

Abstract
Farnesoid X Receptor (FXR) is a member of the nuclear receptor superfamily of transcription factors that plays a key role in the regulation of bile acids, lipid and glucose metabolisms. The regulative function of FXR is governed by conformational changes of the ligand binding domain (LBD) upon ligand binding. Although FXR is a highly researched potential therapeutic target, only a limited number of FXR-agonist complexes have been successfully crystallized and subsequently yielded high resolution structures. There is currently no structural information of any FXR-antagonist complexes publically available. We therefore explored the use of amide hydrogen/deuterium exchange (HDX) coupled with mass spectrometry for characterizing conformational changes in the FXR-LBD upon ligand binding. Ligand-specific deuterium incorporation profiles were obtained for three FXR ligand chemotypes: GW4064, a synthetic non-steroidal high affinity agonist; the bile acid chenodeoxycholic acid (CDCA), the endogenous low affinity agonist of FXR; and Z-guggulsterone (GG), an in vitro antagonist of the steroid chemotype. Comparison of the HDX profiles of their ligand-bound FXR-LBD complexes revealed a unique mode of interaction for GG. The conformational features of the FXR-LBD-antagonist interaction are discussed.
Keywords: Farnesoid X receptor, hydrogen/deuterium exchange, mass spectrometry, conformation, dynamics, ligand interaction, guggulsterone
1. Introduction
The farnesoid X receptor (FXR) is a therapeutic target with the potential to modulate metabolic pathways associated with diverse liver and metabolic disorders. FXR’s prominence stems from its role as a key regulator of bile acid homeostasis, and glucose and lipoprotein metabolisms [1, 2]. FXR has been identified as a bile acid sensor and is expressed mainly in the liver, intestine, and kidney. Many reviews have discussed the physiologically relevant role of FXR regulation and the potential of FXR as a target for synthetic ligands to prevent, manage or treat metabolic diseases, such as hyperlipidemia and type 2 diabetes [3–5].
FXR is a member of the nuclear receptor (NR) superfamily and a ligand-activated transcription factor. Since chenodeoxycholic acid (CDCA) was identified as the endogenous agonist of FXR that modulates the expression of target genes related to bile acid metabolism, FXR activation rapidly became the objective of intense research efforts. Synthetic FXR agonists have been developed with the potential to treat cholestatic liver diseases, including primary biliary cirrhosis, and metabolic disorders [6]. For instance, FXR agonists have been identified that improve myocardial fatty acid metabolism in obese (fa/fa) Zucker rats, and to counteract pro-atherogenic lipoprotein profiles and thereby confer protection against aortic plaque formation in (ApoE−/−) mice, a model of accelerated atherosclerosis [7–10]. Ursodeoxycholic acid (UDCA), an isomer of CDCA, was the first FXR-targeting drug approved by the FDA to treat primary biliary cirrhosis [11].
The X-ray structures of agonist-bound FXR complexes show the classical NR fold, consisting of 12 helices that form a three-layer sandwich harboring the LBC. Ligand binding is primarily facilitated by interactions with residues located in helices 3, 5, 6, 7, and 11. Ligand binding changes the position of the AF-12 helix and facilitates recruitment of co-activator proteins. Despite FXR s importance as a potentially therapeutically relevant receptor, only a limited number of X-ray structures of the FXR-LBD bound to high affinity synthetic ligands are available that may guide structure-activity relationship (SAR) studies. The published X-ray crystal structures of FXR-ligand complexes illustrate binding modes of five different ligand chemotypes: three steroid-like agonists [12, 13], ten stilben-based agonists (GW4064 and derivatives) [14–18], seven benzimidazole-based agonists [19, 20], two azepino [4,5-b] indole agonists (XL335 and FXR 450) [21, 22], and one fexaramide y-shaped ligand [23]. To date, there are no published crystal structures available for antagonist-bound FXR-LBD complexes or the apo-protein. The conformational plasticity of FXR s LBD has been discussed as one of the possible reasons for the limited success of X-ray structural analyses of FXR-LBD ligand complexes [12].
HDX-MS has been previously used successfully for studying the conformational properties of diverse nuclear receptors and their conformational changes upon binding to diverse ligands [24–27]. In the current study, we used HDX-MS for studying conformational changes of the FXR-LBD upon binding to different classes of ligands: (1) CDCA, a primary bile acid ligand and a low affinity agonist; (2) GW4064, a synthetic non-steroidal stilben-based high affinity agonist; and (3) Z-guggulsterone (GG), a natural steroidal FXR ligand and one of the few ligands described as an in vitro FXR antagonist (Kd > 5μM) [28]. Comparison of the deuterium incorporation profiles obtained in absence and presence of the chemically diverse ligands enabled the identification of distinct regions of the FXR-LBD that exhibit ligand-specific exchange behaviors. Insights into the ligand-dependent modulation of the conformational properties within the LBD may aid the development of selective bile acid receptor modulators (SBARMs) which show promise to manipulate FXR s pleiotropic regulation of metabolic networks.
2. Materials and Methods
2.1. Materials
Deuterium oxide (D2O, 99.9% deuterium) was from Sigma-Aldrich Chemical Co. (St. Louis, MO). GG was purchased from ChromaDex™ Corporate (Irvine, CA). GW4064 and CDCA were obtained from Tocris (Ellisville, MO). All ligands were prepared as 10 mM stock solution in dimethylsulfoxide (DMSO). All other materials were obtained from standard commercial sources.
2.2. Protein expression and purification
The pET 15B vector containing hFXR-LBD, residues 193–472 was transformed into Escherichia coli BL21 (DE3) pLysS and grown on LB agar plates. A single colony was used to inoculate 100 mL of 2XYT medium with antibiotics (Carbenicillin 100 ug/mL and Chloramphenicol 35 μg/mL) and grown overnight at 37°C. The overnight culture was centrifuged for 10 min at room temperature and then the pellet was resuspended in 6 mL of fresh 2XYT medium. Each liter of fresh 2XYT medium with antibiotics (ampicillin 150 μg/mL and Chloramphenicol 35 μg/mL) was inoculated with 1 mL of the resuspended cells (total six liter) and grown at 37°C to A600=0.6. Protein expression was induced with 0.8 mM IPTG (isopropyl-β-D-thiogalactopyranoside) and the cells were allowed to continue growing for 4 hours at 20°C. The cell pellets were harvested by centrifugation (4000 RPM, 25 min, 4°C), resuspended in cell wash solution (150 mM NaCl, 50 mM Tris and 10% w/v sucrose), centrifuged again and frozen at −80°C.
The frozen cell pellets were resuspended in buffer solution (50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 15 mM imidazole, 0.5 M sucrose, pH 7.3) and centrifuged. The His6-tagged FXR-LBD was mixed with Clonetech Talon Co2+ polyhistidine affinity resin (equilibrated with the above buffer) at 4°C for 45 min. The proteins were eluted into a solution containing 50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 200 mM imidazole, 0.5 M sucrose, pH 7.3. The His tag was removed by thrombin digestion at 4°C (48 hours) followed by purification on a column packed with Co2+ resin to yield purified human FXR-LBD monomer which was used for all experiments. The protein concentration was determined spectrophotometrically at 280 nm, and the purity (over 95%) was judged by sodium dodecyl sulfate acrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry (Fig. S1).
2.3. Fluorescence titration experiment
Steady state fluorescence measurements were performed with a Perkin Elmer LS 50 luminescence spectrophotometer. The protein sample was excited at 295 nm, and the sample temperature was maintained at 25°C. The ligand solutions (1×10−3 M) were titrated into a fixed volume (400 μl) of FXR-LBD (1×10−5 M) until the ligand/protein ratio reached 5 to 1. The fluorescence intensity was measured over a wavelength range of 305–480 nm. The fluorescence spectra were corrected (Fcor) for background fluorescence, instrument-dependent monochromator and photomultiplier response emission and dilution factor effects: Fcor = (F0–FB) × 100×A/FS, where F0 is the measured fluorescence intensity, FB is the background fluorescence of the specific ligand, A is the dilution factor of the protein sample, and FS is the emission correction factor. Based on the FXR-LBD-GG binding data, the dissociation constant (Kd) of GG was derived by fitting a curve to the Hill equation [29] using the GraphPad Prism software program.
2.4. HDX-MS analysis
The purified FXR-LBD protein (15 μL, 98 μM, in 50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 1 mM TCEP, 0.5 M sucrose and 10% glycerol, pH=7.4) was equilibrated for 30 min in the presence of the respective DMSO solution (0.5 μL, ±10 mM ligand). The ligand/monomeric protein molar ratio was 3.2:1. The small amount of DMSO (3.3% v/v) did not affect protein binding as indicated by fluorescence titration data as well as by intact protein HDX data. The D2O buffer (50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 1 mM TCEP, 0.5 M sucrose and 10% glycerol, pH 7.3) was equilibrated for 30 min in the presence of the same percentage of DMSO. The exchange reaction was initiated by adding 10-times D2O buffer (150 μL). At different reaction time points, 0.5, 1, 2, 5, 10, 30 and 60 min, aliquots (15 μL) were added to pre-chilled vials containing the same volume of quenching solution (15 μL, 0.42% phosphoric acid, pH 2.5). The quenched sample was immediately frozen in liquid nitrogen for subsequent LC-ESI-MS analysis. Experiments were performed in triplicate.
The experimental conditions for the ligand binding studies in combination with the HDX approach were chosen to ascertain that both the FXR-LBD protein and the respective low affinity ligand retained solubility throughout the period of labeling. Even in presence of structure promoting and stabilizing additives (CHAPS, sucrose, glycerol, DMSO) the FXR-LBD protein showed a high tendency to precipitation in presence of low affinity ligands. This limited our flexibility in using large excess of low affinity ligands. We believe that the absence of X-ray structures of FXR-LBD in complex with low affinity ligands is at least in part caused by this shortcoming. The reported dissociation constant/binding affinity values for GW4064, CDCA, and GG are 0.06 μM [30], 10 μM [31], and GG> 5 μM [28], respectively. The bound fraction of FXR-LBD in each ligand binding study before initiation of the HDX reaction is ~100% as estimated by applying equation 1 (Eq 1) [32]:
| (Eq. 1) |
where LT and PT are the total ligand concentration and protein concentration used, respectively, and is the dissociation constant. In the present experimental protocol we kept the mole ratio between protein and ligand constant (being 3.2:1) during the time course of labeling. The evaluation of the mass isotope distributions and elution time features of all peptides evaluated indicated that no mixed populations were detected in accordance with that the fraction of protein bound was the predominant population present in solution (Fig. S5).
For intact protein HDX-MS, the protein was eluted through a Micro Trap™ C4 column (1×8 mm) with a steep 10–90% (v/v) B gradient within 10 min (mobile phase A: 0.1% HCOOH in H2O, mobile phase B: 0.1% HCOOH in CH3CN, flow rate 40 μL/min) and eluted into a Waters LCT ESI-ToF mass spectrometer. HDX information at the peptide level was obtained by employing a nanoUPLC system equipped with a Waters “chilled” LC module, and coupled to a Waters Synapt HDMS instrument. Protein was digested by an online Poroszyme immobilized pepsin column (2.1×30 mm) with a 100 μL/min solvent (0.05% HCOOH in H2O). The proteolytic products were captured on a Acquity BEH C18 VanGuard Pre-column (1.7 μm, 2.1 × 5 mm), eluted and separated on an Acquity UPLC BEH C18 column (1.7 μm, 1.0×100 mm) using the following elution program: solvent B, increase from 8 to 40% within 6 minutes, hold at 40% for 1 minute, then increase from 40 to 85% in 0.5 minute, hold at 85% for 1 minute, then decrease from 85% to 8% in 0.5 minute, hold solvent B at 8% for 3 minutes. The elution protocol was completed in 12 minutes. In order to minimize contamination of the mass spectrometer with additives and detergents a Rheodyne MX Series II 6-port valve was installed between the nanoAcquity UPLC and the ESI source. The details of the UPLC setup used in this study are provided in the supplementary materials (Fig. S3). All solvents were immersed in an ice bath.
2.5. Mass spectral data analysis
The deuterium incorporation levels for protein or peptic peptides were directly derived from the observed mass difference between the deuterated and nondeuterated proteins/peptides without back exchange correction. The back-exchange correction was omitted due to the comparative nature of the present study [33–38]. The intact protein molecular mass and the centroid mass of each peptide, respectively, were obtained using Waters MassLynx 4.0 and HDX Browser software in combination with the HX express program described by Weis et al [39].
To identify pepsin-generated peptides, mass spectral data were acquired on a Synapt HDMS system equipped with a ESI source and operated in the MSE mode [40]. The MSE dataset was input to the ProteinLynx Global Server 2.4 software program (Waters) for searching against an “in house” protein database to which the human FXR-LBD sequence was added. The sequence of the current construct and the peptic map of FXR-LBD are depicted in Fig. S4.
2.6. HDX kinetics
Under our experimental conditions, the deuterium volume percentage is over 90%. In the applied protocol pH and temperature were kept constant at 7.3 and 25 °C, respectively. The total D content in a specific peptide can be described by the equation 2 (Eq. 2) because HDX will follow a first-order kinetics [41].
| (Eq. 2) |
where D is the deuterium content in a peptide containing N amide hydrogens, ki is the rate constant for each exchangeable amide hydrogen, and t is the exchange reaction time. The rate constant ki can be determined by applying a curve fitting software [42, 43]. In the present study, we applied a fixed-rate-constant binning model for fitting the deuterium incorporation curves of all peptides by minimizing the sum of square error in R statistical software. In this model, deuterium uptake can be plotted according to equation 3 derived by equation 2, where Ai is the amplitude which means the number of amide hydrogen located in a specific bin and ki is the rate constant in the i-th kinetic phase which is fixed to six different bins (10, 1, 0.1, 0.01, 0.001, 0.0001 min−1).
| (Eq. 3) |
The HDX data of three replicates for each sample was fitted together. The best fitted curve was plotted using as data points the average D value of the three trials including ± standard deviation as error bar for each exchange time point (Fig. S6).
3. Results
3.1. Ligand binding studies by intrinsic protein fluorescence measurements
The X-ray crystal structure of the FXR-LBD-GW4064 complex (PBD: 3DCT) showed that the two tryptophan residues in FXR-LBD, W454 and W469, located in helix 11 and helix 12, respectively, are in proximity to GW4064. Therefore, we chose intrinsic fluorescence titration experiments to test if the ligands interact and bind to our FXR-LBD construct under the current experimental conditions used. We monitored intensity changes in protein fluorescence emission (Fig. 1) using an excitation wavelength of λexc = 295 nm to minimize fluorescence contributions of the six tyrosine residues present in the FXR-LBD construct. The fluorescence spectra of apo FXR-LBD showed a strong fluorescence emission with a maximum at 343 nm. Binding of the GW4064 to FXR-LBD caused a red-shift in the emission spectrum confirming that under the solution conditions used the mode of binding of GW4064 is consistent with the binding interactions that were observed in the crystal structure of the FXR-LBD-GW4064 complex [18]. Only a slight change (± 3%) of the FXR-LBD fluorescence intensity in presence of CDCA was observed, suggesting that CDCA binding induces rather subtle changes within the microenvironment of the two tryptophan residues. These results were consistent with recent studies by Han et al [44], who reported that the fluorescence spectra of FXR-LBD showed little increase of intensity at a molar ratio of CDCA to FXR-LBD of 100:1. The in vitro antagonist GG [45] caused a concentration-dependent quenching of FXR-LBD fluorescence with no changes in either the maximum emission wavelength or shape of the spectra. The ligand-specific differences observed in the fluorescence titration experiment suggested that each ligand, GG, CDCA and GW4064, adopted distinct modes of binding. From the titration curve of GG (Fig. S2) a dissociation constant of 5.9 ± 0.3 μM was determined implying that GG is a low affinity ligand in accord with previous reports by Burris et al [28].
Figure 1.

Fluorescence spectra of FXR-LBD with structures of the ligands used in the current study: GW4064, CDCA, and GG (Z)
3.2. Global HDX profiles of FXR-LBD
To investigate to what extent interaction of the different ligands affect the global conformational plasticity of the FXR-LBD, we compared the deuterium incorporation profile of apo FXR-LBD with the exchange profiles of the FXR-LBD in the presence of the diverse ligands. Fig. 2 shows the overall deuterium levels of intact FXR-LBD in absence and presence of the ligands at different exchange-in periods ranging from 0.5, 1, 2, 5, 10, 30 to 60 min. Comparison of the global exchange profiles observed for the ligand-bound FXR-LBD revealed that exchange was significantly more retarded for the complex with the high affinity agonist GW4064 compared to the protein complexes formed with the two low affinity binders, CDCA and GG. The FXR-LBD contains a total of 274 exchangeable backbone amide hydrogens. The reduced deuterium incorporation levels observed for the protein-ligand complexes suggested that ligand binding resulted in increased structural compactness, loss of conformational flexibility and/or decreased overall solvent accessibility. HDX-MS analysis of the apo FXR-LBD after 60 min incubation indicated that 144 backbone amide hydrogens were exchanged with deuterium. In contrast, exchange-in deuterium incorporation levels for the ligand-bound FXR-LBD were 108, 132 and 136 (± <10% RSD) in presence of the respective ligand, GW4064, CDCA and GG (323 μM ligand and ligand/protein ratio > 3). Overall conformational stabilization upon ligand binding was also reported for other nuclear receptor LBDs [25, 26].
Figure 2.

Deuterium levels observed for the FXR-LBD with and without ligands after the following incubation periods: 0.5, 1, 2, 5, 10, 30, 60min.
3.3. Peptide-level HDX-MS analysis
Forty peptides covering 91% of the FXR-LBD sequence were generated under the current experimental conditions (Fig. S4). A subset of 20 peptides was selected for which mass spectral data was consistently of high enough quality to allow determining the time-dependent isotopic compositions and deuterium incorporation levels for the different protein systems (Fig. S5). For all deuterium labeled peptides evaluated, the observed mass spectral isotope patterns at all time points were consisted with EX2 kinetics (Fig. S5).
3.3.1. HDX-MS of the apo FXR-LBD
Fig. S7 summarizes the HDX-MS data for apo-FXR-LBD. Both, the composite plot of the deuterium levels determined for peptide after different incubation times (Fig. S7A) and the derived heat map presentation (Fig. S7B), let us to postulate that large parts of the apo-FXR-LBD show exchange behaviors commonly associated with solvent exposed, disordered and/or conformationally flexible regions. For instance, the N-terminal regions, 201–241 (peptides 201–222 and 229–241) and 256–278 (peptides 256–269 and 269–278), and the C-terminal peptide covering residues 454–465, showed high deuterium levels with little time dependency. This type of exchange behaviors is usually observed for highly solvent exposed and/or disordered regions which render little protection against exchange. The peptides encompassing the partial sequences 288–298, 320–328, 328–336, 348–360, 361–367, 368–375 showed exchange profiles that are were indicative of gradual exchange over time in concurrence with exchange properties associated with conformationally flexible regions that undergo multiple partial folding and unfolding reactions and variable solvent exposure which collectively result in some level of exchange retardation. A similar time dependency of the deuterium levels was observed for the peptides encompassing the residues 386–396, 406–412, 419–433 and 434–440. The peptide covering the partial sequence 440–451 was detected with the lowest deuterium levels and little variation of time; after 30 min exposure to D2O deuterium levels barely reached 20%. Overall, these HDX-MS results led us to speculate that apo FXR LBD adopts a state of considerable conformational flexibility and openness. FXR’s plasticity was discussed by Soisson et al. and put forward as possible reason that allows the FXR-LBD to bind structurally diverse ligands but also severely hampers attempts to obtain crystal structural information of the apo protein and FXR-LBD complexes with low affinity ligands [12].
3.3.2 Peptide-level HDX profiles of FXR-LBD in presence of different ligands
To dissect the impact of the different ligands on the conformational dynamics of FXR-LBD, we performed HDX-MS studies of the ligand-bound FXR-LBD complexes. HDX-MS analysis at the peptide level provides an approach to determine the regions in the FXR-LBD that experience conformational changes induced by ligand binding. Fig. 3 compiles heat map presentations of the deuterium uptake for each of three ligand-bound forms of the FXR-LBD. In the heatmaps, regions that show high levels of deuterium incorporation are colored red and regions that showed low levels of deuterium incorporation are colored blue. The comparative presentations of the heatmaps of the deuterium incorporation levels observed for the FXR-LBD in presence of the different ligands allowed us to visualize regions that show disparate deuterium level characteristics dependent on the ligand present. The most pronounced ligand-dependent differences in deuterium uptake were observed for the following peptides: 288–298, 320–328, 328–336, 348–360, 361–367, and 368–375.
Figure 3.

Comparative presentation of the HDX heat maps of FXR-LBD in the presence of ligands. Each horizontal color block represents an analyzed peptic peptide, and each block contains seven time points (from top: 0.5, 1, 2, 5, 10, 30, 60 min).
To further evaluate how the interactions of the ligands affect the conformational properties of the FXR LBD we evaluated the deuterium incorporation dynamics at the peptide level by applying a six-fixed-rate-constant binning model as described in the method section. The results of the fits for each peptide exchange-in curve are compiled in Table S2. The curve fitting plots for the peptide that displayed disparate exchange-in levels dependent on the ligand present are depicted in Figure 4A. In Figure 4B we used the X-ray structure of GW4064-bound FXR LBD to visualize that the peptide that showed the ligand-dependent exchange-in characteristics, i.e. peptides encompassing the partial sequences 288–298, 320–328, 328–326, 348–360, 361–367, and 368–375, are partaking in forming the LBC in the FXR LBD. For instance, the peptide 288–298 with 10 exchangeable amide hydrogens displayed the highest retardation against exchange-in in each protein-ligand system studied. The applied binning model revealed that this peptide has 4, 2, and 4 exchangeable sites in the apo FXR-LBD conforming to category I (fast exchanging sites, k = 10 or 1 min−1), category II (medium exchanging sites, k = 0.1 or 0.01 min−1), and category III (slow exchanging sites, k = 0.001 or 0.0001 min−1), respectively. In presence of the ligand the distribution of the amide hydrogens conforming to the category II and category III increased. For GW4064 the deuterium incorporation profiles showed that the exchanging sites in category I were reduced to 1, whereas 3 and 6 sites conformed to category II and III, respectively. For the low affinity ligand CDCA the distribution of exchanging sites conforming to category I, II and III was 3, 2, and 5, respectively. For GG, the binning modeling resulted in 3, 1 and 6 sites that met the requirements for the exchange characteristics grouped under category I, II and III, respectively. Similar changes in the distribution of sites within each category dependent on the ligand present were observed for the other LBC peptides as well: the number of sites that conform to category I decreased while the number of sites in category II and III increased upon ligand interaction. The retardation of the exchange-in dynamics of these six peptides is consistent with that ligand interactions stabilize the LBC and reduces the dynamical flexibility of the regions that are involved in forming the LBC compared to the respective regions in the apo FXR-LBD. The disparate exchange-in behavior observed for these six peptides also suggests that despite that the ligands studied have different chemotypes all seem to interact and stabilize the same region of the FXR-LBD but each ligand interaction causes a ligand-specific modulation of the conformational properties of the regions forming the cavity.
Figure 4.


Exchange-in plots of peptides that showed disparate exchange-in characteristics. (A) Exchange-in kinetics: apo-FXR-LBD, red line; GW4064, blue line; CDCA, green line; GG, yellow line. (B) X-ray structure of FXR-LBD (green) bound to GW4064 (orange) (PDB ID 3DCT); peptides that showed disparate exchange-in characteristics in presence of the diverse ligands are colored blue.
Differential HDX-MS analysis was used to spatially resolve the impact of the diverse chemotypes on the conformational dynamics of the FXR-LBD. The average differences in percentage of deuterium exchange-in for the three ligand-bound FXR complexes relative to the apo-protein were computed. Peptides showing statistically significant differential deuterium level (p < 0.001) were colored and overlaid on the X-ray crystal structure of the FXR-LBD bound to GW4064 and 6-ECDCA, respectively (Fig. 5, data compiled in Table S1). Comparison of the HDX-MS results for the GW4064-bound protein with the crystallographic data of the FXR-LBD-GW4064 complex (PDB ID 3DCT) revealed that regions containing the contact residues and forming the LBC displayed reduced HDX rates, which included the peptides representing the helices H3 (288–298), H4–H5 (320–328), H5 (328–326), H6 (348–360), and H7 (361–367 and 368–375). As observed in the active conformation of the FXR-LBD GW4064 complex, the carboxylic acid group of GW4064 forms a hydrogen bond with Arg-331 (helix 5), while the stilbenoid moiety interacts with Leu-287 (helix 3), Met-290 (helix 3), Met-328 (helix 5), Ile-335 (helix 5), and Leu-348 (helix 6), and the dichlorophenyl group occupies a hydrophobic core formed by residues Phe-329 (helix 5), Ile-352 (helix 6), Ile-357 (helix 6), Met-365 (helix 7), and Tyr-369 (helix 7) [18]. The hydrogen bonding and hydrophobic interaction network between GW4064 and the LBC of the FXR-LBD resulted in significant exchange protection in these regions. Remarkably, the peptides 299–316 (helix 3-loop-helix 4), 388–405 (helix 9), 406–412 (helix 9) and 434–440 (helix 10), which all encompass regions not involved in GW4064 binding, exhibited reduced exchange-in rates as well. No changes in exchange-in kinetics upon GW4064 binding were observed for the peptides 256–268 (helix 1–helix 2), 269–278 (helix 2), 337–347 (helix 5–helix 6) and 454–465 (helix 11–helix 12).
Figure 5.
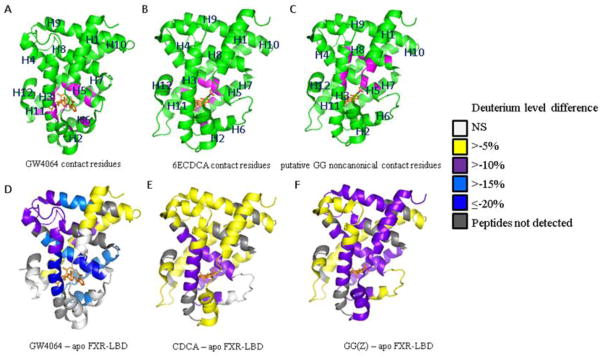
Average differences in deuterium levels (in %) of FXR-LBD with or without ligand overlaid onto crystallographic structures. The N-terminal region which is covered by the peptides 201–222 and 229–241 is not visible in the crystal structures. Top row: Co-crystal structure of FXR-LBD ligand complex, PDB ID 3DCT for (A) and PDB ID 1OSV for (B) and (C). Contact residues between ligand and protein are colored in pink. The ligand itself is colored in orange. Bottom row: Average differences in deuterium levels (D%) of seven time points (0.5, 1, 2, 5, 10, 30, and 60 min) are mapped onto the crystal structure of FXR-LBD-ligand complex, PDB ID 3DCT for (D) and PDB ID 1OSV for (E) and (F). Regions that were not covered by the current experiments are depicted in grey. NS, “not significant”.
To rationalize our current HDX MS data on CDCA binding to FXR in solution we used the crystal structure of FXR-LBD bound to a close synthetic high affinity analog of CDCA, 6-ethylchenodeoxycholic acid (6ECDCA) (PDB ID 1OSV) [13]. We hypothesized that both steroidal ligands may share similar binding sites within the FXR-LBD, except for those hydrophobic interactions between the 6ECDCA 6α-ethyl group and residues Ile-362 (helix 7), Phe-366 (helix 7) and Tyr-369 (helix 7), which collectively contribute to the much higher affinity of 6ECDCA (EC50 99 nM) [46] compared to CDCA (EC50 10 μM) [30]. Peptides encompassing the contact residues, including partial sequences 288–298 (helix 3), 320–328 (helix 4–helix 5), 328–336 (helix 5), 348–360 (helix 6) and 361–367 (helix 7), showed reduced levels of exchange-in compared to the corresponding peptides derived from the apo-protein (Fig. 5). Regions remote from the LBC, such as residues 256–268 (helix 1–helix 2), 406–412 (helix 9), and 434–440 (helix 10), exhibited only slight protection against deuterium exchange-in (Fig. 5).
GG is a low affinity in vitro antagonist of FXR [47] and the co-incubation of GG and FXR-LBD resulted in HDX data that indicated exchange-in protection in regions encompassing the LBC (Fig. 4) suggesting that the interaction sites of GG coincide with the recognition sites of typical FXR agonists. The most intriguing feature of this HDX dataset is however one region that exhibited slightly higher extent of protection in presence of GG compared to the other two agonists probed. This region is the most N-terminal region of FXR-LBD, corresponding to the partial sequence 201–278 encompassing helix 1 - loop - helix 2 (256–268). In addition, the region 388–396 was also found as being protected against exchange-in in presence of GG. Interestingly, these two protected regions coincide with a noncanonical binding site that was predicted by docking studies by Meyer et al [48]. In their model, GG s noncanonical interaction involves Tyr-260 (helix 1), the partial sequence Lys-262 to Met-265 (helix 1 – helix 2 loop), Thr-386 (helix 8) and Ile-390 (helix 8) [48].
4. DISCUSSION
We conducted a comparative HDX-MS study of the FXR-LBD in presence of structurally diverse ligands to advance our understanding of the conformational requirements that are potentially relevant to the molecular mechanisms that culminate in the regulation of the FXR activity. We initially conducted HDX-MS studies of the FXR-LBD in presence of the known high affinity non-steroid agonist GW4064. We evaluated our HDX-MS peptide level data using the B-factors associated with the crystal structure of the GW4064-bound FXR LBD (PDB ID 3DCT) (Fig. S10). Structurally flexible regions with high B-factors showed high average deuterium levels. Regions that showed low average deuterium levels aligned well with regions that have low B-factors (< 40), and these regions coincided with ligand binding recognition sites. For four FXR-LBD peptides, namely 299–316 (helix 3 loop - helix 4), 397–405 (helix 9), 406–412 (helix 9) and 434–440 (helix 10), deuterium levels were observed that were much lower in GW4064-bound protein compared to the apo protein (Figures S9 and S10) and, interestingly, these regions did not contain ligand interaction sites in the crystal structure of GW4064-bound FXR LBD. The extent of protection observed for these regions suggested that ligand binding resulted in reduced solvent accessibility of the backbone amide hydrogens and/or had a stabilizing effect on the conformational dynamics of these regions distant from the GW4064-binding cavity (purple colored regions in Figure 5D).
A comparison of the HDX-MS datasets obtained for the FXR-LBD in the presence of GW4064 and CDCA, respectively, highlights differences in the impact of the chemotype on the conformational dynamics between the synthetic stilben-like agonist GW4064 and the bile acid CDCA (Fig. 6C). The most striking differences in protection between these two ligands were detected for the regions encompassing the helices 3 and 5, both of which are part of the LBC. In addition, disparate response to ligand binding was detected for regions distant to the LBC, namely for regions encompassing helix 9 and helix 10; both showed more extensive exchange-in protection for the GW4064-bound state. We may further speculate that the observed differences in HDX kinetics for the FXR-LBD in presence of a stilben- and a steroid-based agonist, respectively, may reflect distinct conformational states that may translate to the ligand-specific recruitment of coregulator proteins and expression of FXR-target genes. Distinct gene expression profiles have been reported for GW4064 and CDCA [23].
Figure 6.

Differential HDX data of FXR-LBD-agonist and FXR-LBD-antagonist. Left: Deuterium level differences in percentage (D %) from one complex to another are shown at different time points (0.5, 1, 2, 5, 10, 30, and 60 min, from left to right) for each peptide. Right: Average differences in deuterium percentage (D %) of seven time points (0.5, 1, 2, 5, 10, 30, and 60 min) were overlaid onto the crystallographic structure, PDB ID 3DCT for (A) and (B), PDB ID 1OSV for (C). Positive numbers indicate higher deuterium levels and thus less protection in the FXR-LBD-GG relative to the FXR-LBD-GW4064. Negative numbers indicate lower deuterium levels and thus more protection in the FXR-LBD-GG relative to the FXR-LBD-GW4064.
Next, we evaluated the HDX profile obtained for the interaction of FXR-LBD with CDCA. Initially, we used the crystal structure of 6ECDCA-bound FXR-LBD as a template (Fig. 5). This allowed us to study the impact of a low affinity ligand interaction on the conformational properties of the FXR-LBD. Compared to the high affinity interaction with GW4064, the amide hydrogen exchange protection was generally less pronounced. The interaction of CDCA with the LBD was evident by the reduced deuterium incorporation levels that were observed for the peptides 288–298, 320–336, 348–375 indicating stabilization of these regions. Mapping onto the 6ECDCA-bound FXR-LBD structure these stabilized regions marked the cognate binding cavity and included helix 3, the small transition segment helix 4–helix 5 and the regions encompassing the helices 5, 6 and 7.
GG was originally identified as a FXR antagonist in vitro [47], but later was classified as a selective bile acid receptor modulator (SBARM) based on the following findings: (1) GG inhibited activation of FXR-LBD in vitro, but only in presence of CDCA in a dose-dependent manner [45]; and (2) GG acted to enhance transcription of the bile salt export pump (BSEP) in a dose-dependent manner in vivo [47]. Although GG has a steroidal scaffold, it has an all trans and quasi planar ABCD ring system in contrast to bile acids in which ring A and B are conjugated in a cis configuration [48]. In the absence of a co-crystal structure of FXR-LBD in complex with GG, the current HDX-MS results advance our understanding of the molecular determinants that govern the interaction of GG with the FXR-LBD. The differential HDX-MS profiles presented in Fig. 6A and B reveal that the N-terminal regions of the FXR-LBD (residues 201–278) showed slightly higher protection against exchange-in in the GG-bound state compared to conformational states induced by the two agonists studied. Interestingly, recent molecular modeling studies have predicted a novel noncanonical binding site for GG near the loop region located between helix 1 and helix 2 [48] with Gln267 playing a key role in binding of the unusual second coactivator peptide which is visible in the 6ECDCA-bound FXR-LBD crystal structure (PDB ID 1OSV). The observed protection against exchange-in in the N-terminal region in presence of GG is consistent with the prediction of a non-canonical ligand binding site in that region of the LBD [48]. The C-terminal fraction of helix 8 and part of the helix 8 – helix 9 loop, corresponding to the partial sequence 388–396, exhibited protection upon binding of GG (Fig. 5). As such, the HDX-MS data is in accord to the docking study of GG with the FXR-LBD that predicted that helix 8 is involved in the stabilization of GG and flanks the non-canonical GG binding site.
Helix 12 is a highly conserved feature in ligand-activated nuclear receptors and acts as a molecular switch to activate or repress receptor functions upon binding of appropriate ligands [49]. Therefore we expected to observe difference in HDX kinetics between the apoprotein and the agonist-bound LBD. However, the HDX-MS data obtained for the C-terminal fraction of the LBD (peptides 440–451 and 454–465) showed only minimal differences in deuterium incorporation levels for the apo protein and the holo systems studied. The lack of disparity in the HDX-MS data for the sequence encompassing the C-terminal fraction of the FXR-LBD upon ligand binding was surprising because a molecular dynamics simulation predicted that the C-terminal region of the protein should experience a gain in conformational motility upon removal of 6ECDCA [50]. The current observation is however consistent with previous HDX-MS studies of some nuclear receptor LBDs. For instance, a lack of change in HDX kinetics of helix 12 upon ligand binding was also observed for the human apo RXRα LBD [25, 51] and for both isoforms of the ER LBD, the ERα and ERβ protein [26]. For other nuclear receptor, including PPARγ, THR, and GR, higher solvent protections was observed for the helices helix 11 and helix 12 with agonists relative to antagonists [24, 52, 53]. Griffin et al. observed that the helices 11 and 12 of the VDR LBD showed less solvent exposure with agonists compared to the apo VDR-LBD [27]. In view of these diverse observations we propose that binding of a coactivator peptide may govern the exchange properties of helix 12. This hypothesis is based on previous findings that showed that inclusion of a coactivator peptide resulted in a decrease of deuterium uptake for the RXRα helix 12 and GR helix 12 in the presence or absence of different ligands [53, 54].
5. Conclusion
The current work demonstrates that HDX experiments in conjunction with mass spectrometry are capable of providing structural dynamics information for studying FXR ligand interactions. The observed differences in HDX kinetics reflect on the disparate modes of conformational modulation elicited by ligands endowed with structurally distinct scaffolds. The protection against deuterium uptake observed for the partial sequences 201–278 and 388–396, which cover the H1–H2 loop and parts of helix H8 including the H8–H9 loop, respectively, supports a model in which in vitro antagonism and modulation of FXR by GG may indeed involve a noncanonical binding site as predicted previously by Meyer et al [48].
Supplementary Material
Highlights.
Solution-phase conformational dynamics of FXR-LBD by HDX-MS
HDX dynamics of FXR-LBD interactions with low affinity ligands
Differential HDX-MS analysis reveals ligand-specific exchange-in kinetics
HDX-MS analysis confirms noncanonical interaction site for (Z) guggelsterone
Acknowledgments
This research was supported in part by Grants S10RR025628 and P30ES000210 from the National Institutes of Health.
Abbreviations
- HDX
hydrogen/deuterium exchange
- LC-ESI-MS
liquid chromatography electrospray ionization mass spectrometry
- FXR-LBD
farnesoid X receptor ligand binding domain
- LBC
ligand binding cavity
- CDCA
chenodeoxycholic acid
- GG
Z-guggulsterone
- 6ECDCA
6-ethylchenodeoxycholic acid
- UDCA
ursodeoxycholic acid
- DMSO
dimethylsulfoxide
- TCEP
Tris (2-carboxyethyl) phosphine hydrochloride
- TOF
time of flight
- PPARγ
peroxisome proliferator-activated receptor
- RXR
retinoid X receptor
- THR
thyroid hormone receptor
- ER
estrogen receptor
- VDR
vitamin D receptor
- GR
glucocorticoid receptor
Footnotes
Supplementary data to this article can be found online and includes: SDS-PAGE image and ESI-MS spectrum of the purified hFXR-LBD (Fig. S1); plot depicting the binding curve of GG (Z) to FXR-LBD (Fig. S2); nanoAcquity UPLC setup with switching valve to divert buffer constituents and CHAPS to waste (Fig. S3); peptide coverage map (Fig. S4); HDX mass spectra of each peptide (Fig. S5); kinetic plots (deuterium level as a function of labeling time) for each peptide for all four systems studied (Fig. S6); compilation of the HDX-MS data for the apo protein (Fig. S7); compiled deuterium level profiles for the FXR-LBD in presence of GW4064, CDCA and GG(Z) (Fig. S8); ligand-dependent deuterium exchange protection plots (Fig. S9); plot depicting the overlay of the B-values extracted from the crystal structure of FXR with GW4064 bound (PDB ID 3DCT) with the deuterium levels of each peptide observed in the presence of GW4064 (Fig. S10); tabular compilation of the HDX data used for the differential HDX analyses (Table S1); tabular compilation of the kinetic results derived by our kinetic model (Table S2).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 2.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med. 2007;13:298–309. doi: 10.1016/j.molmed.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Soldani C, Bottone MG, Pellicciari C, Scovassi AI. Nucleolus disassembly in mitosis and apoptosis: dynamic redistribution of phosphorylated-c-Myc, fibrillarin and Ki-67. Eur J Histochem. 2006;50:273–280. [PubMed] [Google Scholar]
- 5.Westin S, Heyman RA, Martin R. FXR, a therapeutic target for bile acid and lipid disorders. Mini Rev Med Chem. 2005;5:719–727. doi: 10.2174/1389557054553802. [DOI] [PubMed] [Google Scholar]
- 6.Lanzini A, De Tavonatti MG, Panarotto B, Scalia S, Mora A, Benini F, Baisini O, Lanzarotto F. Intestinal absorption of the bile acid analogue Se-75-homocholic acid-taurine is increased in primary biliary cirrhosis, and reverts to normal during ursodeoxycholic acid administration. Gut. 2003;52:1371–1375. doi: 10.1136/gut.52.9.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 8.Guo GL, Santamarina-Fojo S, Akiyama TE, Amar MJA, Paigen BJ, Brewer B, Gonzalez FJ. Effects of FXR in foam-cell formation and atherosclerosis development. Bba-Mol Cell Biol L. 2006;1761:1401–1409. doi: 10.1016/j.bbalip.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanniman EA, Lambert G, McCarthy TC, Sinal CJ. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. Journal of Lipid Research. 2005;46:2595–2604. doi: 10.1194/jlr.M500390-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang YQ, Wang XP, Vales C, Lee FY, Lee H, Lusis AJ, Edwards PA. FXR deficiency causes reduced atherosclerosis in Ldlr(−/−) mice. Arterioscl Throm Vas. 2006;26:2316–2321. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
- 11.Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525–531. doi: 10.1053/jhep.2002.36088. [DOI] [PubMed] [Google Scholar]
- 12.Soisson SM, Parthasarathy G, Adams AD, Sahoo S, Sitlani A, Sparrow C, Cui J, Becker JW. Identification of a potent synthetic FXR agonist with an unexpected mode of binding and activation. Proc Natl Acad Sci U S A. 2008;105:5337–5342. doi: 10.1073/pnas.0710981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, Khorasanizadeh S, Rastinejad F. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003;11:1093–1100. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 14.Akwabi-Ameyaw A, Caravella JA, Chen LH, Creech KL, Deaton DN, Madauss KP, Marr HB, Miller AB, Navas F, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely B. Conformationally constrained farnesoid X receptor (FXR) agonists: Alternative replacements of the stilbene. Bioorganic & Medicinal Chemistry Letters. 2011;21:6154–6160. doi: 10.1016/j.bmcl.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 15.Bass JY, Caravella JA, Chen LH, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Mills WY, Navas F, Parks DJ, Smalley TL, Spearing PK, Todd D, Williams SP, Wisely GB. Conformationally constrained farnesoid X receptor (FXR) agonists: Heteroaryl replacements of the naphthalene. Bioorganic & Medicinal Chemistry Letters. 2011;21:1206–1213. doi: 10.1016/j.bmcl.2010.12.089. [DOI] [PubMed] [Google Scholar]
- 16.Akwabi-Ameyaw A, Bass JY, Caldwell RD, Caravella JA, Chen LH, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Navas F, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely GB. FXR agonist activity of conformationally constrained analogs of GW 4064. Bioorganic & Medicinal Chemistry Letters. 2009;19:4733–4739. doi: 10.1016/j.bmcl.2009.06.062. [DOI] [PubMed] [Google Scholar]
- 17.Bass JY, Caldwell RD, Caravella JA, Chen LH, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Parks DJ, Todd D, Williams SP, Wisely GB. Substituted isoxazole analogs of farnesoid X receptor (FXR) agonist GW4064. Bioorganic & Medicinal Chemistry Letters. 2009;19:29692973. doi: 10.1016/j.bmcl.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 18.Akwabi-Ameyaw A, Bass JY, Caldwell RD, Caravella JA, Chen L, Creech KL, Deaton DN, Jones SA, Kaldor I, Liu Y, Madauss KP, Marr HB, McFadyen RB, Miller AB, FN, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely GB. Conformationally constrained farnesoid X receptor (FXR) agonists: Naphthoic acid-based analogs of GW 4064. Bioorg Med Chem Lett. 2008;18:4339–4343. doi: 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 19.Richter HGF, Benson GM, Blum D, Chaput E, Feng S, Gardes C, Grether U, Hartman P, Kuhn B, Martin RE, Plancher JM, Rudolph MG, Schuler F, Taylor S, Bleicher KH. Discovery of novel and orally active FXR agonists for the potential treatment of dyslipidemia & diabetes. Bioorganic & Medicinal Chemistry Letters. 2011;21:191–194. doi: 10.1016/j.bmcl.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 20.Richter HGF, Benson GM, Bleicher KH, Blum D, Chaput E, Clemann N, Feng S, Gardes C, Grether U, Hartman P, Kuhn B, Martin RE, Plancher JM, Rudolph MG, Schuler F, Taylor S. Optimization of a novel class of benzimidazole-based farnesoid X receptor (FXR) agonists to improve physicochemical and ADME properties. Bioorganic & Medicinal Chemistry Letters. 2011;21:1134–1140. doi: 10.1016/j.bmcl.2010.12.123. [DOI] [PubMed] [Google Scholar]
- 21.Flatt B, Martin R, Wang TL, Mahaney P, Murphy B, Gu XH, Foster P, Li JL, Pircher P, Petrowski M, Schulman I, Westin S, Wrobel J, Yan G, Bischoff E, Daige C, Mohan R. Discovery of XL335 (WAY-362450), a Highly Potent, Selective, and Orally Active Agonist of the Farnesoid X Receptor (FXR) Journal of Medicinal Chemistry. 2009;52:904–907. doi: 10.1021/jm8014124. [DOI] [PubMed] [Google Scholar]
- 22.Lundquist JT, Harnish DC, Kim CY, Mehlmann JF, Unwalla RJ, Phipps KM, Crawley ML, Commons T, Green DM, Xu WX, Hum WT, Eta JE, Feingold I, Patel V, Evans MJ, Lai K, Borges-Marcucci L, Mahaney PE, Wrobel JE. Improvement of Physiochemical Properties of the Tetrahydroazepinoindole Series of Farnesoid X Receptor (FXR) Agonists: Beneficial Modulation of Lipids in Primates. Journal of Medicinal Chemistry. 2010;53:1774–1787. doi: 10.1021/jm901650u. [DOI] [PubMed] [Google Scholar]
- 23.Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer JL, Anisfeld AM, Edwards PA, Rosenfeld JM, Alvarez JG, Noel JP, Nicolaou KC, Evans RM. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell. 2003;11:1079–1092. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan X, Deinzer ML, Schimerlik MI, Broderick D, Leid ME, Dawson MI. Investigation of ligand interactions with human RXRalpha by hydrogen/deuterium exchange and mass spectrometry. J Am Soc Mass Spectrom. 2006;17:1510–1517. doi: 10.1016/j.jasms.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Dai SY, Burris TP, Dodge JA, Montrose-Rafizadeh C, Wang Y, Pascal BD, Chalmers MJ, Griffin PR. Unique ligand binding patterns between estrogen receptor alpha and beta revealed by hydrogen-deuterium exchange. Biochemistry. 2009;48:9668–9676. doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Garcia-Ordonez RD, Pascal BD, Burris TP, Dodge JA, Griffin PR. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 2010;18:1332–1341. doi: 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burris TP, Montrose C, Houck KA, Osborne HE, Bocchinfuso WP, Yaden BC, Cheng CC, Zink RW, Barr RJ, Hepler CD, Krishnan V, Bullock HA, Burris LL, Galvin RJ, Bramlett K, Stayrook KR. The hypolipidemic natural product guggulsterone is a promiscuous steroid receptor ligand. Molecular Pharmacology. 2005;67:948–954. doi: 10.1124/mol.104.007054. [DOI] [PubMed] [Google Scholar]
- 29.Hill AV. The Combinations of Haemoglobin with Oxygen and with Carbon Monoxide. I. Biochem J. 1913;7:471–480. doi: 10.1042/bj0070471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 31.Lew JL, Zhao A, Yu J, Huang L, De Pedro N, Pelaez F, Wright SD, Cui J. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem. 2004;279:8856–8861. doi: 10.1074/jbc.M306422200. [DOI] [PubMed] [Google Scholar]
- 32.Shortridge MD, Hage DS, Harbison GS, Powers R. Estimating protein-ligand binding affinity using high-throughput screening by NMR. J Comb Chem. 2008;10:948–958. doi: 10.1021/cc800122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 34.Sperry JB, Huang RY, Zhu MM, Rempel DL, Gross ML. Hydrophobic Peptides Affect Binding of Calmodulin and Ca as Explored by H/D Amide Exchange and Mass Spectrometry. Int J Mass Spectrom. 2011;302:85–92. doi: 10.1016/j.ijms.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang RY, Rempel DL, Gross ML. HD exchange and PLIMSTEX determine the affinities and order of binding of Ca2+ with troponin C. Biochemistry. 2011;50:5426–5435. doi: 10.1021/bi200377c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang RY, Wen J, Blankenship RE, Gross ML. Hydrogen-deuterium exchange mass spectrometry reveals the interaction of Fenna-Matthews-Olson protein and chlorosome CsmA protein. Biochemistry. 2012;51:187–193. doi: 10.1021/bi201620y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker CH, Morgan CR, Rand KD, Engen JR, Jorgenson JW, Stafford DW. A Conformational Investigation of Propeptide Binding to the Integral Membrane Protein gamma-Glutamyl Carboxylase Using Nanodisc Hydrogen Exchange Mass Spectrometry. Biochemistry. 2014;53:1511–1520. doi: 10.1021/bi401536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 40.Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, Nicholson JK. UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation (vol 20, pg 1989, 2006) Rapid Commun Mass Sp. 2006;20:2234–2234. doi: 10.1002/rcm.2550. [DOI] [PubMed] [Google Scholar]
- 41.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 42.Yan X, Zhang H, Watson J, Schimerlik MI, Deinzer ML. Hydrogen/deuterium exchange and mass spectrometric analysis of a protein containing multiple disulfide bonds: Solution structure of recombinant macrophage colony stimulating factor-beta (rhM-CSFbeta) Protein Sci. 2002;11:2113–2124. doi: 10.1110/ps.0204402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Post CB, Smith DL. Amide hydrogen exchange determined by mass spectrometry: application to rabbit muscle aldolase. Biochemistry. 1996;35:779–791. doi: 10.1021/bi952227q. [DOI] [PubMed] [Google Scholar]
- 44.Han KC, Kim JH, Kim KH, Kim EE, Seo JH, Yang EG. Identification of farnesoid X receptor modulators by a fluorescence polarization-based interaction assay. Anal Biochem. 2010;398:185–190. doi: 10.1016/j.ab.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P, Yan Y, Gonzalez FJ, Heyman RA, Mangelsdorf DJ, Moore DD. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science. 2002;296:1703–1706. doi: 10.1126/science.1072891. [DOI] [PubMed] [Google Scholar]
- 46.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 47.Cui J, Huang L, Zhao A, Lew JL, Yu J, Sahoo S, Meinke PT, Royo I, Pelaez F, Wright SD. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. J Biol Chem. 2003;278:10214–10220. doi: 10.1074/jbc.M209323200. [DOI] [PubMed] [Google Scholar]
- 48.Meyer U, Costantino G, Macchiarulo A, Pellicciari R. Is antagonism of E/Z-guggulsterone at the farnesoid X receptor mediated by a noncanonical binding site? A molecular modeling study. J Med Chem. 2005;48:6948–6955. doi: 10.1021/jm0505056. [DOI] [PubMed] [Google Scholar]
- 49.Novac N, Heinzel T. Nuclear receptors: overview and classification. Curr Drug Targets Inflamm Allergy. 2004;3:335–346. doi: 10.2174/1568010042634541. [DOI] [PubMed] [Google Scholar]
- 50.Costantino G, Entrena-Guadix A, Macchiarulo A, Gioiello A, Pellicciari R. Molecular dynamics simulation of the ligand binding domain of farnesoid X receptor. Insights into helix-12 stability and coactivator peptide stabilization in response to agonist binding. J Med Chem. 2005;48:3251–3259. doi: 10.1021/jm049182o. [DOI] [PubMed] [Google Scholar]
- 51.Yan X, Perez E, Leid M, Schimerlik MI, de Lera AR, Deinzer ML. Deuterium exchange and mass spectrometry reveal the interaction differences of two synthetic modulators of RXRalpha LBD. Protein Sci. 2007;16:2491–2501. doi: 10.1110/ps.073019707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Figueira AC, Saidemberg DM, Souza PC, Martinez L, Scanlan TS, Baxter JD, Skaf MS, Palma MS, Webb P, Polikarpov I. Analysis of agonist and antagonist effects on thyroid hormone receptor conformation by hydrogen/deuterium exchange. Mol Endocrinol. 2011;25:15–31. doi: 10.1210/me.2010-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frego L, Davidson W. Conformational changes of the glucocorticoid receptor ligand binding domain induced by ligand and cofactor binding, and the location of cofactor binding sites determined by hydrogen/deuterium exchange mass spectrometry. Protein Sci. 2006;15:722–730. doi: 10.1110/ps.051781406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia G, Boerma LJ, Cox BD, Qiu C, Kang S, Smith CD, Renfrow MB, Muccio DD. Structure, Energetics, and Dynamics of Binding Coactivator Peptide to the Human Retinoid X Receptor alpha Ligand Binding Domain Complex with 9-cis-Retinoic Acid. Biochemistry. 2010 doi: 10.1021/bi101288y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.