

Abstract
Docosahexaenoic acid (DHA) is one of the major ingredients of fish oil and has been reported to have anti-inflammatory properties mediated through the GPR120 receptor. Whether cytosolic phospholipase A2 (cPLA2) and lipid mediators produced from cPLA2 activation are involved in the anti-inflammatory role of DHA in macrophages has not been reported. We report here that DHA and the GPR120 agonist, GW9508, activate cPLA2 and cyclooxygenase 2 (COX-2), and cause prostaglandin E2 (PGE2) release in a murine macrophage cell line RAW264.7 and in human primary monocyte-derived macrophages. DHA and GW9508 activate cPLA2 via GPR120 receptor, G protein Gαq and scaffold protein β-arrestin 2. Extracellular signal-regulated kinase 1/2 activation is involved in DHA- and GW9508-induced cPLA2 activation, but not p38 mitogen-activated protein kinase. The anti-inflammatory role of DHA and GW9508 is in part via activation of cPLA2, COX-2 and production of PGE2 as a cPLA2 inhibitor or a COX-2 inhibitor partially reverses the DHA- and GW9508-induced inhibition of lipopolysaccharide-induced interleukin-6 secretion. The cPLA2 product arachidonic acid and PGE2 also play an anti-inflammatory role. This effect of PGE2 is partially through inhibition of the nuclear factor-κB signalling pathway and through the EP4 receptor of PGE2 because an EP4 inhibitor or knock-down of EP4 partially reverses DHA inhibition of lipopolysaccharide-induced interleukin-6 secretion. Hence, DHA has an anti-inflammatory effect partially through induction of PGE2.
Keywords: cytokines, inflammation, lipid mediators, signal transduction
Introduction
Omega-3 fatty acids, mainly docosahexaenoic acid (DHA) and ecosapentaenoic acid (EPA), have been shown to play an anti-inflammatory role in a number of human inflammatory diseases, including atherosclerosis, asthma, arthritis and inflammatory bowel diseases. DHA supplementation has been reported to reduce mortality in patients with existing cardiovascular disease, improve symptoms of mild to moderate asthma, protect from ulcerative colitis and reduce the intensity of joint pain and the need for non-steroidal anti-inflammatory drugs in patients with rheumatoid arthritis.1–5 DHA has also been demonstrated to exert beneficial effects in animal models of neuroinflammation6 and diabetes.7
Unsupplemented, the physiological serum level of DHA is about 1–5 μm,8–10 but it is present at higher concentrations in esterified form, such as triglycerides and phospholipids.5 Dietary intake of 0·5–2 g DHA correlates with plasma phospholipid levels of 90–120 μm.11 Recent studies have indicated that the form of dietary supplement is important for the total bioavailability resulting in the final concentration in the serum.12 Oral administration of an omega-3 fatty acid supplement (Epanova, 4 g) during a low-fat consumption period may achieve plasma total DHA levels of about 85 μm.12 Additionally, sepsis patients receiving a fish-oil-based lipid emulsion (Omegaven; DHA, 5·7 g/day), experienced an increase of plasma-free DHA levels up to 65 μm.13
Docosahexaenoic acid may exert anti-inflammatory effects in part through the G-protein-coupled receptor 120 (GPR120).14 G protein-coupled receptors share common structural motifs such as seven transmembrane helices and the ability to activate heterotrimeric G proteins, such as Gαs, Gαi and Gαq. GPR120 is known to couple with the Gαq/11 family of G proteins.15 After ligand binding, GPR120 is phosphorylated and this generates binding sites for β-arrestin 2.16 β-Arrestin 2 binds to GPR120 and then interacts with downstream signalling molecules such as extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK).17,18 This signalling pathway may lead to a variety of anti-inflammatory effects attributed to omega-3 fatty acids.
Cytosolic phospholipase A2 (cPLA2) has been well known to regulate inflammation,19–22 through downstream lipid mediators, as well as having an impact on gene expression.23 Phospholipase A2s (PLA2s) are enzymes that hydrolyse the sn-2 ester bond of phospholipids, releasing a free fatty acid [such as arachidonic acid (AA)] and creating a lysophospholipid.24 PLA2s are classified according to their sequence homology and Ca2+ dependence. The three most described families are Ca2+-dependent secretory PLA2s (sPLA2) and cytosolic PLA2s (cPLA2s), and Ca2+-independent PLA2 (iPLA2).25,26 However, cPLA2α is the only well-characterized PLA2 that is highly selective for phospholipids containing AA at the sn-2 position.27–29 Cytosolic PLA2 is activated by phosphorylation by MAPKs and is translocated to cell membranes by changes in intracellular free calcium levels.27,30–32
Arachidonic acid, the product of cPLA2 activation, an omega-6 fatty acid (C20:4) is the precursor for eicosanoid mediators including prostaglandins, thromboxanes, leukotrienes, hydroxyeicosatetraenoic acids (HETEs) and epoxyeicosatrienoic acids (EETs), which may have a variety of pro-inflammatory and anti-inflammatory effects.33 Prostaglandins are generated from AA by cyclooxygenases, COX-1 and COX-2.34,35 COX-2 is an inducible enzyme and is involved primarily in the regulation of inflammation.36 Prostaglandin E2 (PGE2) is one of the major COX-2-derived prostanoids at inflammatory sites,37 which may play an anti-inflammatory role.38
Therefore, we hypothesize that DHA may regulate cPLA2 activation, change AA release, and modify its downstream lipid mediator production, which may be involved in its anti-inflammatory effect. Whether cPLA2 and its downstream lipid mediators play an anti-inflammatory role for DHA has not been demonstrated. In this study, we first found that cPLA2 is activated by DHA or GW9508, the GPR120 agonist. This led to activation of COX-2 and production of PGE2, which plays an anti-inflammatory role by inhibiting nuclear factor-κB (NF-κB) signalling and through EP4 receptor in murine macrophage RAW264.7 cells and human primary monocyte-derived macrophages. Hence, we propose a new anti-inflammatory mechanism of DHA through cPLA2–AA–COX-2–PGE2–EP4, which may guide the development of novel therapies, such as an EP4 agonist, for treatment of human inflammatory diseases.
Materials and methods
Reagents and antibodies
The cPLA2 inhibitor, N-((2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl)-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl] acrylamide HCl, a 1,2,4-trisubstituted pyrrolidine derivative, U0126 [mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor], and SB203580 (p38 inhibitor) were obtained from Calbiochem (La Jolla, CA). Docosahexaenoic acid, thioetheramide-PC, bromoenol lactone, NS-398 (COX-2 inhibitor), AH6809 (EP2 inhibitor), GW627368X (EP4 inhibitor), and antibodies against EP2 and EP4 were purchased from Cayman Chemical Co (Ann Arbor, MI). GPR120 agonist, GW9508, was purchased from Tocris Bioscience (Ellisville, MO). Prostaglandin E2 was obtained from Sigma Aldrich (St Louis, MO). Antibodies against phospho-ERK1/2, ERK1/2, phospho-Ser505 cPLA2, total cPLA2 and horseradish peroxidase-conjugated secondary antibody were obtained from Cell Signaling Technology (Beverly, MA). Antibody against GPR120 was obtained from Novus Biologicals (Littleton, CO). Antibodies against Gαq and β-arrestin 2 were purchased from Abcam (Cambridge, MA). Lipopolysaccharide (LPS) from Escherichia coli serotype R515 was purchased from Alexis Biochemicals (San Diego, CA). The CellTiter 96® AQueous One Solution Reagent containing a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium, inner salt; (MTS)] was purchased from Promega (Madison, WI).
Cell culture
The murine macrophage cell line RAW264.7 was obtained from the American Type Culture Collection (Manassas, VA). RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA), supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics Penicillin and Streptomycin (100 U/ml and 100 μg/ml; Life Technologies). Elutriated human peripheral blood monocytes were received from the Department of Transfusion Medicine, Clinical Center, NIH (Bethesda, MD), under an institutional review board-approved protocol. The elutriated monocytes were treated with erythrocyte lysis buffer (Qiagen, Valencia, CA) to remove residual red blood cells and were washed twice with PBS. The monocytes were cultured in RPMI1640 (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum at 2 × 106 cells/ml in a 30ml volume in a Nunc 75cm2 tissue culture flask. After 24 hr, cells were counted, centrifuged and changed to DMEM (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum at 1 × 106 cells/ml in a 20ml volume in a Nunc 75cm2 tissue culture flask. On day 5, 10 ml culture medium was replaced with fresh DMEM. On day 8 or 9, monocyte-derived macrophages were fully differentiated. They were treated with trypsin-EDTA for 1 min, scraped, counted and plated into 12-well plates at 2·5 × 105 cells/ml, 1 ml/well 1 day before experiments. Docosahexaenoic acid and PGE2 were dissolved in ethanol; DHA was aliquoted into screw cap tubes to avoid oxidation. GW9508, cPLA2 inhibitor, thioetheramide-PC, bromoenol lactone, U0126, SB203580, NS-398, AH6809 and GW627368X were dissolved in DMSO. Vehicle controls with the appropriate solvents were used throughout the experiments.
Cell cytotoxicity assay
RAW264.7 cells or human primary monocyte-derived macrophages were plated in 96-well plates at 2·5 × 104 cells/well, 100 μl/well. Cells were treated with 100 μm DHA or GW9508 for 7 hr, with ethanol or DMSO as vehicle control. At the end of treatment, 20 μl MTS (1·90 mg/ml in PBS) was added into each well. The cells were incubated for 2 hr (RAW264.7 cells) or 4 hr (human primary monocyte-derived macrophages) at 37°. The formazan product as measured by the amount of absorbance at 490 nm was detected using the SpectroMax 384 plate reader (Molecular Devices, Sunnyvale, CA). The absorbance is directly proportional to the number of viable cells. The background absorbance of medium only was subtracted from the data. The result was shown as percentage of absorbance of DHA- or GW9508-treated cells compared with that of ethanol or DMSO vehicle control.
AA release
Whole cell AA release was performed as described by Pawliczak et al.23 Briefly, RAW264.7 cells were grown in 12-well dishes for 4 hr and then were labelled with 0·5 μCi/ml of 5,6,8,9,11,12,14,15-3H-arachidonic acid (3H-AA, 150–230 Ci/mmol/ml, GE Healthcare, Piscataway, NJ) in 10% fetal bovine serum culture media for 18 hr. Cells were then washed three times with DMEM. Subsequently, cells were stimulated with DHA or GW9508 in the presence or absence of inhibitors of cPLA2, iPLA2, sPLA2, MEK1/2 or p38 at the indicated time intervals. In other experiments, cells transfected with control small interfering RNA (siRNA) or cPLA2 siRNA or GPR120 siRNA or Gαq siRNA or β-arrestin 2 siRNA were labelled with 0·5 μCi/ml 3H-AA, then cells were stimulated with DHA or GW9508 for the indicated times. At the end of incubation, the medium was removed and centrifuged at 1000 g for 5 min to remove any cells. An aliquot of medium was transferred to scintillation vials containing 10 ml of Bio-Safe II scintillation fluid (Research International Products, Mount Prospect, IL) and counted in an LS6500 scintillation counter (Beckman, Fullerton, CA).
Transfection of siRNA
Smart pool: on-target plus siRNA against GPR120 siRNA, β-arrestin 2 siRNA, EP2 siRNA, EP4 siRNA and on-target plus non-targeting control pool were obtained from Dharmacon, Thermo Scientific (Rockford, IL). Silencer select pre-designed siRNAs39,40 against murine cPLA2, Gαq, and negative control siRNA were purchased from Applied Biosystems (Foster City, CA). The siRNAs were transfected into RAW264.7 cells by electroporation using a Nucleofector transfection kit (Lonza, Walkersville, MD). The interference of cPLA2, GPR120, β-arrestin 2, Gαq, EP2 and EP4 was confirmed at the mRNA level by Taqman RT-PCR and at the protein level by Western blot.
Western blot analysis
RAW264.7 cells grown in six-well plates and stimulated with DHA or GW9508 in the absence or presence of MEK1/2 inhibitor, U0126, or cells transfected with siRNAs were lysed in RIPA buffer containing proteinase inhibitor mixture (Roche Applied Science, Indianapolis, IN) and Halt phosphatase inhibitors (Thermo Scientific, Rockford, IL). The cell lysates were centrifuged, and equivalent amounts of protein were loaded onto 4–20% Tris–glycine SDSpolyacrylamide gels (Life Technologies). After electrophoresis, the proteins were transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline/Tween-20 (20 mm Tris–HCl pH 7·4, 154 mm NaCl, and 0·05% Tween-20). The blots were then incubated with primary antibodies, phospho-cPLA2, cPLA2, phospho-ERK1/2 and ERK1/2, GPR120, Gαq, β-arrestin 2, EP2 and EP4 overnight at 4° and washed with Tris-buffered saline/Tween-20. The blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hr. An electrochemiluminescence (ECL) chemiluminescence detection kit (GE Health Care, Piscataway, NJ) was used to develop the blots and the signals were visualized on a Kodak Image Station 440CF.
Real-time PCR
Total RNA was extracted from RAW264.7 cells using QIA Shredder columns and RNeasy kits following the manufacturer's instructions (Qiagen) and was quantified using a NanoDrop spectrophotometer (BioLabNet, Great Falls, VA). Messenger RNA expression for selected genes was measured using real-time PCR performed on an ABI Prism 7900 sequence detection system (Applied Biosystems) using the following commercially available probe and primers sets (Applied Biosystems): GPR120-Mm00725193_m1, cPLA2-Mm00447040_m1, β-arrestin 2-Mm00520665_m1, Gαq-Mm00492381_m1, COX-2-Mm00478374_m1, EP2-Mm00436051_m1, EP4-Mm00436053_m1 and murine GAPDH-Mm99999915_g1. Reverse transcription and real-time PCR were performed with an RT kit and TaqMan Universal PCR master mix (Applied Biosystems) according to the manufacturer's instructions. Gene expression levels were presented as relative mRNA copy numbers compared with the internal control of GAPDH expression level and calculated as percentage of control siRNA.
Measurement of interleukin-6 secretion by ELISA
Murine interleukin-6 (IL-6) ELISA kits and human IL-6 ELISA kits were obtained from Life Technologies. RAW264.7 cells or human primary monocyte-derived macrophages were pre-treated or not with cPLA2 inhibitor (5 μm) or COX-2 inhibitor (3 μm) or EP2 inhibitor (3 μm) or EP4 inhibitor (2 μm) for 1 hr, then stimulated with 100 μm DHA or GW9508 for 1 hr, and finally stimulated with 10 ng/ml LPS for 6 hr. In other experiments, RAW264.7 cells were also pre-treated with 20 μm AA for 1 hr, then stimulated with 10 ng/ml LPS for 6 hr. Additional sets of RAW264.7 cells or human monocyte-derived macrophages were pre-treated with 100 nm PGE2 for 1 hr, then stimulated with 10 ng/ml LPS for 6 hr. Cell culture supernatants were collected and stored at −80°. Cell supernatants were diluted 1 : 10 to 1 : 20 for the assay. The concentration of IL-6 in supernatants was measured according to manufacturer's instructions. The detection limit for mouse and human IL-6 is < 3 pg/ml and < 2 pg/ml, respectively.
Measurement of eicosanoid release by EIA
A PGE2 EIA kit was obtained from Cayman Chemical Co. (Ann Arbor, MI). RAW264.7 cells and human primary monocyte-derived macrophages were pre-treated or not with cPLA2 inhibitor (5 μm) or COX-2 inhibitor (3 μm) for 1 hr, then stimulated with 100 μm DHA for 1 hr or 6 hr. Cell culture supernatants were collected. The PGE2 release was detected according to the manufacturer's instructions. Leukotriene C4, PGD2, and 15-HETE EIA kits were also obtained from Cayman Chemical Co. (Ann Arbor, MI). RAW264.7 cells were stimulated with 100 μm DHA for 6 hr, with vehicle control. The leukotriene C4, PGD2 and 15-HETE release were measured in cell culture supernatants following manufacturer's instructions.
NF-κB promoter luciferase assay
The NF-κB luciferase reporter construct (Promega, Madison, WI) and a Renilla luciferase construct, Rluc/TK (Promega) were co-transfected into RAW264.7 using a Nucleofector transfection kit (Lonza, Walkersville, MD). After 24 hr of transfection, the cells were pre-treated with 100 nm PGE2 or 100 μm DHA for 1 hr, then stimulated with 10 ng/ml LPS for 6 hr. After stimulation, the cells were lysed in passive lysis buffer (Promega), reporter gene activity was measured using the Dual-Luciferase assay reporter system (Promega) and luciferase activity was detected with a Victor 2 plate reader luminometer (PerkinElmer Life Sciences, Santa Clara, CA). Firefly luciferase of NF-κB promoter was normalized to Renilla luciferase activity and the data are presented as the mean ± SEM of fold stimulation.
Statistical analysis
Statistical analyses were performed with one-way analyses of variance and Holm–Sidak post hoc test or unpaired Student's t-test as indicated. P < 0·05 was considered statistically significant.
Results
DHA and the GPR120 agonist, GW9508, cause AA release from macrophages
To determine whether PLA2 is involved in the effect of DHA or GW9508, AA release representing PLA2 activity was assayed. DHA and GW9508 treatment increased AA release from RAW264.7 cells in a dose-dependent manner. RAW264.7 cells were treated with DHA or GW9508 at 10, 25, 50 and 100 μm for 4 hr. RAW264.7 cells showed detectable AA release after 10 μm DHA stimulation, or 25 μm GW9508 stimulation. A significant increase in AA release was noted starting at the concentration of 10 μm of DHA or 25 μm of GW9508. There was a concentration-dependent effect on AA release, with maximal effect at 100 μm (Fig. 1a). RAW264.7 cells were treated with 100 μm DHA or GW9508 for 1 hr, 2 hr and 4 hr. AA release increased at 1 hr, 2 hr and 4 hr compared with vehicle control (Fig. 1b). We chose to use 100 μm DHA based on previous published studies on monocytes and macrphages.14,41–43 To study whether 100 μm DHA or GW9508 might affect cell viability, cell cytotoxicity assay was performed using MTS reagent in RAW264.7 cells and human primary monocyte-derived macrophages. The results showed that 100 μm DHA or GW9508 had no effect on RAW264.7 cell viability (Fig. 1c) and human primary monocyte-derived macrophages (Fig. 9e). Therefore, DHA or GW9508 at 100 μm was used for subsequent studies. Apart from the receptor-mediated effects, free omega-3 fatty acid can be incorporated into the phospholipids of the cell membrane.3 The amount of DHA incorporated into cell membrane of RAW264.7 cell was analysed by total fatty acid panel analysis (LIPID MAPS, Lipidomics Core at University of California, San Diego, La Jolla, CA). Incubation with 100 μm DHA for 1 hr resulted in a membrane concentration of 532·0 ± 53·09 pmol/106 cells compared with vehicle control cells of 201·6 ± 0·33 pmol/106 cells. Treatment of RAW264.7 cells with DHA for 6 hr resulted in a concentration of 1104·9 ± 63·28 pmol/106 cells, compared with vehicle control cells of 301·9 ± 32·06 pmol/106 cells (n = 3, P < 0·01 by Student's t-test).
Figure 1.
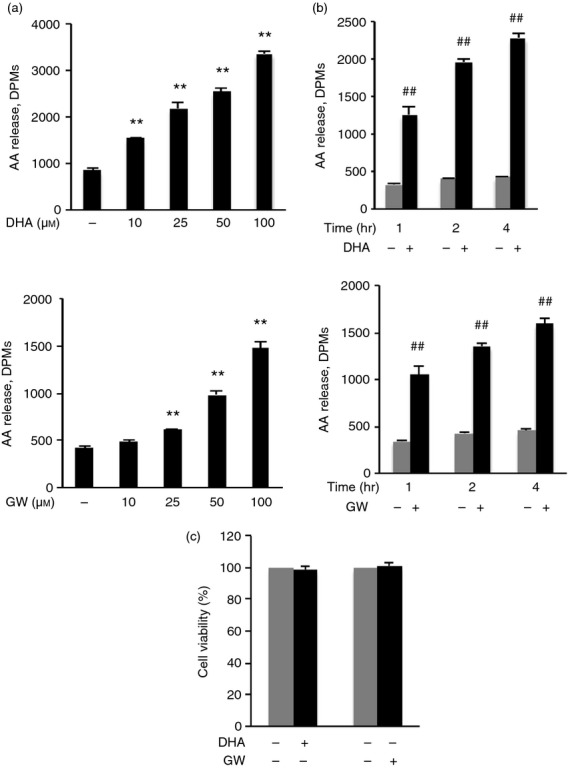
Docosahexaenoic acid (DHA) and GW9508 induce arachidonic acid (AA) release in RAW264.7 cells. (a) The 3H-AA labelled cells were stimulated with indicated doses of DHA or GW9508 for 4 hr. Data are presented as the mean ± SEM (n = 3) of disintegrations per minute (DPMs) and are representative of the results from three independent experiments. **P < 0·01 compared with the vehicle control, as assessed by analysis of variance with the Holm–Sidak post hoc test. There is a linear dose–response of DHA or GW9508 concentration and AA release, as assessed by linear regression model. (b) 3H-AA labelled cells were stimulated with 100 μm DHA or GW9508 at the indicated times. Data are presented as the mean ± SEM (n = 3) of DPMs and are representative of the results from three independent experiments. ##P < 0·01 compared with vehicle control of each time point, as assessed by Student's t-test. (c) Determination of cell viability. Cells were stimulated with 100 μm DHA or GW9508 or vehicle for 7 hr. Data are shown as the mean ± SEM of percentage of absorbance of the formazan product at 490 nm of DHA- or GW9508- treated cells compared with that of vehicle control with four replicates and are representative results from three independent experiments.
Figure 9.
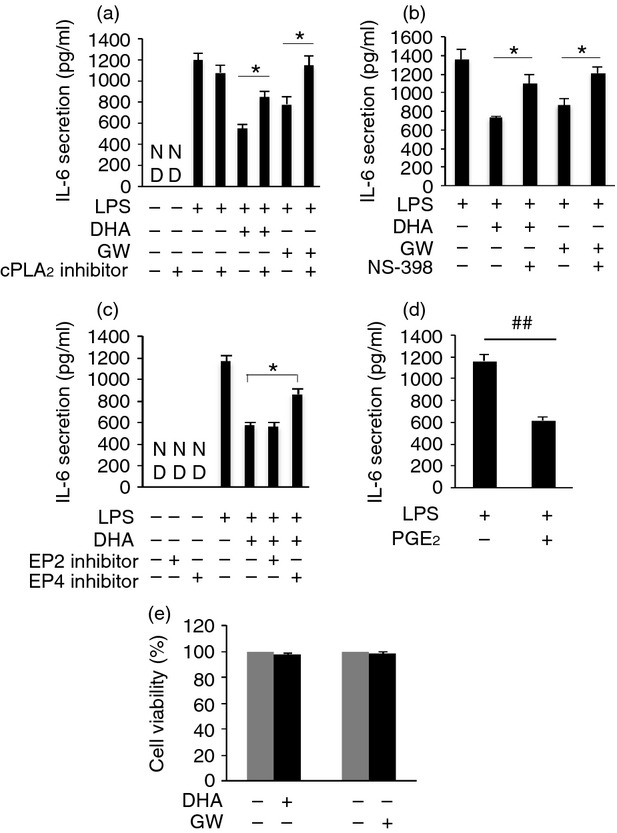
Cytosolic phospholipase A2 (cPLA2) activation, cyclooxygenase 2 (COX-2) activation, prostaglandin E2 (PGE2) induction and EP4 receptor are involved in the anti-inflammatory effect of docosahexaenoic acid (DHA) or GW9508 in human primary monocyte-derived macrophages. (a–c) Human primary monocyte-derived macrophages were pre-treated or not with cytosolic phospholipase A2 (cPLA2) inhibitor (5 μm) (a) or COX-2 inhibitor (3 μm) (b) or EP2 inhibitor (3 μm), or EP4 inhibitor (2 μm) (c) for 1 hr, then stimulated with DHA or GW9508 (100 μm) for 1 hr and finally stimulated with lipopolysaccharide (LPS) (10 ng/ml) for 6 hr. Cell culture supernatants were collected and assayed for interlukin-6 (IL-6) secretion. Data are presented as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of the results from three different donors. *P < 0·05 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test. ND = non-detectable. (d) Human primary monocyte-derived macrophages were pre-treated with 100 nm PGE2 for 1 hr, and then stimulated with 10 ng/ml LPS for 6 hr. Cell culture supernatants were assayed for IL-6 secretion. Data are shown as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of data from three different donors. ##P < 0·01 as indicated, as assessed by Student's t-test. (e) Determination of cell viability. Human primary monocyte-derived macrophages were stimulated with 100 μm DHA or GW9508 or vehicle for 7 hr. Data are shown as the mean ± SEM of percentage of absorbance of the formazan product at 490 nm of DHA- or GW9508-treated cells as compared with that of vehicle control with four replicates. The data shown are representative results from three different donors.
DHA and GW9508 activate cPLA2 and cause AA release
To determine which type of phosphoslipase A2 is involved in DHA- and GW9508-induced AA release, the cPLA2 inhibitor, N-((2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl)-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl] acrylamide HCl (1 μm), iPLA2 inhibitor, bromoenol lactone (1 μm), or the sPLA2 inhibitor, thioetheramide-PC (10 μm), was used to pre-treat RAW264.7 cells for 1 hr. Cells were then stimulated with DHA or GW9508 (100 μm) for 4 hr and AA release was assayed. We found that only the cPLA2 inhibitor significantly decreased DHA- or GW9508-induced AA release. The cPLA2 inhibitor at 1 μm decreased DHA- or GW-induced AA release by 37·2% and 47·1%, respectively (Fig. 2a), cPLA2 inhibitor at 5 μm further decreased DHA-induced AA release by 75% and decreased GW9508-induced AA release by 88·5% (Fig. 2b), cPLA2 inhibitor or iPLA2 inhibitor or sPLA2 inhibitor alone has no effect on AA release. To determine whether DHA and GW9508 specifically induce AA release, three other fatty acids were tested, the unsaturated fatty acid, linolenic acid and the saturated fatty acids, myristic acid or docosanoic acid. These three fatty acids did not induce AA release (Fig. 2c). This suggests that the effect of DHA and GW9508 on AA release is specific. Moreover, knock-down of cPLA2 with cPLA2 siRNA (100 nm) reduced cPLA2 mRNA and protein level (Fig. 2d) and decreased DHA- and GW9508-induced AA release by 89% and 57%, respectively (Fig. 2e,f), suggesting that cPLA2 activation is involved in DHA- and GW9508-induced AA release.
Figure 2.
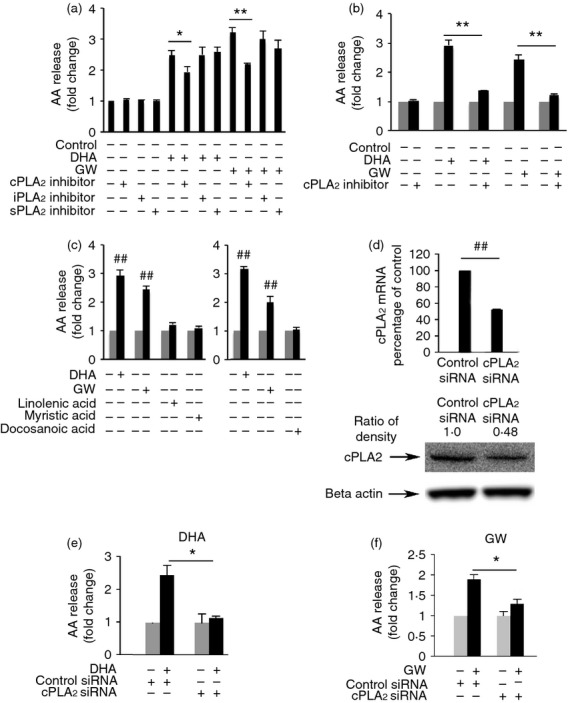
Docosahexaenoic acid (DHA) and GW9508 induce arachidonic acid (AA) release through cytosolic phospholipase A2 (cPLA2) specifically in RAW264.7 cells. (a) Cells were pre-treated with a cPLA2 inhibitor, pyrrolidin derivative (1 μm), iPLA2 inhibitor, bromoenol lactone (1 μm), or the sPLA2 inhibitor, thioetheramide-PC (10 μm) for 1 hr, then stimulated with 100 μm DHA or GW9508 for 4 hr and AA release was assayed. (b) Cells were pre-treated with cPLA2 inhibitor (5 μm) for 1 hr, and then stimulated with 100 μm DHA or GW9508 for 4 hr and AA release was assayed. Data are presented as the mean ± SEM (n = 3) of the fold change, compared with the vehicle-treated cells and are representative of three independent experiments. *P < 0·05, **P < 0·01 as indicated, assessed by analysis of variance with Holm–Sidak post hoc test in (a, b). (c) DHA and GW9508 induced AA release specifically. Cells were treated with DHA, GW9508, unsaturated fatty acid, linolenic acid, or the saturated fatty acid, myristic acid or docosanoic acid for 4 hr, followed by AA release assay. Data are presented as the mean ± SEM (n = 3) of the fold change, compared to the vehicle-treated cells and are representative results of three independent experiments. ##P < 0·01 as compared to the vehicle-treated cells, as assessed by Student's t-test. (d) Knock-down efficiency of cPLA2 siRNA. Cells were transfected with either cPLA2 siRNA (100 nm) or Negative Control siRNA (100 nm) for 24 hr. The mRNA level of cPLA2 expression was determined by Taqman real-time PCR. Data are presented as the mean ± SEM (n = 3) of the percentage change compared with the negative control siRNA. ##P < 0·01 as indicated, as assessed by Student's t-test. The protein level of cPLA2 was determined by Western blot and was quantified. Data are representative from three independent experiments. (e, f) Knock-down of cPLA2 decreases DHA and GW9508 induced AA release. Cells were transfected with either cPLA2 siRNA or Negative Control siRNA for 24 hr, stimulated with 100 μm DHA or GW9508 for 4 hr and AA release was assayed. The results are presented as the mean ± SEM (n = 3) of fold stimulation compared with control and are representative of three independent experiments. *P < 0·05 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test.
DHA and GW9508 induce cPLA2 activity through GPR120 receptor, Gαq subunit and β-arrestin 2 adaptor protein
Omega-3 fatty acids signal through GPR120 receptor and β-arrestin 2.14 β-Arrestin 2 serves as a scaffold or adaptor protein for a wide range of G protein-coupled receptors, associates with ligand-stimulated GPR120 receptor and participates in downstream ERK1/2 MAPK signalling.17,18 To determine whether GPR120 receptor, Gαq and β-arrestin 2 are involved in cPLA2 activation, RAW264.7 cells were transfected with Control siRNA, GPR120 siRNA, Gαq siRNA and β-arrestin 2 siRNA for 48 hr. Transfection of RAW264.7 cells with each of these siRNAs resulted in a significant decrease of the target mRNA and protein level as shown in Fig. 3(a,d,g). Cells were then stimulated with 100 μm DHA or GW9508 for 2 hr and AA release was measured. Knock-down of GPR120, Gαq and β-arrestin 2 significantly decreased AA release compared with transfection of control siRNA. Knock-down of GPR120 decreased DHA- or GW9508-induced AA release by 86% and 81·3%, respectively (Fig. 3b,c). Knock-down of Gαq decreased DHA- or GW9508-induced AA release 61% and 52·5%, respectively (Fig. 3e,f). β-Arrestin 2 siRNA decreased DHA- or GW9508-induced AA release 75·4% and 69·9%, respectively (Fig. 3h,i). The results suggest that DHA and GW9508 activate cPLA2 at least in part through GPR120 receptor, Gαq and β-arrestin 2.
Figure 3.
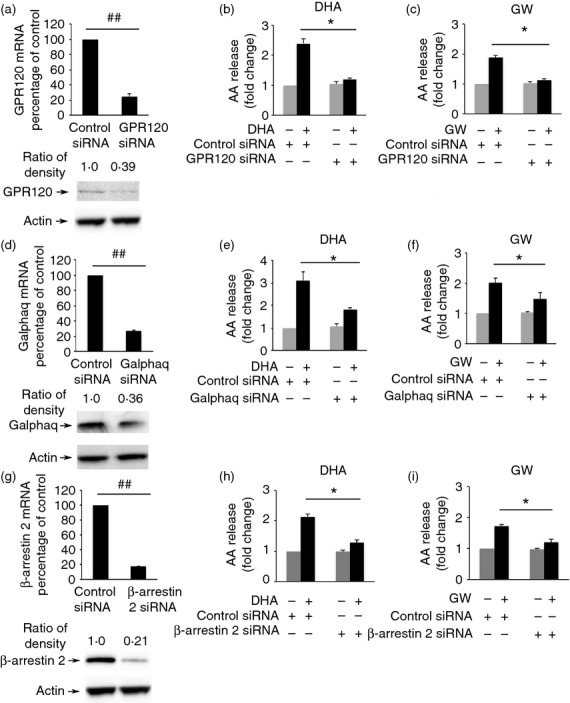
Docosahexaenoic acid (DHA) and GW9508 induce cytosolic phospholipase A2 (cPLA2) activation through GPR120 receptor, Gαq protein and β-arrestin 2. (a, d, g). Knock-down efficiency of GPR120, Gαq and β-arrestin 2 small interfering RNA (siRNA). RAW264.7 cells were transfected with either GPR120 siRNA (100 nm), Gαq siRNA (100 nm), β-arrestin 2 siRNA (100 nm) or Negative Control siRNA (100 nm). The mRNA level of GPR120 (a), Gαq (d) and β-arrestin 2 (g) expression was determined by Taqman real-time PCR. Data are presented as the mean ± SEM (n = 3) of percentage change compared with negative control siRNA. ##P < 0·01 as indicated, as assessed by Student's t-test. The protein level of GPR120, Gαq and β-arrestin 2 was determined by Western blot and was quantified. Data are representative of three independent experiments. Knock-down of GPR120 (b, c), Gαq (e, f) and β-arrestin 2 (h, i) by siRNA decreases DHA- or GW9508-induced arachidonic acid (AA) release. RAW264.7 cells were transfected with either GPR120 siRNA, Gαq siRNA, β-arrestin 2 siRNA or Negative Control siRNA for 48 hr, stimulated with 100 μm DHA or GW9508 for 2 hr and then AA release was detected. Data are shown as the mean ± SEM (n = 3) of fold change compared to control and are representative of the results of three independent experiments. *P < 0·05 as indicated, as assessed by analysis of variance with Holm-Sidak post hoc test.
DHA and GW9508 activate cPLA2 through activation of ERK1/2, but not p38 MAPK
After ligand binding, GPR120 is phosphorylated and this generates binding sites for β-arrestin 2.16 β-Arrestin 2 then interacts with down-stream signalling molecules such as ERK1/2 MAPK.17,18 To determine if MAPKs are involved in cPLA2 activation caused by DHA and GW9508, cells were pre-treated with a MEK1/2 inhibitor or a p38 inhibitor. Treatment with the MEK1/2 inhibitor, U0126 (20 μm), resulted in decreased DHA- or GW9508-induced AA release by 68% and 57·2%, respectively (Fig. 4a). Treatment of cells with the p38 inhibitor, SB203580 (10 μm), had no effect on DHA- or GW9508-induced AA release (Fig. 4b). These results indicate that DHA and GW9508 may activate cPLA2 through activation of ERK1/2. Next, we studied the effect of DHA and GW9508 on phosphorylation of ERK1/2. As shown in Fig. 5(a), DHA increased phosphorylation of ERK1/2 after 10 min stimulation. GW9508 increased phosphorylation of ERK1/2 from 5 min stimulation with a peak effect between 10 and 20 min. Furthermore, we studied whether DHA or GW9508 causes phosphorylation of cPLA2 by Western blot. As shown in Fig. 5(b), DHA and GW9508 increased phosphorylation of cPLA2. DHA increased phosphorylation of cPLA2 at 30 min incubation while GW9508 increased phosphorylation of cPLA2 at 30 min and 1 hr incubation. MEK1/2 inhibitor, U0126, abolished the phosphorylation of ERK1/2 (Fig. 5c) and eliminated the increased phosphorylation of cPLA2 (Fig. 5d) caused by DHA or GW9508 stimulation. The results indicated that cPLA2 activation is caused by ERK1/2 activation.
Figure 4.
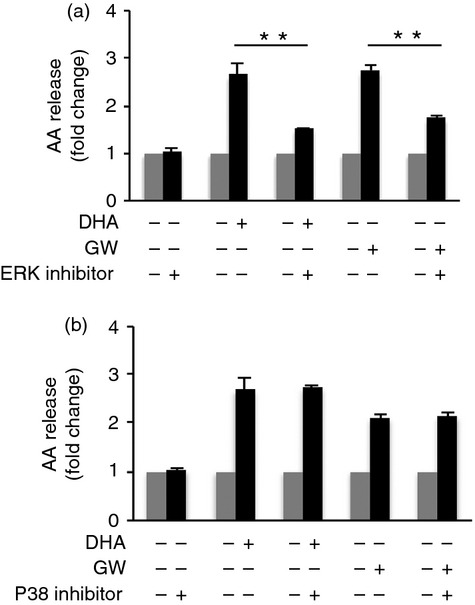
MEK1/2 inhibitor decreases DHA- or GW9508- induced AA release. (a) RAW264.7 cells were pretreated with the MEK1/2 inhibitor, U0126 (20 μM) or (b) the p38 inhibitor, SB203580 (10 μM) for 1 hr, then stimulated with 100 μM DHA or GW9508 for 2 hr and AA release was measured. Data are presented as the mean ± SEM (n=3) of fold change compared to vehicle control and are representative of three independent experiments. **p < 0.01 as indicated, as assessed by ANOVA analysis followed by the Holm-Sidak post hoc test.
Figure 5.
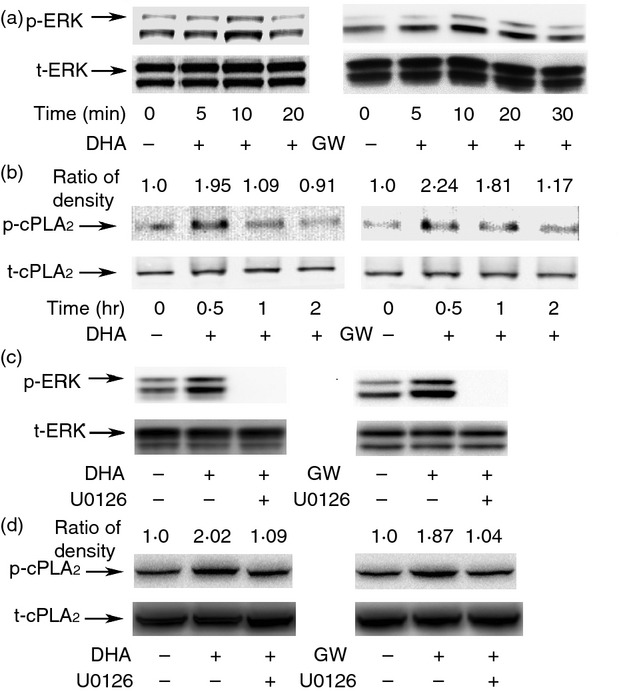
Docosahexaenoic acid (DHA) and GW9508 activate cytosolic phospholipase A2 (cPLA2) through activation of extracellular signal-regulated kinase 1/2 (ERK1/2). (a) RAW264.7 cells were serum starved for 2 hr with Dulbecco's modified Eagle's medium containing 0·2% fetal bovine serum, then stimulated with 100 μm DHA or GW9508 for the indicated time. Phospho-ERK1/2 and total ERK1/2 were detected by immunoblot. (b) DHA and GW9508 cause phosphorylation of cPLA2. Cells were stimulated with 100 μm DHA or GW9508 for the indicated time. Cells were assayed by immunoblot with phospho-cPLA2 and total cPLA2 antibody. The ratio of density of phospho-cPLA2/total cPLA2 of DHA- or GW9508-treated group is compared with that of vehicle control, which is set as 1·0. (c) Cells were pre-treated with mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor, U0126 (20 μm), for 1 hr, then stimulated with 100 μm DHA or GW9508 for 10 min. Phospho-ERK1/2 and total ERK1/2 were detected by immunoblot. (d) Cells were stimulated with U0126 (20 μm) for 1 hr, then stimulated with 100 μm DHA or GW9508 for 0·5 hr. Cells were assayed by immunoblot with phospho-cPLA2 and total cPLA2 antibody. The ratio of density is shown as in 5b. The blots shown are from one of three independent experiments, each showing similar results.
Docosahexaenoic acid and GW9508 inhibit LPS-induced pro-inflammatory cytokine release in part through activation of cPLA2, COX-2 and production of prostaglandin E2, resulting in inhibition of the NF-κB signalling pathway
RAW264.7 cells or human primary monocyte-derived macrophages were pre-treated with 100 μm DHA or GW9508 for 1 hr, then treated with LPS (10 ng/ml) for 6 hr and IL-6 secretion was detected. Both DHA and GW9508 inhibited LPS-induced IL-6 release in RAW264.7 cells (Fig. 6a) and in human primary monocyte-derived macrophages (Fig. 9a). When cells were pre-treated with a cPLA2 inhibitor, the inhibitory effect of DHA or GW9508 on LPS-induced IL-6 production was reduced in RAW264.7 cells (Fig. 6a) or human primary monocyte-derived macrophages (Fig. 9a). We have shown that DHA and GW9508 activate cPLA2, which cleaves the sn-2 fatty acyl bond of phospholipids to produce AA. To determine if the effect of the cPLA2 inhibitor is a result of inhibition of DHA- or GW9508-induced AA release, AA was added to cultures before LPS was added and IL-6 release was assayed. AA also inhibited LPS-induced IL-6 release by 33% (Fig. 6b). To determine whether the AA produced by DHA can be metabolized by COX-2 to form PGE2, COX-2 mRNA expression by RT-PCR and PGE2 release by EIA were studied. DHA significantly activated COX-2 gene expression, while cPLA2 inhibitor reduced this effect by 88·7% (Fig. 6c). When cells were pre-treated with a COX-2 inhibitor, NS-398, the COX-2 inhibitor partially reversed DHA or GW9508 inhibition of LPS-induced IL-6 secretion in RAW264.7 cells (Fig. 6d) and in human primary monocyte-derived macrophages (Fig. 9b). DHA caused PGE2 release after 1 hr incubation in RAW264.7 and human primary monocyte-derived macrophages. The cPLA2 inhibitor and a COX-2 inhibitor partially inhibited this release. The cPLA2 inhibitor decreased PGE2 release in RAW264.7 cells and in human primary monocyte-derived macrophages (Fig. 7a,b). The COX-2 inhibitor inhibited PGE2 release to a similar degree to the cPLA2 inhibitor (Fig. 7c,d). Whether PGE2 inhibits LPS-induced IL-6 secretion in macrophages has not been reported. To test whether PGE2 can inhibit LPS-induced IL-6 secretion, RAW264.7 cells or human primary monocyte-derived macrophages were pre-treated with 100 nm PGE2 for 1 hr, then stimulated with 10 ng/ml LPS for 6 hr and IL-6 ELISA was performed. Prostaglandin E2 inhibited LPS-induced IL-6 secretion by 73% in RAW264.7 cells (Fig. 7e) and inhibited LPS induced IL-6 secretion by 47% (Fig. 9d). To test whether PGE2 or DHA inhibits LPS-induced IL-6 secretion through inhibition of the NF-κB pathway, NF-κB promoter luciferase activity was studied. Prostaglandin E2 or DHA decreased LPS-induced NF-κB promoter activity by 31% and 28%, respectively (Fig. 7f). These data suggest that DHA or PGE2 plays an anti-inflammatory role partially through inhibition of the NF-κB signalling pathway. In addition to PGE2 release, we have assayed for other eicosanoids that may be released from RAW264.7 cells after stimulation with 100 μm DHA for 6 hr. These include leukotriene C4 (vehicle control, 1·15 ± 0·17 pg/ml; DHA 100 μm, 85·80 ± 3·94 pg/ml; n = 3, P < 0·01 by Student's t-test), PGD2 (vehicle control, 121·56 ± 10·54 pg/ml; DHA 100 μm, 279·88 ± 21·84 pg/ml; n = 3, P < 0·01 by Student's t-test), and 15-HETE [vehicle control, ND (non-detectable); DHA 100 μm, 424·09 ± 50·64 pg/ml; n = 3]. However, PGE2 is the most abundant released eicosanoid in our system.
Figure 6.
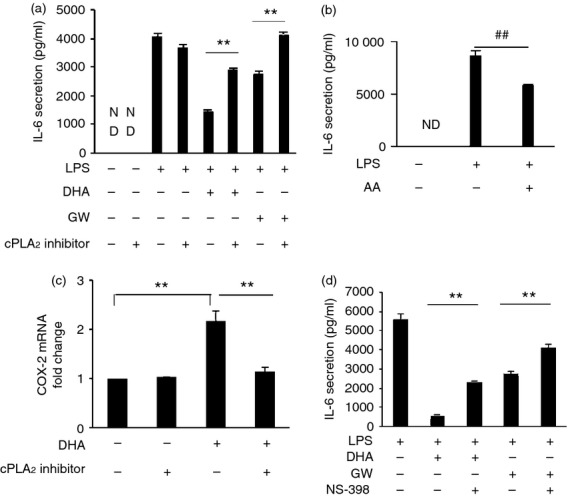
Docosahexaenoic acid (DHA) and GW9508 inhibit lipopolysaccharide (LPS) -induced interleukin-6 (IL-6) secretion in part through activation of cytosolic phospholipase A2 (cPLA2) and COX-2. (a) RAW264.7 cells were pre-treated or not with cPLA2 inhibitor (5 μm) for 1 hr, then stimulated with DHA or GW9508 (100 μm) for 1 hr and finally stimulated with LPS (10 ng/ml) for 6 hr. Cell culture supernatants were collected and IL-6 secretion was determined by ELISA. Data are shown as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of the results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test. ND = non-detectable. (b) Cells were pre-treated with arachidonic acid (20 μm) for 1 hr, then stimulated with LPS (10 ng/ml) for 6 hr. Cell culture supernatants were assayed for IL-6 secretion. Data are shown as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative result from three independent experiments. ##P < 0·01 as indicated, as assessed by Student's t test. ND = non-detectable. (c) Cells were pre-treated with cPLA2 inhibitor (5 μm) or not for 1 hr, then stimulated with DHA (100 μm) for 6 hr. Cyclooxygenase 2 (COX-2) mRNA expression was determined by Taqman real-time PCR. Data are shown as the mean ± SEM (n = 3) of fold change compared with vehicle-treated control and are representative of the results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test. (d) Cells were pre-treated or not with COX-2 inhibitor, NS-398 (3 μm) for 1 hr, then stimulated with DHA or GW9508 and LPS as in 6A. Cell culture supernatants were assayed for IL-6 secretion. Data are presented as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of the results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variance followed by Holm–Sidak post hoc test.
Figure 7.
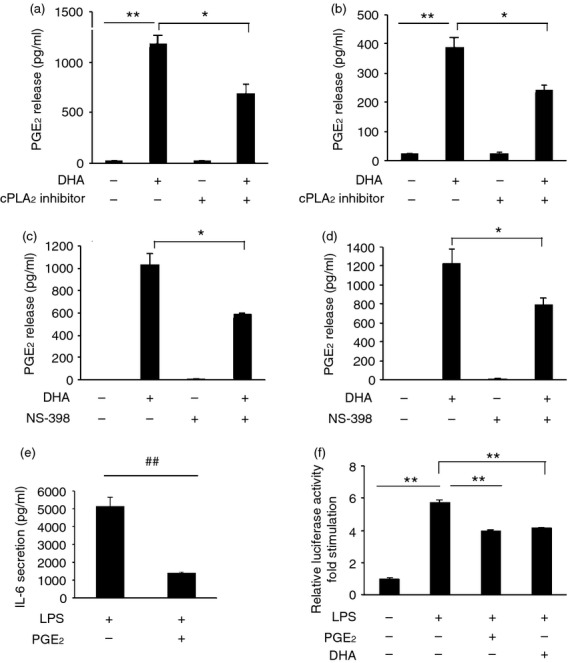
Docosahexaenoic acid (DHA) stimulates production of prostaglandin E2 (PGE2) and PGE2 decreases lipopolysaccharide (LPS) induced interleukin 6 (IL-6) secretion, which is partially through inhibition of nuclear factor (NF-κB) signalling pathway. (a) RAW264.7 cells and (b) human primary monocyte-derived macrophages were pre-treated or not with cytosolic phospholipase A2 (cPLA2) inhibitor (5 μm) for 1 hr, then stimulated with DHA (100 μm) for 6 hr. Cell culture supernatants were collected and PGE2 release was assayed. Data are shown as the mean ± SEM (n = 3) of prostaglandin E2 (PGE2) release (pg/ml) and are representative results of three independent experiments in RAW264.7 cells and of three different donors in primary monocyte-derived macrophages. **P < 0·01 or *P < 0·05 as indicated, as assessed by analysis of variance with Holm–Sidak post-hoc test. (c) RAW264.7 cells and (d) human primary monocyte-derived macrophages were pre-treated or not with cyclooxygenase 2 (COX-2) inhibitor, NS-398 (3 μm), for 1 hr followed by stimulation with DHA (100 μm) for 1 hr. Cell culture supernatants were collected and PGE2 release was assayed. Data are shown as in (a, b). *P < 0·05 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test. (e) RAW264.7 cells were pre-treated with PGE2 (100 nm) for 1 hr and then stimulated with 10 ng/ml LPS for 6 hr. Cell culture supernatants were assayed for IL-6 secretion. Data are shown as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of data from three independent experiments. ##P < 0·01 as indicated, as assessed by Student's t-test. (f) RAW264.7 cells were co-transfected with an NF-κB luciferase reporter construct and a Renilla luciferase construct. After 24 hr, cells were pre-treated with 100 nm PGE2 or 100 μm DHA for 1 hr, then stimulated with 10 ng/ml LPS for 6 hr. After stimulation, the cells were lysed in passive lysis buffer and reporter gene activity was measured. Firefly luciferase of NF-κB promoter was normalized to Renilla luciferase activity and data are presented as the mean ± SEM of fold stimulation and are representative results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variancefollowed by Holm-Sidak post hoc test.
The anti-inflammatory effect of DHA is partially through EP4 receptor
We have shown that DHA activates cPLA2, resulting in the production of PGE2 and that PGE2 can suppress LPS-induced IL-6 production. Next, we studied whether DHA and PGE2 exert this effect via PGE2 receptor activation. We used EP2 or EP4 inhibitors and knock-down of EP2 or EP4 by siRNA. RAW264.7 cells or human primary monocyte-derived macrophages were pre-treated with the EP2 inhibitor, AH6809 (3 μm), or the EP4 inhibitor, GW627368X (2 μm), for 1 hr, then stimulated with DHA (100 μm) for 1 hr and finally stimulated with 10 ng/ml LPS for 6 hr. Cell culture supernatants were collected and IL-6 ELISA was performed. AH6809 antagonizes EP1, EP2 and EP3. EP2 and EP4 are primarily expressed in RAW264.7 cells with very little EP3 expression and no EP1 expression.44 Therefore, AH6809 is an antagonist of the EP2 receptor in RAW264.7 cells. The EP4 inhibitor, GW627368X, antagonizes the EP4 receptor specifically. The EP4 inhibitor partially reversed DHA inhibition of LPS-induced IL-6 secretion. AH6809 had no effect on DHA inhibition in RAW264.7 cells (Fig. 8a) or human primary monocyte-derived macrophages (Fig. 9c). Neither AH6809 nor GW627368X alone had an effect on LPS-induced IL-6 secretion. To determine if knock-down of EP2 or EP4 had an effect on DHA inhibition. RAW264.7 cells were transfected with 100 nm EP2 or EP4 siRNA for 48 hr, then stimulated with 100 μm DHA for 1 hr, and then stimulated with 10 ng/ml LPS for 6 hr, and IL-6 ELISA was performed. Knock-down of EP2 siRNA or EP4 siRNA decreased mRNA and protein expression of EP2 or EP4 (Fig. 8b). Knock-down of EP4 partially reversed DHA inhibition compared with cells transfected with control siRNA. However, knock-down EP2 had no effect on DHA inhibition (Fig. 8c). These data suggest that DHA plays an anti-inflammatory role partially through production of PGE2 and via EP4 binding.
Figure 8.
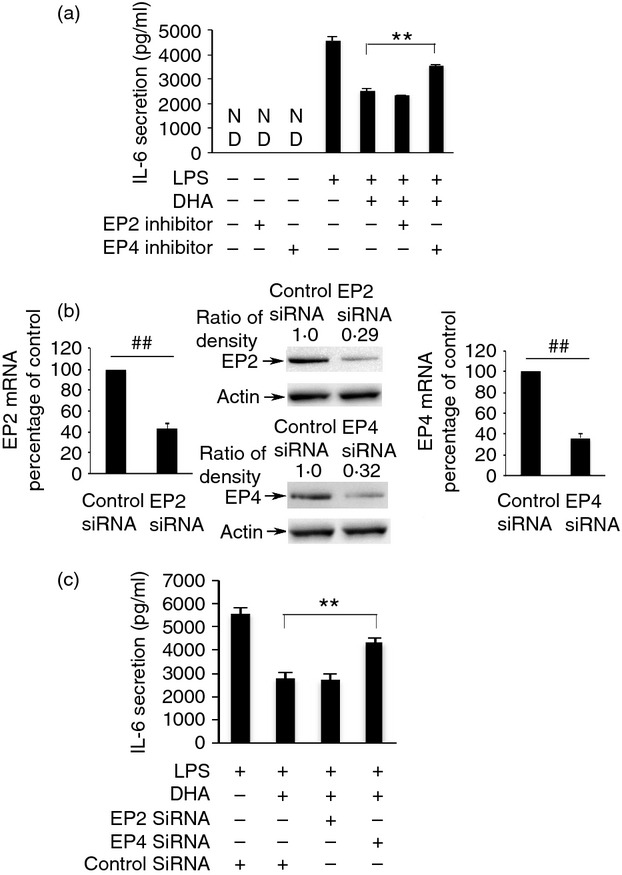
EP4 receptor of ptostaglandin E2 (PGE2) is involved in the anti-inflammatory effect of docosahexaenoic acid (DHA). (a) RAW264.7 cells were pre-treated with an EP2 inhibitor, AH6809 (3 μm), or an EP4 inhibitor, GW627368X (2 μm) for 1 hr, then stimulated with 100 μm DHA for 1 hr and finally stimulated with 10 ng/ml lipopolysaccharide (LPS) for 6 hr. Cell culture supernatants were collected and assayed for IL-6 secretion. Data shown are the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of the results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variance with Holm–Sidak post hoc test, ND = non-detectable. (b) Knock-down efficiency of EP2 or EP4 siRNA. RAW264.7 cells were transfected with EP2 siRNA (100 nm), EP4 siRNA (100 nm) or Negative Control siRNA (100 nm). Total RNA was isolated from transfected cells. The mRNA level of EP2 and EP4 expression was determined by Taqman real time PCR. The results are presented as the mean ± SEM (n = 3) of percentage change compared to negative control siRNA. ##P < 0·01 as indiated, as assessed by Student's t-test. The protein level of EP2 and EP4 was detected by Western blot and was quantified. Data are representative results of three independent experiments, each showing similar results. (c) RAW264.7 cells were transfected with either EP2 siRNA (100 nm), EP4 siRNA (100 nm) or Negative Control siRNA (100 nm) for 48 hr, stimulated with 100 μm DHA for 1 hr, then stimulated with lipopolysaccharide (LPS; 10 ng/ml) for 6 hr. Cell culture supernatants were assayed for IL-6 secretion. Data are presented as the mean ± SEM (n = 3) of IL-6 secretion (pg/ml) and are representative of the results of three independent experiments. **P < 0·01 as indicated, as assessed by analysis of variance and Holm–Sidak post hoc test.
Discussion
In this study, we demonstrated that DHA, and the GPR120 agonist, GW9508, activate cytosolic phospholipase A2 (cPLA2), and cause COX-2 activation in the murine macrophage cell line, RAW264.7 and result in PGE2 release in RAW264.7 cells and human primary monocyte-derived macrophages. We also showed that DHA-induced PGE2 release plays an anti-inflammatory role by inhibiting LPS-induced IL-6 release and by inhibiting the NF-κB signalling pathway and via the EP4 receptor. The cPLA2 plays an important role in modulating inflammation.19–22 Whether cPLA2 activation and its downstream lipid mediators are involved in the anti-inflammatory effect of DHA has not been studied previously. We are the first to indicate that cPLA2 activation and its downstream lipid mediator PGE2 are involved in the anti-inflammatory effect of DHA.
The DHA serum level under physiological conditions is about 1–5 μm.8–10 Dietary intake of 0·5–2 g DHA correlates with plasma phospholipid levels of 90–120 μm.11 DHA supplement (Epanova, 4 g/day) may achieve a plasma total DHA level close to 100 μm.12 There are some clinical situations when the high dose supplementation of DHA is highly beneficial. In sepsis patients receiving fish-oil-based lipid emulsion, the plasma-free DHA level can reach 65 μm.13 Patients with hypertriglyceridaemia can take DHA + EPA supplement 2–4 g/day to lower triglyceride.45 Supplements of 1·7–9·6 g/day DHA + EPA were given to patients with rheumatoid arthritis with promising results in reducing joint pain in patients.3,5 There is some increase at the concentration of non-esterified fatty acid with DHA supplementation.46 Most in vitro studies use free DHA at concentrations 10–100-fold higher than that of the physiological level, which may equate to total plasma concentrations of omega-3 fatty acid achievable with supplementation. However, the esterified form of DHA may not have the same profile of bioavailability as free DHA.5 We used 100 μm DHA in our study based on previous publications on monocytes and macrophages.14,42,43 However, we did not demonstrate similar responses at concentrations < 10 μm in vitro in our system.
The organized chain of observed events and their reversibility by specific inhibitors or siRNAs, suggest that our results are specific and are GPR120 receptor-mediated. Although some DHA is incorporated into membrane, which might suggest additional non-receptor-mediated events, we observed similar effects on AA release, ERK1/2 and cPLA2 phosphorylation, as well as inhibition of LPS-induced IL-6 secretion exerted by GW9508, the GPR120 receptor agonist. Moreover, it has been shown that at the site of inflammation in the tissue, during oedema, vascular leakage and cellular infiltration, there is a temporary, local increase of several cytokines, lipid mediators and the family of specialized pro-resolving mediators, which are derivatives of EPA and DHA.47,48 Hence, there may be a possibility that under these conditions tissue macrophages might be subjected to a higher concentration of free DHA than that reflected by the systemic plasma concentration.
We found that DHA and GW9508 increase AA release in the murine macrophage cell line, RAW264.7. A specific cPLA2 inhibitor, pyrrolidin derivative, and knock-down of cPLA2 decreased DHA- and GW9508-induced AA release, whereas sPLA2 and iPLA2 inhibitors had no effect on DHA- and GW9508-induced AA release. These results indicate that cPLA2 is activated by DHA and GW9508. Our findings are consistent with previous studies that DHA increases AA release in astrocytes.49,50 Moreover, DHA increased activation of cPLA2 in a prion-infected neuronal cell line, ScGT1.51 Our study showed that DHA and GW9508 can cause phosphorylation of ERK1/2 and phosphorylation of cPLA2 in macrophages. A MEK1/2 inhibitor partially inhibited DHA- or GW9508-induced AA release, whereas a p38 inhibitor had no effect. Moreover, MEK1/2 inhibitor, U0126 abolished phosphorylation of ERK1/2 and eliminated the increased phosphorylation of cPLA2 caused by DHA or GW9508. These results indicated that cPLA2 activation is caused by ERK1/2 MAPK activation.
It has been reported that DHA exerts its anti-inflammatory effects through the G protein coupled receptor 120 (GPR120) and β-arrestin 2.14 We explored whether cPLA2 activation is through GPR120 receptor, Gαq and β-arrestin 2, which binds to GPR120. Knock-down of GPR120, Gαq and β-arrestin 2 decreased DHA- and GW9508-induced AA release. These results suggest that cPLA2 activation by DHA and GW9508 is driven partially through the GPR120, Gαq and β-arrestin 2 pathway.
Both DHA and GW9508 inhibit LPS-induced IL-6 secretion and so play an anti-inflammatory role in macrophages. Interestingly, cPLA2 inhibitor significantly reversed DHA or GW9508 inhibition of LPS-induced IL-6 secretion, suggesting that cPLA2 is involved in the anti-inflammatory role of DHA. The cPLA2 product AA also inhibited LPS-induced IL-6 release. Therefore, AA down-stream signalling was studied. COX-2 inhibitor partially reversed DHA or GW9508 inhibition of LPS-induced IL-6 secretion. Prostaglandin E2 also reduces LPS-induced IL-6 secretion. The cPLA2 inhibitor or COX-2 inhibitor partially decreased DHA-induced PGE2 release in RAW264.7 cells and human primary monocyte-derived macrophages. These results indicate that cPLA2, the cPLA2 product AA, and its downstream COX-2-dependent lipid mediator, PGE2, are involved in the DHA or GW9508 anti-inflammatory effect. Moreover, we showed that DHA inhibits inflammation partially through EP4 receptor as an EP4 inhibitor or knock-down of EP4 by siRNA partially reverses DHA inhibition of LPS-induced IL-6 secretion. PGE2 or DHA also partially inhibits the NF-κB signalling pathway. These findings indicate that DHA plays an anti-inflammatory role through a cPLA2-AA-COX-2-PGE2-EP4 receptor pathway that results in the inhibition of the NF-κB signalling pathway.
Prostaglandin E2 exerts its biological functions mainly via four G-protein-coupled receptors EP1 to EP4.52 Macrophages primarily express EP4 and EP2.44 Several studies support a role for PGE2 as an inhibitor of inflammation. Targeted disruption of the EP4 receptor gene in mice decreased PGE2-mediated inhibition of IL-12 and tumour necrosis factor-α production in LPS-activated macrophages.38 The PGE2 also plays an anti-inflammatory role in T cells by suppressing the release of some cytokines and chemokines from T cells and inhibits the proliferation and activation of T cells and the induction of T-cell apoptosis.38 Prostaglandin E2 also reduces inflammation in atherosclerosis, aneurysm lesions and myocardial ischaemia.53–55 The PGE2 may also play an anti-inflammatory role in other inflammatory disorders including colitis and gastric ulcers.56,57 Prostaglandin E2 pre-treatment selectively inhibits LPS-induced NF-κB 1, p105 phosphorylation and degradation in mouse bone-marrow-derived macrophages through EP4-dependent mechanisms.58 Our results are consistent with these observations in that PGE2 inhibits LPS-induced IL-6 secretion and the anti-inflammatory role is partially through inhibition of NF-κB signalling pathway.
Other mechanisms are also involved in the anti-inflammatory effect of DHA. The DHA may inhibit inflammation through an Nrf2-HO-1 pathway.59 DHA may reduce inflammation by decreasing adhesion molecules on leucocytes and on endothelial cells and decreasing chemotactic responses of leucocytes.60–62 DHA may also prevent inflammation through inhibition of NLRP3 inflammasome activation.63 Further, metabolites of DHA, resolvin D1, protectin D1 and maresins, also possess potent anti-inflammatory and inflammation-resolving activity and may also participate in this response.64–67 Changes in membrane phospholipid fatty acid composition by stimulation cells with omega-3 fatty acid can also influence the function of cells involved in inflammation.3,41
In conclusion, DHA and GW9508 activate cPLA2 through the GPR120 receptor, Gαq protein and the scaffold protein β-arrestin 2. The anti-inflammatory effect of DHA is, in part, through cPLA2 activation, COX-2 activation and PGE2 production and through EP4 receptor activation. PGE2 or DHA decreases inflammation partially through inhibition of the NF-κB signalling pathway. Our study demonstrates that cPLA2 and its down-stream lipid mediator, PGE2, are involved in the anti-inflammatory effect of DHA. It adds a possible new mechanism to the existing mechanisms of DHA inhibition of inflammation.
Studies in human clinical trials have suggested that omega-3 fatty acids play an anti-inflammatory role in several human inflammatory diseases including atherosclerosis, asthma, arthritis and inflammatory bowel disease.1–5 Therefore, deeper understanding of the anti-inflammatory mechanisms may support the development of novel therapies, such as an EP4 agonist, for treatment of human inflammatory diseases.
Disclosures
All authors declare no competing financial interests.
References
- Fritsche K. Fatty acids as modulators of the immune response. Annu Rev Nutr. 2006;26:45–73. doi: 10.1146/annurev.nutr.25.050304.092610. [DOI] [PubMed] [Google Scholar]
- Zhang MJ, Spite M. Resolvins: anti-inflammatory and proresolving mediators derived from omega-3 polyunsaturated fatty acids. Annu Rev Nutr. 2012;32:203–27. doi: 10.1146/annurev-nutr-071811-150726. [DOI] [PubMed] [Google Scholar]
- Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2:355–74. doi: 10.3390/nu2030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. Long-chain fatty acids and inflammation. Proc Nutr Soc. 2012;71:284–9. doi: 10.1017/S0029665112000067. [DOI] [PubMed] [Google Scholar]
- Yates CM, Calder PC, Ed Rainger G. Pharmacology and therapeutics of omega-3 polyunsaturated fatty acids in chronic inflammatory disease. Pharmacol Ther. 2014;141:272–82. doi: 10.1016/j.pharmthera.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Orr SK, Palumbo S, Bosetti F, et al. Unesterified docosahexaenoic acid is protective in neuroinflammation. J Neurochem. 2013;127:378–93. doi: 10.1111/jnc.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Periz A, Horrillo R, Ferre N, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23:1946–57. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychogios N, Hau DD, Peng J, et al. The human serum metabolome. PLoS ONE. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawood AL, Ding R, Napper FL, et al. Eicosapentaenoic acid (EPA) from highly concentrated n-3 fatty acid ethyl esters is incorporated into advanced atherosclerotic plaques and higher plaque EPA is associated with decreased plaque inflammation and increased stability. Atherosclerosis. 2010;212:252–9. doi: 10.1016/j.atherosclerosis.2010.05.022. [DOI] [PubMed] [Google Scholar]
- Kusunoki M, Tsutsumi K, Nakayama M, et al. Relationship between serum concentrations of saturated fatty acids and unsaturated fatty acids and the homeostasis model insulin resistance index in Japanese patients with type 2 diabetes mellitus. J Med Invest. 2007;54:243–7. doi: 10.2152/jmi.54.243. [DOI] [PubMed] [Google Scholar]
- Andersen LF, Solvoll K, Drevon CA. Very-long-chain n-3 fatty acids as biomarkers for intake of fish and n-3 fatty acid concentrates. Am J Clin Nutr. 1996;64:305–11. doi: 10.1093/ajcn/64.3.305. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Johnson J, Rooney MW, Kyle ML, Kling DF. A novel omega-3 free fatty acid formulation has dramatically improved bioavailability during a low-fat diet compared with omega-3-acid ethyl esters: the ECLIPSE (Epanova® compared with Lovaza®) in a pharmacokinetic single-dose evaluation) study. J Clin Lipidol. 2012;6:573–84. doi: 10.1016/j.jacl.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Mayer K, Gokorsch S, Fegbeutel C, Hattar K, Rosseau S, Walmrath D, Seeger W, Grimminger F. Parenteral nutrition with fish oil modulates cytokine response in patients with sepsis. Am J Respir Crit Care Med. 2003;167:1321–8. doi: 10.1164/rccm.200207-674OC. [DOI] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–98. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa A, Tsumaya K, Awaji T, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–4. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Burns RN, Moniri NH. Agonism with the omega-3 fatty acids α-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun. 2010;396:1030–5. doi: 10.1016/j.bbrc.2010.05.057. [DOI] [PubMed] [Google Scholar]
- Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009;21:462–9. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WE, Lefkowitz RJ. Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol. 2001;13:139–45. doi: 10.1016/s0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]
- Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–5. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- Uozumi N, Kume K, Nagase T, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–22. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- Sapirstein A, Bonventre JV. Specific physiological roles of cytosolic phospholipase A2 as defined by gene knockouts. Biochim Biophys Acta. 2000;1488:139–48. doi: 10.1016/s1388-1981(00)00116-5. [DOI] [PubMed] [Google Scholar]
- Fujishima H, Sanchez Mejia RO, Bingham CO, 3rd, Lam BK, Sapirstein A, Bonventre JV, Austen KF, Arm JP. Cytosolic phospholipase A2 is essential for both the immediate and the delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc Natl Acad Sci U S A. 1999;96:4803–7. doi: 10.1073/pnas.96.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawliczak R, Logun C, Madara P, et al. Cytosolic phospholipase A2 Group IVα but not secreted phospholipase A2 Group IIA, V, or X induces interleukin-8 and cyclooxygenase-2 gene and protein expression through peroxisome proliferator-activated receptors γ1 and 2 in human lung cells. J Biol Chem. 2004;279:48550–61. doi: 10.1074/jbc.M408926200. [DOI] [PubMed] [Google Scholar]
- Six DA, Dennis EA. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–42. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–59. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–51. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- Hanel AM, Schuttel S, Gelb MH. Processive interfacial catalysis by mammalian 85-kilodalton phospholipase A2 enzymes on product-containing vesicles: application to the determination of substrate preferences. Biochemistry. 1993;32:5949–58. doi: 10.1021/bi00074a005. [DOI] [PubMed] [Google Scholar]
- Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–78. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- Kramer RM, Roberts EF, Um SL, Borsch-Haubold AG, Watson SP, Fisher MJ, Jakubowski JA. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem. 1996;271:27723–9. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- Sharp JD, White DL, Chiou XG, et al. Molecular cloning and expression of human Ca2+-sensitive cytosolic phospholipase A2. J Biol Chem. 1991;266:14850–3. [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–5. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- Smith WL. Prostanoid biosynthesis and mechanisms of action. Am J Physiol. 1992;263:F181–91. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- Smith WL, Meade EA, DeWitt DL. Pharmacology of prostaglandin endoperoxide synthase isozymes-1 and -2. Ann N Y Acad Sci. 1994;714:136–42. doi: 10.1111/j.1749-6632.1994.tb12037.x. [DOI] [PubMed] [Google Scholar]
- Giuliano F, Warner TD. Origins of prostaglandin E2: involvements of cyclooxygenase (COX)-1 and COX-2 in human and rat systems. J Pharmacol Exp Ther. 2002;303:1001–6. doi: 10.1124/jpet.102.041244. [DOI] [PubMed] [Google Scholar]
- Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E2 that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–35. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woszczek G, Chen LY, Nagineni S, Shelhamer JH. IL-10 inhibits cysteinyl leukotriene-induced activation of human monocytes and monocyte-derived dendritic cells. J Immunol. 2008;180:7597–603. doi: 10.4049/jimmunol.180.11.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasinsig L, Pizzirani C, Skerlavaj B, Pellegatti P, Gulinelli S, Tossi A, Di Virgilio F, Zanetti M. The human cathelicidin LL-37 modulates the activities of the P2X7 receptor in a structure-dependent manner. J Biol Chem. 2008;283:30471–81. doi: 10.1074/jbc.M802185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PC, Dennis EA. Omega-3 fatty acids cause dramatic changes in TLR4 and purinergic eicosanoid signaling. Proc Natl Acad Sci U S A. 2012;109:8517–22. doi: 10.1073/pnas.1200189109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon SM, Mullen AC, Loscher CE, Hurley LA, Roche HM. Docosahexaenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J Nutr Biochem. 2007;18:250–8. doi: 10.1016/j.jnutbio.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Southon S, Pinder AC. (n-3) Polyunsaturated fatty acids modulate the expression of functionally associated molecules on human monocytes in vitro. J Nutr. 1996;126:603–10. doi: 10.1093/jn/126.3.603. [DOI] [PubMed] [Google Scholar]
- Hubbard NE, Lee S, Lim D, Erickson KL. Differential mRNA expression of prostaglandin receptor subtypes in macrophage activation. Prostaglandins Leukot Essent Fatty Acids. 2001;65:287–94. doi: 10.1054/plef.2001.0327. [DOI] [PubMed] [Google Scholar]
- Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23:e20–30. doi: 10.1161/01.atv.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- Conquer JA, Holub BJ. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J Lipid Res. 1998;39:286–92. [PubMed] [Google Scholar]
- Spite M, Claria J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19:21–36. doi: 10.1016/j.cmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, Toner M, Serhan CN. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J Immunol. 2008;181:8677–87. doi: 10.4049/jimmunol.181.12.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva M, Strokin M, Wang H, Ubl JJ, Reiser G. Arachidonic acid and docosahexaenoic acid suppress thrombin-evoked Ca2+ response in rat astrocytes by endogenous arachidonic acid liberation. J Neurochem. 2002;82:1252–61. doi: 10.1046/j.1471-4159.2002.01052.x. [DOI] [PubMed] [Google Scholar]
- Sergeeva M, Strokin M, Reiser G. Regulation of intracellular calcium levels by polyunsaturated fatty acids, arachidonic acid and docosahexaenoic acid, in astrocytes: possible involvement of phospholipase A2. Reprod Nutr Dev. 2005;45:633–46. doi: 10.1051/rnd:2005050. [DOI] [PubMed] [Google Scholar]
- Bate C, Tayebi M, Diomede L, Salmona M, Williams A. Docosahexaenoic and eicosapentaenoic acids increase prion formation in neuronal cells. BMC Biol. 2008;6:39. doi: 10.1186/1741-7007-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–29. [PubMed] [Google Scholar]
- Tang EH, Libby P, Vanhoutte PM, Xu A. Anti-inflammation therapy by activation of prostaglandin EP4 receptor in cardiovascular and other inflammatory diseases. J Cardiovasc Pharmacol. 2012;59:116–23. doi: 10.1097/FJC.0b013e3182244a12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang EH, Shimizu K, Christen T, et al. Lack of EP4 receptors on bone marrow-derived cells enhances inflammation in atherosclerotic lesions. Cardiovasc Res. 2011;89:234–43. doi: 10.1093/cvr/cvq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang EH, Shvartz E, Shimizu K, et al. Deletion of EP4 on bone marrow-derived cells enhances inflammation and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:261–9. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–93. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang GL, Im WB, Donde Y, Wheeler LA. EP4 agonist alleviates indomethacin-induced gastric lesions and promotes chronic gastric ulcer healing. World J Gastroenterol. 2009;15:5149–56. doi: 10.3748/wjg.15.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Shimizu K, Okamoto Y, Folco E, Ilasaca ML, Feinberg MW, Aikawa M, Libby P. Prostaglandin E receptor type 4-associated protein interacts directly with NF-κB1 and attenuates macrophage activation. J Biol Chem. 2008;283:9692–703. doi: 10.1074/jbc.M709663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y, Kong AN. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm. 2010;7:2185–93. doi: 10.1021/mp100199m. [DOI] [PubMed] [Google Scholar]
- Collie-Duguid ES, Wahle KW. Inhibitory effect of fish oil N-3 polyunsaturated fatty acids on the expression of endothelial cell adhesion molecules. Biochem Biophys Res Commun. 1996;220:969–74. doi: 10.1006/bbrc.1996.0516. [DOI] [PubMed] [Google Scholar]
- Miles EA, Wallace FA, Calder PC. Dietary fish oil reduces intercellular adhesion molecule 1 and scavenger receptor expression on murine macrophages. Atherosclerosis. 2000;152:43–50. doi: 10.1016/s0021-9150(99)00446-3. [DOI] [PubMed] [Google Scholar]
- Sperling RI, Benincaso AI, Knoell CT, Larkin JK, Austen KF, Robinson DR. Dietary omega-3 polyunsaturated fatty acids inhibit phosphoinositide formation and chemotaxis in neutrophils. J Clin Invest. 1993;91:651–60. doi: 10.1172/JCI116245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Jiang W, Spinetti T, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–63. doi: 10.1016/j.immuni.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Novel omega-3-derived local mediators in anti-inflammation and resolution. Pharmacol Ther. 2005;105:7–21. doi: 10.1016/j.pharmthera.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl. 1):S200–15. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Gotlinger K, Hong S, et al. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176:1848–59. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206:15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]