

Abstract
Bisphenol-A (4,4′-dihydroxy-2,2-diphenylpropane, BPA, or BPA-A) and its derivatives, when exposed to humans, may affect functions of multiple organs by specific binding to the human estrogen-related receptor γ (ERRγ). We carried out atomistic molecular dynamics (MD) simulations of three ligand compounds including BPA-A, 4-α-cumylphenol (BPA-C), and 2,2-diphenylpropane (BPA-D) binding to the ligand binding domain (LBD) of a human ERRγ to study the structures and energies associated with the binding. We used the implicit Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) method to estimate the free energies of binding for the phenyl based compound/ERRγ systems. The addition of hydroxyl groups to the aromatic ring had only a minor effect on binding structures and a significant effect on ligand/protein binding energy in an aqueous solution. Free binding energies of BPA-D to the ERRγ were found to be considerably less than those of BPA-A and BPA-C to the ERRγ. These results are well correlated with those from experiments where no binding affinities were determined in the BPA-D/ERRγ complex. No conformational change was observed for the helix 12 (H-12) of ERRγ upon binding of these compounds preserving an active transcriptional conformation state.
1. Introduction
A bisphenol A (4,4′-dihydroxy-2,2-diphenylpropane, BPA, or BPA-A) is a chemical used to make epoxy resin and daily polycarbonate plastic products to modify their hardness. It is also used in the epoxy resin lining of metal food cans, dental sealants, carbonless paper/receipt, and some children’s toys. BPA is detected in human urine at an average concentration of 2.6 μg/L.1 During metabolism, some derivatives of BPA, such as 4-α-cumylphenol (BPA-C) and 2,2-diphenylpropane (BPA-D), may be generated. Recent studies showed that BPA exposure may cause human prostate and breast health problems.2 It was also found that BPA-A can strongly interact with a human nuclear estrogen-related receptor γ (ERRγ, also known as 2E2R), which is one of the 48 nuclear receptors.3,4 The ERRγ receptor belongs to the subclass of estrogen receptors (ER) and can be considered as a eukaryotic or an intracellular receptor.5 Nuclear receptors exhibit a high degree of homology in structures that usually consist of three functional domains including a ligand binding domain (LBD). The ERRγ receptor has a close sequence homology with two other members of estrogen-related receptors (ERα and ERβ).5−7
Many studies have shown that nuclear receptors play a vital role in many aspects of human physiological development and functions including embryonic development, reproduction, and cell formation.8−11 The functions of a nuclear receptor in the development of cancer cells has been a subject for scientific investigations.12 A special characteristic of a nuclear receptor is its transcriptional activity triggered by binding of ligands to the LBD. Two ERRγ conformations (H-12 helix displacement) were determined as agonist and antagonist for estrogen receptor β (ERβ), corresponding to the active and inactive transcriptional states, respectively.6 Ligand-free transcriptional activation was also reported for ERRγ.13 Meanwhile, a few organic compounds have been shown to influence the transcriptional activities of ERRγ. There are four distinct ligand activities associated with ligand binding responses. These activities can be classified as agonist, antagonist, inverse agonist, and inverse antagonist.14 Early studies had shown that the 4-hydroxytamoxifen compound functions as a deactivator due to its strong binding to ERRγ.15 Reorientation of phenylalanine (Phe345) and displacement of the H-12 helix were observed in the presence of 4-hydroxytamoxifen. This ligand was referred to as an inverse agonist. 2,2-Bis(4-hydroxyphenyl) propane A compound was also found to bind to the ERRγ receptor. Strong intermolecular interactions between BPA-A and ERRγ were observed from an X-ray diffraction experiment indicating that BPA is an active endocrine disruptor.16,17 However, the binding of BPA-A to EERγ did not change the helix conformation of EERγ.18
The binding configurations of BPA-C and BPA-D to ERRγ were also studied by X-ray diffraction.19 It was observed that one of the cumylphenol rings interacts with the Tyr326 amino residue via OH/π intermolecular interactions. The hydrophobic isobutyl group Leu309 was also found participating in the interaction with the aromatic ring of BPA-C, while the BPA-D compound did not show any specific interactions with ERRγ. These compounds did not change an active conformation of ERRγ and were not considered as agonist, antagonist, or inverse agonist bound compounds. The half-maximum inhibitory concentrations (IC50) have been determined for 4-hydroxytamoxifen, BPA-C, 2,2-bis(4-hydroxyphenyl) propane A, BPA-A, and other derivatives.17 No inhibitory concentrations were determined for phenol and BPA-D compounds.
Ligand/protein flexible docking was performed to predict binding sites of three ligand (BPA-A, BPA-C, and BPA-D) model compounds. Next, we have performed molecular dynamics simulations of these ligand/protein (ERRγ) complexes in aqueous solutions to predict the effect of hydroxyl (−OH) groups on intermolecular interactions and binding. Ligand/water and ligand/protein intermolecular interactions were studied by calculating a liquid structure and solvation/binding free energies via molecular dynamics (MD) simulations. This article further discusses the methods and results of docking and MD simulations.
2. Docking and MD Simulation Methods
2.1. Molecular Binding and Docking Predictions
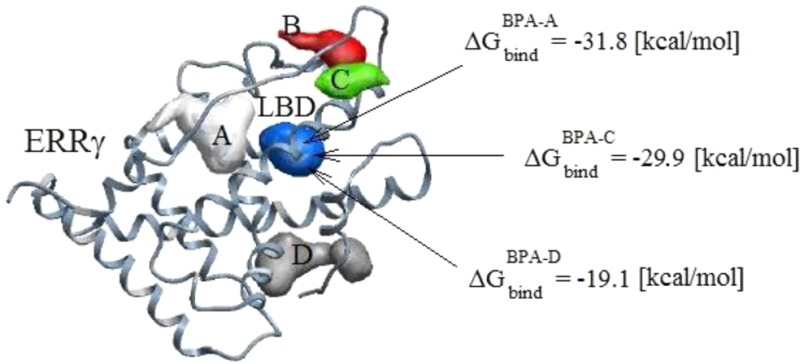
We used the MolSoft ICM-Pro20 software package to predict the binding sites of BPA-A, BPA-C, and BPA-D to ERRγ. The rigid and flexible docking analysis was performed on the ligand/protein systems. The most probable docking configurations were found by implementing stochastic global optimization procedures.20 All ligands were found docking in a similar manner inside LBD. The predicted docking configurations are in agreement with experimental results and MD simulations, which will be described in the Ligand/Protein Intermolecular Interactions section. The ligand chemical structures are given in Figure 1, and the predicted binding pockets of BPA-A are shown in Figure 2.
Figure 1.
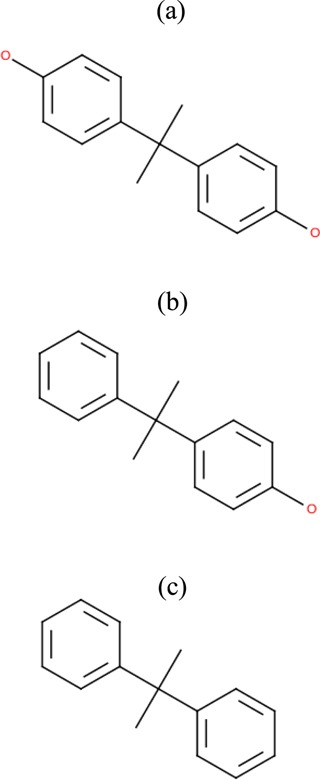
Chemical structures of (a) 4,4′-dihydroxy-2,2-diphenylpropane (BPA-A), (b) 4-α- cumylphenol (BPA-C), and (c) diphenylpropane (BPA-D).
Figure 2.
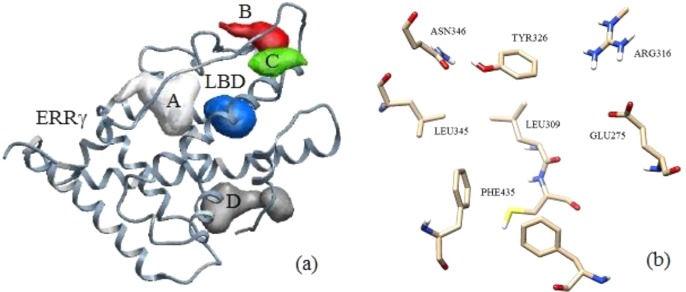
Human estrogen-related receptor γ (ERRγ) is shown with five predicted binding pockets that are marked as A, B, C, D, and ligand binding domain (LBD) (a). Zoomed in view of the LBD is shown with marked domain residues (b).
2.2. Molecular Dynamics Simulation
Molecular dynamics simulations can provide valuable insights into the structure and thermodynamics of complex biological systems. AmberTools21 have been used to set up ligand/protein/ion/water systems for our simulation. Specifically, the xleap setting tool was used to setup the ligand/protein dimer, Na+ counterions were added to make a system charge neutral, and TIP4P-Ew22 water molecules were added to solvate systems. The Amber 1121 software simulation package was used to carry out MD simulations using a cubic simulation cell to apply periodic boundary conditions. The Shake algorithm23 was employed to constrain the bond length and geometry of TIP4P-Ew water. Initially, all systems were minimized for 3000 steps to move the system from a higher energy state. Minimized systems were heated up by using a stepwise approach. First, solutions were heated up to 278 K for a period of 200 ps to ensure a smooth temperature transition, followed by heating up to 298 K. Heated systems were equilibrated in the isothermal–isobaric ensembles until an average pressure of 1 [Atm] and a density of ∼1 [g/cm3] were established. Production runs were carried out for over 25 ns. The cutoff radius of 8.5 Å was used for nonbonded and electrostatic interactions. The Velocity Verlet algorithm was implemented to solve the equations of motion. The temperature of the systems was maintained using a Langevin thermostat24 with a collision frequency of γ = 1.0 ps–1. Berendsen barostat25 was implemented for pressure control with a relaxation rate of τ = 2.0 ps–1. Charge–charge long-range electrostatic interactions were solved using the particle mesh Ewald approach.26 The free energy calculations were performed using the well established MM/PBSA method.27 Three ligand/water structures were analyzed via pairs of correlation functions, and the differences were compared. The ligand/protein binding structures were also analyzed by calculating probability distributions of distances between the aromatic carbons (CA) and oxygen atoms (O) of amino residues in the LBD.
2.3. Force Field Parameters
The force fields for molecular dynamics simulations of various compounds are under continuous improvements. There are nonpolarizable28,29 and polarizable30 force fields for simulation of proteins and organic molecules. One of the advanced force fields is AMOEBA31 as it employs the multipolar electrostatic model and induced atomic polarization. However, we choose the nonpolarizable Amber protein ff99SB32 force field for ERRγ and the GAFF33 force field using AM1-BCC partial charges for the phenyl based ligands to reduce computational time. The performance of the ff99SB force field was validated against available experimental data including structural and relaxation data that makes this force field a good model for the simulations of proteins.32
There are many different types of water models available for molecular dynamics simulations.34 These water models reproduce most of the physical properties well, but still there is a lot of room for improvement. One of the most advanced water models has been recently introduced that employs an electron density based concept.35 However, we chose the TIP4P-Ew (a modified transferrable intermolecular potential with 4 interaction sites and Ewald) water model to represent a solvent.22 This water model was selected among a number of nonpolarizable water potentials including TIP3P/TIP4P36 and SPC37 due to a better prediction (the reported error is less than 1%) of enthalpies of vaporization ΔHVAP, liquid densities ρ, and self-diffusion coefficients DW within the temperature ranging from 235.5 to 400 K. The previous studies of the ff99SB force field had also shown a better agreement with experimental NMR scalar couplings using the TIP4P-Ew in comparison with the TIP3P water model.38
2.4. MD Simulations of Ligand/Protein Aqueous Solutions
The reported coordinates from X-ray analysis of the BPA-A/ERRγ crystal structure were taken from the RCSB protein data bank as an initial input where X-ray calculations were performed with a 1.6 Å resolution.39 All water and glycerol (precipitant) molecules were deleted from the original pdb file. We set up BPA-X/ERRγ/Na+, ERRγ/Na+/TIP4P-Ew, and BPA-X/ERRγ/Na+/TIP4P-Ew systems at 298 K where X stands for A, C, and D. ERRγ was taken as a monomer to reduce computational cost. No homodimer simulations were performed. Eleven Na+ ions were added to the system to make it charge neutral. Molecular dynamics simulations were performed for BPA-X/ERRγ/Na+ sets in vacuum. Aqueous solutions comprised BPA-X/ERRγ, sodium ions, and water molecules with a total of ∼45000 atoms. Molecular dynamics simulations were also performed for the ERRγ/Na+/TIP4P-Ew system for a comparison of ligand in/out effects on protein structure.
2.5. Thermodynamics of Ligand/Protein Binding
There are several methods that can be implemented to calculate the binding free energy including thermodynamic integration (TI) and free energy perturbation (FEP) methods.40,41 These methods are accurate and rigorous but computationally expensive due to explicit solvent calculations.42 We employed the continuum MM/PBSA method,27 which was implemented in the Amber 11 simulation package. A thermodynamic cycle associated with ligand solvation and binding free energy was well-defined in previous publications.42 The free energy of ligand/protein binding in the cycle can be evaluated by eq 1:
| 1 |
where ΔGgasLP is dimer (ligand/protein) free energy in the gas phase, ΔGsolv is free energy of solvation for the ligand (L) and protein (P) complex, while ΔGsolvL and ΔGsolv are free energy of solvation for a single ligand and protein in solution, respectively. TΔSconf is the configuration entropy term. Each term should be further decomposed according to the Poisson–Boltzmann continuum approach where free energy of solvation can be estimated as a sum of polar and nonpolar terms as indicated by eq 2:
| 2 |
Free energy of solvation is a sum of energies due to electrostatic interactions ΔGsolvele and nonpolar interactions ΔGsolv. Free energy change due to electrostatic interactions can be solved by implementing the Poisson–Boltzmann approach. The free energy change due to nonpolar interactions can be solved using a fitting function as indicated by eq 3:
| 3 |
where SASA is a solvent accessible surface area of atom type i, and γ and α are adjustable parameters. The adjustable parameter γ represents a surface tension, and it has units of kcal/mol/Å2, while α represents a constant that does not include hydration effects due to changes in surface accessible area. Entropy contribution has not been considered while calculating free energies of binding.
3. Results and Discussion
3.1. Ligand Docking
The chemical structures of three ligands are shown in Figure 1. Ligand binding can be divided into specific and nonspecific binding. Specific binding can be referred to as the binding of a ligand to the LBD, and it was found to be ∼80% of the total ligand binding.16,17 X-ray crystal structure of ERRγ was used to predict binding pockets.39 Five potential binding pockets were predicted as shown in Figure 2a. Four binding pockets are marked as A, B, C, and D. Another binding pocket is marked as LBD. The LBD consists of Phe435, Leu345, Tyr326, Arg316, and Glu275 amino residues as shown in Figure 2b. Docking analysis has shown that the binding energies in those pockets are significantly higher than the binding energies in LBD. Therefore, the most stable protein/ligand complex is formed when we dock BPA based derivatives inside the LBD. All other potential binding pockets have not been considered for further binding optimization search and docking calculations. We find that binding configurations for BPA based ligands are similar to the configurations reported in refs (19 and 39). These binding configurations correspond to the lowest binding energies as calculated by the docking approach. We have found that the most energetically favorable configuration for BPA-C is that when it forms a hydrogen bond with the Glu275 amino residue and not with Asn346. Performing docking analysis on the phenol shows a similar tendency by preferentially making a hydrogen bond with the Glu275 residue. As a result of this docking analysis, we conclude that the most energetically favorable binding site is the Glu275 residue. BPA-D is docked in a similar orientation as BPA-A and BPA-C ligands inside LBD having no hydrogen bonds with domain residues. The details of these calculations are further discussed in sections 3.3–3.5.
3.2. Ligand Solvation Thermodynamics
We began our studies with determining the solvation energetic penalties associated with ligand/protein binding. The SASA and free energies of solvation are vital characteristics of the organic compounds especially in pharmaceutical drug design that incorporates structure/energy relationships. Ligand/protein binding is a multistep process that involves ligand solvation and binding steps. Each step is associated with a free energy change. The corresponding ligand free energies of solvation were calculated and compared with available results from MD simulations and experimental data. We also considered the free energy of solvation for benzene and phenol as a reference, having about a two times smaller SASA than BPA based compounds. Therefore, we have calculated the free energy of solvation for benzene, phenol, and BPA based compounds for comparison.
The free energy of solvation results employing MM/PBSA method are given in Table 1. MM/PBSA calculations resulted in ∼1.5 kcal/mol more favorable energy of solvation for the benzene and ∼1.9 kcal/mol for the phenol in comparison with those from the experiment.43 Free energy of solvation for 2,2-diphenylpropane (ΔG = −3.06 kcal/mol) is comparable with the free energy of solvation for biphenyl (ΔG = −2.66 kcal/mol). Addition of another aromatic ring and two methyl groups to the benzene resulted in a 0.7 kcal/mol energy gain, which is less than a 1.7 kcal/mol energy gain for biphenyl compared to that of benzene. Free energy of solvation for 4-α-cumylphenol is ΔG = −11.59 kcal/mol, which is close to the experimental value of ΔG = 12.00 kcal/mol for 1,4-dihydroxybenzene.44 On the basis of the assumption of additivity, the free energy of solvation for BPA-A should be around ∼15.68 kcal/mol. The free energy of BPA-A in this study is ΔG = −14.63 kcal/mol.
Table 1. Dipole moments (μ), SASA, and Free Energies of Ligand Solvation (ΔGsolv) at 298 Ka.
| ligand | μ (D) | SASA (Å2) | ΔGsolv (kcal/mol) |
|---|---|---|---|
| benzene (BNZ) | 0.00 | 135.86 | –2.35 ± 0.1 |
| –1.93b | |||
| biphenyl | 0.00 | 243.41 | –2.64b |
| 2,2-diphenylpropane (BPA-D) | 0.22 | 337.93 | –3.06 ± 0.20 |
| phenol (PHN) | 1.35 | 147.21 | –8.43 ± 0.25 |
| 1,4-dihydroxybenzene | 0.00 | 158.58 | –9.60b |
| 3,5-xylenol | 1.17 | 211.44 | –5.45b |
| 4,4-biphenol | 2.54 | 265.18 | |
| 2,2-bis(4-hydroxyphenyl) propane (BPA-A) | 2.31 | 359.42 | –14.63 ± 0.87 |
| 4-α-cumylphenol (BPA-C) | 1.46 | 348.84 | –11.59 ± 0.82 |
Notes: Measurements of SASA were obtained from http://www.chemicalize.org. Calculations of dipole moments were performed using the B3LYP/6-31G(d) level of theory.
Ref (44).
We further analyzed radial distribution functions for (CA-O) and (H-O) atomic pairs to study the effect of solvent accessible surface area and a number of hydroxyl groups on ligand solvated structures. The comparison of ligand/water solvation structures for benzene, phenol, BPA, and its derivatives is given in Figure 3. Despite having larger solvent accessible surface areas for BPA and its derivatives, the probability of finding water molecules around aromatic rings is significantly less than that for a single benzene or phenol compounds; see Figure 3a. This effect can be attributed to the spatial arrangements of aromatic rings on BPA based ligands. Aromatic rings are stabilized through π–π and CH-π hydrophobic interactions corresponding to their lowest energy conformation as was predicted from quantum chemistry calculations.
Figure 3.

(a) Solute/solvent radial distribution functions between the aromatic carbon and oxygen of water, (b) the hydrogen of hydroxyl group and oxygen of water, and the oxygen of hydroxyl group and hydrogen of water.
Substitution of aromatic hydrogen with a hydroxyl group has a minor effect on ligand/water solvation structures; see Figure 3a. Fewer number of water molecules can be found in the first hydration shell for phenol and BPA-A. These results well associate with previous studies, as hydrophobic solutes tend to have more water molecules in the first hydration shell due to water ordering.45 We have also performed hydrogen bond analysis for the phenol, BPA-A, and BPA-C ligands; see Figure 3b. No effects on hydrogen bonding were found due to the change in the solvent accessible area or a number of hydroxyl groups.
3.3. Ligand/Protein Intermolecular Interactions
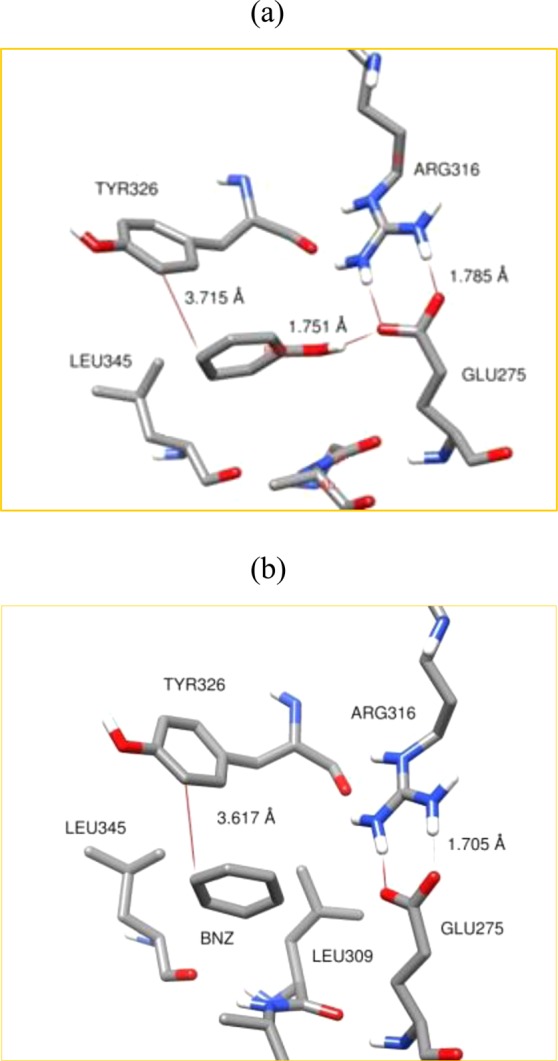
The LBD of ERRγ mainly comprised Glu, Arg, Asn, Tyr, Leu, and Phe residues as discussed in section 3.1. Glu, Arg, and Asn amino acid residues can be considered as hydrophilic, having COO– or NH2+ side groups. The Tyr side group is neutral having one phenyl ring with an attached hydroxyl (−OH) group. Phe and Leu residues are hydrophobic due to the presence of an aromatic ring and two methyl groups, respectively. Binding structures obtained from flexible docking analyses and MD simulations are in agreement with experimental results. Intermolecular interactions between ligand and constituent residues of the LBD are shown in Figure 4. These intermolecular interactions can be characterized as hydrophilic or hydrophobic interactions. Hydrophilic interactions come from the formation of hydrogen bonds between hydroxyl groups of the phenyl rings and Glu275, Arg316, and Asn346 amino residues. The hydroxyl group of BPA-A forms a hydrogen bond with oxygen atoms of Glu275 (1.816 Å), and the Arg316 also forms a hydrogen bond with Glu275; see Figure 4a. Another hydroxyl group makes a hydrogen bond with the Asn346 protein residue at a distance of 1.698 Å. Similar intermolecular interactions can be seen for BPA-C. It is also hydrogen bonded to hydrophilic residues Arg316 and Glu275; see Figure 4b. It should be noticed that Arg316 and Glu275 are mutually hydrogen bonded despite the presence of a ligand. Binding of the ligands to Arg316 and Glu275 does not disturb the hydrogen bond network between those protein residues. However, the Asn346 residue moves toward the hydroxyl group of BPA-A upon binding. The hydroxyl group of Tyr326 residues interchangeably forms hydrogen bonds with the oxygen of Asn326 or the oxygen of the hydroxyl group on the aromatic ring of BPA-A.
Figure 4.
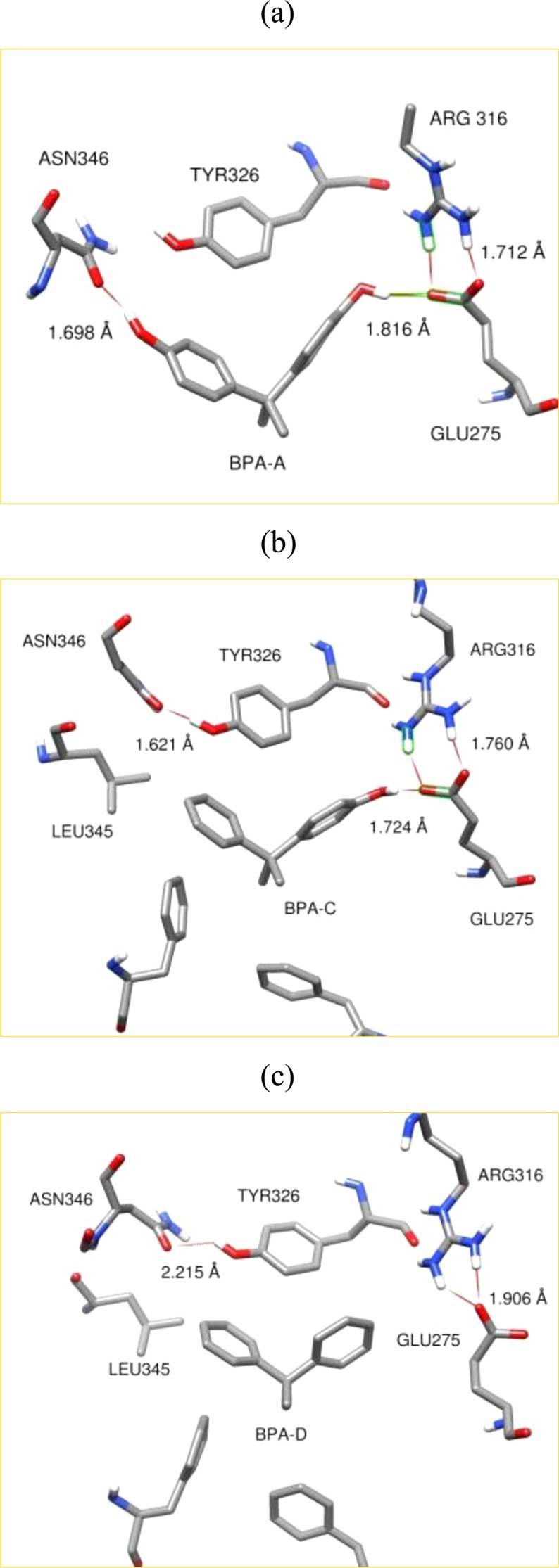
Comparison of intermolecular interactions of BPA and its derivatives with the receptor residues inside the LBD. Intermolecular interactions of BPA-A (a), BPA-C (b), and BPA-D (d) with the receptor residues inside the LBD.
Hydrophobic interactions of BPA-A and BPA-C involve π–π and CH3–π intermolecular interactions. These interactions include interactions of Tyr326 and Phe435 residues with aromatic rings (π–π) and a methyl group on sp3 carbon (CH3–π) of the ligands, respectively. Leu309 (not shown) and Leu345 residues are also involved in CH3–π hydrophobic interactions giving additional intermolecular stability for BPA based ligands. BPA-D was found to align in a similar manner as BPA-A and BPA-C inside the LBD; see Figure 4c. No reorientation of BPA-D was observed during the simulation runs. This particular alignment has been kept due to hydrophobic interactions only including π–π and CH3–π interactions. Therefore, a combination of hydrogen bonding, π–π, and CH3–π interactions are responsible for the binding and stability of BPA-A and BPA-C ligands, while BPA-D is bound through hydrophobic interactions only.
We also performed studies of binding configurations for the phenol and benzene rings. These binding configurations are given in Figure 5. We can see that the phenol ring closely resembles binding configurations of BPA-A and BPA-C within the LBD; see Figure 5a. The binding of the phenol ring also comprised of hydrophobic and hydrophilic interactions. A hydroxyl group on the phenol ring makes hydrogen bonds with Glu275 and Arg316 residues without disturbing their hydrogen bond network. The benzene molecule is found to be well fitted in the LBD in a manner similar to that of the phenol ring; see Figure 5b. It is caged by the hydrophobic constituents of the LBD. However, docking results show that the most favorable position of benzene resembles the position of a phenol ring. Phenol and benzene rings are also stabilized through the π–π and CH3–π hydrophobic interactions. Similar results are obtained from molecular dynamics simulations. No ligand/solvent exchange was observed during the simulation runs.
Figure 5.
Comparison of intermolecular interactions of phenol (a) and benzene (b) with the receptor residues inside the LBD.
We further analyzed ligand/protein interactions by calculating intermolecular distances between aromatic carbons (CA), hydrogen of hydroxyl groups (H), and oxygen atoms (O) of ligand binding constituents. The resulting distances were fitted to the Gaussian distribution functions for each ligand. These distributions are summarized in Figure 6. Probability distributions of intermolecular distances are obtained between ligands and constituents of the LBD; see Figure 6a. The most probable CA-O distance for BPA-A is 3.75 Å. We can clearly see the change in ligand binding due to the dehydroxylation of BPA-A on one side. The most probable CA-O intermolecular distance for BPA-C is 3.5 Å, which is shorter than the distance for BPA-A. These results show tighter intermolecular structure for BPA-C than for BPA-A having narrower distribution, which is comparable with the distribution for the phenol binding. These results also well correlate with distributions for hydrogen bonds; see Figure 6b. Shorter hydrogen bond distances correspond to the phenyl and BPA-C binding. Broader distribution for hydrogen bond lengths is obtained for BPA-A. The most probable hydrogen bond distance is also shifted to the longer distance of ∼1.770 Å. The most evident effect on intermolecular structure is observed on the benzene and BPA-D. Intermolecular interactions are shifted to the longer distances of 3.9 and 4.1 Å. Wider distributions are also obtained for the benzene and BPA-D in comparison with distributions for BPA-A, BPA-C, and phenol ligands. These intermolecular interactions can be found in a range between 2.1 Å to 5.5 Å indicating a dominance of weak hydrophobic interactions within the LBD. It can be concluded that BPA-A is moving more freely within the LBD than phenol or BPA-C ligands despite having two hydroxyl groups. Having no hydroxyl groups will more likely result in the displacement of a ligand from LBD due to ligand/solvent dissociation.
Figure 6.

Distribution distances between aromatic carbons and oxygen atoms of residues inside the LBD of ERRγ (a). Distribution of the hydrogen bond distances inside the LBD of ERRγ for BPA-A, BPA-C, and phenol compounds (b).
3.4. Ligand/ERRγ and ERRγ Structures
Experimental results have shown that the ERRγ receptor is kept in active conformation upon binding of the BPA-A and BPA-C ligands.19 We have superimposed protein structures obtained from molecular dynamics simulations and the experiment structure for the BPA-A/ERRγ complex. The position and conformation of helix 12 was conserved after a 25 ns run. These results support the experimental observations that the ERRγ is preserved in a transcriptionally active state upon the binding of ligands. The binding of BPA-C and BPA-D did not have any observable effects on the position of helix 12.
3.5. Free Energy of Ligand/Protein Binding
We calculated free energies of binding for benzene, phenol, and BPA based ligands. All binding energies were found to be favorable despite variations in the ligand chemical structure. Inhibitor concentrations of BPA-A and BPA-C were determined as 9.78 ± 0.87 nM and 10.60 ± 0.87 nM from experiments.17 No IC50 concentrations were detected for the phenol or BPA-D and phenol compounds indicating very low binding affinity. We converted experimental IC50 concentrations to the free energies of binding using the Cheng–Prusoff equation.46 We also include binding free energies obtained from docking analysis for comparison. Results of these calculations are given in Table 2.
Table 2. Free Energies of Ligand/Protein Binding at T = 298 Ka.
| MM/PBSAb |
ICMc | experimentd | |||
|---|---|---|---|---|---|
| kcal/mol |
|||||
| ligand/protein | ΔGbind | ΔGg | ΔGg | ΔGbind | IC50e (nM) |
| BNZ/ERRγ | –14.26 ± 1.32 | –17.93 ± 1.43 | –25.97 | ND | |
| PHN/ERRγ | –16.49 ± 1.97 | –25.57 ± 2.12 | –32.27 | ND | |
| BPA-A/ERRγ | –31.82 ± 2.04 | –53.30 ± 2.12 | –65.35 | –10.88 | 9.78 ± 0.87 |
| BPA-C/ERRγ | –29.88 ± 2.28 | –52.59 ± 2.12 | –63.51 | –10.83 | 10.6 ± 0.87 |
| BPA-D/ERRγ | –19.14 ± 1.87 | –36.49 ± 1.73 | –53.79 | ND | |
3.5.1. Ligand/Protein Binding in Vacuum
Calculated free energies of ligand/protein binding (ΔGg) are given in Table 2 for the MM/PBSA and ICM methods. The most favorable free energy of binding was calculated for the BPA-A/protein complex that would correspond to the maximum binding affinity. It can be seen that the free energy of binding for BPA-C is only 1 kcal/mol less favorable than the free energy of binding for BPA-A. Significant reduction in the free energy of binding can be seen for BPA-D. According to the gas phase calculations, free energy of binding is 17 kcal/mol less favorable in comparison with the free energy of BPA-A. A more pronounced energy change can be seen for the phenol and benzene rings. Free energies of binding for the phenol and benzene are around 30 kcal/mol less favorable than those for BPA-A. As a result, the order of the gas phase free energy of binding is ΔGBPA-A < ΔGBPA-C < ΔGBPA-D < ΔGPHN < ΔGBNZ. Similar results are obtained from molecular docking analysis. However, the binding energies are ∼10 kcal/mol more favorable in comparison with energies obtained from MM/PBSA calculations.
3.5.2. Ligand/Protein Binding in Solvent
Binding free energies (ΔGbind) in aqueous solutions are also found to be favorable for all the compounds; see Table 2. The energy of binding is more favorable for BPA-A and BPA-C ligands in comparison with that of BPA-D. However, binding energies are less favorable, by a factor of 2, in comparison with free energies from the gas phase. This difference arises due to the ligand/solvent and protein/solvent interactions. It can be seen that the order of free energy of binding is conserved for both states. Previous studies have shown a significant change in free energies of binding associated with water displacement from the LBD.47 It was ensured that there is no water in the LBD for our calculations. Therefore, the free energy change associated with the displacement of water was not taken into account.
4. Conclusions
Ligand/protein flexible docking and MD simulations provide unique insights into the effects of the hydroxylation of phenyl based ligands on the solvation and thermodynamics of ligand/ERRγ binding. The hydrophilic/hydrophobic intermolecular interactions, solvent accessible surface areas, molecular structure, and the number of hydroxyl groups affect the solvation and binding thermodynamics of BPA and its derivatives. The free energies of binding for BPA-A and BPA-C were the most favorable among all ligands studied. The binding energy for BPA-D was found to be comparable with the free energy of binding for the phenol ring. Our simulation results show that the most stable ligand/protein structure is BPA-A/ERRγ. Its binding energy is 1 kcal/mol more favorable than that for BPA-C/ERRγ and 12.7 kcal/mol more than that for BPA-D/ERRγ.
Acknowledgments
We acknowledge LONI for providing us with HPC allocation time. We thank Drs. R. M. Uppu and S. Babu for some very helpful discussions.
Glossary
Abbreviations
- ERRγ
estrogen related receptor γ
- BPA-A
4,4′-dihydroxy-2,2-diphenylpropane
- BPA-C
4-α-cumylphenol
- BPA-D
2,2-diphenylpropane
- LBD
ligand binding domain
- MM/PBSA
molecular mechanics/Poisson–Boltzmann surface area
- ER
estrogen receptor
- IC50
maximum inhibitory concentration
- Phe
phenylalanine
- Tyr
tyrosine
- Leu
leucine
- Glu
glutamic acid
- Arg
arginine
- Asn
asparagine
- OH
hydroxyl group
- CH
alkane bond
- COO–
carboxylate group
- COOH
carboxyl group
- NH2+
amine group
- π
bond
- Amber
assisted model building with energy refinement
- MD
molecular dynamics
- TIP4P-Ew
transferrable interaction potential with 4 points and Ewald
- TIP3P
transferrable interaction potential with 3 points
- SPC
simple point charge
- CA
aromatic carbon
- O
oxygen atom
- AMOEBA
atomic multipole optimized energetics for biomolecular applications
- GAFF
general Amber force field
- NMR
nuclear magnetic resonance
- TI
thermodynamic integration
- FEP
free energy perturbation
- SASA
solvent accessible surface area
- H-12
helix 12
This work was partially supported by grants from the National Center for Research Resources (5P20RR016456-11) and the National Institute of General Medical Sciences (8 P20GM103424-11) from the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Calafat A. M.; Ye X.; Wong L. Y.; Reidy J. A.; Needham L. L. (2008) Exposure of the US opulation to bisphenol a and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 116, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E.; Bourguignon J. P.; Giudice L. C.; Hauser R.; Prins G. S.; Soto A. M.; Zoeller R. T.; Gore A. C. (2009) Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr. Rev. 30, 293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi S.; Tokunaga T.; Liu X.; Okada H.; Matsushima A.; Shimohigashi Y. (2006) Endocrine disruptor bisphenol a strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol. Lett. 167, 95–105. [DOI] [PubMed] [Google Scholar]
- Arase S.; Ishii K.; Igarashi K.; Aisaki K.; Yoshio Y.; Matsushima A.; Shimohigashi Y.; Arima K.; Kanno J.; Sugimura Y. (2011) Bisphenol a exposure during adulthood alters expression of aromatase and 5α-reductase isozymes in rat prostate. Biol. Reprod. 84, 734–742. [DOI] [PubMed] [Google Scholar]
- Hong H.; Yang L.; Stallcup M. R. (1999) Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. J. Biol. Chem. 274, 22618–22626. [DOI] [PubMed] [Google Scholar]
- Nilsson S.; Makela S.; Treuter E.; Tujague M.; Thomsen T. J.; Andersson G.; Petterson K.; Warner M.; Gustafsson J. (2001) Mechanisms of estrogen action. Physiol. Rev. 81, 1535–1565. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Mani S. (2010) Orphan nuclear receptors as targets for drug development. Pharm. Res. 27, 1439–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère V. (2002) To ERR in the estrogen pathway. Trends Endocrinol. Metab. 13, 220–225. [DOI] [PubMed] [Google Scholar]
- Hentschke M.; Süsens U.; Borgmeyer U. (2002) Domains of ERRγ that mediate homodimerization and interaction with factors stimulating DNA binding. Eur. J. Biochem. 269, 4086–4097. [DOI] [PubMed] [Google Scholar]
- Greene G. L. (2003) In vivo imaging reveals estrogen receptor’s hidden personality. Nat. Med. 9, 22–23. [DOI] [PubMed] [Google Scholar]
- Hashimoto S.; Okada K.; Imaoka S. (2008) Interaction between bisphenol derivatives and protein disulphide isomerase (PDI) and inhibition of PDI functions: Requirement of chemical structure for binding to PDI. J. Biochem. 144, 335–342. [DOI] [PubMed] [Google Scholar]
- Hu P.; Kinyamu H. K.; Wang L.; Martin J.; Archer T. K.; Tend C. T. (2008) Estrogen induces estrogen-related receptor alpha gene expression and chromatin structural changes in estrogen receptor (ER)-positive and ER-negative breast cancer cells. J. Biol. Chem. 283, 6752–6763. [DOI] [PubMed] [Google Scholar]
- Greschik H.; Wurtz J. M.; Sanglier S.; Bourguet W.; Dorsselaer A.; Moras D.; Renaud J. P. (2002) Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol. Cell 9, 303–313. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zuercher W. J.; Consler T. G.; Lambert M. H.; Miller A. B.; Orband-Miller L. A.; McKee D. D.; Willson T. M.; Nolte R. T. (2006) X-ray crystal structures of the estrogen-related receptor-γ ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J. Biol. Chem. 281, 37773–37781. [DOI] [PubMed] [Google Scholar]
- Coward P.; Lee D.; Hull M. V.; Lehmann J. M. (2001) 4-hydroxytamoxifen binds to and deactivates the estrogen-related receptor γ. Proc. Natl. Acad. Sci. U.S.A. 98, 8880–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima A.; Kakuta Y.; Teramoto T.; Koshiba T.; Liu X.; Okada H.; Tokunada T.; Kawabata S.-i.; Kimura M.; Shimohigashi Y. (2007) Structural evidence for endocrine disruptor bisphenol a binding to human nuclear receptor ERRγ. J. Biochem. 142, 517–524. [DOI] [PubMed] [Google Scholar]
- Okada H.; Tokunaga T.; Liu X.; Takayanagi S.; Matsushima A.; Shimohigashi Y. (2008) Direct evidence revealing structural elements essential for the high binding ability of bisphenol a to human estrogen-related receptor-γ. Environ. Health Perspect. 116, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad M. C.; Askari H.; O’Neill J.; Klinger A. L.; Milligan C.; Lewandowski F.; Springer B.; Spurlino J.; Rentzeperis D. (2008) Structural determination of estrogen-related receptor γ in the presence of phenol derivative compounds. J. Steroid Biochem. Mol. Biol. 108, 44–54. [DOI] [PubMed] [Google Scholar]
- Matsushima A.; Teramoto T.; Okada H.; Liu X.; Tokunaga T.; Kakuta Y.; Shimohigashi Y. (2008) ERRγ tethers strongly bisphenol a and 4-α-cumylphenol in an induced-fit manner. Biochem. Biophys. Res. Commun. 373, 408–413. [DOI] [PubMed] [Google Scholar]
- http://www.molsoft.com. The ICM-Pro v.3.8 software was used in all of our ligand/protein dockings.
- Case D. A., Darden T. A., Cheatham T. E. I., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Wang B., Hayik S., Roitberg A., Seabra G., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., and Kollman P. A. (2010) AMBER 11, University of California, San Francisco, CA, http://ambermd.org/doc11/Amber11.pdf.
- Horn H. W.; Swope W. C.; Pitera J. W.; Madura J. D.; Dick T. J.; Hura G. L.; Head-Gordon T. (2004) Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- Ryckaert J. P.; Cicotti G.; Berendsen H. J.C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Grest G. S.; Kremer K. (1986) Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 33, 3628–3631. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh ewald: An n-log(n) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Srinivasan J.; Cheatham T. E.; Cieplak P.; Kollman P. A.; Case D. A. (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate–DNA helices. J. Am. Chem. Soc. 120, 9401–9409. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179. [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. (1996) Development and Testing of the OPLS All-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. [Google Scholar]
- Starovoytov O. N.; Borodin O.; Bedrov D.; Smith G. D. (2011) Development of a polarizable force field for molecular dynamics simulations of poly (ethylene oxide) in aqueous solution. J. Chem. Theory Comput. 7, 1902–1915. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; Schnieders M. J.; Haque I.; Mobley D. L.; Lambrecht D. S.; DiStasio R. A. Jr.; Head-Gordon M.; Clark G. N.; Johnson M. E.; Head-Gordon T. (2010) Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Stockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Bioinf. 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Guillot B. (2002) A Reappraisal of what we have learnt during three decades of computer simulations on water. J. Mol. Liq. 101, 219–260. [Google Scholar]
- Duke R. E.; Starovoytov O. N.; Piquemal J. P.; Cisneros G. A. (2014) GEM*: A molecular electronic density-based force field for molecular dynamics simulations. J. Chem. Theory Comput. 10, 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., Van Gansteren W. F., and Hermans J. (1981) Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces (Pullman B., Ed.) p 331, Reidel Publishing Co., Dordrecht, The Netherlands. [Google Scholar]
- Wickstrom L.; Okur A.; Simmerling C. (2009) Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys. J. 97, 853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima A.; Kakuta Y.; Teramoto T.; Koshiba T.; Liu X.; Okada H.; Tokunaga T.; Kawabata S.; Kimura M.; Shimohigashi Y. (2007) Crystal structure of human estrogen-related receptor gamma ligand binding domain complex with bisphenol A. J. Biochem. 142, 517–524. [DOI] [PubMed] [Google Scholar]
- Mezei M.; Beveridge D. L. (1986) Free energy simulations. Ann. N.Y. Acad. Sci. 482, 1–23. [DOI] [PubMed] [Google Scholar]
- Kollman P. (1993) Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 93, 2395–2417. [Google Scholar]
- Homeyer N.; Gohlke H. (2012) Free energy calculations by the molecular mechanics poisson–boltzmann surface area method. Mol. Inf. 31, 114–122. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Huo S.; Lee M.; Kollman P. A. (2001) Solvation model based on weighted solvent accessible surface area. J. Phys. Chem. B 105, 5055–5067. [Google Scholar]
- Fennell C. J.; Dill K. A. (2011) Physical modeling of aqueous solvation. J. Stat. Phys. 145, 209–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Gao J.; Ravimohan C. (1985) Monte Carlo simulations of alkanes in water: Hydration numbers and the hydrophobic effect. J. Phys. Chem. 89, 3470–3473. [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Michel J.; Tirado-Rives J.; Jorgensen W. L. (2009) Energetics of displacing water molecules from protein binding sites: Consequences for ligand optimization. J. Am. Chem. Soc. 131, 15403–15411. [DOI] [PMC free article] [PubMed] [Google Scholar]