

Abstract
Background
The bacterial genus Salmonella contains thousands of serotypes that infect humans or other hosts, causing mild gastroenteritis to potentially fatal systemic infections in humans. Pathogenically distinct Salmonella serotypes have been classified as individual species or as serological variants of merely one or two species, causing considerable confusion in both research and clinical settings. This situation reflects a long unanswered question regarding whether the Salmonella serotypes exist as discrete genetic clusters (natural species) of organisms or as phenotypic (e.g. pathogenic) variants of a single (or two) natural species with a continuous spectrum of genetic divergence among them. Our recent work, based on genomic sequence divergence analysis, has demonstrated that genetic boundaries exist among Salmonella serotypes, circumscribing them into clear-cut genetic clusters of bacteria.
Methodologies/Principal Findings
To further test the genetic boundary concept for delineating Salmonella into clearly defined natural lineages (e.g., species), we sampled a small subset of conserved genomic DNA sequences, i.e., the endonuclease cleavage sites that contain the highly conserved CTAG sequence such as TCTAGA for XbaI. We found that the CTAG-containing cleavage sequence profiles could be used to resolve the genetic boundaries as reliably and efficiently as whole genome sequence comparisons but with enormously reduced requirements for time and resources.
Conclusions
Profiling of CTAG sequence subsets reflects genetic boundaries among Salmonella lineages and can delineate these bacteria into discrete natural clusters.
Introduction
Since the first isolation of a Salmonella pathogen from a typhoid patient in 1881, more than 2500 different Salmonella types have been documented [1], [2]. Based on their differences in the somatic (O) and flagellar (H) antigens, the Salmonella bacteria are classified into serotypes by the Kauffmann-White scheme [3]. Initially, the Salmonella serotypes were treated as individual species each having a Latinized scientific name such as Salmonella typhi and Salmonella typhimurium, but in the 1980s all Salmonella serotypes were combined into one species (Salmonella enterica [4]) or two species (Salmonella enterica and Salmonella bongori [5]) as serological variants (serovars [6]) due largely to the extraordinarily high genetic similarity among them, which has caused confusion in research and clinical settings. Indeed, all Salmonella serotypes have very similar genetic backgrounds as revealed by DNA-DNA re-association [7], comparison of genome structures [8], [9] and genomic sequencing [10]–[12], but on the other hand they may differ radically in pathogenic properties. For example, whereas many Salmonella serotypes may cause self-limited gastroenteritis (such as S. typhimurium, S. enteritidis, etc.) or may be virtually non-pathogenic to humans, a few may elicit potentially fatal systemic infections, such as S. typhi that causes typhoid [13]. The dynamic and confusing Salmonella taxonomy reflects long lasting uncertainties about the phylogenetic status of Salmonella: do they dwell in nature as discrete genetic clusters of organisms or as phenotypic variants of a single (or two) natural species with a continuous spectrum of genetic divergence among them?
To examine this issue, we have tested two hypotheses: first, that all Salmonella serotypes form a common gene pool in which DNA exchange occurs readily so that each member has an equal chance to become a different pathogen (e.g., infecting a different host species or causing a different disease) by acquiring appropriate genetic material and incorporating it into the genome; and second, that each Salmonella type (e.g. a serological or pathogenic type) is already an established biological unit, members of which have a common and highly stable genome structure as a result of natural selection over long evolutionary time.
If the first hypothesis is correct, all Salmonella serotypes should be combined into just one species. If the second hypothesis is correct, each Salmonella type is a genetically well-defined natural species. The first hypothesis would be supported by demonstration of a continuous spectrum of genetic divergence among different Salmonella types and, conversely, the second hypothesis would be validated by demonstration of clear-cut genetic boundaries among different Salmonella types as a result of genetic isolation and independent accumulation of mutations over long evolutionary time. Findings that support either hypothesis will lead to novel insights into the population structure of Salmonella and the mechanisms of divergence that have occurred during their adaptation to different environments (e.g., a particular host) during their evolution. A key step towards an answer is to elucidate whether the individual Salmonella types can be grouped into discrete, well separated genetic clusters. The classical method for Salmonella differentiation is serological typing, but a serotype may be polyphyletic. For example, the antigenic formula of 6,7∶c∶1,5 is common to multiple distinct pathogens, e.g., S. paratyphi C, S. choleraesuis and S. typhisuis, which infect different hosts or cause different diseases. Furthermore, based on serotyping only, one cannot judge whether the Salmonella serotypes are genetically well isolated from one another or whether some might be genetic “intermediates” between other serotypes.
Recently, we provided evidence showing that Salmonella exist in discrete genetic clusters isolated by clear-cut genetic boundaries [14]. However, that work was based on whole genome analysis. To further test the robustness of the genetic boundary concept in delineating Salmonella into clearly defined natural lineages (e.g., species), we sampled a small subset of conserved genomic DNA sequences, i.e., the endonuclease cleavage sites that contain the CTAG sequence such as TCTAGA for XbaI. As enteric bacteria tend to eliminate the short sequence CTAG by the Very Short Patch (VSP) repair mechanism [15], endonuclease cleavage sites containing CTAG are scarce and highly conserved in Salmonella. We found that profiling of the CTAG-containing cleavage sequences could resolve the genetic boundaries as reliably and efficiently as whole genome analyses but with enormously reduced requirements for time and resources.
Results
Monophyletic Salmonella serotypes have highly conserved cleavage patterns by CTAG-containing endonucleases
It has been well documented that wild type strains of a monophyletic Salmonella serotype exhibit highly similar endonuclease cleavage patterns for XbaI and BlnI/AvrII on PFGE, such as S. typhimurium [16], S. typhi [17] or S. paratyphi A [18], in comparison with the diverse cleavage patterns seen in polyphyletic serotypes such as S. paratyphi B [19]. However, when we looked at S. gallinarum, known as a monophyletic Salmonella serotype, we saw considerable diversity of cleavage patterns among wild type strains for the endonucleases that have CTAG in the cleavage sites, as illustrated by AvrII cleavage in Figure 1. To determine whether the diversity of cleavage patterns was created by nucleotide base changes (leading to addition or deletion of cleavage sites) or by genomic rearrangements (changing the lengths of DNA fragments between the cleavage sites), we compared the genome structures of these strains. Analysis of incomplete I-CeuI cleavage products of the bacterial genomes showed that these strains had their genomes rearranged in several ways by recombination between rrn operons (Figure 2; for details about I-CeuI and rrn-mediated genomic rearrangements, see [8], [20]), suggesting that at least part of the diverse cleavage patterns have resulted from genomic rearrangements.
Figure 1. Diversity of cleavage patterns with AvrII among S. gallinarum wild type strains.
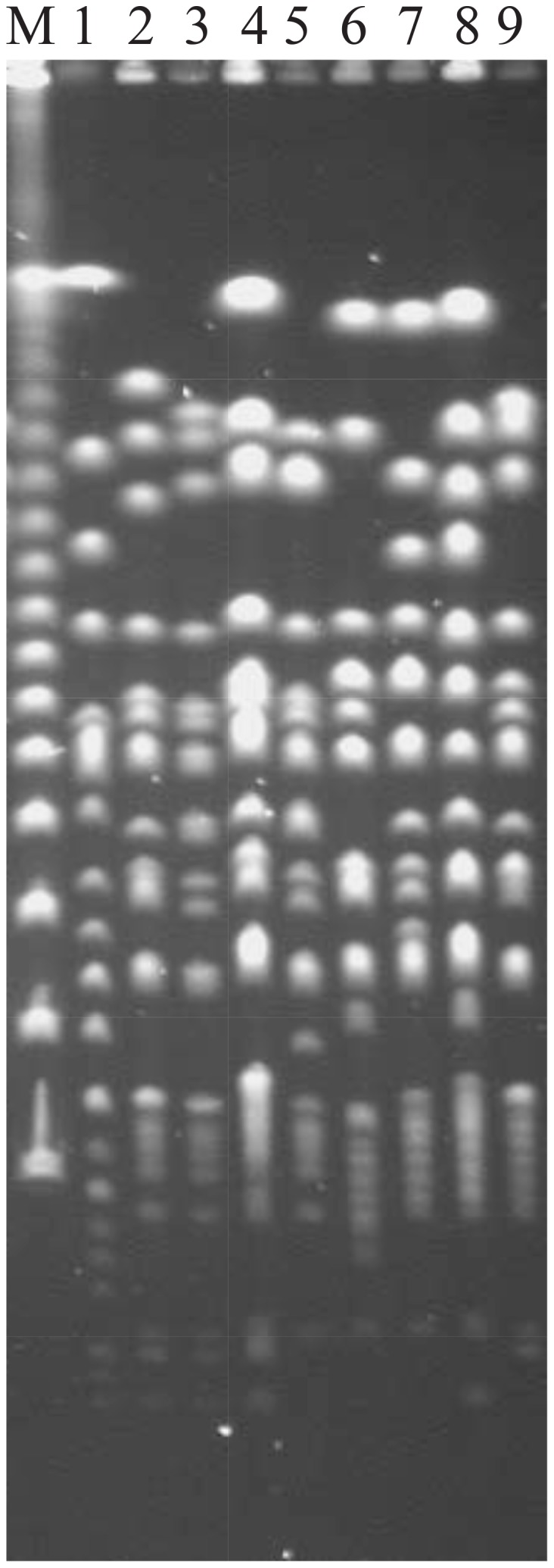
Lanes: 1, molecular size marker (λDNA concatemer); 2, RKS5078; 3, SGSC2423; 4, SGSC2292; 5, SGSC2293; 6, R1481; 7, R1482; 8, R1483; 9, SARB21; 10, 287/91. S. pullorum RKS5078 (Lane 2) is included here for a comparison with the S. gallinarum strains.
Figure 2. Genomic rearrangements of S. gallinarum strains.

(A), PFGE patterns of incomplete I-CeuI cleavage cleaved genomic DNA. Lanes: same as in Figure 1; (B) Genome maps based on I-CeuI data in (A). As seen here, wild type strains have the seven I-CeuI fragments organized differently, with six genome types being resolved among the 8 strains of S. gallinarum. The map of S. pullorum RKS5078 is presented here for a comparison with the S. gallinarum strains.
Next, we needed to determine whether the genomic rearrangements have just altered the lengths between pairs of AvrII sites or might have disrupted any of the AvrII cleavage sites (it is highly unlikely that genomic rearrangements may create new CTAG-containing cleavage sites). For this, we compared two representative S. gallinarum strains, SARB21 and 287/91 (Figure 3), which were previously mapped [21] or sequenced [22], respectively. We analyzed the genome maps of the two strains by matching the homologous cleavage sites between them for XbaI and AvrII, in addition to I-CeuI. We found that, as expected, most of the cleavage pattern differences between S. gallinarum SARB21 and 287/91 could be accounted for by two inversions (one between rrnH and rrnG and one between rrnD and rrnC) and one translocation (I-CeuI Fragment D), all of which massively altered the lengths of homologous genomic DNA segments flanked by the CTAG-containing endonuclease cleavage sites (Figure 4). The rrnH-rrnG inversion made XbaI Fragments C and I to join, forming Fragments C′+I′ and ‘C+’I (XbaI C391 and I248 missing and XbaI C′+I′ 614 and ‘C+’I 25 appearing in strain SARB21 relative to 287/91), along with corresponding changes in AvrII cleavage (See Figures 3 and 4). The I-CeuI Fragment D translocation and rrnD-rrnC inversion resulted in XbaI Fragment B533 splitting to B′ and ‘B, with B’ joining H′ to become B′+H′488, and a truncated ‘B160 fusing with ‘H+F to create a 483 kb segment, along with corresponding changes in AvrII cleavage (See Figures 3 and 4). The only unique AvrII cleavage site is present in strain 287/91 at about 3250 kb from gene thrL (Figure 4, indicated by the open arrowhead), probably as a result of a mutation in the corresponding AvrII cleavage site in strain SARB21 rather than creation of an AvrII cleavage site in strain 287/91.
Figure 3. XbaI and AvrII cleavage patterns of S. gallinarum strains 287/91 and SARB21 after PFGE separation.

(A) XbaI cleavage. Lanes: 1, SARB21; 2, 287/91; 3, λDNA as molecular size marker. (B) AvrII cleavage. Lanes: 1, λDNA as molecular size marker; 2, SARB21; 3, 287/91. Letter designations are for strain 287/91; the same letters are used for homologous fragments in strain SARB21. In the designation of fragments in SARB21, C′ means a fragment homologous to C in 287/91 but truncated on the right-hand part by genomic rearrangement, and ‘C means truncation on the left-hand part of the fragment.
Figure 4. Physical map comparison between S. gallinarum strains 287/91 and SARB21.

The map of SARB21 was reported previously [21]; here letter designations for the cleavage fragments of SARB21 have been changed according to the homologues in strain 287/91 for the convenience of comparison. Note that all XbaI, I-CeuI and AvrII (maps from top to bottom) cleavage sites are conserved in the two strains except the AvrII site between fragments F and J in 287/91 (open arrow), which is missing from SARB21. Lines with solid arrowheads at both ends indicate the ranges of genomic inversions via rrn-mediated recombination between the two strains and filled arrows indicate recombination sites that have resulted in the translocation of I-CeuI fragment D.
Conservation of CTAG-containing endonuclease cleavage sites within other representative Salmonella serotypes
To assess the extent of conservation of the CTAG-containing endonuclease cleavage sites, we conducted systematic comparisons of the cleavage locations on the genome for XbaI among strains of representative Salmonella serotypes, numbering the cleavage sites sequentially according to their locations on the genome of S. typhimurium LT2. Cleavage sites present in any strains but not in LT2 were not numbered. As exemplified by the six S. typhimurium strains, the XbaI cleavage sites were highly conserved within a Salmonella lineage, consistent with the findings by the PFGE techniques. LT2 has 27 XbaI cleavage sites numbered XbaI 1–27 (Table 1), most of which were conserved among all six compared S. typhimurium strains. Of particular significance, as many as over one third of the 27 XbaI cleavage sites fell in intergenic sequences, strongly suggesting the potential importance of these sequences. Among the six S. typhimurium strains, we found two kinds of differences in XbaI sites: presence/absence and presence/degeneracy. The non-conserved XbaI cleavage sites have largely resulted from recent insertions such as prophages or phage remnants (Supplementary Table S1). The sequence degeneracy of the XbaI cleavage sites can be illustrated by XbaI 9, which was present in LT2 but not in any of the other five S. typhimurium strains due to nucleotide substitution, changing the XbaI cleavage site TCTAGA to TCCAGA and leading to the replacement of leucine in LT2 by proline in the other five S. typhimurium strains.
Table 1. Comparison of XbaI cleavage sites among six S. typhimurium strains.
| No.a | Gene IDb | Startc | Endd | LT2 | 14028S | D23580 | SL1344 | ST474 | UK1 |
| 406086 | |||||||||
| XbaI 1 | STM0557 | 614804 | 616456 | 615218 | 615912 | 654028 | 614706 | 614706 | 615911 |
| XbaI 2 | STM1331–STM1332 | 1409765 | 1410086 | 1409947 | 1419923 | 1406459 | 1366800 | 1366800 | 1368113 |
| XbaI 3 | STM1377–STM1378 | 1459616 | 1459925 | 1459627 | 1469603 | 1456139 | 1416479 | 1416479 | 1417794 |
| XbaI 4 | STM1622–STM1623 | 1711483 | 1711701 | 1711569 | 1721546 | 1704084 | 1668422 | 1668422 | 1669738 |
| XbaI 5 | STM2093 | 2174120 | 2175112 | 2174259 | 2225811 | 2197914 | 2171974 | 2171974 | 2174004 |
| XbaI 6 | STM2292 | 2399433 | 2400215 | 2399521 | 2451074 | 2423187 | 2397237 | 2397237 | 2399266 |
| XbaI 7 | STM2394–STM2394 | 2505894 | 2506054 | 2506054 | 2557606 | 2529720 | 2503770 | 2503770 | 2505799 |
| XbaI 8 | STM2584–STM2585 | 2730470 | 2731355 | 2730547 | 2782100 | 2754927 | 2728264 | 2728264 | 2730311 |
| XbaI 9 | STM2616 | 2762064 | 2762627 | 2762167 | |||||
| XbaI 10 | STM2658 | 2799958 | 2800030 | 2800018 | 2818163 | 2790985 | 2800431 | 2800431 | 2802188 |
| 2824797 | 2824797 | ||||||||
| 2853935 | 2826674 | ||||||||
| 2877794 | 2877794 | ||||||||
| 2888767 | 2888767 | ||||||||
| XbaI 11 | STM2742 | 2880444 | 2881637 | 2880896 | 2901121 | 2873861 | 2903461 | 2903461 | 2849374 |
| XbaI 12 | STM2747 | 2887401 | 2888219 | 2887451 | 2907676 | 2880416 | 2910016 | 2910016 | 2855929 |
| XbaI 13 | STM2767 | 2908420 | 2910402 | 2908601 | 2928836 | 2901586 | 2931177 | 2931177 | 2877089 |
| XbaI 14 | STM2767 | 2908420 | 2910402 | 2909761 | 2929996 | 2902746 | 2932337 | 2932337 | 2878249 |
| XbaI 15 | STM3397 | 3570307 | 3570379 | 3570365 | 3584223 | 3593463 | 3591847 | 3591847 | 3531830 |
| XbaI 16 | STM3405–STM3406 | 3576805 | 3576935 | 3576883 | 3590741 | 3599981 | 3598365 | 3598365 | 3538348 |
| XbaI 17 | STM3443–STM3444 | 3597851 | 3597922 | 3597875 | 3611733 | 3620973 | 3619357 | 3619357 | 3559340 |
| XbaI 18 | STM3594–STM3595 | 3765983 | 3766163 | 3766042 | 3779743 | 3788912 | 3787367 | 3787368 | 3727350 |
| XbaI 19 | STM3646 | 3833687 | 3834661 | 3834463 | 3848154 | 3857340 | 3855778 | 3855779 | 3795761 |
| XbaI 20 | STM3714–STM3715 | 3910705 | 3910806 | 3910806 | 3924498 | 3933683 | 3932120 | 3932121 | 3872105 |
| XbaI 21 | STM3785 | 3984439 | 3985182 | 3984523 | 3998215 | 4007398 | 4005836 | 4005837 | 3945822 |
| XbaI 22 | STM3846–STM3847 | 4053276 | 4054076 | 4053903 | 4067596 | 4076779 | 4075214 | 4075215 | 4015203 |
| XbaI 23 | STM3890 | 4101759 | 4101831 | 4101769 | 4115462 | 4124645 | 4123076 | 4123077 | 4063068 |
| XbaI 24 | STM4178 | 4396303 | 4396375 | 4396313 | 4409893 | 4419063 | 4417645 | 4417646 | 4357499 |
| XbaI 25 | STM4285 | 4525350 | 4527497 | 4526230 | 4539772 | 4548929 | 4547522 | 4547523 | 4487378 |
| XbaI 26 | STM4362 | 4604955 | 4606235 | 4606231 | 4619062 | 4628219 | 4626812 | 4626813 | 4566668 |
| XbaI 27 | STM4524 | 4777606 | 4779015 | 4778149 | 4790982 | 4800127 | 4798732 | 4798733 | 4738586 |
Notes:
, XbaI cleavage sites numbered according to their appearance order in LT2 starting from the “beginning” of the genome, i.e., gene thrL.
, One gene ID, such as STM0557, means that the XbaI cleavage site falls in a gene; two gene IDs, such as STM1331–STM1332, mean that the XbaI cleavage site falls in an intergenic region between two genes.
, start and end nucleotide positions, respectively of a gene or an intergenic region between two genes.
Within each of the other Salmonella serotypes analyzed, the CTAG-containing cleavage sites were also highly conserved, with the main differences among the wild type strains being additional cleavage sites in prophages or genomic islands (Supplementary Table S1). For example, S. heidelberg SL476 had three large genomic islands, 58, 30 and 42 kb in size, respectively, all containing multiple XbaI cleavage sites; the 42 kb island, present in S. heidelberg SL476 but not in S. heidelberg B182, contained as many as seven additional XbaI cleavage sites within a 20 kb region (Supplementary Table S1). Other endonucleases (e.g., SpeI) having CTAG in the cleavage sites had similar situations as XbaI (data not shown). The overall conservation of the CTAG-containing endonuclease cleavage sequences in the Salmonella genomes makes it possible to use these endonucleases for the identification of Salmonella isolates. For this, the distinctness of cleavage patterns of endonucleases with CTAG in the cleavage sequences across different Salmonella serotypes (or lineages; a monophyletic Salmonella serotype is equivalent to “a Salmonella lineage” but a polyphyletic Salmonella serotype contains two or more Salmonella lineages) would have to be documented.
CTAG endonuclease cleavage patterns are distinct across Salmonella lineages
Across the 13 Salmonella serotypes analyzed, cleavage patterns for the endonucleases that contain CTAG in the cleavage sites were drastically different and the sites at different genomic locations also had different levels of conservation; here we take XbaI cleavage as an example to illustrate the levels of conservation of the CTAG-containing sequences at different genomic locations. First of all, the XbaI cleavage sites within the tRNA encoding sequences had the highest level of conservation among the 13 Salmonella serotypes and even E. coli strain K12 as illustrated previously (Fig. 4 in [23]). Of great interest, XbaI 3 within an intergenic sequence (between STM1377–STM1378) is also conserved among the 13 Salmonella serotypes and E. coli strain K12; the potential biological function encoded by this genomic region is now under scrutiny. XbaI 4, 16 and 17, located in intergenic sequences between STM1622–STM1623, STM3405–STM3406 and STM3443–STM3444, respectively, are conserved in all analyzed Salmonella strains; characterization of these intergenic sequences for their potential roles in bacterial biology might provide novel insights into the evolution of bacteria. XbaI 26 in STM4362 (hflX) is conserved in all analyzed Salmonella strains, and XbaI 7, located in an intergenic sequence between STM2394–STM2394, is conserved in all Salmonella subgroup I strains analyzed here. Most other XbaI cleavage sites are specific either to one or a subset of Salmonella lineages (Supplementary Table S1). SpeI and other endonucleases having CTAG in the cleavage sites had similar general patterns as XbaI (data not shown). The distinct profiles of the CTAG-containing endonuclease cleavage sequences among the Salmonella serotypes make it possible to use these enzymes for delineating Salmonella into genetically well defined natural clusters, which would have to be further validated by comparisons between CTAG-containing cleavage site profiling and genome sequence information.
Distinct CTAG-containing cleavage profiles to delineate Salmonella into natural lineages: correlation with core genome-based phylogenetics
The high levels of conservation of the CTAG-containing cleavage sequences as exemplified by the distinct XbaI cleavage patterns in different Salmonella lineages suggest that profiling of such sequences may be used to delineate Salmonella into discrete natural lineages. To validate this, we conducted hierarchical clustering analysis on the XbaI cleavage profiling data among the Salmonella strains (Supplementary Table S2). Based on this analysis, we constructed a phylogenetic tree (Figure 5) and compared it to the core genome-based tree (Figure 6); the two trees revealed essentially the same phylogenetic relationships among the Salmonella strains.
Figure 5. Phylogenetic tree constructed with the XbaI cleavage data based on numbers of conserved sites shared by subsets of the bacteria; B.

Figure 6. Phylogenetic tree constructed with concatenated core genome sequences, with the numbers beside the nodes indicating bootstrap values.

Discussion
In this study, we sampled a tiny portion of highly conserved sequences of the Salmonella genome, i.e., the CTAG-containing endonuclease cleavage sequences, as genomic signatures to probe the genetic uniqueness of individual Salmonella lineages and further test our hypothesis that bacteria dwell in nature as discrete genetic clusters. Findings from this may help evaluate and validate the genetic boundary concept, which is the core of our hypothesis. The highly similar genetic backgrounds in sharp contrast to the radical pathogenic differences among Salmonella make this genus of bacteria an ideal model for testing the hypothesis and for the studies of pathogenic evolution that turns benign organisms into infectious agents.
The topic on bacterial diversification, evolution and speciation has been a focus of extensive discussions, especially by investigators viewing from different angles and using different methods [24]–[32]. Originally, we initiated this work on the comparison between S. typhimurium and S. typhi, the former causing self-limiting gastroenteritis but the latter eliciting deadly typhoid fever in humans, to look for distinct genomic features that can be used to unambiguously divide them into discrete bacterial clusters, which, if demonstrated to exist, we call “natural species”, as they should be clusters of bacteria (“species”) formed by natural selection. We recently recognized and characterized clear-cut genomic divergence between them [33], which we defined as the genetic boundary. Such genetic boundaries have been documented in a broad range of bacteria, such as Yesinia and Staphylococcus [14]. In this study, we demonstrate that the selected subset of highly conserved sequences could reveal the genetic boundaries as clearly and reliably as whole genome analyses.
Compared to the whole genome strategies, CTAG-containing sequence profiling for Salmonella has several advantages. First, CTAG-containing cleavage sequence profiling by PFGE requires much less time and resources than genome sequencing strategies but still provides adequate information to delineate Salmonella into discrete genetic clusters, which is especially important when very large numbers of bacterial strains are involved; and second, the collection and analysis of CTAG-containing sequence data profiled by PFGE can be conducted in virtually any molecular biology laboratory equipped with the PFGE apparatus. Additionally, like whole genome sequences, the CTAG-containing cleavage sequence profiles are also objective and can be compared between laboratories and between platforms used. One case to be pointed out here is that monophyletic Salmonella serotypes like S. gallinarum may have diverse PFGE patterns (Fig. 1) of cleavage by XbaI or other endonucleases that have CTAG-containing cleavage sites, which may reduce the value of CTAG-containing endonuclease cleavage sequence profiling. However, even in such cases, well over 50% of the cleavage bands on PFGE are similar among the wild type strains, so creating no ambiguity.
We chose profiling the CTAG-containing endonuclease cleavage sequences to probe the Salmonella genomes for their genetic distinction also because it is a very useful and efficient method for a broad range of studies. For example, in addition to delineating the bacteria into discrete genetic clusters (i.e., natural species), which is our primary objective of this study, the profiling has a particular advantage in tracking the evolutionary scenarios of the Salmonella lineages, because the CTAG-containing sequences, though highly conserved in Salmonella, have been in the process of being eliminated from the genome by the VSP repair mechanism [15]. Assuming that all remaining CTAG-containing sequences through natural selection should be very important, we anticipated to see the gradual degeneracy processes of the CTAG-containing sequences among Salmonella as a whole. Specifically, the levels of conservation of the CTAG-containing sequences can be stratified by comparing their presence and degeneracy status (substitution of any of the CTAG nucleotides by transition or transversion) among the Salmonella lineages. For example, five XbaI cleavage sites are conserved not only across all Salmonella lineages compared in this study but also in E. coli (Supplementary Table S1). Other XbaI cleavage sites are either conserved among the Salmonella lineages but not in E. coli, or among Salmonella subgroup I lineages but not in other subgroups, or among strains of the same lineage, or specific to only particular strains of even the same lineage (in such cases, they are mostly in prophages or genomic islands). The differential profiles of the CTAG-containing cleavage sequences make each of the Salmonella lineages unique for identification, and the different patterns of sequence degeneracy among the Salmonella lineages (Supplementary Table S2) may provide important clues for their strategies in adapting to different environments (e.g., different host species).
Based on our results, we speculate the following evolutionary scenario that makes a small subset of highly conserved sequences to remain as a reliable and informative genetic signature of individual lineages. During the long process of CTAG elimination [15], each Salmonella lineage (dwelling in its own gene pool, [32]) accumulates nucleotide substitutions independently, leading to gradual degeneracy of the CTAG sequences in a particular way specific to each of the Salmonella lineages. Detailed analysis of the substituting and substituted nucleotides during the process of CTAG sequence degeneracy should provide novel insights into the strategy and mechanisms during the adaptation process of individual Salmonella pathogens, especially regarding their interaction with the host that they infect. We conclude that CTAG-containing sequence profiling can be used to unambiguously and efficiently delineate Salmonella into distinct genetic lineages, which are equivalent to the natural species of bacteria.
Materials and Methods
Bacterial strains
Bacterial strains used in this study along with the accession numbers of the sequenced genomes, are listed in Table 2; more detailed information on these bacteria can be found at the Salmonella Genetic Stock Center (http://www.ucalgary.ca/~kesander/). Bacteria were grown overnight at 37°C with shaking in Luria-Bertani (LB) broth or on LB plates. Stock cultures were stored at −70°C in LB broth with 25% glycerol.
Table 2. Bacterial strains used in this studya.
| Strain | Accession numberb | Reference |
| S. typhimurium LT2 | AE006468 | [23] |
| S. typhimurium 14028S | CP001363 | |
| S. typhimurium SL1344 | FQ312003 | |
| S. typhimurium D23580 | FN424405 | |
| S. typhimurium ST4/74 | CP002487 | |
| S. typhimurium UK/1 | CP002614 | |
| S. typhi Ty2 | AE014613 | [17] |
| S. typhi CT18 | NC_003198 | |
| S. typhi P-stx-12 | NC_016832 | |
| S. paratyphi A ATCC9150 | CP000026 | [18] |
| S. paratyphi A AKU_12601 | FM200053 | |
| S. paratyphi C RKS4594 | CP000857 | [12], [36] |
| S. agona SL483 | CP001138 | |
| S. dublin CT_02021853 | CP001144 | |
| S. dublin SD3246 | CM001151 | |
| S. enteritidis P125109 | AM933172 | |
| S. pullorum RKS5078 | CP003047 | [21], [37] |
| S. gallinarum 287/91 | AM933173 | [22] |
| S. gallinarum SGSC2423 | N/A | |
| S. gallinarum SGSC2292 | N/A | |
| S. gallinarum SGSC2293 | N/A | |
| S. gallinarum R1481 | N/A | |
| S. gallinarum R1482 | N/A | |
| S. gallinarum R1483 | N/A | |
| S. gallinarum SARB21 | N/A | |
| S. choleraesuis A50 | CM001062 | |
| S. choleraesuis SC-B67 | AE017220 | |
| S. heidelberg B182 | NC_017623 | |
| S. heidelberg SL476 | CP001120 | |
| S. newport SL254 | CP001113 | |
| S. schwarzengrund CVM19633 | CP001127 | |
| S. arizonae RKS2980 | CP000880 | |
| S. arizonae RKS2893 | CP006693 | |
| S. bongori NCTC 12419 | FR877557 | |
| S. bongori RKS3044 | CP006692 |
See more detailed information on these bacterial strains at www.ucalgary.ca/~kesander.
N/A means that the bacterial strain is not sequenced and the genome sequence was not needed for this study.
Reagents and PFGE analyses of genomic DNA
I-CeuI, XbaI and AvrII were purchased from New England Biolabs, and proteinase K was from Roche. Most other reagents were from Sigma. Bacterial genomic DNA isolation, endonuclease cleavage with I-CeuI, XbaI and AvrII, and separation of the cleavage fragments were described previously [8], [17], [34]. Briefly, PFGE was used to separate DNA fragments cleaved by the endonucleases, and I-CeuI partial cleavage was used to lay out the overall genome structure of bacteria. PFGE was done in a CHEF DR II electrophoresis system (BioRad) at 5.6 V/cm with 0.5×TBE buffer as the running buffer.
Genomic and statistics analysis tools
We determined the phylogenetic relationships of the bacteria based on their differences in the numbers of conserved CTAG-containing endonuclease cleavage sites common to subsets of Salmonella strains or sequence identity of genes common to them using the neighbor-joining (NJ) method, and the tree construction was done with MEGA4.0.2 [35] and CLUSTALW. The statistical analyses were performed by using software SPSS v20.
Supporting Information
Profiles of XbaI cleavage sites in representative Salmonella genomes.
(XLS)
Numbers of XbaI cleavage sites common to pairs of the Salmonella genomes.
(XLS)
Funding Statement
This work was supported by a Heilongjiang Innovation Endowment Award for graduate studies (YJSCX2012-197HLJ) to LT; a National Natural Science Foundation of China (NSFC30970078) and a grant of Natural Science Foundation of Heilongjiang Province of China to GRL; a grant of Canadian Institutes of Health Research to RNJ; and grants of the National Natural Science Foundation of China (NSFC30970119, 81030029, 81271786, NSFC-NIH 81161120416) to SLL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Popoff MY, Bockemuhl J, Gheesling LL (2004) Supplement 2002 (no. 46) to the Kauffmann-White scheme. Research in microbiology 155: 568–570. [DOI] [PubMed] [Google Scholar]
- 2.Popoff MY, Le Minor LE (2005) Genus XXXIII. Salmonella. In: D. JBrenner, N. RKrieg and J. TStanley, editors. Bergey's Mannual of Systematic Bacteriology. Springer. pp. 764–799. [Google Scholar]
- 3. Edwards PR, Kauffmann F (1952) A simplification of the Kauffmann-White schema. Am J Clin Pathol 22: 692–697. [DOI] [PubMed] [Google Scholar]
- 4. Le Minor L, Popoff MY (1987) Designation of Salmonella enterica sp. nov., nom. rev., as the type and only species of the genus Salmonella. International journal of systematic bacteriology 37: 465–468. [Google Scholar]
- 5. Reeves MW, Evins GM, Heiba AA, Plikaytis BD, Farmer JJ 3rd (1989) Clonal nature of Salmonella typhi and its genetic relatedness to other salmonellae as shown by multilocus enzyme electrophoresis, and proposal of Salmonella bongori comb. nov. Journal of clinical microbiology 27: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Minor L (1984) Genus III. Salmonella. In: N. RKrieg, editor editors. Bergey's Manual of Systematic Bacteriology. Baltimore: Williams & Wilkins. pp. 427–458. [Google Scholar]
- 7. Crosa JH, Brenner DJ, Ewing WH, Falkow S (1973) Molecular relationships among the Salmonelleae. Journal of bacteriology 115: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu SL, Hessel A, Sanderson KE (1993) Genomic mapping with I-Ceu I, an intron-encoded endonuclease specific for genes for ribosomal RNA, in Salmonella spp., Escherichia coli, and other bacteria. Proc Natl Acad Sci U S A 90: 6874–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu SL, Schryvers AB, Sanderson KE, Johnston RN (1999) Bacterial phylogenetic clusters revealed by genome structure. Journal of bacteriology 181: 6747–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, et al. (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413: 852–856. [DOI] [PubMed] [Google Scholar]
- 11. Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, et al. (2001) Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413: 848–852. [DOI] [PubMed] [Google Scholar]
- 12. Liu WQ, Feng Y, Wang Y, Zou QH, Chen F, et al. (2009) Salmonella paratyphi C: genetic divergence from Salmonella choleraesuis and pathogenic convergence with Salmonella typhi. PLoS ONE 4: e4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parry CM, Hien TT, Dougan G, White NJ, Farrar JJ (2002) Typhoid fever. N Engl J Med 347: 1770–1782. [DOI] [PubMed] [Google Scholar]
- 14. Tang L, Li Y, Deng X, Johnston RN, Liu GR, et al. (2013) Defining natural species of bacteria: clear-cut genomic boundaries revealed by a turning point in nucleotide sequence divergence. BMC Genomics 14: 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhagwat AS, McClelland M (1992) DNA mismatch correction by Very Short Patch repair may have altered the abundance of oligonucleotides in the E. coli genome. Nucleic Acids Res 20: 1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu SL, Sanderson KE (1995) I-CeuI reveals conservation of the genome of independent strains of Salmonella typhimurium. Journal of bacteriology 177: 3355–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu SL, Sanderson KE (1995) Genomic cleavage map of Salmonella typhi Ty2. Journal of bacteriology 177: 5099–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu SL, Sanderson KE (1995) The chromosome of Salmonella paratyphi A is inverted by recombination between rrnH and rrnG. Journal of bacteriology 177: 6585–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu SL, Hessel A, Cheng HY, Sanderson KE (1994) The XbaI-BlnI-CeuI genomic cleavage map of Salmonella paratyphi B. Journal of bacteriology 176: 1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marshall P, Lemieux C (1991) Cleavage pattern of the homing endonuclease encoded by the fifth intron in the chloroplast large subunit rRNA-encoding gene of Chlamydomonas eugametos. Gene 104: 241–245. [DOI] [PubMed] [Google Scholar]
- 21. Wu KY, Liu GR, Liu WQ, Wang AQ, Zhan S, et al. (2005) The genome of Salmonella enterica serovar gallinarum: distinct insertions/deletions and rare rearrangements. Journal of bacteriology 187: 4720–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, et al. (2008) Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res 18: 1624–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu SL, Hessel A, Sanderson KE (1993) The XbaI-BlnI-CeuI genomic cleavage map of Salmonella typhimurium LT2 determined by double digestion, end labelling, and pulsed-field gel electrophoresis. Journal of bacteriology 175: 4104–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plucain J, Hindre T, Le Gac M, Tenaillon O, Cruveiller S, et al. (2014) Epistasis and allele specificity in the emergence of a stable polymorphism in Escherichia coli. Science (New York, NY 343: 1366–1369. [DOI] [PubMed] [Google Scholar]
- 25. Barrick JE, Lenski RE (2013) Genome dynamics during experimental evolution. Nat Rev Genet 14: 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Covert AW 3rd, Lenski RE, Wilke CO, Ofria C (2013) Experiments on the role of deleterious mutations as stepping stones in adaptive evolution. Proc Natl Acad Sci U S A 110: E3171–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wielgoss S, Barrick JE, Tenaillon O, Wiser MJ, Dittmar WJ, et al. (2013) Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc Natl Acad Sci U S A 110: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP (2009) The bacterial species challenge: making sense of genetic and ecological diversity. Science (New York, NY 323: 741–746. [DOI] [PubMed] [Google Scholar]
- 29. Cohan FM (2001) Bacterial species and speciation. Systematic biology 50: 513–524. [DOI] [PubMed] [Google Scholar]
- 30. Cohan FM (2002) What are bacterial species? Annual review of microbiology 56: 457–487. [DOI] [PubMed] [Google Scholar]
- 31. Koeppel AF, Wertheim JO, Barone L, Gentile N, Krizanc D, et al. (2013) Speedy speciation in a bacterial microcosm: new species can arise as frequently as adaptations within a species. ISME J 7: 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang L, Liu SL (2012) The 3Cs provide a novel concept of bacterial species: messages from the genome as illustrated by Salmonella. Antonie Van Leeuwenhoek 101: 67–72. [DOI] [PubMed] [Google Scholar]
- 33. Tang L, Wang CX, Zhu SL, Li Y, Deng X, et al. (2013) Genetic boundaries to delineate the typhoid agent and other Salmonella serotypes into distinct natural lineages. Genomics 102: 331–337. [DOI] [PubMed] [Google Scholar]
- 34. Liu SL (2007) Physical mapping of Salmonella genomes. Methods Mol Biol 39–58. [DOI] [PubMed] [Google Scholar]
- 35. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
- 36. Liu WQ, Liu GR, Li JQ, Xu GM, Qi D, et al. (2007) Diverse genome structures of Salmonella paratyphi C. BMC Genomics 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng Y, Xu HF, Li QH, Zhang SY, Wang CX, et al. (2012) Complete genome sequence of Salmonella enterica serovar pullorum RKS5078. Journal of bacteriology 194: 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Profiles of XbaI cleavage sites in representative Salmonella genomes.
(XLS)
Numbers of XbaI cleavage sites common to pairs of the Salmonella genomes.
(XLS)