

Abstract
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer causing approximately 660,000 deaths worldwide annually. The preferred treatment of HCC is surgical resection or orthotopic liver transplantation (OLT) for patients meeting specific criteria. For patients outside these criteria, options are limited including medical therapy, radiofrequency ablation, chemoembolization, or palliative measures, and result in poor outcomes.
Goals
Our centers are elucidating the genomics of HCC to improve treatment options with a focus on three etiologies: hepatitis C virus, hepatitis B virus, and non-viral.
Methods
Through collaborative efforts, we established an effective specimen biobanking protocol. Currently we are using several techniques to analyze HCC including whole genome sequencing, whole exome sequencing, gene specific analysis, gene expression, and epigenetic analysis.
Results
We have completed whole genome sequencing on two patient samples, whole exome sequencing on 47 patient samples, gene specific analysis on 94 patient samples, gene expression on four patient samples, and epigenetic analysis on one patient sample.
Discussion
We hope to define novel therapeutic strategies by aiming towards genetic therapies that may work in conjunction with surgical approaches to improve long-term patient and graft survival in patients with HCC. We also strive to provide a functional framework of a comprehensive program for genomic analysis that may be imitated by other institutions and for other tumors in the global quest towards personalized genomic medicine.
Introduction
Hepatocellular carcinoma (HCC) accounts for 85-90% of primary liver cancers, has an incidence in the United States of 9,000 to 18,000, a worldwide incidence of 1,000,000 annually, and is the cause of approximately 660,000 deaths worldwide per year [1, 2, 3]. HCC is predominantly found in Asian countries; however, increases in HCC have been reported recently in the United States where it is one of the fastest growing causes of cancer-related deaths in men [3].
The treatment of HCC is complex and many variables must be considered. Surgical resection of HCC is preferred; however, it is offered to patients without cirrhosis, or in rare cases in patients with Child's Class A cirrhosis or Model for End Stage Liver Disease score (MELD) ≤ 10, a single lesion, and no evidence of metastasis. If surgical resection it not appropriate, then the patient may be evaluated for orthotopic liver transplantation (OLT). OLT revolutionized the survival of patients with HCC. Studies continue to demonstrate increased survival of patients with HCC after adaptation of the MELD score and the Milan criteria [4, 5]. Milan criteria states that a patient must have one lesion smaller than five centimeters in diameter or three or less lesions all less than three centimeters in diameter, no evidence of metastatic disease, and no vascular invasion [6]. OLT under the guide of Milan criteria provides similar survival expectations for patients with HCC versus those without neoplastic processes [6]. As even more support for OLT, surgical resection was recently found to be inferior to OLT with respect to patient survival due to recurrence in patients with HCC outside the Milan criteria [7]. OLT treats the malignancy and possibly the cause of malignancy, theoretically increasing life expectation. If neither surgical criterion is met, the patient is relegated to medical therapy, radiofrequency ablation, chemoembolization, or palliative care. The prognosis of untreated HCC is poor but any treatment of HCC will generally prolong a patient's life, although the extension of survival can not be predicted.
Despite our best treatment strategies, it is obvious that the current treatment of HCC is inadequate. While cirrhosis is not always concurrently present with HCC, it is present in approximately 70% of patients with HCC. Oftentimes HCC develops in a stepwise fashion in patients with viral hepatitis, leading from acute to chronic infection, resulting in cirrhosis, thereby increasing the chance for development of HCC (Figure 1). Moreover, if cirrhosis is secondary to viral hepatitis, a patient can develop cirrhosis in the graft after OLT. Recurrence of cirrhosis may lead to recurrence of HCC in the transplanted liver. Approximately 20% of patients will have a recurrence of HCC after OLT, although literature reveals a range from 6.4 to 40% [4, 5, 8].
Figure 1. Progression of HCC from Hepatitis Infection.
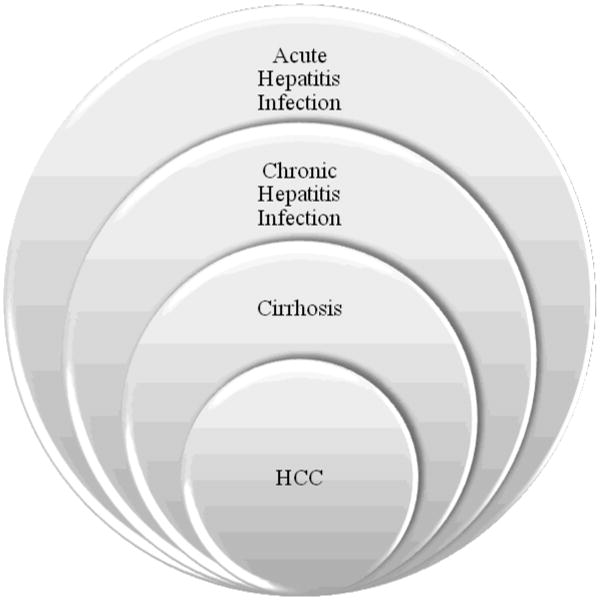
Although not all HCC develop from viral infections, viral hepatits infections contribute to development of HCC. Through this pathway, a patient acquires acute viral hepatitis. A small percentage of those patients will then progress to chronic hepatitis infection. Likewise, a small percentage of patients with chronic hepatitis infection will develop cirrhosis, and approximately 3-6% of patients with cirrhosis will develop HCC [1]. This entire process may take upwards of 30 to 40 years.
Project Goals
Due to the treatment strategy issues and the worldwide impact of HCC, our center has strived to learn as much as possible about this deadly disease. Since 2005, the Michael E. DeBakey Department of Surgery at Baylor College of Medicine in conjunction with the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine, have built a partnership with a goal to better treat HCC. We have begun to identify relevant clinical questions; establish a sustainable mechanism for tissue collection, de-identification, and short- and long-term storage; identify target molecular mechanisms for analysis; identify appropriate techniques for analysis; and correlate genomic and clinical data. This process will allow us to proceed from biobanking, genetic sequencing, data analysis and evaluation of mutations, towards personalized genomic medicine (Figure 2), which has not been undertaken with regards to HCC.
Figure 2. Steps towards Personalized Genomic Medicine with HCC.
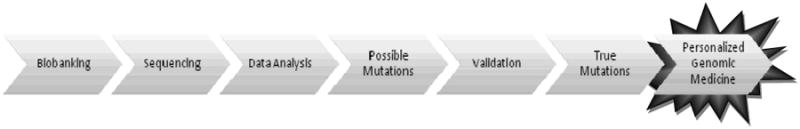
Our centers' schematic towards personalized genomic medicine: 1) an effective specimen biobanking protocol must be instituted; 2) genomic sequencing takes place after adequate tissue is confirmed by independent pathologists; 3) data analysis of sequencing reveals 4)possible mutations; 5) validation of possible mutations is performed by inspection and on another sequencing platform to reveal 6) true mutations; 7) by targeting true mutations, new therapeutics may be created and customized, therefore realizing the goal of personalized genomic medicine.
We have focused on HCC due to three major etiologies: hepatitis C virus (HCV), hepatitis B virus (HBV), and non-viral causes. To attain this goal, we are collecting fifty high-quality tissue samples from each of the three etiological groups and sequencing the tissue samples at the HGSC. At the time of surgical resection or transplantation, tissue is de-identified and processed for storage. A portion of the sample is sent to an independent pathologist for review to ensure adequate tumor tissue prior to sequencing. The Cancer Genome Atlas (TCGA), a project of the National Cancer Institute to understand the genetics of cancer, standards for acceptable tumor levels (≥80% tumor nuclei, ≤20% necrosis, and ≤20% stroma) are used to determine if a sample is appropriate for sequencing.
Interestingly, HCC is one of the cancers that remains largely treated in the United States prior to surgical intervention, providing unique opportunities related to genomic sequencing. Many tissue banks, including the TCGA bank, require all tumor tissue to be non-treated. In the case of HCC, many times the tumor will be treated as a bridge to transplantation until an organ is available, or to qualify the patient for transplantation based on Milan criteria. Many opponents would argue that this pre-treatment of approximately 90% of our patients would disrupt our analysis; however, since many of these cancers are pre-treated in the United States, we believe that we will find mutations crucial to long-term survival. These pre-treated tumor mutations may in fact be the key to understanding recurrence and survival rates in our patient population and throughout the country.
The first step of any sequencing is acquiring appropriate tissue. Within our institution, the relationship between the liver transplant surgical team and the HGSC is vital. Our mechanism of specimen biobanking is described in A Primer on a Hepatocellular Carcinoma Bioresource Bank Using the Cancer Genome Atlas Guidelines: Practical Issues and Pitfalls. Our institutions have developed and maintained a functional biobank protocol resulting in high quality tissue for DNA and RNA extraction for analysis. Briefly, tissue is sectioned from the explanted or resected liver and flash-frozen or immersed in a solution to preserve RNA. These tissues are then stored at -80 degrees Celsius until an independent pathologist can verify the clinical diagnosis and ensure adequate quality. Only after verification is the tissue then transported to the HGSC for extraction of DNA and RNA and eventual sequencing.
Methods
Multiple platforms including Sanger sequencers to next-generation sequencers are available at the HGSC. These platforms coupled with different methods allow our centers to perform whole genome sequencing, whole exome sequencing, gene specific sequencing, gene expression analysis, and epigenetic analysis (Table 1). Descriptions of exact methods and the respective sequencing platforms have been clearly illustrated throughout the medical literature, including publications from our own institutions [9, 10]. This publication does not aim to re-describe these methods, but instead demonstrate how these methods are being used to find novel therapeutic approaches to an otherwise deadly disease. To understand sequencing strategies, however; one must learn basics about the history and technologies of genomic sequencing.
Table 1. Type of Sequencing Performed on HCC at the HGSC.
| Type of Sequencing | Number of Samples Sequenced | Platform |
|---|---|---|
| Whole Genome Sequencing | 2 | Applied Biosystems SOLiD, Illumina Solexa, Pacific BioSciences |
| Whole Exome Sequencing | 47 | Applied Biosystems SOLiD |
| Gene Specific Analysis | 94* | Sanger |
| Gene Expression | 4 | Applied Biosystems SOLiD, Illumina Solexa |
| Epigenetic Analysis | 1 | Illumina Solexa |
During gene specific analysis, 28 genes were analyzed in our 94 paired samples.
Genomic sequencing started with Frederick Sanger in 1975 [11], and it has grown as a technology steadily since, with recent explosive expansion (Figure 3). The Human Genome Project required 13 years to produce an entire human genome in 2003 at a cost of $3 billion dollars [12]. Since then, other projects have decreased the time and the cost to sequence the human genome, including the Venter genome sequenced on Sanger technology, which required 4 years and $100 million dollars [13], and the Watson genome, which required 4.5 months and $2 million dollars and was performed at the HGSC on 2nd-generation sequencers [14]. Next-generation technology now promises even quicker sequencing with even lower prices. Currently companies are offering individual whole human genome sequencing for approximately $20,000 to $50,000. In June 2010, Illumina began offering whole genome sequencing for $19,500 per individual, $14,500 for groups of five individuals or more, and $9,500 for individuals with serious medical conditions. Over the next several years, prices are expected to decrease substantially to $1,000 per genome. There are even projections by private companies, such as a statement by Pacific Biosciences in March 2010, stating that whole human genome sequencing will be available in less than fifteen minutes at the cost of $100 by 2015. Additionally, GnuBio, a startup company from the Weitz lab at Harvard, has promised genome sequencing for approximately $30, but has not stipulated when this will be available. Although there will be a surplus of information, many clinicians are concerned that it will still be years until analysis is available to support the amount of data received from sequencing to provide useful clinical information.
Figure 3. Timeline of Genomic Costs.

A partial timeline of the genomic discovery and the associated time and money costs. Sanger sequencing was the first platform discovered by Fred Sanger in 1975 [11]. The Human Genome Project, performed on Sanger sequencers, was started in 1990 and was completed in 2003, requiring $3 billion [12]. The Venter genome, also performed on Sanger sequencers, took 4 years and $100 million and was completed in 2007 [13]. The Watson genome, performed on 2nd-generation sequencers, took 4.5 months and $2 million for the HGSC to complete [14]. Currently, companies are offering whole genome sequencing for approximately $20,000 and it is projected that by 2015, whole genome sequencing will be available in less than fifteen minutes for $100.
The final step of sequencing is validation. At the HGSC, data analysis experts first visually validate all mutations in a lengthy and time-consuming process. Then, another sequencing method is used to re-examine the same specimens to make sure that the mutations seen previously are seen in the new sequence pattern, virtually eliminating the errors from each individual sequencing method. Although theoretically this process may discard some true positive mutations, it almost ensures that false positive mutations are completely expunged.
Results
Whole genome sequencing determines the complete DNA sequence of an organism's genome at one time, including all of chromosomal and mitochondrial DNA. Sanger sequencing platforms utilize a chain-termination method and require single-stranded DNA template, DNA primer, DNA polymerase, radioactive or fluorescently labeled nucleotides, and modified nucleotides that terminate DNA strand elongation [15]. Although this method returns long read lengths, it also has a high cost. At the HGSC, whole genome sequencing is performed instead on 2nd-generation sequencing platforms including Applied Biosystems SOLiD and Illumina Solexa sequencers. In the very near future, we will be performing whole genome sequencing on next-generation platforms, including technologies from Pacific BioSciences. Due to length of time and cost required, we have completed whole genome sequencing on two patients: one with HCV and one with HBV. Validation results are pending at the time of the writing of this manuscript.
To combat high costs of whole genome sequencing, whole exome sequencing may be preferable. Whole exome sequencing selectively sequences the exons, the coding region of the human genome, which comprises approximately 1.5% of the total genome. It is estimated that the exons contain approximately 85% of disease-causing mutations. Therefore, whole exome sequencing provides a method for sequencing that requires less time and money than whole genome sequencing. In this technology, genomic DNA is hybridized with probes for over 17,000 genes and then sequenced using Roche NimbleGen Sequence Capture and Applied Biosytems SOLiD. Currently, we have 47 patient samples that have whole exome sequencing data including 7 patients with HBV, 33 patients with HCV, and 7 patients with non-viral causes of HCC. This data has now been validated by Roche 454 sequencers and abstracts have been accepted to the Association for Academic Surgery conference to be held in February 2011.
Since Sanger sequencing technology is not cost effective in sequencing whole genomes, our center has utilized this technology to perform gene specific DNA analysis instead. Using 28 genes, including known tumor suppressor genes and oncogenes implicated in HCC, 94 tumor tissue and matched normal samples, 36 HCV, 52 HBV and six non-viral, were analyzed with the intent to directly compare the three etiologies of HCC and their mutation profiles. Results from this sequencing are currently pending validation with Roche 454 sequencers.
Other methods to evaluate the genomic structure of HCC lead us to gene expression and epigenetic analysis. To evaluate gene expression, RNA is extracted from tumor and non-tumor liver tissue and cDNA libraries are created. Sequencing of these libraries occurs on Illumina Solexa and Applied Biosystems SOLiD platforms. Currently, four patient samples, two HBV and two HCV, have been analyzed with this technology. Lastly, in epigenetic analysis, which may be due to mutational effects or may be independent of any mutations, we are able to enrich methylated DNA from tumor and non-tumor liver samples. The selected fragments are then sequenced through the Illumina Solexa sequencing platform. Our center has performed this analysis on one HCV patient sample.
Discussion
Our centers have embarked on a relationship to elucidate the nature of HCC as this is a deadly and progressive disease with few effective treatment options. We have described our collaborative efforts to sequence HCC using various methods to achieve different levels of interpretation of genomic influences. By pinpointing these modifications, whether they are genomic, exomic, due to specific genes, or by gene expression or epigenetic influences, we will then be able to use them for treatment strategies. Our goal is to eventually find genetic modifications that are pivotal in the progression of HCC to prevent, stifle growth, or shrink disease. Therapies may be tailored to individuals displaying certain genomic characteristics, and the response of genetic populations to different therapies may provide even more information. From here, we will continue our efforts in sequencing, examine pathways and functional analysis, and then correlate the genomic information with clinical data. Additionally, we strive to collaborate with other centers to increase our specimen biobanking repository and the potential to further these projects. Lastly, we hope that our project may influence other centers to establish their own genomic program so as to collaboratively work towards a cure of HCC. In the future, these efforts may allow us to find individualized treatment strategies to unite with or to replace OLT to even further patient and graft survival. Even though our center is proud to be a leader in OLT, we hope that through this research we can provide improved and focused treatment options with personalized genomic targets, thereby decreasing the need for whole organ transplantation.
Acknowledgments
This work was presented at the Molecular Surgeon Symposium on Personalized Genomic Medicine and Surgery – Development of Clinical Model of Genomic Studies at the Baylor College of Medicine, Houston, Texas, USA, on May 7, 2010. Acknowledgement goes to the personnel at the Human Genome Sequencing Center at Baylor College of Medicine. Special acknowledgement also goes to the surgical team of The Liver Center at Baylor College of Medicine, and the residents from the Michael E. DeBakey Department of Surgery.
Grant support: This work was supported by a grant to the Human Genome Sequencing Center (U54 HG003273).
Footnotes
Conflict of Interest Statement: The authors report no potential or real conflicts of interest.
References
- 1.Geller DA, Goss JA, Tsung A. Chapter 31. Liver. In: Brunicardi FC, Andersen DK, Billiar TR, Dunn DL, Hunter JG, Matthews JB, Pollock RE, editors. Schwartz's Principles of Surgery. 9th. The McGraw-Hill Companies; 2009. http://www.accesssurgery.com/content.aspx?aID=5018142. [Google Scholar]
- 2.Jarnagin WR. CURRENT Diagnosis & Treatment: Surgery. 13th. The McGraw-Hill Companies; 2009. Chapter 24. Liver & Portal Venous System. http://www.accesssurgery.com/content.aspx?aID=5216497. [Google Scholar]
- 3.El-Serag HB, Rudolph KL. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 4.Tsoulfas G, Kawai T, Elias N, et al. Long-term experience with liver transplantation for hepatocellular carcinoma. J Gastroenterol. 2010 doi: 10.1007/s00535-010-0302-9. Online First™. [DOI] [PubMed] [Google Scholar]
- 5.Island ER, Pomposelli J, Pomfret EA, et al. Twenty-Year Experience with Liver Transplantation for Hepatocellular Carcinom. Arch Surg. 2005;140:353–358. doi: 10.1001/archsurg.140.4.353. [DOI] [PubMed] [Google Scholar]
- 6.Mazzaferro V, Regalia E, Doci R, et al. Liver Transplantation for the Treatment of Small Hepatocellular Carcinomas in Patients with Cirrhosis. N Engl J Med. 1996;334:693–699. doi: 10.1056/NEJM199603143341104. [DOI] [PubMed] [Google Scholar]
- 7.Fan HL, Chen TW, Hsieh CB, et al. Liver transplantation is an alternative treatment of hepatocellular carcinoma beyond the Milan criteria. Am J Surg. 2010;200:252–257. doi: 10.1016/j.amjsurg.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman MA, Ghobrial RM, Tong MJ, et al. Recurrence of Hepatocellular Carcinoma Following Liver Transplantation: A Review of Preoperative and Postoperative Prognostic Indicators. Arch Surg. 2008;143:182–188. doi: 10.1001/archsurg.2007.39. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez JA, Guiteau JJ, Nazareth L, et al. Sequencing the Full-Length of the Phosphatase and Tensin Homolog (PTEN) Gene in Hepatocellular Carcinoma (HCC) Using the 454 GS20 and Illumina GA DNA Sequencing Platforms. World J Surg. 2009;33:647–652. doi: 10.1007/s00268-008-9852-x. [DOI] [PubMed] [Google Scholar]
- 10.Voidonikolas G, Kreml SS, Chen C, et al. Basic Principles and Technologies for Deciphering the Genetic May of Cancer. World J Surg. 2009;33:615–629. doi: 10.1007/s00268-008-9851-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 12.The International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 13.Levy S, Sutton G, Ng PC, et al. The Diploid Genome Sequence of an Individual Human. PLoS Biol. 2007;5:2113–2144. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–877. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]