

Abstract
Menin is an essential co-factor of oncogenic MLL fusion proteins and the menin-MLL interaction is critical for development of acute leukemia in vivo. Targeting the menin-MLL interaction with small molecules represents an attractive strategy to develop new anticancer agents. Recent developments, including determination of menin crystal structure and development of potent small molecule and peptidomimetic inhibitors, demonstrate feasibility of targeting the menin-MLL interaction. On the other hand, biochemical and structural studies revealed that MLL binds to menin in a complex bivalent mode engaging two MLL motifs, and therefore inhibition of this protein-protein interaction represents a challenge. This review summarizes most recent achievements in targeting the menin-MLL interaction as well as discusses potential benefits of blocking menin in cancer.
Menin as an oncogenic co-factor of MLL fusion proteins in leukemia
Chromosomal rearrangements of the MLL gene located at chromosome band 11q23 are found in patients with de novo acute myeloid (AML) and acute lymphoblastic (ALL) leukemias [1, 2], and in therapy related leukemias or myelodysplastic syndrome (MDS) [3]. As a consequence of chromosomal translocations, the MLL gene is fused with one of over 60 different protein partners [4], such as the most frequent AF4, AF9, ENL, AF6, ELL, and AF10 (Figure 1A) [5, 6]. Disruption of MLL by gene fusions upregulates expression of HOXA9 and MEIS1 genes that are critical to leukemogenesis [7–10]. The role of HOXA genes in leukemic transformation has been verified in both, in vitro and in vivo models [11–13], demonstrating that MLL fusion protein mediated upregulation of HOXA9 and MEIS1 genes results in enhanced proliferation and blockage of hematopoietic differentiation, ultimately leading to acute leukemia [7, 14, 15]. Patients with leukemias harboring MLL translocations have very unfavorable prognosis (20% event free survival at 3 years) and respond poorly to available treatments [16], demonstrating a clear need for new therapies.
Figure 1.
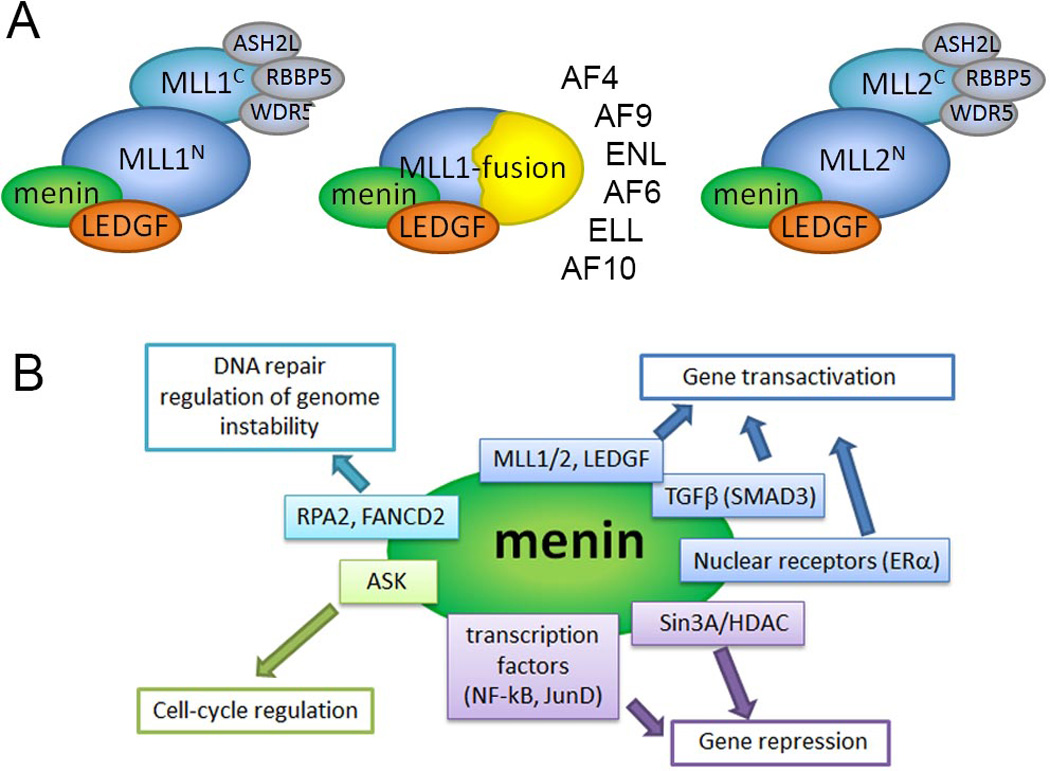
Menin is involved in a diverse network of protein-protein interactions. A. Cartoon showing menin as a conserved component of MLL1, MLL fusion and MLL2 complexes. The most common MLL fusion partners are listed. B. Major classes of menin binding partners and their function. For simplicity, only selected proteins are shown. More extensive network of menin interactions is reviewed elsewhere [60–62].
The oncogenic function of MLL fusion proteins is critically dependent on their direct interaction with menin [17, 18]. Menin is a 67 kDa protein encoded by the MEN1 (Multiple Endocrine Neoplasia I) gene localized on chromosome 11q13 [19]. Menin is an ubiquitously expressed protein, predominantly localized in the nucleus [20]. Menin directly binds to the N-terminus of MLL [17, 21, 22] that is retained in all MLL fusion proteins and plays an important role in recruitment of MLL and MLL fusions to target genes, including HOXA9 [17, 18, 21, 23]. Loss of menin binding by MLL fusion proteins abolishes their oncogenic properties in vitro and in vivo [17, 21]. Mutations within the N-terminus of MLL-ENL oncoprotein, resulting in protein unable to associate with menin, abolish its potential to upregulate Hox gene expression and induce leukemia in mice [17]. Expression of a dominant-negative inhibitor composed of the amino terminal MLL sequence inhibits growth of the MLL-AF9 transformed bone marrow cells and blocks leukemogenic transformation [21]. Our group has recently developed potent small molecule inhibitors that bind to menin and disrupt its interaction with MLL fusion proteins [24, 25]. These compounds strongly inhibit proliferation and induce differentiation of MLL leukemia cells [24]. Overall these results emphasize that blocking the menin-MLL interaction might represent a viable approach to reverse the oncogenic activity of MLL fusion proteins in leukemia and may lead to novel therapeutics.
Menin as an integral component of MLL1 and MLL2 histone methyltransferase complexes
Biochemical studies revealed that menin interacts with trithorax family histone methyltransferases (HMT) MLL1 and MLL2 [23, 26]. MLL1 and MLL/2 function as large macromolecular complexes composed of more than 30 subunits, including several core components such as WDR5, PbBP5, Ash2L associated with HMT activity [23, 26–31]. Menin binds to the N-terminus of MLL and therefore it is found as a common component of the wild type MLL1, MLL2 as well as MLL1-fusion protein complexes (Figure 1A) [17, 23, 26]. Although the exact role of menin in these complexes is not known, multiple studies demonstrate that menin is required for the transcriptional activity of MLL1 and MLL2 most likely via facilitating their recruitment to target genes [17, 18, 21, 23, 32–34]. Menin is required for maintenance of homeotic genes regulated by MLL1 and MLL2, such as Hoxa9, Hoxc6, Hoxc8 [23, 26], and conditional menin knockout significantly reduces binding of MLL1 to the Hoxa9 locus [18]. Menin is required for MLL1 to bind to the p27Kip1 and p18Ink4c loci to induce expression of p27 and p18 cyclin-dependent kinase (CDK) inhibitors [32]. Recruitment of MLL1 to the GATA3 locus to regulate GATA3 expression and Th2 cytokine production also requires menin [33], strongly suggesting that menin plays broader role in recruitment of the methyltransferase complex to target genes. Mechanistically, menin might function to link MLL with the chromatin associated protein LEDGF (lens epithelium-derived growth factor) [35]. Functional studies revealed that LEDGF plays a role in co-localization of menin and wild type MLL1 or MLL fusions to relevant target genes such as Hox and CDKIs [35]. LEDGF is a component of both MLL1 and MLL2 complexes [36], and biochemical and structural studies demonstrated that menin simultaneously interacts with the N-terminus of MLL and the IBD domain of LEDGF [35, 37]. Considering that menin is an ubiquitously expressed nuclear protein, it is very likely that menin exists as an integral component of the MLL1 and MLL2 complexes, and is required for H3K4 methylation at target genes [38]. Genome-wide analysis found that menin and MLL1 co-localize to promoters of thousands of human genes but do not always bind together [39]. Despite multiple studies it is still not clear whether function of MLL1 and MLL2 is entirely dependent on menin. For example, it has been recently found that menin and MLL1 regulate distinct pathways and act independently during normal hematopoiesis [40].
Menin as a tumor suppressor in MEN1
Menin is a tumor suppressor, which directly controls cell growth in selected endocrine organs, including parathyroid, pancreatic islets, and the pituitary gland [41]. Mutations in MEN1 occur with an estimated frequency of one in 30,000 individuals, and more than 95 % of MEN1 patients develop tumors of the parathyroid glands, pancreatic islet cells and anterior pituitary gland by the fifth decade of life [42, 43]. MEN1 tumors are frequently associated with loss of heterozygosity (LOH) of MEN1 locus [20], and menin follows the classic “two-hit” model of tumor suppressor proposed by Knudson [44]. Studies of menin knockout further support the role of menin as a tumor suppressor [45, 46]. Mice with homozygous MEN1 deletion die at embryonic day 11.5 to 13.5 due to developmental defects. Heterozygous mice develop endocrine tumors, which closely resemble MEN1 syndrome observed in humans [45]. To date, more than 1336 mutations have been reported in patients with MEN1 syndrome [47]. Approximately 80% are nonsense mutations, deletions, insertions and splice-site mutations and 20% are missense mutations [47]. The majority of these mutations result in expression of truncated protein or affect protein stability and lead to rapid degradation [48, 49]. Furthermore, recent structural studies of menin revealed that MEN1 missense mutations are predominantly found in the hydrophobic core of menin and they likely destabilize the entire protein [37, 50]. Therefore, the majority of MEN1 mutations might result in loss of menin rather than impair menin function.
Menin is involved in multiple protein-protein interactions
Menin is an ubiquitously expressed nuclear protein [20] that is engaged in a complex network of interactions (Figure 1B). The best understood is the role of menin as a component of the MLL1 and MLL2 complexes that maintain gene expression by methylation of H3K4 [23, 26]. The interaction of menin with transcription factors such as JunD [51], NF-κB [52], SMAD3 [53] may lead to gene transactivation or repression (Figure 1B). Menin was reported to function as a co-repressor by recruiting histone deacetylases via mSin3A [54], and as a co-activator of nuclear receptors via linking them to MLL1 and MLL2 methyltransferases [55, 56]. Menin also interacts with the activator of S-phase kinase (ASK) involved in cell-cycle regulation [57], the DNA-damage-repair protein FANCD2 [58], and the replication protein A subunit RPA2 [59]. The high diversity among menin binding partners strongly suggests that menin functions to bridge various proteins and plays a role in multiple biological pathways, including cell growth regulation, cell cycle control, genomic stability, bone development and hematopoiesis. While the list of proteins interacting with menin is constantly growing [60–62], additional studies are necessary to validate these interactions and understand their biological implications.
Structural studies of menin reveal that menin is a “druggable” target
The need for structural characterization of menin has been emphasized in several studies [55, 63]. However, crystallization of menin turned out to be very challenging, and the structure of the full length protein has not been obtained to date. Menin is a highly conserved protein among species [64], but it is absent in the Nematodes and in yeasts [65]. The size of the human menin is 615 amino acids, and it varies between different homologs from 539 (Nematostella vectensis) to 763 (Drosophila melanogaster) amino acids. Bioinformatics analysis and NMR studies revealed that menin is a globular protein with several disordered fragments (Figure 2A) [66]. Truncations of these flexible fragments turned out to be essential for successful crystallization of menin. We determined the first crystal structure of menin from Nematostella vectensis by deleting a single internal disordered fragment and the C-terminus of the protein [50]. More recently, the crystal structure of human menin was reported, and in order to obtain diffraction quality crystals, it was necessary to delete a part of the internal flexible fragment [37]. By more extensive deletions of loop regions, we engineered human menin to yield crystals diffracting to high resolution and solved the structure at 1.3Å resolution [25].
Figure 2.
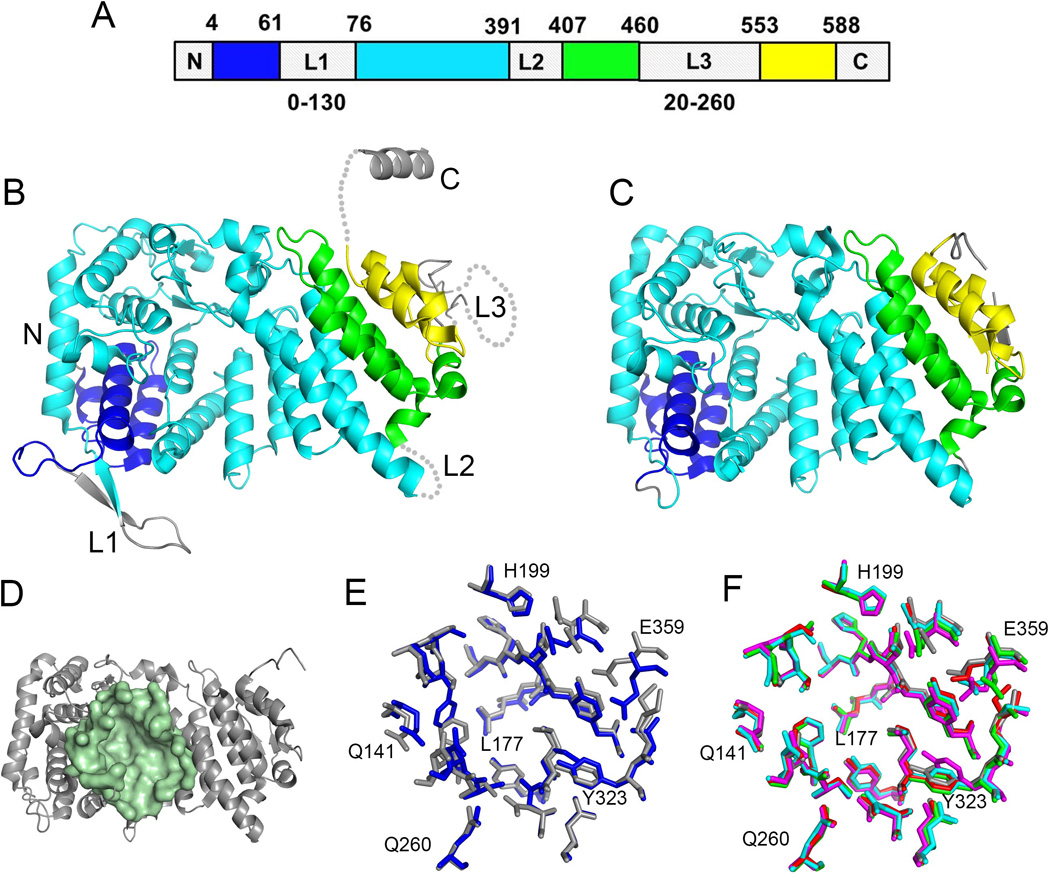
Structure of menin and the central cavity. A. Schematics of a sequence of human menin showing the location of elements of the folded core (structured segments are shown in blue, cyan, green and yellow) and location of three loops L1 to L3. The sequences and lengths of these loops vary significantly among menin homologs and the length for the most variable loops L1 and L3 is shown at the bottom. B. Crystal structure of human menin (PDB code 3U84) with color coding the same as in A. C. High resolution crystal structure of human menin obtained upon deletion or truncation of loops L1 – L3 and the C-terminus (4GPQ). D. Structure of human menin showing a central cavity; residues lining the cavity are shown in pale green. E. Superposition of residues in the central cavity of human menin (4GPQ, gray) and Nematostella menin (3RE2, blue). F. Comparison of conformations of residues lining the central cavity in apo menin (4GPQ, gray; 3U84, magenta) and menin fromthe complexes with bound protein ligands (4GQ6, red; 3U86, green; 3U85, cyan); selected residues are labeled.
Structural studies revealed that menin is predominantly an α-helical protein with the fold based on three tertatricopeptide (TRP) repeats (Figure 2B, C) [25, 37, 50]. Sequence alignment of menin homologs shows that the amino acid sequences of the structured core are highly conserved with sequence identity exceeding 50%. As a consequence, the structure of menin is highly conserved between human and even one of the most distant species like Nematostella [50]. To the contrary, the three loops and the C-terminus in menin can vary significantly between species and contain posttranslational modification sites (e.g. phosphorylation [67, 68], sumoylation [69]) as well as other motifs modulating protein function such as nuclear localization sites [20].
The very unique feature of meninis the presence of a large central cavity, which is formed by the TRP repeats and is enclosed by the helical subdomains (Figure 2D). The size of this cavity is striking as in human menin it reaches approximately 5,000 Å3 (calculated using CASTp) [70]. The central cavity is well defined and is lined predominantly by the side chains of hydrophobic and acidic amino acids [25]. The architecture of this cavity is highly conserved between Nematostella and human menins and likely among other species (Figure 2E). Mutational and structural studies indicate that in human menin the central cavity represents the binding site for the protein-protein interactions with MLL and JunD [24, 25, 37]. Comparison of the menin structures of free protein and in complex with MLL or JunD clearly shows the lack of conformational changes upon binding of large peptide ligands (Figure 2F). In summary, the central cavity on menin has well defined architecture, is relatively rigid and does not change conformation upon binding of protein ligands. Overall, these features distinguish the central cavity as a highly suitable site for targeting by small molecule compounds to inhibit menin interactions with MLL.
Characterization of the menin-MLL interaction
Biochemical characterization of the menin-MLL interaction revealed that relatively short N-terminal fragment of MLL encompassing RWRFP sequence is essential for binding to menin [17]. Subsequent studies established that the high affinity menin-MLL interaction requires a longer, approximately 46 amino acid N-terminal fragment of MLL [21]. Based on biophysical characterization of the menin-MLL interaction in our group we determined that MLL binds to menin with high affinity (Kd=10nM). Interestingly, we identified two short motifs in the N-terminus of MLL, which contribute to binding to menin (Figure 3A) [22]. The menin binding motif 1 (MBM1) encompasses MLL residues 6–13 and represents the high affinity menin binding motif (Kd=56nM). The second motif, MBM2, involving residues 24–40, binds to menin with lower affinity (Kd=1µM). Interestingly, MBM1 and MBM2 simultaneously bind to menin with negative cooperativity and they compete with each other for binding to menin [22]. This strongly suggests that both motifs bind to the proximal sites on menin [22]. Recent structural studies showed that MBM1 binds to the central cavity in menin in a U-shaped conformation and the complex is maintained by hydrophobic and electrostatic interactions [25, 37]. Alanine scanning mutagenesis mapped three hydrophobic residues in MBM1, F9, P10 and P13, which have the most significant contribution to the binding affinity [22], and side chains of these residues bind to well defined hydrophobic pockets on menin (Figure 3B, C).
Figure 3.
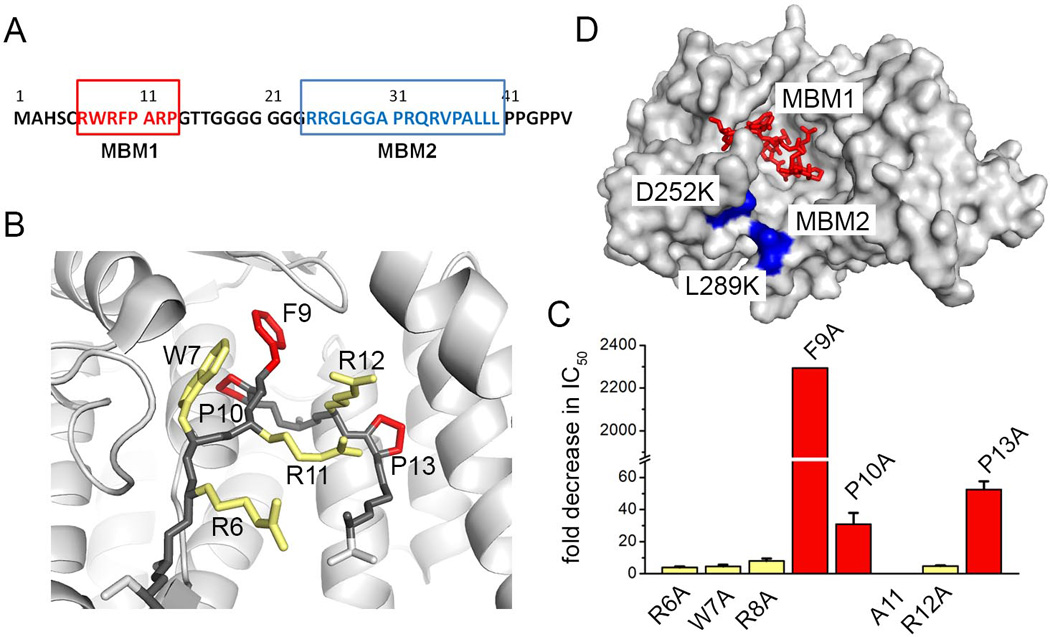
Mapping of the menin-MLL interaction. A. Sequence of MLL N-terminus with menin binding motifs MBM1 and MBM2 marked in red and blue, respectively. B. Structure of MBM1 bound to menin (PDB code 4GQ6); side chains of residues most critical for binding are red and less essential residues are yellow. C. Alanine scanning mutagenesis demonstrating the effect of mutations on inhibition of the menin-MBM1interaction (colors are the same as in B) [22]. D. Surface representation of menin with bound MBM1 (red) and mapped MBM2 site (blue).
The binding site for the lower affinity motif MBM2 is less understood. The MBM2 carries a strong positive charge due to the presence of four arginine residues (Figure 3A) and is likely involved in electrostatic interactions with the central cavity on menin, which is negatively charged [25]. Indeed, we identified two surface point mutations in menin, D252K and L289K (Figure 3D), which significantly reduce binding of MBM2 without affecting protein stability or the interaction with MBM1 [25]. Therefore, MBM2 may represent an electrostatic anchor which enhances the interaction of MBM1 with menin.
In addition to the well-characterized interaction of menin with the N-terminus of MLL, co-immunoprecipitation experiments revealed a possibility for additional, more distant and much weaker menin interacting motif within MLL, involving a region spanning residues 640–1251 of MLL [17]. Importantly, previous studies demonstrated that expression of the N-terminus of MLL encompassing residues 2–44 (corresponding to MBM1 and MBM2 fragments) is sufficient to inhibit growth of MLL-AF9 transformed cells [21]. These results strongly suggests that inhibition of the MBM1 and MBM2 is going to be sufficient to abolish activity of MLL fusion proteins and very likely the third motif is much less important.
MBM1 pocket on menin represents a major site for targeting by small molecule inhibitors
Targeting protein-protein interactions using small molecule inhibitors is a challenging task [71], and careful selection of binding site plays an essential role in development of potent small molecule inhibitors. The N-terminus of MLL binds to a very large central cavity on menin, and therefore blocking the entire MLL binding site with small molecules is not feasible. Structural studies revealed that part of the central cavity where MBM1 binds might represent the most promising and “druggable” site for inhibitor development. However, due to the complex mode of interaction involving two MLL fragments, it was necessary to establish whether targeting the MBM1 site alone is sufficient for efficient disruption of the menin-MLL complex. Studies from Yokoyama et al. provided the crucial evidence that MBM1 represents the hot-spot for disruption of the menin-MLL interaction, as deletion of the 5 amino acid motif RWRFP from the MLL-ENL fusion protein abrogates development of leukemia in vivo [17]. Alanine scanning mutagenesis identified the side chain of F9 as the most critical for high affinity interaction of MBM1 with menin, and the mutation of F9 to alanine in MBM1 has a very pronounced impact on the menin-MLL interaction. The MLL1–160 fragment bearing F9A mutation binds to menin with Kd=1.5µM, representing ~150 fold weaker affinity as compared to the wild type MLL1–160 [22]. Furthermore, the binding affinity of the MLL1–160 F9A mutant is comparable to MBM2 alone and the F9A mutation is consistent with nearly complete loss of MBM1 binding [22]. Using competition experiments we demonstrated that a peptide corresponding to the MBM1 fragment can efficiently disrupt the interaction of menin with MLL fragment encompassing both MBM1 and MBM2 [22]. Altogether, MBM1 constitutes a hot-spot for the menin-MLL interaction and represents the primary site for small molecule binding. Nevertheless, the presence of MBM2 represents a challenge in targeting the menin-MLL interaction as it will reduce the activity of any small molecule inhibitor solely targeting the MBM1 site.
Development of small molecules disrupting menin-MLL interaction
To date several small molecule inhibitors targeting the menin-MBM1 interaction have been reported (Figure 4) [24, 25, 72, 73]. We developed the first small molecule inhibitors of the menin-MLL interaction, which belong to the thienopyrimidine class. This class of compounds was identified by HTS (High Throughput Screening) using a competition-based fluorescence polarization assay with the fluorescein-labeled MBM1 peptide. The most potent compound identified in HTS, MI-1, inhibits the menin-MBM1 interaction with IC50=1.9 µM (Figure 4) [24]. Further medicinal chemistry optimization resulted in several analogs with improved activities, including MI-2 (Figure 4). Structural studies revealed that MI-2 binds to the central cavity on menin in the MBM1 pocket and closely mimics the key interactions of MBM1 with menin (Figure 5A). The aliphatic group on the thienopyrimidine ring binds to the pocket occupied by F9of MLL and the thiazoline moiety fits into the P13 pocket (Figure 5A). The piperazine linker further mimics the backbone of A11 and R12 from MLL. Binding of MI-2 is stabilized by hydrophobic interactions and two hydrogen bonds with the side chains of Y276 and N282. MI-2 is a relatively rigid compound and its potent binding affinity likely arises from high shape complementarity to the binding pocket on menin. Furthermore, binding of the inhibitor does not cause any conformational changes in menin, emphasizing that the binding site is relatively rigid.
Figure 4.
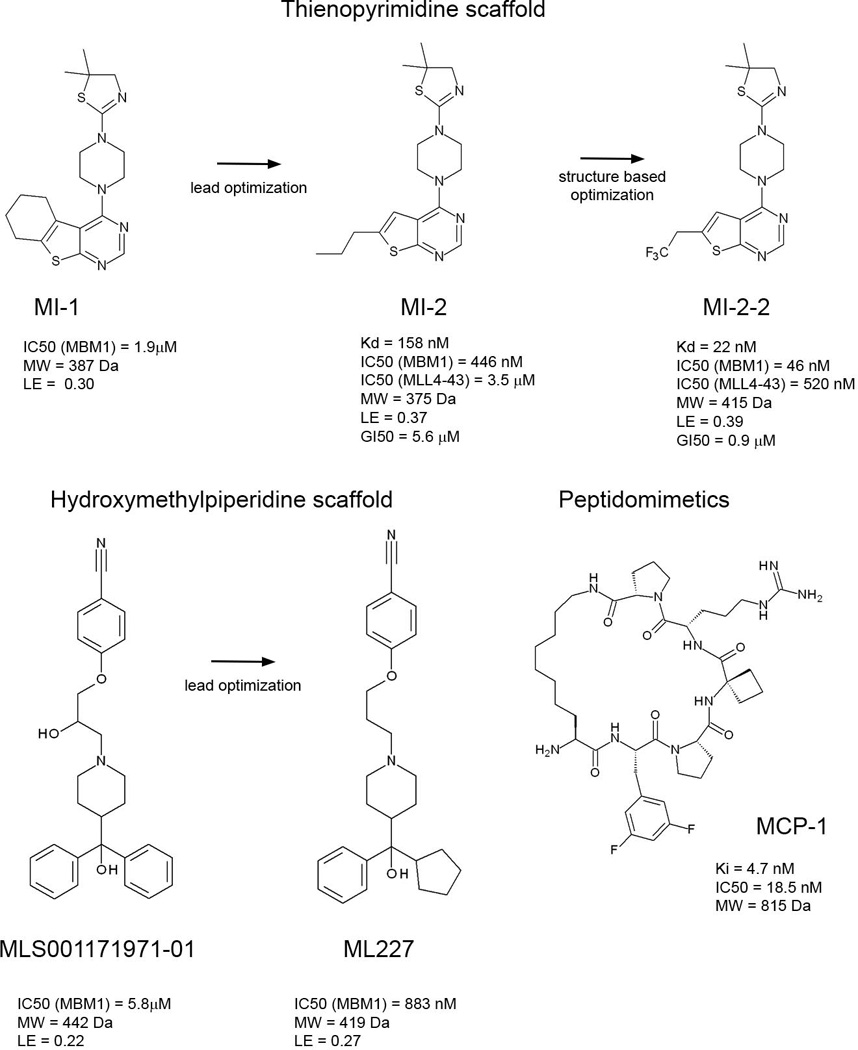
Small molecule and peptidomimetic inhibitors of the menin-MLL interaction [24, 25, 72, 73].
Figure 5.
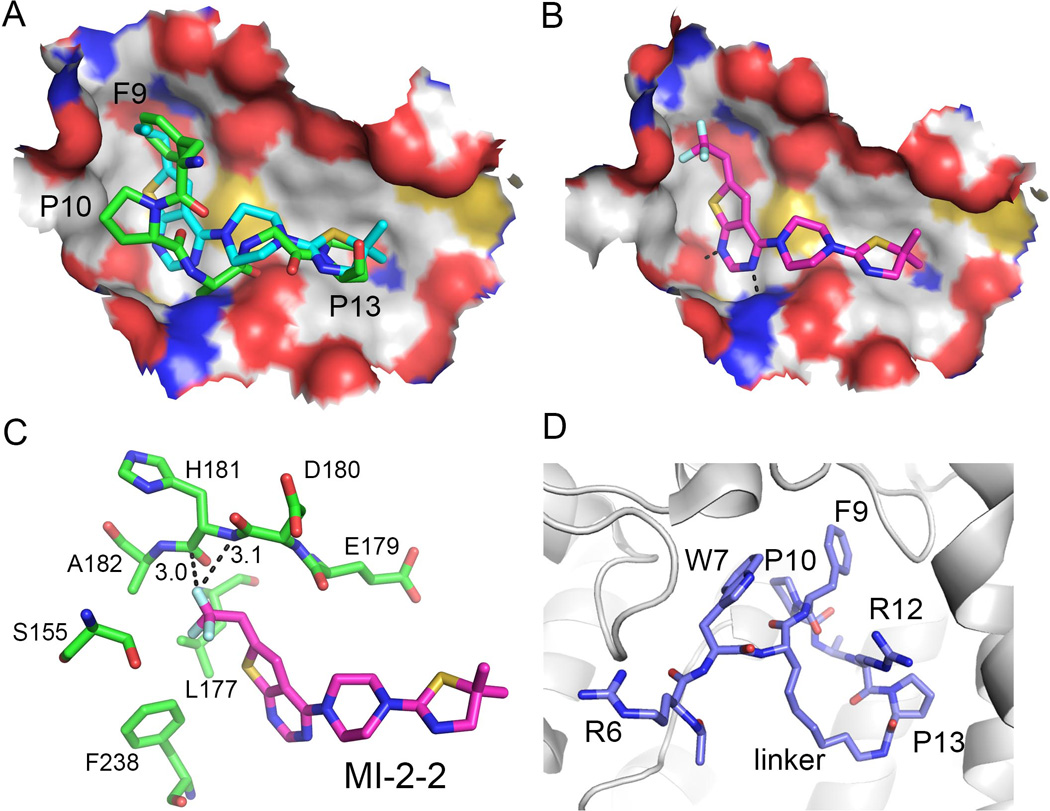
Structures of small molecule inhibitors bound to menin. A. MI-2 (cyan carbon atoms, PDB code 4GQ3) inhibitor mimics key interaction of MBM1 (green carbon atoms, PDB code 4GQ6) with menin. F9, P10 and P13 pockets are labeled. B. Binding mode of MI-2-2 to menin (4GQ4). C. Trifluoroethyl group in MI-2-2 forms favorable interactions with menin backbone; short distances between fluorine atom and backbone are labeled. D. Structure of the macrocyclic MLL peptide (blue carbon atoms, PDB code 4I80) bound to menin (shown as gray ribbon). Position of the linker used for cyclization of residues 8 and 13 is labeled.
By employing the structural information for the menin-MI-2 complex we developed a more potent analog MI-2-2 via substitution of n-propyl with trifluoroethyl group, which enhanced the affinity by 7 to 9 fold (Figure 4) [25]. The high resolution crystal structure of MI-2-2 bound to menin rationalized a significant gain in the affinity, revealing that the fluorine atom in the trifluoroethyl group is located in a close proximity to the protein backbone and is involved in favorable orthogonal dipolar interactions with the menin backbone (Figure 5 B, C). Such interactions have been observed in several protein-ligand complexes and have significant contribution to the binding affinity [74].
Altogether, the thienopyrimidine class of menin-MLL inhibitors represents a very potent and promising class of protein-protein interaction inhibitors. These compounds have favorable drug-like properties, including relatively low molecular weight (Mw < 450 Da), high target potency and ligand efficiency (Figure 4), representing a very promising lead class for further development. Structural information for these compounds bound to menin will facilitate their further optimization. For example, these compounds do not occupy the P10 pocket on menin (Figure 5A, B) and therefore, further modifications could be focused on substitutions to reach this pocket.
The second class of small molecule inhibitors of the menin-MLL interaction has recently been identified by HTS of ~280,000 compounds at the NIH MLPCN (Molecular Libraries Probe Production Centers Network) [73]. These compounds have unrelated structures to the thienopyrimidine class and are based on the hydroxymethylpiperidine scaffold (Figure 4). The HTS hit, MLS001171971, showed low micromolar activity in inhibiting the menin-MLL interaction (IC50 = 5.8 µM, Figure 4). Medicinal chemistry optimization resulted in several fold improvement in binding affinity of this class of menin-MLL inhibitors, leading to ML227 with IC50 = 883nM for the inhibition of the menin-MBM1 interaction (Figure 4). Despite sub-micromolar in vitro inhibition of the menin-MLL interaction, it remains to be tested whether this class of compounds will show activity in MLL leukemia cells and has a potential for further development into therapeutically useful agents.
Peptidomimetics as menin inhibitors
Menin binds a peptide fragment of MLL, and peptide-based inhibitors may offer another attractive strategy to target the menin-MLL interaction. Recently, potent peptidomimetics mimicking the MLL MBM1 fragment have been developed via structure-based design (Figure 5D) [72]. A number of short peptides have been designed based on the structure of the menin-MLL complex and were tested for binding to menin. MLL binds to menin in a relatively unusual, U-shaped conformation with the R8 side chain positioned in close proximity to the backbone carbonyl group of P13. Significantly improved peptidic inhibitors have been developed by designing macrocyclic analogs, in which the side chain of R8 was replaced with a 7-carbon aliphatic amine and coupled through the amino group to the carboxylic acid group in P13 to form an amide. Additional optimization of the side chains yielded peptidomimetic analog MCP-1, which strongly binds to menin with a Ki value of 4.7 nM (Figure 4). Interestingly, the MCP-1 is >600 times more potent than the corresponding acyclic peptide. To date the cellular activity of the peptidomimetic inhibitors of the menin-MLL interaction has not been reported. Further optimization of MCP-1 may ultimately yield a class of cell-permeable peptidomimetic inhibitors of the menin–MLL1 interaction. Importantly, peptidomimetics may offer a potential advantage to develop inhibitors simultaneously targeting the MBM1 and MBM2 sites on menin.
Inhibitors of menin-MLL need to disrupt bivalent protein-protein interaction
MLL binds to menin in a complex mode involving bivalent interactions, which represents a challenge for targeting by small molecules. The MBM1 site represents a “druggable” pocket on menin and high affinity ligands that bind to this site have already been developed [25]. Despite targeting the MBM1 site only, these compounds are able to displace MLL from menin and reverse the leukemogenic activity of MLL fusion proteins [24, 25]. To understand the mechanism of inhibition by these compounds, competition of MI-2-2 against various MLL fragments, including MBM1, MBM2 and MLL4–43 encompassing the intact menin binding fragment, was tested [25]. MI-2-2 binds to menin with Kd=22nM and very efficiently inhibits the menin-MBM1 interaction (IC50=46nM); however, it has almost no effect on the menin-MBM2 interaction (Figure 6A, B). Therefore, MBM2 can still interact with menin in the presence of bound MI-2-2. Interestingly, MI-2-2 is capable of inhibiting the interaction of menin with the intact MLL binding fragment MLL4–43 (IC50=520nM), but inhibitory activity is ~10-fold reduced when compared with the shorter MBM1. We propose a two-step mechanism to explain the inhibition of the menin-MLL interaction. In the first step MI-2-2 displaces MBM1 and the menin-MLL complex is held together via MBM2. Subsequently, because of a relatively low affinity of MBM2 to menin (Kd=1µM) the MBM2 alone is not sufficient to maintain the complex and MLL dissociates from menin (Figure 6). Consistently with this mechanism, we observed that inhibition of the MLL4–43-menin interaction using small molecule inhibitors has slower kinetics and requires longer incubation time when compared to inhibition of MBM1 [25]. In summary, inhibitors solely targeting MBM1 interaction are sufficient to block menin-MLL interaction; however, the presence of the second MBM2 motif reduces the activity of such inhibitors.
Figure 6.
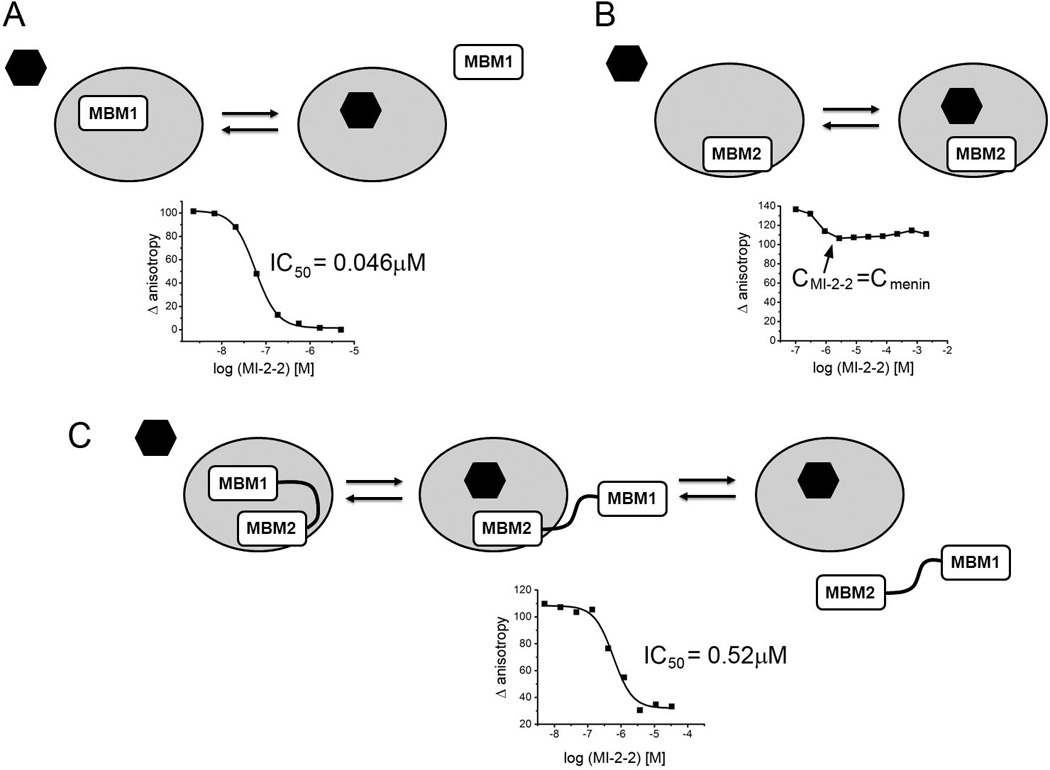
Mechanism of inhibition of menin-MLL by small molecules targeting MBM1 site on menin. A. MI-2-2 binds to menin and directly competes with MBM1 peptide. B. MI-2-2 does not compete with MBM2 peptide. C. Proposed two-step mechanism of inhibition of MLL containing both MBM1 and MBM2 via MI-2-2. Graphs represent fluorescence polarization experiments showing inhibition of binding of different MLL fragments to menin via MI-2-2 [25].
The requirement to disrupt the bivalent menin-MLL interaction has pronounced consequences on small molecule inhibitor development. The thienopyrimidine inhibitors bind to the MBM1 site and, as expected, when tested for disruption of the menin interaction with MLL4–43 they exhibit 7–10 fold reduced potency (Figure 6C) [25]. In consequence, development of compounds targeting the menin-MLL interaction via binding to the MBM1 site might require very potent compounds to demonstrate in vivo efficacy.
Small molecules targeting menin reverse oncogenic activity of MLL fusion proteins in leukemia
The leukemogenic activity of MLL fusion proteins is dependent on their interaction with menin and disruption of this interaction using genetic methods reversed the oncogenic activity of MLL fusion proteins and abrogated development of acute leukemia in vivo [17, 18, 21]. Therefore, inhibition of the protein-protein interaction between menin and MLL fusions should result in a similar outcome and might represent an attractive therapeutic strategy for MLL leukemia patients. Extensive characterization of the thienopyrimidine class revealed that these compounds inhibit the menin-MLL fusion protein interaction in mammalian cells, selectively block proliferation, and induce apoptosis and differentiation in the MLL leukemia cells [24, 25]. Furthermore, the thienopyrimidine inhibitors of menin-MLL downregulate expression of HOXA9 and MEIS1 genes, which are downstream targets of MLL fusion proteins required for their oncogenic activity [7], demonstrating a very specific mechanism of action. Consequently, small molecule inhibitors targeting menin block the immortalization potential of MLL fusion proteins and reverse their oncogenic activity. Furthermore, investigation of several thienopyrimidine compounds shows a very good correlation between in vitro inhibition of the menin-MLL interaction and cellular activities [24]. The more potent MI-2-2 [25] demonstrated approximately 4-fold improvement in cellular activity as compared to MI-2 and MI-3 compounds [24]. This clearly indicates that development of potent small molecule inhibitors of the menin-MLL interaction is feasible. However, it remains to be established whether menin-MLL inhibitors with favorable drug like properties will demonstrate efficacy in animal models.
Menin-MLL interaction as a target for drug discovery – potential applications
The requirement of menin for leukemogenic activity of MLL fusion proteins has been well validated [17, 18, 21] and is sufficient to rationalize efforts to develop small molecule inhibitors to target the menin-MLL interaction. Since menin is likely to function as a more general co-factor of wild type MLL1 and MLL2 histone methyltransferases, it is worth considering new potential applications of menin-MLL inhibitors beyond MLL leukemia. The activity of both MLL1 and MLL2 has been linked to various diseases and menin inhibitors might have broader therapeutic potential (Figure 7). This has not been explored to date and prospective applications of the menin-MLL inhibitors are outlined below.
Figure 7.
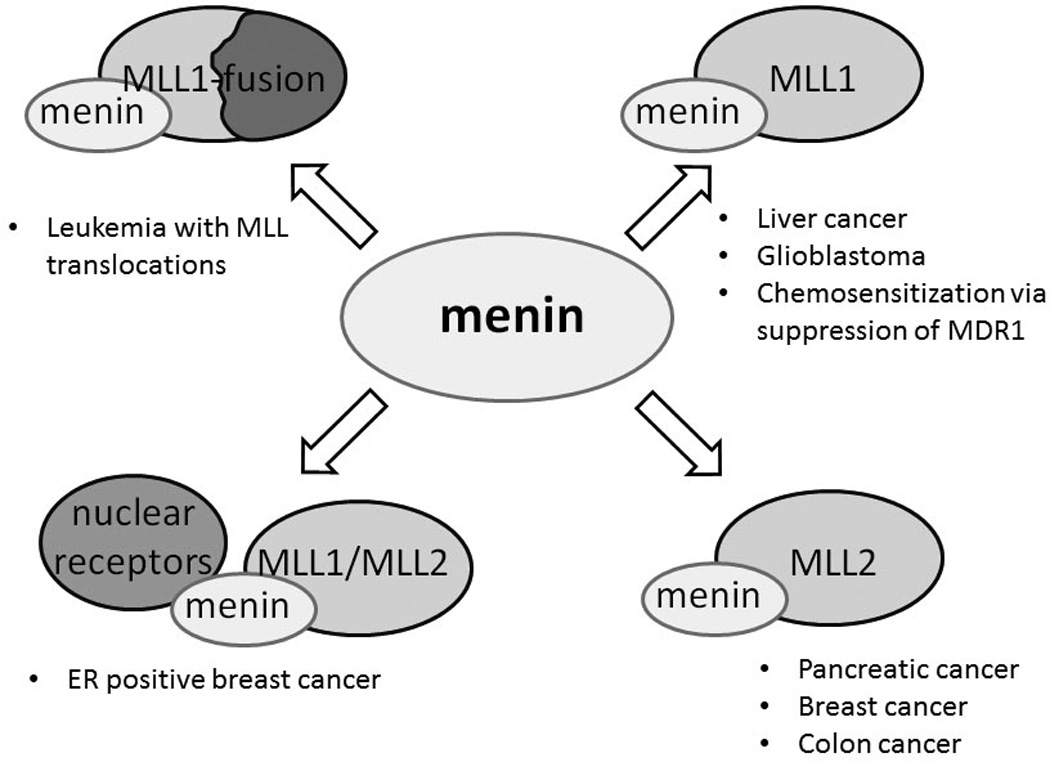
Potential applications of menin-MLL inhibitors in cancer via targeting menin interactions with MLL-fusion proteins, wild type MLL1 and MLL2.
MLL1 in cancer
MLL1 is a general epigenetic regulator of gene transcription and one of the key regulators of HOX genes [75, 76]. MLL1 is implicated in embryogenesis and development [75, 77], in cell-cycle regulation and response to stress [78–80]. Down-regulation of MLL1 results in cell-cycle arrest at G2/M phase [78] and inhibition of the MLL1complex may have therapeutic effect in multiple cancers. For example, knockdown of MLL1 induced apoptosis in cultured tumor cancer cells and suppressed the growth of xenografted HeLa tumors in vivo [80]. Recent studies demonstrated that MLL1 is required for invasion and metastatic growth of hepatocellular carcinoma cell lines [81, 82]. Silencing of MLL1 severely compromised metastatic growth of HepG2 cells in mouse xenografts [81]. It has recently been demonstrated that menin-MLL up-regulates Yap1 transcription in hepatocellular carcinoma cell lines and, importantly, knockdown of menin significantly reduced tumor volume in HepG2 xenografts [82]. MLL1 maintains cancer stem cell characteristics in human glioblastoma by activation of homeobox genes including HOXA10 [83]. Furthermore, MLL1 enhances hypoxic responses in glioblastoma and loss of MLL1 expression via RNA interference results in reduction of self-renewal, growth, and tumorigenicity of glioma stem cells [84]. In addition, there is evidence that wild type MLL1 cooperates with MLL1 fusion proteins and might be required for their recruitment and leukemogenic transformation [34, 85]. Therefore, simultaneous targeting of menin-MLL1 and menin-MLL1-fusion protein interactions might be required to achieve therapeutic effect in MLL leukemias.
Interestingly, MLL1 as well as MLL-AF4, has been shown to regulate expression of multidrug resistance 1 gene (MDR1), which encodes P-glycoprotein (Pgp), a broad range drug transporter conferring resistance to many chemotherapeutic agents [86]. Knockdown of MLL1 resulted in decreased MDR1 expression and increased retention of chemotherapeutic agents [86]. Therefore, menin-MLL inhibitors may have beneficial activity in sensitization of cancer cells to chemotherapy. Altogether, MLL1 may represent an attractive target for drug discovery. However, therapeutic potential of inhibiting the activity of MLL1 via targeting the menin-MLL1 interaction remains to be explored.
MLL2 in cancer
MLL2is a close homolog of MLL1, both belong to the Trithorax family of methyltransferases, and are expressed during embryogenesis and development [87], although they regulate distinct sets of genes [88]. Of note, due to the existing confusion in the nomenclature, MLL2 is frequently referred as MLL4 and vice versa [89]. The important difference is that menin binds to MLL2 but not to MLL4. The role of MLL2 is less understood than MLL1 and several studies relate MLL2 activity to various malignancies. For example, the MLL2 gene is amplified in pancreatic carcinoma [90] and elevated levels of MLL2 were found in human colon and breast cancer cells [91]. MLL2 knockdown severely affects cell-cycle progression and induces apoptotic cell death in cultured colon and breast cancer cells [92]. Furthermore, knockdown of MLL2 in colon cancer SW480 xenografts suppressed tumor growth in vivo [92]. Therefore, pharmacological suppression of MLL2 activity may have therapeutic potential in solid cancers and further studies are needed to establish whether such an effect can be achieved via inhibition of the menin-MLL2 interaction.
Menin as co-activator of nuclear receptors
Menin plays a role as a co-activator of nuclear receptors via direct interaction with estrogen and vitamin D receptors [55]. Menin interacts with hormone activated estrogen receptor alpha (ERα) and links ERα to MLL1 and MLL2 methyltransferase complexes [55]. This results in H3K4 methylation of target genes, such as the estrogen regulated TFF1 gene [55]. Independent studies implicated menin as a direct activator of ERα function in MCF-7 breast cancer cells [56], and it has been hypothesized that the expression of menin in breast cancer cells might be associated with resistance to antiestrogen therapy. The analysis of ER-positive breast cancer samples revealed that patients with menin-positive tumors had a worse outcome than those with menin-negative tumors [56, 93]. Therefore, inhibition of the menin interaction with MLL methyltransferases may impair co-activation of the estrogen receptor and may offer a therapeutic benefit for patients with breast cancers dependent on ER signaling.
Challenges in targeting menin-MLL protein-protein interaction: conclusions and future perspectives
Targeting the menin-MLL interaction using small molecules may represent an attractive strategy to develop new therapeutics for MLL leukemia. The activity of MLL fusion proteins is dependent on binding to menin and abolishing this interaction via small molecules is expected to lead to an effective therapy for this incurable disease. Furthermore, inhibition of the menin interaction with wild type MLL1 and MLL2 may have a potential therapeutic effect in a number of solid cancers, such as liver [81], brain [83], colon [92], breast [56] and may chemo-sensitize cancer cells to chemotherapeutic agents via downregulation of MDR1 [86] (Figure 7). Although biochemical studies indicate that menin is an integral component of the MLL1 and MLL2 methyltransferase complexes, it remains to be established whether blocking their interactions with menin impairs global recruitment and activity of these HMT complexes. Favorably, recent studies confirmed that menin is not an exquisite co-factor of MLL1 in normal hematopoiesis, and therefore no related on-target toxicity in the hematopoietic system is expected upon blocking the menin-MLL interaction [40]. The major concern of targeting menin relates to its tumor suppressor activity in endocrine tissues [41, 62]. Further studies will be required to establish whether abolishing menin binding to MLL, while preserving its other protein-protein interactions, will impair menin function as a tumor suppressor.
Although targeting protein-protein interactions is considered a challenging and risky task, significant progress has been made in this field and a number of protein-protein interaction inhibitors have entered clinical trials. Multiple reasons distinguish menin as a feasible target for inhibitor development: 1) two classes of small molecule inhibitors and potent peptidomimetics have already been reported, demonstrating a proof of concept that targeting menin is feasible; 2) the most potent small molecule inhibitors from the thienopyrimidine class binds to menin with low nanomolar affinity, demonstrating that very potent inhibitors can be developed [25]; 3) the high resolution crystal structure of menin and menin complexes with MLL and small molecule inhibitors have been obtained [25]; 4) the MBM1 binding site on menin is well defined and will likely constitute a primary site for inhibitor development; 5) the structure of the binding site on menin is not affected by ligand binding, which may facilitate structure-based design of inhibitors using computational methods; 6) the menin-MLL interaction is highly conserved and it is unlikely that mutations in menin will confer resistance to small molecule inhibitors while preserving the interaction with MLL. In the long term, this may allow to avoid the resistance to menin-targeting drugs.
On the other hand, development of drugs targeting menin might constitute a significant challenge for drug discovery, because: 1) menin binds a relatively long, ~40 amino acid long fragment of MLL, which consists of two motifs: MBM1 and MBM2 that interact with menin in a complex bivalent mode and with high affinity; 2) The MBM1 site on menin is likely the most suitable for small molecule inhibitor development; however, the presence of a second MBM2 motif in MLL will decrease the activity of inhibitors when assessed against the full length MLL. In order to efficiently disrupt the menin-MLL interaction via targeting the MBM1 site a very potent inhibitor (Kd<1nM) might be required; 3) development of very effective inhibitors of the menin-MLL interaction may require reaching the MBM2 pocket, which will lead to larger molecules with significantly increased molecular weight and compromised drug-like properties; 4) The MLL binding site on menin is very large (~5,000A3) and blocking this site with small molecules represents a challenge. As recently reviewed, concise and tight binding protein-protein interactions are most amenable to inhibition by small molecules [94]. The menin-MLL interaction represents a more challenging system involving a large binding interface. 5) Finally, menin is involved in multiple protein-protein interactions and it remains to be demonstrated whether selective inhibition of the menin-MLL interaction is feasible.
In summary, more potent inhibitors of the menin-MLL interaction with favorable drug-like properties are needed in order to establish the therapeutic potential of targeting menin. Significant progress towards achieving this goal has already been made and future studies will reveal whether pharmacologic inhibition of this important drug target will lead to novel drugs for acute leukemias and possibly other cancers.
Executive summary.
Background
Benefits in targeting menin
The interaction of menin with MLL-fusion proteins is a well-validated therapeutic target in leukemia
The menin-MLL interaction is not necessary in normal hematopoiesis
Menin is a component of MLL1 and MLL2 histone methyltransferase complexes and targeting menin-MLL may have potential therapeutic effects in various cancers
Concerns in targeting menin
Menin is a tumor suppressor in endocrine tissues
Challenges in targeting menin-MLL protein-protein interaction
Feasibility of targeting menin
First class of small molecule inhibitors with nanomolar activities has been developed
High resolution crystal structures of menin with MLL and thienopyrimidine inhibitors are available
The MBM1 site on menin is a “druggable” site for targeting by small molecule inhibitors
Difficulty in targeting menin-MLL
Inhibitors need to disrupt bivalent interaction of MLL with menin
MLL binding site on menin is very large (~5,000Å3), representing a challenge for targeting by small molecules
Very potent inhibitors might be needed to efficiently inhibit menin-MLL interaction
Key terms
- Bivalent interaction
type of interaction where two independent fragments of one protein simultaneously interact with the binding partner.
- Fusion protein
chimeric protein, which is a product of fusion of two genes resulting from chromosomal translocation. Fusion protein frequently leads to transformation of cells in cancer.
- “Druggable” target
biological target such as a protein, which is capable of binding small molecules (drugs) with high affinity. “Druggability” may also refer to a particular site on a protein.
- Histone methyltransferase
enzyme, that catalyzes transfer of one, two, or three methyl groups to lysine or arginine residues on histone proteins.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Pui CH, Gaynon PS, Boyett JM, et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet. 2002;359(9321):1909–1915. doi: 10.1016/S0140-6736(02)08782-2. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, Cheng EH, Hsieh JJ. MLL fusions: pathways to leukemia. Cancer Biol. Ther. 2009;8(13):1204–1211. doi: 10.4161/cbt.8.13.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley JD, Olney HJ. International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: overview report. Genes Chromosomes Cancer. 2002;33(4):331–345. doi: 10.1002/gcc.10040. [DOI] [PubMed] [Google Scholar]
- 4.Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J. Cell. Biochem. 2005;95(2):234–242. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 5.Meyer C, Schneider B, Jakob S, et al. The MLL recombinome of acute leukemias. Leukemia. 2006;20(5):777–784. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- 6.Dou Y, Hess JL. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int. J. Hematol. 2008;87(1):10–18. doi: 10.1007/s12185-007-0009-8. [DOI] [PubMed] [Google Scholar]
- 7.Zeisig BB, Milne T, Garcia-Cuellar MP, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell. Biol. 2004;24(2):617–628. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer. Res. 2005;65(24):11367–11374. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 9.Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc. Natl. Acad. Sci. U S A. 2005;102(41):14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21(21):2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owens BM, Hawley RG. HOX and non-HOX homeobox genes in leukemic hematopoiesis. Stem Cells. 2002;20(5):364–379. doi: 10.1634/stemcells.20-5-364. [DOI] [PubMed] [Google Scholar]
- 12.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 13.Rice KL, Licht JD. HOX deregulation in acute myeloid leukemia. J. Clin. Invest. 2007;117(4):865–868. doi: 10.1172/JCI31861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002;30(1):41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 15.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17(18):2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slany RK. When epigenetics kills: MLL fusion proteins in leukemia. Hematol. Oncol. 2005;23(1):1–9. doi: 10.1002/hon.739. [DOI] [PubMed] [Google Scholar]
- 17. Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–218. doi: 10.1016/j.cell.2005.09.025. ** This paper descibed the discovery and validation of menin as a critical co-factor of MLL fusion proteins in leukemia and validated that interaction of MLL fusions with menin is required for development of leukemia in vivo in animal models.
- 18.Chen YX, Yan J, Keeshan K, et al. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc. Natl. Acad. Sci. U S A. 2006;103(4):1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 20.Guru SC, Goldsmith PK, Burns AL, et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. U S A. 1998;95(4):1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer. Res. 2007;67(15):7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. * Studies demonstrating that short N-terminal fragment of MLL may function as a dominant negative inhibitor of the activity of MLL fusion proteins.
- 22. Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J. Biol. Chem. 2010;285(52):40690–40698. doi: 10.1074/jbc.M110.172783. * First characterization of the menin-MLL interaction using biochemical and biophysical methods and finding that there are two menin binding motifs within the N-terminus of MLL.
- 23.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell. 2004;13(4):587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 24. Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012;8(3):277–284. doi: 10.1038/nchembio.773. ** Development of a first class of small molecule inhibitors of menin-MLL interaction and characterization of their activities in MLL leukemia cells.
- 25. Shi A, Murai MJ, He S, et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120(23):4461–4469. doi: 10.1182/blood-2012-05-429274. * Study reporting a first high resolution crystal structure of human menin with bound small molecule inhibitor targeting the MLL binding site.
- 26.Yokoyama A, Wang Z, Wysocka J, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004;24(13):5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosgrove MS, Patel A. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J. 2010;277(8):1832–1842. doi: 10.1111/j.1742-4658.2010.07609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94(7):984–993. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dou Y, Milne TA, Ruthenburg AJ, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006;13(8):713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 31.Dou Y, Milne TA, Tackett AJ, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121(6):873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 32.Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U S A. 2005;102(3):749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onodera A, Yamashita M, Endo Y, et al. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J. Exp. Med. 2010;207(11):2493–2506. doi: 10.1084/jem.20100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiel AT, Blessington P, Zou T, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17(2):148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14(1):36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013;33(10):2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang J, Gurung B, Wan B, et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482(7386):542–546. doi: 10.1038/nature10806. * Study reporting the first crystal structure of human menin bound to MLL.
- 38.Agarwal SK, Jothi R. Genome-wide characterization of menin-dependent H3K4me3 reveals a specific role for menin in the regulation of genes implicated in MEN1-like tumors. PLoS One. 2012;7(5):e37952. doi: 10.1371/journal.pone.0037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scacheri PC, Davis S, Odom DT, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2(4):e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122(12):2039–2046. doi: 10.1182/blood-2013-03-486647. * Study demonstrating that menin is not a requisite cofactor for MLL1 during normal hematopoiesis and drugs disrupting menin-MLL1 interaction should not impair hematopoiesis.
- 41.Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat. Rev. Cancer. 2005;5(5):367–375. doi: 10.1038/nrc1610. [DOI] [PubMed] [Google Scholar]
- 42.Trump D, Farren B, Wooding C, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM. 1996;89(9):653–669. doi: 10.1093/qjmed/89.9.653. [DOI] [PubMed] [Google Scholar]
- 43.Pannett AA, Thakker RV. Multiple endocrine neoplasia type 1. Endocr Relat. Cancer. 1999;6(4):449–473. doi: 10.1677/erc.0.0060449. [DOI] [PubMed] [Google Scholar]
- 44.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U S A. 2001;98(3):1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 2003;17(9):1880–1892. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 47.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008;29(1):22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 48.Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2004;24(15):6569–6580. doi: 10.1128/MCB.24.15.6569-6580.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimazu S, Nagamura Y, Yaguchi H, Ohkura N, Tsukada T. Correlation of mutant menin stability with clinical expression of multiple endocrine neoplasia type 1 and its incomplete forms. Cancer. Sci. 2011;102(11):2097–2102. doi: 10.1111/j.1349-7006.2011.02055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 2011;286(36):31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agarwal SK, Guru SC, Heppner C, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96(1):143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 52.Heppner C, Bilimoria KY, Agarwal SK, et al. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene. 2001;20(36):4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- 53.Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc. Natl. Acad. Sci. U S A. 2001;98(7):3837–3842. doi: 10.1073/pnas.061358098. Epub 2001 Mar 3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer. Res. 2003;63(19):6135–6139. [PubMed] [Google Scholar]
- 55.Dreijerink KM, Mulder KW, Winkler GS, Hoppener JW, Lips CJ, Timmers HT. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer. Res. 2006;66(9):4929–4935. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- 56.Imachi H, Murao K, Dobashi H, et al. Menin, a product of the MENI gene, binds to estrogen receptor to enhance its activity in breast cancer cells: possibility of a novel predictive factor for tamoxifen resistance. Breast Cancer Res. Treat. 2010;122(2):395–407. doi: 10.1007/s10549-009-0581-0. [DOI] [PubMed] [Google Scholar]
- 57.Schnepp RW, Hou Z, Wang H, et al. Functional interaction between tumor suppressor menin and activator of S-phase kinase. Cancer. Res. 2004;64(18):6791–6796. doi: 10.1158/0008-5472.CAN-04-0724. [DOI] [PubMed] [Google Scholar]
- 58.Jin S, Mao H, Schnepp RW, et al. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer. Res. 2003;63(14):4204–4210. [PubMed] [Google Scholar]
- 59.Sukhodolets KE, Hickman AB, Agarwal SK, et al. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol. Cell. Biol. 2003;23(2):493–509. doi: 10.1128/MCB.23.2.493-509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balogh K, Patocs A, Hunyady L, Racz K. Menin dynamics and functional insight: take your partners. Mol. Cell. Endocrinol. 2010;326(1–2):80–84. doi: 10.1016/j.mce.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 61.Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013;38(8):394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4) Mol. Cell. Endocrinol. 2013 doi: 10.1016/j.mce.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol. Cell Endocrinol. 2007:265–266. 34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karges W, Maier S, Wissmann A, Dralle H, Dosch HM, Boehm BO. Primary structure, gene expression and chromosomal mapping of rodent homologs of the MEN1 tumor suppressor gene. Biochim. Biophys. Acta. 1999;1446(3):286–294. doi: 10.1016/s0167-4781(99)00089-5. [DOI] [PubMed] [Google Scholar]
- 65.Papaconstantinou M, Maslikowski BM, Pepper AN, Bedard PA. Menin: the protein behind the MEN1 syndrome. Adv. Exp. Med. Biol. 2009;668:27–36. doi: 10.1007/978-1-4419-1664-8_3. [DOI] [PubMed] [Google Scholar]
- 66.Gray FL, Murai MJ, Grembecka J, Cierpicki T. Detection of disordered regions in globular proteins using (1)(3)C-detected NMR. Protein Sci. 2012;21(12):1954–1960. doi: 10.1002/pro.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macconaill LE, Hughes CM, Rozenblatt-Rosen O, Nannepaga S, Meyerson M. Phosphorylation of the menin tumor suppressor protein on serine 543 and serine 583. Mol. Cancer Res. 2006;4(10):793–801. doi: 10.1158/1541-7786.MCR-06-0123. [DOI] [PubMed] [Google Scholar]
- 68.Francis J, Lin W, Rozenblatt-Rosen O, Meyerson M. The menin tumor suppressor protein is phosphorylated in response to DNA damage. PLoS One. 2011;6(1):e16119. doi: 10.1371/journal.pone.0016119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng ZJ, Gurung B, Jin GH, Yang XL, Hua XX. SUMO modification of menin. Am. J. Cancer Res. 2013;3(1):96–106. [PMC free article] [PubMed] [Google Scholar]
- 70.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White AW, Westwell AD, Brahemi G. Protein-protein interactions as targets for small-molecule therapeutics in cancer. Expert Rev. Mol. Med. 2008;10:e8. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- 72.Zhou H, Liu L, Huang J, et al. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J. Med. Chem. 2013;56(3):1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
- 73.Manka J, Daniels RN, Dawson E, et al. Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction. Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD) 2013 [Google Scholar]
- 74.Olsen JA, Banner DW, Seiler P, et al. Fluorine interactions at the thrombin active site: protein backbone fragments H-C(alpha)-C=O comprise a favorable C-F environment and interactions of C-F with electrophiles. Chembiochem. 2004;5(5):666–675. doi: 10.1002/cbic.200300907. [DOI] [PubMed] [Google Scholar]
- 75.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378(6556):505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 76.Guenther MG, Jenner RG, Chevalier B, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. U S A. 2005;102(24):8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc. Natl. Acad. Sci. U S A. 1998;95(18):10632–10636. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mishra BP, Ansari KI, Mandal SS. Dynamic association of MLL1, H3K4 trimethylation with chromatin and Hox gene expression during the cell cycle. FEBS J. 2009;276(6):1629–1640. doi: 10.1111/j.1742-4658.2009.06895.x. [DOI] [PubMed] [Google Scholar]
- 79.Ansari KI, Hussain I, Das HK, Mandal SS. Overexpression of human histone methylase MLL1 upon exposure to a food contaminant mycotoxin, deoxynivalenol. FEBS J. 2009;276(12):3299–3307. doi: 10.1111/j.1742-4658.2009.07055.x. [DOI] [PubMed] [Google Scholar]
- 80.Ansari KI, Kasiri S, Mandal SS. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene. 2013;32(28):3359–3370. doi: 10.1038/onc.2012.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takeda S, Liu H, Sasagawa S, et al. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Invest. 2013;123(7):3154–3165. doi: 10.1172/JCI65566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu B, Li SH, Zheng R, et al. Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates Yap1 transcription. Proc. Natl. Acad. Sci. U S A. 2013;110(43):17480–17485. doi: 10.1073/pnas.1312022110. * Study validating that menin promotes development of hepatocellular carcinoma and that drugs inhibiting the menin-MLL interaction might demonstrate activity in hepatocellular carcinoma.
- 83.Gallo M, Ho J, Coutinho FJ, et al. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer. Res. 2013;73(1):417–427. doi: 10.1158/0008-5472.CAN-12-1881. [DOI] [PubMed] [Google Scholar]
- 84.Heddleston JM, Wu Q, Rivera M, et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell. Death Differ. 2012;19(3):428–439. doi: 10.1038/cdd.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Milne TA, Kim J, Wang GG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol. Cell. 2010;38(6):853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huo H, Magro PG, Pietsch EC, Patel BB, Scotto KW. Histone methyltransferase MLL1 regulates MDR1 transcription and chemoresistance. Cancer. Res. 2010;70(21):8726–8735. doi: 10.1158/0008-5472.CAN-10-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Glaser S, Schaft J, Lubitz S, et al. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133(8):1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- 88.Lubitz S, Glaser S, Schaft J, Stewart AF, Anastassiadis K. Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol. Biol. Cell. 2007;18(6):2356–2366. doi: 10.1091/mbc.E06-11-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bogershausen N, Bruford E, Wollnik B. Skirting the pitfalls: a clear-cut nomenclature for H3K4 methyltransferases. Clin. Genet. 2013;83(3):212–214. doi: 10.1111/cge.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huntsman DG, Chin SF, Muleris M, et al. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene. 1999;18(56):7975–7984. doi: 10.1038/sj.onc.1203291. [DOI] [PubMed] [Google Scholar]
- 91.Natarajan TG, Kallakury BV, Sheehan CE, et al. Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer. Cell Int. 2010;10:13. doi: 10.1186/1475-2867-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ansari KI, Kasiri S, Mishra BP, Mandal SS. Mixed lineage leukaemia-4 regulates cell-cycle progression and cell viability and its depletion suppresses growth of xenografted tumour in vivo. Br. J. Cancer. 2012;107(2):315–324. doi: 10.1038/bjc.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imachi H, Yu X, Nishiuchi T, Miyai Y, Masugata H, Murao K. Raloxifene inhibits menin-dependent estrogen receptor activation in breast cancer cells. J. Endocrinol. Invest. 2011;34(11):813–815. doi: 10.1007/BF03346730. [DOI] [PubMed] [Google Scholar]
- 94.Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert. Rev. Mol. Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]