chemoresistance.8 Several lines of evidence have established a defective feedback regulation of NF-κB underlying pSS,9–15 so this study was designed to assess the effect of RTX treatment on the expression of RKIP inhibitor protein and on the NF-κB-dependent cytokines/chemokines released by SGEC derived from pSS patients in a co-culture system with pSS lymphocytes.
Materials and methods
Patient population
Twenty pSS patients fulfilling the American–European consensus group criteria for pSS that displayed salivary gland focus scores of at least three were enrolled in the study.16 All patients had the clinical symptoms of dry eyes and mouth, a positive Schirmer's test (< 5 mm wetting of a strip of filter paper per 5 min) and Rose Bengal staining (increased uptake of Rose Bengal dye in devitalized areas in the conjunctiva and cornea) along with the presence of at least one of the following autoantibodies: anti-Ro/SSA, anti-La/SSB, antinuclear antibodies, rheumatoid factor. None of the patients studied had received glucocorticoid or immunosuppressive drug treatment before the biopsy. Furthermore, none of the patients had received biological therapy at the time of the lip biopsy. The patients had all given their written consent, the study was approved by the local ethics review committee and the experiments were conducted according to the tenets of the Declaration of Helsinki. After obtaining informed consent, biopsy samples were taken from the minor salivary glands of pSS patients. Minor salivary glands were harvested from the lower lip under local anaesthesia through normal mucosa, according to the explant outgrowth technique.17 Healthy control subjects (n = 8) were employed in these experiments, enrolled from studies evaluating the salivary gland function of healthy control subjects.
Lymphocyte isolation
Peripheral blood mononuclear cells were isolated from venous blood of pSS patients by Ficoll gradient separation. Blood samples were collected in heparinized test tubes using a vacutainer system and diluted (1 : 1) with Hanks’ balanced salt solution (Sigma Aldrich, St Louis, MO), layered over Ficoll-Paque Plus (Amersham Biosciences, Chalfont St Giles, UK) and centrifuged at 1000 g for 15 min. After a short centrifugation at room temperature, lymphocytes, together with monocytes and platelets, were harvested from the interface between the Ficoll-Paque PLUS and sample layers. This material was then centrifuged twice in Hanks’ balanced salt solution to wash the lymphocytes and monocytes and to remove the platelets. The cells were collected and washed in RPMI-1640 supplemented with 10% heat-inactivated (56° for 30 min) fetal calf serum. Peripheral blood lymphocytes were separated from the peripheral blood mononuclear cell population by removing the monocytes that adhered to plastic: the peripheral blood mononuclear cells were washed twice and resuspended at a concentration of 5 × 106/ml in RPMI-1640 supplemented with 2 mm glutamine, 10% heat-inactivated (56° for 30 min) fetal calf serum, 100 μg/ml streptomycin and 100 IU/ml penicillin (complete medium). Then, the cell suspension was distributed in four-well microculture plates (Nunc, Roskilde, Denmark). After 2 hr incubation at 37°, 5% CO2, the non-adherent lymphocytes were collected by gentle washing with medium. The cell population obtained in these conditions contained < 0·2% non-lymphocyte cells by fluorescence-activated cell sorter analysis after incubation with monoclonal antibodies (data not shown).
Salivary gland epithelial cells cultures and co-cultures
In order to obtain long-term SGEC cultures, the cells were isolated from the minor salivary glands by microdissection and collagenase (Worthington Diagnostic Division, Millipore, Freehold, NJ) digestion in physiological saline containing 1 mm Ca2+. Following dispersal, cells were re-suspended in McCoy's 5a modified medium supplemented with 10% heat-inactivated (56° for 30 min) fetal calf serum, 1% antibiotic solution, 2 mm l-glutamine, 50 ng/ml epidermal growth factor (Promega, Madison, WI), 0·5 μg/ml insulin (Novo, Bagsvaerd, Denmark) and incubated at 37°, 5% CO2 in air. Contaminating fibroblasts were selectively removed by treatment of the cultures with 0·02% EDTA. The epithelial origin of cultured cells was routinely confirmed by staining with monoclonal antibodies against epithelial-specific markers, including the various cytokeratins and epithelial membrane antigens and the absence of myoepithelial, fibroblastoid and lymphoid markers, using immunocytochemistry as previously described.18 In the co-culture experiments, pSS SGEC were seeded at 5 × 105 cells into six-well flat-bottomed culture plates (Nunc, Kamstrup, Denmark). After 4 hr allowing cells to attach, the pSS lymphocytes were co-cultured for 48 hr with SGEC. To address the implication of the anti-CD20 antibody in pro-inflammatory cytokine secretion by pSS SGEC, RTX at 20 μg/ml was added or not in the co-culture for 24–48 hr. Each analysis was performed after extensive washing to remove lymphocytes; this was validated by FACS, reporting that CD20-negative cells were over 98% (data not shown).
Quantitative real-time PCR and PCR array analysis
Total RNA was extracted and purified from healthy and pSS SGEC using Trizol reagent (Life Technologies, Carlsbad, CA). Both RNA isolation and cDNA synthesis were performed according to the manufacturers’ instructions. RKIP mRNA quantification was determined by quantitative real-time PCR using gene-specific probes as described previously.11 Glyceraldehyde 3-phosphate dehydrogenase was used as a normalizing control. PCR array analysis was performed and the relative expression of 84 inflammatory cytokines, chemokines and their receptors was examined. Total cellular RNA was isolated using Trizol (Qiagen, Valencia, CA) and isopropanol precipitation or RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Real-time PCR for signalling pathway genes were analysed in total RNA using the Human Inflammatory Response and Autoimmunity SuperArray RT2 Profiler (SABiosciences Corporation, Frederick, MD,) according to the manufacturer's protocol. The SuperArray combines SYBR Green-based real-time quantitative RT-PCR technology with a multi-gene array plate format to simultaneously analyse 84 human cytokine and chemokine genes. The cDNA was prepared from 1 μg total RNA using an RT2 PCR array first-strand kit. The reactions were conducted in a 25-μl mixture, which included Master mix, nuclease-free H2O and 1 μl of template cDNA. The PCR amplification was conducted with an initial 10-min step at 95° followed by 40 cycles of 95° for 15 seconds and 60° for 1 min. Real-time quantifications were carried out using the ABI Prism 7700 fast real-time PCR system (Applied Biosystems, Foster City, CA). The ΔΔCt method was used to analyse the expression level of each gene in comparison with five endogenous control housekeeping genes, present on the PCR array, that were used for normalization. Results were expressed as fold changes in gene expression.
Cytokine proteins array analysis
A human cytokine Multi-Analyte ELISArray Kit (SABiosciences) was used to measure the cytokine production of interleukin-1α (IL-1α), IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17A, interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α) and granulocyte–macrophage colony-stimulating factor in supernatants of untreated and RTX-treated pSS SGEC. The arrays were performed according to the manufacturer's instructions. The absorbance levels of the cytokines were measured on a plate reader [VERSAmax microplate reader (Molecular Devices Corp, Silicon Valley, CA)] at 450 nm. Sampling was performed in triplicates including standards. Standard errors (SE) of the mean were calculated from three biological repeats.
Western blot
Protein lysates, obtained from healthy control SGEC, untreated and RTX-treated pSS SGEC were subjected to SDS–PAGE. Membranes were incubated for 90 min with goat anti-human RKIP polyclonal antibody (R&D Systems, Minneapolis, MN) as first antibody. Tween-20-diluted secondary antibody was IRDye-labelled (680/800CW) (LI-COR Biosciences, Lincoln, NE). For immunoblot analysis, the Li-cor Odyssey infrared imaging system was used (LI-COR). The Western blot images were analysed by imaging densitometry using Quantity One Software (Bio-Rad Laboratories, Hercules, CA) and compared with β-actin. The data are expressed as optical density × mm2.
FACS analysis
For flow cytometric analysis, healthy control SGEC, untreated and RTX-treated pSS SGEC were permeabilized with PBS containing 0·5% BSA (Sigma) and 0·5% saponin (Sigma) and incubated with goat anti-human RKIP polyclonal antibody (R&D Systems) and with secondary antibody conjugated with AlexaFluor 488 (Life Technologies). The protein expression was analysed by a Becton Dickinson (BD, Heidelberg, Germany) FACSCanto™ II flow cytometer and bd facs diva software. Values are given as percentages of positive cells and the mean fluorescence intensity (MFI) is reported.
Small interfering RNA transfection
The RKIP-specific small interfering RNAs (siRNAs) were designed by Ambion (Austin, TX).The glyceraldehyde 3-phosphate dehydrogenase-positive control siRNA (used in experiments to optimize the siRNA transfection efficiency) was also from Ambion. The negative control siRNA was from Ambion and comprised an siRNA sequence not corresponding to any eukaryotic gene. Healthy and pSS SGEC were transfected with siRNAs using the siPORT NeoFX transfection agent (Ambion). The highest efficiencies in silencing the target gene were obtained using mixtures of siRNA duplexes targeting different regions of the gene of interest. The procedure was performed according to the manufacturer's instructions. Silencing was observed by semi-quantitative RT-PCR.
NF-κB DNA binding assays
We used the ELISA-based TransAM Flexi NF-κB p65 Kit (Active Motif, Carlsbad, CA) to monitor the activity of NF-κB p65 family members in healthy control SGEC, untreated pSS SGEC and RTX-treated pSS SGEC. Nuclear fractions were harvested from cells using the nuclear extraction kit following the manufacturer's suggestions (Active Motif). The protein concentration was determined using the Bradford's method. NF-κB DNA binding activity was quantitatively assessed using the TransAM NF-κB p65 kit (Active Motif) according to the manufacturer's protocol. To each well containing an NF-κB consensus binding site (5′-GGGACTTTCC-3′), 10 μg of nuclear extract in cell binding and cell lysis buffer was added to each well in triplicate. We used 5 μg of nuclear extract of Raji cells (a Burkitt's lymphoma cell line) as positive control (Active Motif). To assess DNA binding specificity, excess wild-type NF-κB consensus oligonucleotide was added (20 pmol/well) to compete for binding, as compared with other wells, to which an inactive mutated consensus oligonucleotide was added. The absorbance was measured at 450 nm by the VERSAmax microplate reader (Molecular Devices Corp, Silicon Valley, CA).
Statistics
The data were analysed for normality using the Wilks–Shapiro test. Differences in means for paired observations were analysed by Student's t-test. In all instances values of P < 0·05 were considered statistically significant.
Results
RTX alters the gene expression profile of inflammatory cytokines and chemokines
To identify specific molecular changes in RTX-treated pSS SGEC derived from a co-culture system with pSS lymphocytes, we performed array-based PCR transcriptional profiling of a variety of inflammatory cytokines, chemokines and their respective receptor genes. The pSS SGEC were co-cultured with pSS lymphocytes for 2 days and then RNA was extracted. Among a total of 84 genes screened (see Supporting information, Table S1), 30 genes were measurable in all the samples analysed (Fig. 1a); the other genes were either undetectable or no technically satisfactory amplification was achieved. This in vitro study demonstrated that RTX-treated pSS SGEC exhibited differences compared with untreated pSS SGEC and RTX treatment significantly decreased the expression of almost all the measurable genes. The under-expressed genes were classified into two categories. The largest group consisted of down-regulated interleukins, e.g. IL-1α, IL-1β, IL-1F8, IL-6, IL-8, IL-9, IL-13, IL-17, IL-22, TNF-α, IFN-γ and their receptors (IL-1RN, IL-10Rα, IL-13Rα, CCR1, CCR2, CCR3, CCR4, CCR5, CCR8, CCR9 and CXCR1). Interestingly, the second group of down-regulated genes encodes proteins that attracted lymphocytes and were involved in the inflammatory response [some of the CXC chemokines (CXCL1, CXCL6, CXCL9, CXCL12, CXCL13) and CC chemokines (CCL2, CCL13)]. The decline in expression levels of pro-inflammatory chemokine and cytokine genes was unexpected because RTX targets CD20, which is restricted to B cells. To estimate the effect of RTX treatment on cytokine production at protein level in co-culture experiments performed in vitro associating pSS SGEC and pSS lymphocytes, a human cytokine ELISA array was used to evaluate various cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17A, IFN-γ, TNF-α) in supernatants of co-culture-derived pSS SGEC. The levels of all inflammatory cytokines were decreased after treatment with RTX, showing statistically significant differences (P < 0·01) compared with untreated pSS SGEC (Fig. 1b). Interestingly, pro-inflammatory chemokines and cytokines produced by pSS SGEC may be affected by the lymphocytic infiltrate as they were decreased in supernatants after RTX treatment and we hypothesized that B cells might influence production of these factors from SGEC.
Figure 1.

(a) Genes profile showing altered regulation of genes induced by Rituximab (RTX) in primary Sjögren syndrome salivary gland epithelial cells (pSS SGEC). RTX treatment determines significant differential pro-inflammatory cytokines/chemokine and receptor gene expression in pSS SGEC derived from a co-culture system with pSS lymphocytes compared with untreated pSS SGEC. The pooled total RNAs isolated from untreated and RTX-treated pSS SGEC [indicated respectively as pSS SGEC (co-culture) and pSS SGEC+RTX (co-culture)] were used and real-time PCR arrays were repeated twice. (Data represent the mean ± SE of three independent experiments). As depicted by histograms, blockade of lymphocyte activation through the use of the anti-CD20 mAb RTX reduces the pro-inflammatory cytokines/chemokine gene induction in pSS SGEC. (b) Comparison of protein levels of inflammatory mediators upon RTX treatment of pSS SGEC. A human cytokine Multi-Analyte ELISA array was used to measure cytokine production by pSS SGEC co-cultured with pSS lymphocytes treated or not with RTX [indicated respectively as pSS SGEC (co-culture) and pSS SGEC+RTX (co-culture)]. Values represent the optical density (OD) decrease versus untreated pSS SGEC cultures from triplicate wells. (Data represent the mean ± SE of three independent experiments). The statistical analysis was performed using the Student's t test (*P < 0·05; **P < 0·01). For both gene and protein arrays, untreated and RTX-treated pSS SGEC cultures without the addition of lymphocytes were used as experimental controls.
RTX treatment diminished NF-κB activation in pSS SGEC: evaluation of RKIP inhibitory protein expression
One of the mechanisms by which RTX inhibits the NF-κB signalling pathway involves RKIP induction and RKIP was identified as a negative regulator of the NF-κB activation,19 so in the first part of our study we performed the TransAM Flexi NF-κB p65 assay on pSS SGEC following treatment with RTX (20 μg/ml) to evaluate the effect of the addition of RTX on NF-κB activation in vitro. As shown in Fig. 2(a), the findings demonstrate that RTX antibody treatment decreases the ability of NF-κB to bind to its DNA binding site in a significant manner (P < 0·01). As RKIP expression is unknown in pSS SGEC, we then examined RKIP protein levels by Western blot analysis in pSS SGEC in comparison with healthy SGEC. β-Actin was chosen as loading control because of its uniform expression in the cells. Expression levels were densitometrically determined, the ratio RKIP : β-actin was calculated and subsequently normalized to a calibrator sample, which was set to a value of 100%. Western blot results revealed that the quantity of RKIP protein expressed in pSS SGEC was significantly lower than that of healthy SGEC (P < 0·01) (Fig. 2b,c). Flow cytometric analysis confirmed this result, detecting low, but definite, intracellular expression of RKIP protein by pSS SGEC whereas healthy SGEC exhibited high RKIP staining. Accordingly, a decreased significant difference in the MFI of RKIP was identified for pSS SGEC compared with healthy SGEC (P < 0·01) (Fig. 2d,e). Immunohistochemical results were concordant with those obtained with the above described techniques. A dissimilar expression pattern by the epithelial cells was detected in minor salivary gland tissues obtained from healthy controls that show a high RKIP expression level and minor salivary glands from pSS patients that revealed low expression of RKIP in ductal and acinar epithelial cells (Fig. 2f). To assess whether RKIP protein loss observed in pSS SGEC correlates with decreased mRNA levels, we performed quantitative real-time PCR and found that mRNA levels presented statistically significant differences (from twofold to threefold less RKIP mRNA, P < 0·01) among the pSS SGEC and healthy SGEC (Fig. 2g). The protein and gene analysis results seem to concur that RKIP is down-regulated in pSS SGEC and this prompted us to try to understand the biological role of RKIP down-regulation in the mechanism of RTX action in pSS SGEC.
Figure 2.

(a) Rituximab (RTX) treatment decreases nuclear factor-κB (NF-κB)-DNA-binding activity in primary Sjögren syndrome salivary gland epithelial cells (pSS SGEC). Nuclear fractions were harvested from untreated and RTX-treated pSS SGEC and the binding capacity of p65 NF-κB subunit to a plate-immobilized oligonucleotide containing a κB binding site was measured by TransAM NF-κB assay in the nuclear extracts. Data are means ± SE of three independent experiments. (b, c) Loss of Raf-1 kinase inhibitor protein (RKIP) expression in pSS SGEC. Representative Western blot analysis of healthy and pSS SGEC, respectively, showing that the expression levels of RKIP were down-regulated in pSS SGEC. Graph demonstrates the x-fold change in RKIP protein expression compared with β-actin control as measured by densitometry. (d, e) Flow cytometric analysis of RKIP expression in healthy and pSS SGEC. The graph summarizes the results of mean fluorescence intesnity (MFI) evaluation of at least three independent experiments. Data are expressed as mean values ± SE and P-values have been calculated using Student's t-test (**P < 0·01). (f) Representative immunohistochemical staining for RKIP protein on normal healthy minor salivary glands and pSS minor salivary glands. RKIP protein expression was visualized by brown DAB staining. The nuclei were stained with haematoxylin (blue). Scale bar = 20 μm. (g) The endogenous levels of RKIP mRNA in pSS SGEC as measured by real-time quantitative PCR (mean ± SE of three independent experiments; **P < 0·01).
RKIP gene silencing activates the NF-κB signalling pathways in pSS SGEC
We estimated the effect of RTX treatment on RKIP expression in co-culture experiments performed associating pSS SGEC and pSS lymphocytes, observing an increased expression of RKIP in RTX-treated pSS SGEC in comparison with untreated pSS SGEC (Fig. 3a–d). Hence, we examined if RKIP induction was associated with RTX-mediated inhibition of the NF-κB pathway observed in pSS SGEC, assuming that RTX-mediated up-regulation of RKIP expression enhances its association with signalling molecules, interfering with the activation of the NF-κB signalling pathway. We therefore performed a series of directed in vitro experiments to examine the role of RKIP on NF-κB signalling. By inhibiting the endogenous RKIP activity by RKIP gene knock-down and by monitoring the NF-κB activation status in healthy SGEC and untreated pSS SGEC, we found that the activity of the p65 family member of NF-κB was increased as a result of RKIP gene silencing in SGEC tested (Fig. 3e) supporting the evidence that RKIP exerts its inhibitory function on NF-κB activation pathway.
Figure 3.

Effects of Rituximab (RTX) treatment on Raf-1 kinase inhibitor protein (RKIP) expression in primary Sjögren syndrome salivary gland epithelial cells (pSS SGEC). (a, b) Protein lysates were harvested at the indicated time from untreated and RTX-treated pSS SGEC and subjected to Western blot analysis for RKIP protein determination. β-Actin was used as protein loading control. (c, d) Flow cytometry profiles (c), and quantification of the cellular fluorescence shown via mean fluorescence intensity (MFI) (d) of RKIP-positive pSS SGEC before and after RTX treatment. (e) DNA binding capacity of p65 nuclear factor-κB (NF-κB) subunit measured by TransAM NF-κB assay in the nuclear extracts from pSS SGEC after transfection with small interfering (siRNA) specific for RKIP. Healthy SGEC were used as control. Data are means ± SE of three independent experiments. (f) RTX diminishes the NF-κB activity in pSS SGEC through the over-expression of RKIP. DNA binding capacity of the p65 NF-κB subunit in the nuclear extracts from untransfected pSS SGEC and pSS SGEC following specific down-regulation of RKIP expression by RKIP siRNA. The graph is representative of two independent experiments (mean ± SE). (g) Role of RKIP in the RTX-mediated regulation of the pro-inflammatory cytokines release by pSS SGEC. RKIP gene expression was specifically inhibited using siRNA technology. In the presence of RTX treatment, transfectants showed an increased production of all the pro-inflammatory cytokines tested by Multi-Analyte ELISA array (mean ± SE of two independent experiments). The statistical analysis was performed using the Student's t-test (*P < 0·05; **P < 0·01).
RTX inhibits NF-κB activation in pSS SGEC via induction of RKIP
Because RTX-treated cells exhibited decreased NF-κB activity as described above, we analysed the effect of RTX on NF-κB in response to RKIP gene silencing in pSS SGEC to investigate the possible mechanism by which RTX inhibits the NF-κB signalling pathway. In Fig. 3(f), we reported an increased NF-κB activation in RTX-treated pSS SGEC following RKIP gene knockdown, showing that RTX-mediated up-regulated RKIP expression could be involved in the suppression of the NF-κB activation observed in response to anti-CD20 treatment of pSS SGEC.
RTX-mediated induction of RKIP: role of RKIP in the regulation of the pro-inflammatory cytokines secretion by pSS SGEC
In the light of the reported effects of RTX on RKIP-mediated NF-κB signalling inhibition, we considered the possibility that RTX could modulate the pro-inflammatory cytokine secretion by pSS SGEC through the over-expression of RKIP and the subsequent inhibition of the NF-κB activation. The use of siRNA technology demonstrated a direct role of RKIP in the RTX-mediated regulation of the release of pro-inflammatory cytokines by pSS SGEC. In fact, as shown in Fig. 3(g), transfectants showed increased production of all the pro-inflammatory cytokines tested in the presence of RTX treatment. Taken together, these results denote the involvement of NF-κB in the regulation of cytokine expression by pSS SGEC and the ability of RTX to decrease NF-κB-dependent cytokine gene transcription through the RKIP over-expression.
Discussion
The study of disorders that affect the salivary glands necessitates the investigation of the physiology and pathophysiology of SGEC. Cultured cell systems provide a means for such investigation, including the influence of infectious agents, hormones, toxins and drugs. The in vitro analysis of cultured SGEC is particularly advantageous for the study of pSS. Studies performed on pSS patients have demonstrated the aberrant in situ expression of various molecules by SGEC, including pro-inflammatory cytokines,20 MHC class II molecules,21 co-stimulatory and adhesion molecules,18 oncogenes,22 autoantigens21 and markers of apoptosis.23–25 These phenomena may be due to the epithelial activation by cytokines locally produced by the lymphocytic infiltrates, or to intrinsically operating processes that may occur in the epithelia of pSS patients. Evidence from our laboratory had indicated that pSS SGEC, cultured in vitro, are capable of expressing various immune-response proteins such as cytokines, chemokines and their receptors.13,26–28 Prompted by such protein production, we sought to establish a co-culture system for the functional assessment of SGEC capacity to interact with lymphoid cells in vitro. In this study, the conditions for the long-term maintenance and analysis of human pSS SGEC, as well as a co-culture system, studying the interactions between epithelial cells and lymphocytes, were used to assess the effect of RTX, successfully used in the treatment of patients with follicular low-grade and aggressive diffuse large B-cell lymphoma,29,30 on pSS SGEC release of pro-inflammatory mediators. The potential clinical utility of RTX therapy has recently been investigated in pSS,31,32 owing to its proven efficacy in other chronic inflammatory diseases, such as rheumatoid arthritis33 and systemic vasculitis,34 and its effects on potential disease-inciting B cells. As B cells are involved in the pathogenesis of pSS, and B-cell down-regulation may be a target of treatment, RTX is then a putative therapy for both sicca syndrome and SS-related B-cell lymphoproliferation.29,35 Preliminary experiences of RTX therapy in pSS patients, with or without a lymphoproliferative disorder, suggest that pSS patients with more residual exocrine gland function might better benefit from RTX.29,35 However, the efficacy of RTX in pSS remains an open issue. The study reported here provides evidence for the first time that RTX treatment determines a significantly decreased release of inflammatory proteins, cytokines and growth factors by pSS SGEC in the co-culture system with pSS lymphocytes. In response to stimulation with RTX, which causes a depletion of B cells, the secretion of all of the pro-inflammatory mediators tested was drastically reduced at both gene and protein levels. This effect suggests that, under chronic inflammatory conditions, lymphocytes regulate the cytokine and chemokine expression by SGEC located in their environment. This exacerbates the inflammation, influencing the intrinsic activation status of SGEC to further modulate the release of inflammatory mediators by post-translational pathways. To clarify the mechanisms suspected of mediating the therapeutic effects of RTX, we tested in vitro whether RTX-mediated pro-inflammatory protein down-regulation could be a result of the inhibition of the constitutive activity of the NF-κB pathway in pSS SGEC. Results of our previous studies and those of other authors have shown that pSS SGEC are characterized by persistent NF-κB activation, which could explain the deregulated cytokine production observed in pSS.9–15 These reported findings are consistent with those demonstrating that defects in the regulation of NF-κB-dependent gene expression contribute to a variety of inflammatory and autoimmune diseases, neurological disorders and cancer.36,37 Here, we reported that the treatment of pSS SGEC with RTX decreases the NF-κB DNA binding activity in the cells, suggesting that the down-regulation of NF-κB activity could be involved in the inhibition of the secretion of pro-inflammatory mediators that is observed following RTX treatment. The underlying mechanism by which RTX interferes with the constitutive activity of the NF-κB pathway might be due to the modulation of RKIP expression, which inhibits the NF-κB. RKIP, which is expressed in various tissues and cell types, participates in the modulation and control of intracellular G protein-coupled receptor signalling pathways and in the NF-κB signal channel.38 Recent studies have suggested that RKIP is likely to play an important role in various inflammatory diseases and cancers8 and RTX could promote RKIP expression in a non-Hodgkin's lymphoma cell line, which increased sensitivity to the drug.19 These findings raise important questions about the potential role of RKIP in the pathogenesis of pSS. However, there are no reports on the role of RKIP in pSS. Hence, the present in vitro study was performed to investigate, as a second approach, the expression and function of RKIP in pSS SGEC. We demonstrated that RKIP expression was significantly reduced in pSS SGEC compared with healthy cells at both the mRNA and protein levels, suggesting that RKIP is constitutively under-expressed in pSS SGEC and that it controls the expression of inflammatory factors by regulating the NF-κB activation. As the NF-κB transcription factor plays a critical role in coordinating the expression of a large number of pro-inflammatory mediators in pSS,9–15 the under-expression of RKIP could increase the NF-κB activity, leading to a persistent inflammatory condition as observed in pSS. We confirmed that the function of RTX as a negative regulator of the NF-κB pathway in pSS SGEC is a result of the modulation of RKIP expression, demonstrating that RTX up-regulates RKIP expression in pSS SGEC. Our results corroborate the findings that RTX-mediated RKIP induction diminishes the phosphorylation of the components of the NF-κB pathway,19 providing a new mechanism induced by RTX that could regulate inflammation. To confirm that the RTX-mediated decreased NF-κB activity occurs through the increased expression of the inhibitory protein RKIP, we performed a set of experiments demonstrating that RKIP gene silencing resulted in augmentation of the pro-inflammatory cytokine secretion by pSS SGEC through preventing NF-κB inhibition. These findings provide a possible mechanism induced by RTX that regulates the inflammatory processes characterizing pSS through the induction of RKIP and subsequent inhibition of the activity of the NF-κB pathway. Mechanisms involved in RTX-mediated regulation of the inflammatory processes characterizing pSS through the modulation of the NF-κB activity by interaction with pSS lymphocytes and SGEC need further investigation. Data in the literature indicate that in patients with pSS, treatment with RTX led to effective depletion of blood B cells.29 There are several postulated mechanisms for the action of RTX in B-cell depletion. The mechanism of the antibody-dependent cell-mediated cytotoxicity predominates in the blood and its effects in pSS would be dependent on the membrane receptors for the Fc portion of immunoglobulin G class antibodies (FcγR) that display a variable affinity for IgG1.39 Evidence suggests that Fc/FcγR interactions are critical, as determined in both animal models and humans.39 Cross-linking of these receptors by aggregated IgG, in the form of antigen–antibody complexes, triggers a very wide array of responses important for host defence and for modulation of the immune response.40 There is a great deal of interest in understanding the signalling mechanisms that lead to the various cell responses. Very little is known about the signal transduction pathway from FcγR to nuclear factors in the cell nucleus.
To explain the anti-inflammatory role of RTX in SGEC from pSS patients reported in this work, we could postulate a hypothetical mechanism based on the recently demonstrated over-expression of FcγRI, FcγRII, and FcγRIII on pSS SGEC.41 This hypothesis involves the formation of the IgG immune complexes between B lymphocytes and RTX that could engage specific FcγR on pSS SGEC, resulting in a decreased NF-κB activity and interruption of the NF-κB signalling pathway through the up-regulation of the RKIP protein. It is known from the literature that the signalling pathway from FcγR leading to expression of different pro-inflammatory genes, initiates with tyrosine kinases and requires mitogen-activated protein kinase activation. Therefore, we have reason to think that the missing link between the activation of the FcγR and the inhibition of the secretion of pro-inflammatory cytokines by RTX could involve mitogen-activated protein kinase signalling. However, the molecular events that follow and that transduce signals from these receptors to NF-κB activation in the nucleus are still poorly defined and this hypothesis needs to be validated through future experiments to obtain a greater knowledge of the mechanisms in which they are involved. A schematic model of RTX-mediated inhibition of the NF-κB pathway in pSS SGEC is reported in Fig. 4.
Figure 4.
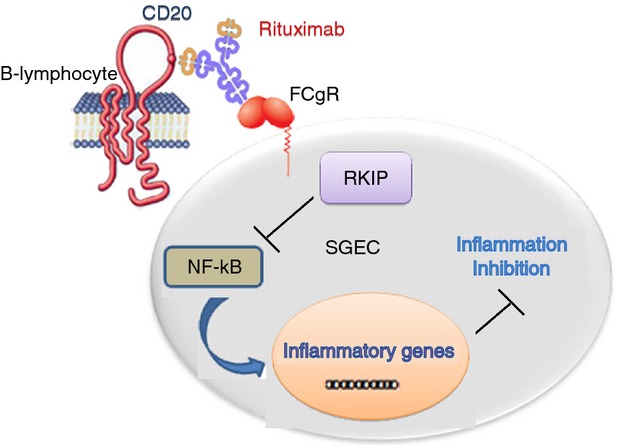
Proposed model of rituximab (RTX)-mediated inhibition of the nuclear factor-κB (NF-κB) pathway in primary Sjögren syndrome salivary gland epithelial cells (pSS SGEC). The NF-κB signalling pathway is constitutively active in pSS SGEC and these cells express low levels of Raf-1 kinase inhibitor protein (RKIP). In a co-culture system with pSS lymphocytes, on ligation to CD20, RTX up-regulates RKIP expression. RKIP inhibits the activity of the NF-κB pathway, culminating in decreased expression of NF-κB-dependent pro-inflammatory genes.
The above studies reveal a role for RTX as a negative regulator of major inflammatory pathways in pSS, identifying new targets for therapeutic intervention and providing a rational molecular basis that opens new therapeutic perspectives in pSS.
Acknowledgments
This work was supported by a grant (No: 20216000056) from the Italian Ministry for Universities and Research. We are grateful to M.V.C. Pragnell, for critical reading of the manuscript.
Disclosures
The authors declare that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
List of genes on the RT2 Profiler PCR Array.
References
- Fox RI. Sjögren's syndrome. Lancet. 2005;366:321–31. doi: 10.1016/S0140-6736(05)66990-5. [DOI] [PubMed] [Google Scholar]
- Adamson TC, Fox RI, Frisman DM, Howell FV. Immmunohistologic analysis of lymphoid infiltrates in primary Sjögren's syndrome using monoclonal antibodies. J Immunol. 1983;130:203–8. [PubMed] [Google Scholar]
- Mitsias DI, Kapsogeorgou EK, Moutsopoulos HM. The role of epithelial cells in the initiation and perpetuation of autoimmune lesions: lessons from Sjögren's syndrome (autoimmune epithelitis) Lupus. 2006;15:255–61. doi: 10.1191/0961203306lu2290rr. [DOI] [PubMed] [Google Scholar]
- Hansen A, Lipsky PE, Dörner T. Immunopathogenesis of primary Sjögren's syndrome: implications for disease management and therapy. Curr Opin Rheumatol. 2005;17:558–65. doi: 10.1097/01.bor.0000172801.56744.c3. [DOI] [PubMed] [Google Scholar]
- Zintzaras E, Voulgarelis M, Moutsopoulos HM. The risk of lymphoma development in autoimmune diseases: a meta-analysis. Arch Intern Med. 2005;165:2337–44. doi: 10.1001/archinte.165.20.2337. [DOI] [PubMed] [Google Scholar]
- Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104:1793–800. doi: 10.1182/blood-2004-01-0039. [DOI] [PubMed] [Google Scholar]
- Jazirehi AR, Huerta-Yepez S, Cheng G, Bonavida B. Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-κB signalling pathway in non-Hodgkin's lymphoma B-cell lines: role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005;65:264–76. [PubMed] [Google Scholar]
- Al-Mulla F, Bitar MS, Taqi Z, Yeung KC. RKIP: much more than Raf kinase inhibitory protein. J Cell Physiol. 2013;228:1688–702. doi: 10.1002/jcp.24335. [DOI] [PubMed] [Google Scholar]
- Peng B, Ling J, Lee AJ, et al. Defective feedback regulation of NF-κB underlies Sjögren's syndrome in mice with mutated κB enhancers of the IκBα promoter. Proc Natl Acad Sci USA. 2010;107:15193–8. doi: 10.1073/pnas.1005533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa-Geffen A, Wolf Y, Hanin G, et al. Activation of the alternative NFκB pathway improves disease symptoms in a model of Sjögren's syndrome. PLoS ONE. 2011;6:e28727. doi: 10.1371/journal.pone.0028727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisto M, Lisi S, Lofrumento DD, Ingravallo G, Maiorano E, D'Amore M. A failure of TNFAIP3 negative regulation maintains sustained NF-κB activation in Sjögren's syndrome. Histochem Cell Biol. 2011;135:615–25. doi: 10.1007/s00418-011-0821-3. [DOI] [PubMed] [Google Scholar]
- Lisi S, Sisto M, Lofrumento DD, D'Amore M. Altered IκBα expression promotes NF-κB activation in monocytes from primary Sjögren's syndrome patients. Pathology. 2012;44:557–61. doi: 10.1097/PAT.0b013e3283580388. [DOI] [PubMed] [Google Scholar]
- Lisi S, Sisto M, Lofrumento DD, D'Amore M. Sjögren's syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab Invest. 2012;92:615–24. doi: 10.1038/labinvest.2011.190. [DOI] [PubMed] [Google Scholar]
- Sisto M, Lisi S, Lofrumento DD, D'Amore M, Frassanito MA, Ribatti D. Sjögren's syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012;13:411–20. doi: 10.1038/gene.2012.9. [DOI] [PubMed] [Google Scholar]
- Sisto M, Lisi S, Lofrumento DD, Ingravallo G, De Lucro R, D'Amore M. Salivary gland expression level of IκBα regulatory protein in Sjögren's syndrome. J Mol Histol. 2013;44:447–54. doi: 10.1007/s10735-013-9487-6. [DOI] [PubMed] [Google Scholar]
- Vitali C, Bootsma H, Bowman SJ, et al. Classification criteria for Sjögren's syndrome: we actually need to definitively resolve the long debate on the issue. Ann Rheum Dis. 2013;72:476–8. doi: 10.1136/annrheumdis-2012-202565. [DOI] [PubMed] [Google Scholar]
- Sens DA, Hintz DS, Rudisill MT, Sens MA, Spicer SS. Explant culture of human submandibular gland epithelial cells: evidence for ductal origin. Lab Invest. 1985;52:559–67. [PubMed] [Google Scholar]
- Kapsogeorgou EK, Dimitriou ID, Abu-Helu RF, Moutsopoulos HM, Manoussakis MN. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjögren's syndrome: high expression of intercellular adhesion molecule-1 (ICAM-1) in biopsy specimens and cultured cells. Clin Exp Immunol. 2001;124:126–33. doi: 10.1046/j.1365-2249.2001.01500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazirehi AR, Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxan, anti-CD20 mAb) in non-Hodgkin's lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene. 2005;24:2121–43. doi: 10.1038/sj.onc.1208349. [DOI] [PubMed] [Google Scholar]
- Youinou P, Pers JO. Disturbance of cytokine networks in Sjögren's syndrome. Arthritis Res Ther. 2011;13:227–31. doi: 10.1186/ar3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routsias JG, Tzioufas AG. Autoimmune response and target autoantigens in Sjögren's syndrome. Eur J Clin Invest. 2010;40:1026–36. doi: 10.1111/j.1365-2362.2010.02342.x. [DOI] [PubMed] [Google Scholar]
- Masaki Y, Sugai S. Lymphoproliferative disorders in Sjögren's syndrome. Autoimmun Rev. 2004;3:175–82. doi: 10.1016/S1568-9972(03)00102-2. [DOI] [PubMed] [Google Scholar]
- Sisto M, Lisi S, Lofrumento D, D'Amore M, Scagliusi P, Mitolo V. Autoantibodies from Sjögren's syndrome trigger apoptosis in salivary gland cell line. Ann N Y Acad Sci. 2007;1108:418–25. doi: 10.1196/annals.1422.044. [DOI] [PubMed] [Google Scholar]
- Sisto M, D'Amore M, Caprio S, Mitolo V, Scagliusi P, Lisi S. Tumor necrosis factor inhibitors block apoptosis of human epithelial cells of the salivary glands. Ann N Y Acad Sci. 2009;1171:407–14. doi: 10.1111/j.1749-6632.2009.04688.x. [DOI] [PubMed] [Google Scholar]
- Varin MM, Guerrier T, Devauchelle-Pensec V, Jamin C, Youinou P, Pers JO. In Sjögren's syndrome, B lymphocytes induce epithelial cells of salivary glands into apoptosis through protein kinase Cδ activation. Autoimmun Rev. 2012;11:252–8. doi: 10.1016/j.autrev.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Lisi S, Sisto M, Lofrumento DD, Cucci L, Frassanito MA, Mitolo V, D'Amore M. Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J Autoimmun. 2010a;35:160–70. doi: 10.1016/j.jaut.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Lisi S, Sisto M. Effects of biological drug adalimumab on tumour necrosis factor-α-converting enzyme activation. Immunol Cell Biol. 2010b;88:297–304. doi: 10.1038/icb.2009.97. [DOI] [PubMed] [Google Scholar]
- Sisto M, Lisi S, Lofrumento DD, Ingravallo G, Mitolo V, D'Amore M. Expression of pro-inflammatory TACE-TNF-α-amphiregulin axis in Sjögren's syndrome salivary glands. Histochem Cell Biol. 2010;134:345–53. doi: 10.1007/s00418-010-0735-5. [DOI] [PubMed] [Google Scholar]
- Devauchelle-Pensec V, Pennec Y, Morvan J, et al. Improvement of Sjögren's syndrome after two infusions of rituximab (anti-CD20) Arthritis Rheum. 2007;57:310–7. doi: 10.1002/art.22536. [DOI] [PubMed] [Google Scholar]
- Pers JO, Devauchelle V, Daridon C, et al. BAFF-modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab-treated patients with Sjögren's syndrome. Arthritis Rheum. 2007;56:1464–77. doi: 10.1002/art.22603. [DOI] [PubMed] [Google Scholar]
- Meijer JM, Meiners PM, Vissink A, et al. Effectiveness of rituximab treatment in primary Sjögren's syndrome: a randomized, double-blind, placebo controlled trial. Arthritis Rheum. 2010;62:960–8. doi: 10.1002/art.27314. [DOI] [PubMed] [Google Scholar]
- Abdulahad WH, Meijer JM, Kroese FG, Meiners PM, Vissink A, Spijkervet FK, Kallenberg CG, Bootsma H. B cell reconstitution and T helper cell balance after rituximab treatment of active primary Sjögren's syndrome: a double-blind, placebo-controlled study. Arthritis Rheum. 2011;63:1116–23. doi: 10.1002/art.30236. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijpe J, van Imhoff GW, Spijkervet FK, et al. Rituximab treatment in patients with primary Sjögren's syndrome: an open-label phase II study. Arthritis Rheum. 2005;52:2740–50. doi: 10.1002/art.21260. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB. Role of the NF-κB pathway in the pathogenesis of human disease states. Curr Mol Med. 2001;1:287–96. doi: 10.2174/1566524013363816. [DOI] [PubMed] [Google Scholar]
- Karin M. The IκB kinase-a bridge between inflammation and cancer. Cell Res. 2008;18:334–42. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- Zeng L, Imamoto A, Rosner MR. Raf kinase inhibitory protein (RKIP): a physiological regulator and future therapeutic target. Expert Opin Ther Targets. 2008;12:1275–87. doi: 10.1517/14728222.12.10.1275. [DOI] [PubMed] [Google Scholar]
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- Hatjiharissi E, Xu L, Santos DD, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood. 2007;110:2561–4. doi: 10.1182/blood-2007-01-070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi S, D'Amore M, Lofrumento D, Mitolo V, Frassanito MA, Dammacco F, Scagliusi P, Sisto M. Modulation of the Fcγ receptors induced by anti-Ro and anti-La autoantibodies: observations in salivary gland cells. Rheumatol Int. 2008;28:43–8. doi: 10.1007/s00296-008-0536-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genes on the RT2 Profiler PCR Array.