

Abstract
Personalized medicine is at the forefront of cancer diagnosis and therapy. Molecularly targeted therapies such as trastuzumab and tamoxifen have enhanced prognosis of patients with cancers expressing ERBB2 and the estrogen receptor, respectively. One obstacle to targeted therapy is the development of resistance. A targeted peptide that could distinguish resistance-susceptible cancer would aid in treatment. BT-474 human breast cancer cells can be resistant to both tamoxifen and trastuzumab, and may serve as a model for malignancies in which targeted therapy may not work. Bacteriophage (phage) display is a combinatorial technology that has been used to isolate peptides that target a specific cancer subtype. It was hypothesized that in vivo phage display could be used to select a peptide for SPECT imaging of BT-474 human breast cancer xenografts. A phage library displaying random 15 amino acid peptides was subjected to four rounds of selection, after which 14 clones were analyzed for BT-474 binding and specificity. One phage clone, 51, demonstrated superior binding and specificity, and the displayed peptide was synthesized for in vitro characterization. Peptide 51 bound specifically to BT-474 cells with an EC50 = 2.33 µM and was synthesized as a DOTA-conjugated peptide and radiolabeled with 111In for in vitro and in vivo analysis. The radiolabeled peptide exhibited an IC50 = 16.1 nM to BT-474 cells and its biodistribution and SPECT imaging in BT-474 xenografted mice was analyzed. Although tumor uptake was moderate at 0.11% ID/g, SPECT imaging revealed a distinct tumor vasculature binding pattern. It was discovered that peptide 51 had an identical 5 amino acid N-terminal sequence to a peptide, V1, which bound to Nrp1, a tumor vasculature protein. Peptide 51 and V1 were examined for binding to target cells, and 51 bound both target and endothelial cells, while V1 only bound endothelial cells. Truncated versions of 51 did not bind BT-474 cells, demonstrating that the targeting ability of 51 was independent of the homologous V1 sequence. These results demonstrate that in vivo phage display can effectively identify a peptide that specifically targets a breast cancer cell line that is susceptible to targeted therapy resistance.
Keywords: Peptide, phage display, molecular imaging, breast cancer
Introduction
The ability to detect malignant tissue non-invasively remains an important factor in diagnosing and treating carcinomas, including breast cancer. Traditionally, methods of diagnosis, such as mammography and 18F-fluorodeoxyglucose positron emission tomography, are used to identify areas of malignancy. Other targeted imaging agents, including radiolabeled peptides for single photon emission computed tomography/x-ray computed tomography (SPECT/CT), have been incorporated in diagnostic procedures to not only precisely pinpoint tumors, but also provide biologically relevant information about the malignancy non-invasively [1]. Additionally, molecular characterization of the tumor often dictates the course of treatment. For example, breast cancers that express the estrogen receptor and the receptor tyrosine kinase ERBB2 are often treated with the estrogen receptor antagonist tamoxifen and anti-ERBB2 antibody trastuzumab in combination with a chemotherapeutic agent [2,3]. Despite the success accomplished using these treatment strategies, a major obstacle is the occurrence of resistance to the targeted therapies, with 30% developing tamoxifen resistance, and greater than 50% developing trastuzumab resistance [4,5]. A targeting agent with the ability to distinguish cancers prone to develop resistance would greatly aid in the direction of treatment strategies.
Currently, there is no predictive diagnostic agent to assess resistance to targeted therapies. In order to develop an imaging agent capable of detecting carcinomas susceptible to therapeutics such as tamoxifen and trastuzumab, a proper animal model of breast cancer expressing the estrogen receptor and ERBB2 that can be investigated in vivo and accurately mimicked in vitro is needed. A breast cancer cell line that forms reliable tumors in a mouse, expresses both the estrogen receptor and ERBB2 at physiologically relevant levels, and has been shown to develop resistance to therapies such as tamoxifen and trastuzumab could potentially serve as a template for resistance-susceptible breast cancer. When choosing an applicable cell line for targeted therapy resistance, the most often used human breast cancer cell lines that form tumors in mice can be surveyed for estrogen receptor and ERBB2 status. It is well established that T47D and MCF7 cell lines, which are estrogen receptor positive, do not express ERBB2 [6,7]. Likewise, SK-BR-3 and MDA-MB-453 cells over-express ERBB2 but lack detectable estrogen receptor [7]. However, BT-474 human breast cancer cells are estrogen-dependent, over-express ERBB2 and form tumors in mice [8,9]. Although estrogen dependant, BT-474 tumors are naturally resistant to tamoxifen, a widely used antiestrogen therapy [10]. Tamoxifen resistance is thought to be mediated by the over-expression of ERBB2, a major driver of breast cancer [4]. Trastuzumab, a humanized monoclonal antibody, has been successfully used for treating approximately 50% of cancers that over-express ERBB2 [2]. Interestingly, BT-474 cells have been demonstrated to develop resistance to trastuzumab [5]. The BT-474 cell line, therefore, offers a unique opportunity as a target for developing an imaging agent capable of detecting breast carcinomas susceptible to resistance to multiple targeted therapies, namely tamoxifen and trastruzumab. A targeted agent specific for BT-474 breast cancer may offer a novel method of identifying resistance susceptible cancers prior to treatment.
Bacteriophage (phage) display has been used to successfully select imaging agents, such as peptides, with the affinity and specificity to image human cancer in vivo [11]. Following the discovery that phages tolerate insertion of foreign peptide sequences while retaining the functions of infection and replication, phage display has been used to identify peptides from a library of random sequences based on a desired target [12]. The power of phage display is derived from the ability to test up to 109 unique phage displayed sequences simultaneously for the optimal peptide based on a desired function. Phages are incubated with a target, allowing a portion to bind, while the unbound phages are removed. Although only a small portion may bind to the target, the recovered phages are exponentially increased by propagation in a host bacterial cell. The enriched subpopulation is then subjected to a subsequent round of selection, providing an enhanced level of competition due to the increased number of target-avid clones represented in the total phage population. Following a number of rounds of selection, the output of phage generally represents the fittest clones for the desired function. This process can be used to identify peptides which bind specifically to an antigen which is expressed or over-expressed in cancer, in addition to identifying peptides specific for breast cancer cell lines [13]. Phage display has been used in vitro to identify peptides which bind to a host of antigens and image tumor cells in vivo, including integrins, receptor tyrosine kinases, and carbohydrate antigens [14-16].
In vivo phage display provides the combinatorial power of a traditional selection, while offering unique advantages. By probing a tumor in the context of a living system, antigens are more likely to be presented in the manner in which they would be found in a patient. The selection is favorably biased towards antigens that are accessible to the tumor vasculature, which may differ from those identified ex vivo or in vitro [17]. An additional benefit is that peptides must successfully avoid binding to antigens displayed in non-target organs in order to be captured, decreasing the likelihood of non-target organ uptake. In vivo phage display has been demonstrated to select peptides that bind human tumors and specifically target the vasculature of most organs, including tumor vasculature [18,19].
In vivo phage display is especially useful when a specific protein target is not known. For example, BT-474 cells are known to express ERBB2 and estrogen receptor, however, neither marker by itself is predictive of susceptibility to resistance [5,9,10]. It is extremely likely, however, that many potential targets on BT-474 cells may serve not only as novel breast cancer antigens, but also as predictors of therapeutic resistance. In vivo phage display can serve as an initial screen for peptides which specifically target BT-474 cells, providing the basis for development an imaging agent for further refinement and characterization. It was hypothesized that a novel BT-474 targeted peptide could be selected by in vivo phage display, which would possess the capability of detecting human breast tumors in xenografted mice. To test this, 4 rounds of in vivo selection were performed, and individual phages were characterized for their ability to bind BT-474 cells in vitro. A peptide corresponding to a phage with high specificity and affinity for the target cells was synthesized as a biotinylated conjugate and tested for cell binding using fluorescent confocal microscopy, flow cytometry and colorimetric binding assays. Retained affinity and specificity of the biotinylated peptide in vitro warranted analysis of the peptide as an 111In-radiolabeled SPECT imaging agent. Following confirmation of specificity and high affinity of the radiolabeled peptide, in vivo biodistribution was assessed. Finally, the peptide was tested as for the ability to detect BT-474 human breast tumors by SPECT/CT.
Materials and methods
Materials
Materials for cell culture were obtained from Invitrogen (Carlsbad, CA). Unless otherwise specified, all other materials were purchased from Sigma Chemical Co. (St. Louis, MO).
Cell lines
BT-474 cells were grown in RPMI-1640 with 10% heat-inactivated fetal bovine serum (FBS), 4.5 g/L D-glucose, 2.83 g/L HEPES buffer, L-glutamine, 1.5 g/L sodium bicarbonate, 110 mg/L sodium pyruvate, and 48 µg/ml gentamicin at 37°C in 5% CO2. 184A.1 and human umbilical vein endothelial (HUVEC) cells were grown in RPMI 1640 with 10% FBS and 48 µg/ml gentamicin. Cell lines were examined for viability and presence of pathogens prior to injection into mice.
Mouse strains and handling
Four- to 6-week-old athymic nude mice were purchased from Harlan (Indianapolis, IN) and maintained in approved pathogen-free institutional housing. Animal studies were conducted as outlined in the NIH Guidelines for the Care and Use of Laboratory Animals and the Policy and Procedures for Animal Research of the Harry S. Truman Veterans Memorial Hospital. To establish solid tumors, BT-474 human breast cancer cells (5 × 106) were subcutaneously injected into the rear flank of athymic nude mice. Time-release 17β-estradiol pellets (Innovative Research, Sarasota, FL) were implanted subcutaneously to supplement tumor growth. Visible tumors formed after approximately 5 weeks. Mice injected with either phage or radiolabeled peptide were euthanized prior to excision of tumors and organs of interest.
in vivo phage display selection
A library of phage displaying 15 random amino acids from the N-terminal tip of cpIII, in the fUSE5 vector, was a generous gift from Dr. George P. Smith. In order to remove phages with a propensity to bind normal tissues and vasculature, 1 × 1012 transducing units (TU) of library was injected into non-tumor bearing mice and unbound phages were recovered from the blood 15 min after initial injection, amplified and purified, as previously described [20]. The pre-cleared library was used for the ensuing rounds of selection in BT-474 tumor bearing mice. Briefly, 1 × 1012 TU of pre-cleared phage was injected into BT-474 xenografted mice and allowed to circulate for 4 h. Mice were anesthetized and tumors excised and frozen in liquid nitrogen. The tumors were manually homogenized and washed 10x with Tris buffered saline with 0.1% Tween-20 (0.1% TBST) in order to remove non-specifically bound phages. The tumor homogenate was then incubated with 2.5% (w:v) 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) for 1 h to elute bound phages and disrupt cells for recovery of any phage that had been internalized by cells. Eluted phages were used to infect log phase K91BK Escherichia coli (E. coli) for amplification, followed by purification by polyethylene glycol as described previously [http://www.biosci.missouri.edu/smithGP/PhageDisplayWebsite/PhageDisplayWebsiteIndex.html]. The tumor-avid, amplified library was used for the subsequent round of selection, which proceeded exactly as the first round. A total of four rounds of selection were performed in BT-474 human breast tumor bearing mice.
Analysis of selected phages
Following the fourth round of selection, 96 individual phages were sequenced in order to ascertain their displayed peptides. The sequences were analyzed for multiple occurrences, partial sequence multiple occurrences, amino acid frequency and by algorithms including basic local alignment search tool (BLAST) and the scanner and reporter of target-unrelated peptides algorithm (SAROTUP) [21,22]. Phage displayed peptides found in previous unrelated selections were removed from consideration and 14 phages were chosen for cell binding assays. Each purified phage clone was diluted to 1 × 108 TU/mL in RPMI and 100 µL was incubated with either 1 × 105 BT-474 human breast cancer or 184A.1 normal breast epithelial cells for 1 h at 37°C. Cells were washed three times with 0.1% TBST and bound phage were eluted by incubation with 2.5% CHAPS at 4°C for 1 h. Eluted phage were quantified by titration and infection of E. coli.
Peptide synthesis
The amino acid sequence corresponding to the displayed peptide of clone 51, in addition to the N-terminal 7 amino acid sequence (51N) and C-terminal 8 amino acid sequence (51C) and a vascular endothelial growth factor (VEGF) inhibiting peptide (V1) were chemically synthesized. Synthesis occurred using an Advanced Chem Tech 396 multiple peptide synthesizer (Advanced Chem Tech, Louisville, KY) by solid phase fluorenylmethyloxycarbonyl (FMOC) chemistry. Biotin was covalently coupled to each peptide at the n-terminus with a tripeptide glycine-serine-glycine tripeptide (GSG) spacer. The full length 51 peptide was also conjugated to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) (Macrocyclic, Inc. Dallas, TX) by a GSG spacer.
Fluorescent microscopy
BT-474, 184A.1 and HUVEC human endothelial cells were fixed in 4% paraformaldehyde and dried onto microscope slides. Following rehydration with TBS, cells were blocked with 6% (w:v) bovine serum albumin (BSA) for 1 h at room temperature. Peptides 51, 51N and V1 were diluted to 10 µM in 0.1% TBST. After blocking, 100 µL of the appropriate peptide solution was added to cells and allowed to bind for 1 h at room temperature. Slides were washed three times with 0.1% TBST and 100 µL of α-Biotin-Alexafluor 488 conjugated monoclonal antibody diluted 1:1000 in 0.1% TBST was added to cells and incubated at room temperature for 1 h. Slides were washed 3x with 0.1% TBST and analyzed by an epifluorescent-equipped Nikon T1-SM inverted microscope (Nikon, Melville, NY).
Flow cytometry analysis of peptide binding
In the same manner as fluorescent microscopy, BT-474, 184A.1 and HUVEC cells were fixed and diluted to 1 × 106 cells/mL in RPMI and preincubated with a 1:1000 dilution of α-Biotin-Alexafluor 488 antibody. Peptides (51, 51N, V1) were diluted from 100 nM to 100 µM in 0.1% TBST and incubated with cells for 1 h at 37°C. Cells were washed three times with 0.1% TBST, counted and fluorescence quantified per cell by a BD FACScan flow cytometer (BD Biosciences, San Jose, CA).
96 well colorimetric cell binding assay
BT-474 and 184A.1 cells were grown to 80% confluency in TPP 96 well flat bottom tissue culture plates and fixed with 4% paraformaldehyde. Biotinylated 51, 51N and 51C were diluted from 10 nM to 100 µM in RPMI and incubated with cells for 1 h at room temperature. Cells were washed three times with 0.1% TBST and 100 µL horseradish peroxidase-conjugated streptavidin (1 µg/mL) was added to cells and allowed to bind for 1 h at room temperature. Again, washing occurred in the same manner and 100 µL of 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) was incubated with cells for 15 min at room temperature. Color development was terminated by the addition of 1% (w:v) sodium dodecyl sulfate and absorbance at 405 nm was quantified using a µQuant Universal Microplate Spectrophotometer (Bio-Tek Instruments, Winooski VT).
Radiolabeling and peptide cell binding assays
DOTA-conjugated 51 peptide was diluted to 1 mg/mL in water and 20 µL was added to 200 µL of 0.1 M ammonium acetate and 18.5 MBq of 111InCl3. The reaction was incubated at 85°C for 1 h and terminated by the addition of 10 µL of 0.1 M ethylenediaminetetraacetic acid. Reversed phase HPLC using a linear gradient from 5-95% acetonitrile and 0.1% (v:v) trifluoroacetic acid was used to purify the radiolabeled peptide. For cell binding analysis, BT-474 cells were diluted to 1 × 107 cells/mL in RPMI with 1% (w:v) BSA. Purified, radiolabeled 111In-DOTA-51 peptide was diluted to 1 × 106 CPM/mL in RPMI plus 1% BSA and 100 µL were added to 200 µL cells. Unlabeled DOTA-51 was serial diluted and added to radiolabeled peptide and cells at concentrations from 100 pM to 1 µM and incubated at 37°C for 1 h. Cells were washed with ice cold PBS with 0.1% BSA three times and counted by gamma counter (Perkin Elmer, Santa Clara, CA).
Radiolabeled peptide biodistribution
111In-DOTA-51 peptide was radiolabeled and purified as described in in vitro cell binding. Radiolabeled peptide was then diluted with sterile PBS to 1.85 MBq/mL. Four mice bearing BT-474 tumors were intravenously injected with 100 µL of 111In-DOTA-51 and sacrificed at 2 h post-injection. Following sacrifice, pertinent organs and tissues were excised, weighed and counted via gamma counter. Uptake was normalized by weight as percentage of injected dose per gram of tissue (%ID/g).
MicroSPECT/CT imaging
111In-DOTA-51 was radiolabeled, purified and diluted to 11.1 MBq in 100 µL of sterile PBS. The radiolabeled peptide was injected intravenously into a mouse bearing a BT-474 human breast tumor xenograft. After allowing the peptide to circulate for 2 h post-injection, the mouse was sacrificed and imaged at the Biomolecular Imaging Center at the Harry S. Truman Veterans Memorial Hospital. Acquisition of the image proceeded for 7 h using a Siemens Inveon Micro-SPECT/CT (Siemens, Knoxville, TN) outfitted with mouse whole body 1.0 mm collimators. Processing of the image data was accomplished using Inveon Research Workplace processing software. Fan beam (Feldkamp) filtered back projection algorithms were employed to reconstruct the CT tomographic image.
Results
In vivo selected phage characterization
Following the completion of four rounds of in vivo selection, 96 individual phages were isolated and their relevant DNA sequenced to obtain the displayed peptide amino acid sequence. Selection of phages for cell binding characterization was first accomplished by analyzing peptides for the presence of target unrelated peptides. Sequences were compared to those from previous published selections with unrelated targets using the SAROTUP algorithm [22]. Of the 96 total sequences, 28 were reported in previous selections. Phages that bound targets unrelated to breast cancer, including the blood-brain barrier, hemagglutinin A, polyclonal rabbit antibody, and normal tissue were excluded from consideration [23-26]. Instead, 14 phages unique to the selection, and listed in Figure 1A, were chosen because they were found multiple times, or a portion of the sequence was present in multiple phages. Phages were purified and analyzed for BT-474 specificity and apparent affinity.
Figure 1.
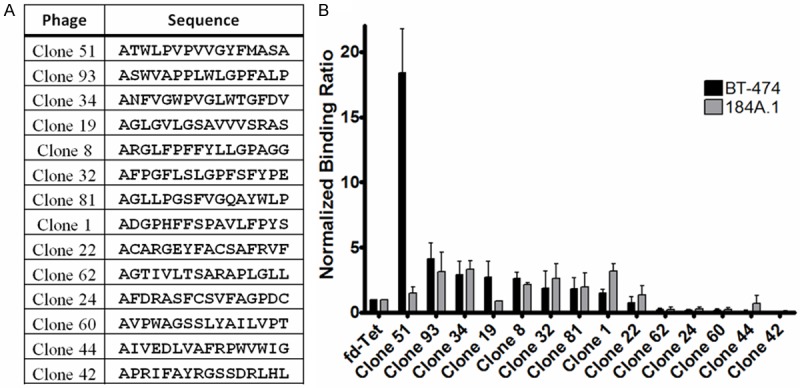
Analysis of Selected Phage Clones. Round 4 of the in vivo phage display selection was sequenced and 14 individual phages were chosen for cell binding analysis. A: Individual phage and the corresponding amino acid sequence are depicted. B: Individual phage were incubated with either target BT-474 cells or normal 184A.1 breast epithelial cells. Total bound phages were quantified and normalized to wild-type phage. Shaded bars represent a meanof three replicate experiments, error bars denote standard deviation.
Individual phages were tested for their ability to selectively bind BT-474 human breast cancer cells while not binding 184A.1 cells, a normal breast epithelial cell line. Recovered phages were normalized to the binding of insertless wild-type phage as an internal control between experiments. The relative binding ratio of each phage in comparison to the wild-type phage was used to assess the relative specificity and affinity of each displayed peptide (Figure 1B). Of the 14 phages analyzed, 5 bound 2-fold greater or higher than wild-type to the target BT-474 cell line. However, when accounting for specificity by assessing the binding to 184A.1 breast epithelial cells, only 2 clones appeared to be specific for BT-474 cells. Clones 51 and 19 bound 18.4 and 2.7 times higher to BT-474 cells than the wild-type phage, respectively. Additionally, the phages preferentially bound breast cancer cells, with binding ratios to BT-474/184A.1 cells of 1.48 for Clone 51 and 0.78 for Clone 19. Clone 51, which bound nearly 7 times more to BT-474 cells than any other phage and did not bind normal breast epithelial binding, was chosen for investigation of its displayed peptide outside of the phage scaffolding.
Peptide 51 in vitro cell binding
Peptide 51 (ATWLPVPVVGYFMASA) was covalently linked to biotin for detection in cell binding assays. The peptide was first analyzed by fluorescent microscopy as a qualitative assessment of binding. Fluorescent images demonstrated that the peptide bound to BT-474 human breast cancer cells and had no detectable binding to normal breast epithelial cells (Figure 2A). A control peptide chosen from a target unrelated phage was used as a control and demonstrated no binding to either cell line. For confirmation of the results of fluorescent microscopy and to attempt to quantify peptide 51 binding, flow cytometry was performed. Peptide 51 bound with moderate affinity for BT-474 cells, with a calculated fifty-percent effective concentration (EC50) = 2.33 ± 0.66 µM (Figure 2B). Binding to BT-474 cells was also significantly higher than 184A.1 cells at all peptide concentrations analyzed, and peptide 51 binding to 184A.1 cells did not display a sigmoid dose response, indicating a lack of specificity for the cells. A satisfactory affinity for BT-474 cells, in addition to minimal normal breast tissue binding, indicated that the peptide would be a suitable candidate for development as a radiolabeled imaging agent.
Figure 2.
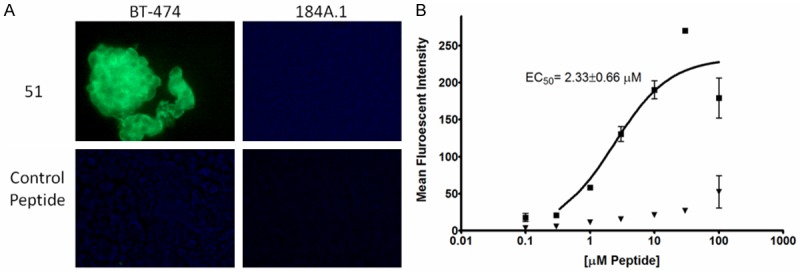
Peptide 51 In Vitro Cell Binding Assays. A: Biotinylated peptide 51 and a control peptide were incubated with BT-474 human breast cancer and 184A.1 normal breast epithelial cells fixed onto microscope slides. Following washing, bound peptides were detected by addition of an anti-biotin Alexafluor 488-conjugated antibody. Strong binding is observed for 51 with the target BT-474 cells, but not normal breast epithelial cells. The control peptide does not exhibit binding to either cell line. B: Peptide 51 was analyzed for BT-474 and 184A.1 specificity and affinity by flow cytometry. Peptides were diluted from 100 nM to 100 µM in 0.1% TBST. Following incubation of peptide with cells, bound peptide was detected by anti-biotin Alexafluor 488. Squares represent the mean of 3 BT-474 replicates at the indicated peptide concentration, triangles represent the mean of 3 184A.1 replicates. Error bars represent the standard deviation.
In vitro and in vivo 111In-DOTA-51 analysis
For radiolabeled peptide assessment, peptide 51 was conjugated to DOTA through an N-terminal GSG spacer and radiolabeled with 111In. 111In-DOTA-51 was first tested for a retained affinity for BT-474 cells in vitro using unlabeled peptide in a competition assay. The relative fifty-percent inhibition concentration (IC50) was calculated at 16 ± 7 nM, an affinity comparable to previous peptides used for in vivo analysis (Figure 3) [15,27]. 111In-DOTA-51 was injected into mice bearing BT-474 human breast cancer xenografts for pharmacokinetic analysis. The biodistribution of 111In-DOTA-51 revealed tumor uptake of 0.12 ± 0.02 %ID/g (Figure 4). The tumor to blood ratio was determined to be 2.3 and the tumor to muscle ratio was 7.1, indicating specificity of the peptide for BT-474 human breast tumors. Tumor uptake in all other organs was low, further confirming tumor specificity. In particular, organs that could produce background signal for breast cancer imaging, including the heart (0.04 ± 0.01 %ID/g), and lung (0.13 ± 0.03 %ID/g) were low (Figure 4). Radiosensitive organ uptake, such as bone (0.03 ± 0.02 %ID/g) was also minimal (Figure 4), which is important in the development of a safe and effective radiolabeled imaging agent. SPECT/CT image analysis revealed high peptide uptake surrounding the tumor, in addition to kidney uptake, which coincided with the measured kidney retention of 30.4 %ID/g (Figure 5). The pattern of tumor uptake was similar to known vasculature targeting agents such as radiolabeled anti-VEGF antibody and arginine-glycine-aspartic aciced tripeptide (RGD) [28,29]. Although the sequences did not contain any well known vasculature targeting motifs, such as RGD, peptide 51 sequence was rigorously compared to known vasculature targeting peptides in order to determine if they shared any other partial homology.
Figure 3.
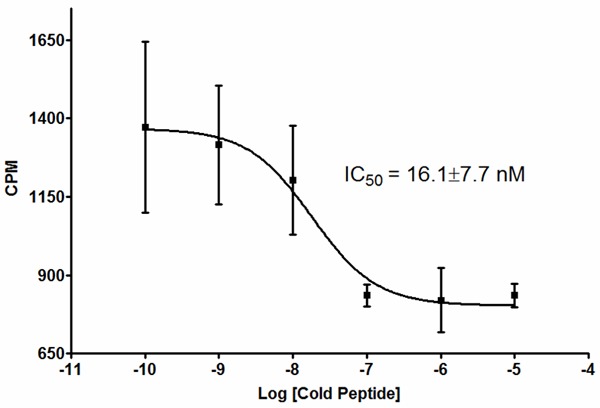
111In-DOTA-51 Cell Binding Competition. 111In-DOTA-51 was diluted to 1 × 106 CPM/mL in RPMI and 1% BSA and 100 µL was added to 1 million BT-474 cells suspended in 100 µL of the same buffer. Unlabeled DOTA-51 was serially diluted from 100 pM to 1 µM and added to labeled peptide and cells. Following a 1 h incubation at 37°C, cells were washed and bound radioactivity quantified. Square boxes reperesent the mean of 3 replicates and error bars represent standard deviation. CPM - Counts per minute.
Figure 4.
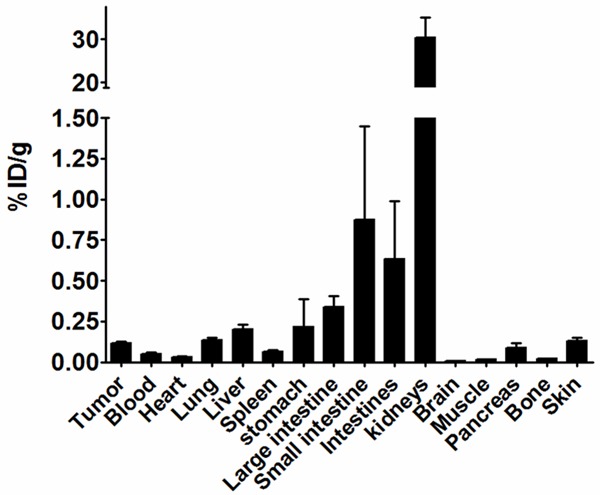
111In-DOTA-51 Biodistribution. 111In-DOTA-51 was prepared at 1.85 Mbq/mL in sterile PBS and injected into BT-474 xenograft bearing mice. At 2 h post-injection, animals were sacrificed, organs removed, and total radioactivity counted by gamma counter. Each bar represents the average of 4 mice, and error bars denote standard deviation.
Figure 5.
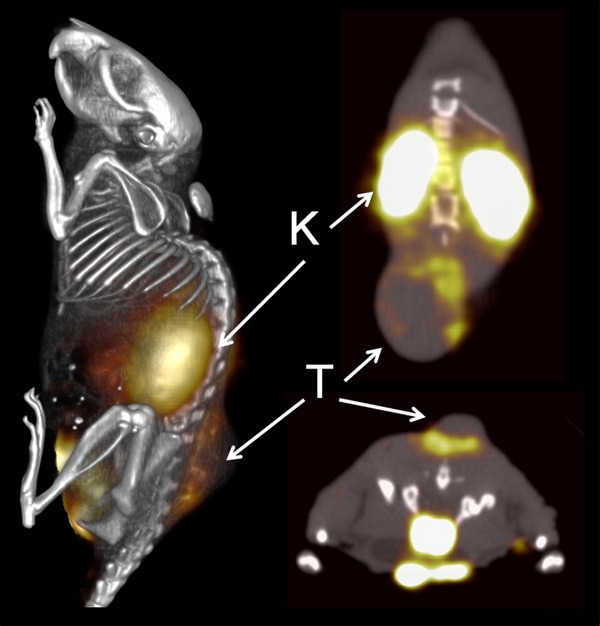
111In-DOTA-51 SPECT Imaging. A BT-474 tumor bearing mouse was injected with 11.1MBq of 111In-DOTA-51 and peptide was allowed to circulate for 2 h prior to imaging. SPECT/CT images were acquired overnight. All images are from the same mouse. T - Tumor, K - Kidney.
Upon extensive literature review, it was noticed that peptide 51 shared a 5 amino acid homology with a peptide that had been previously selected by phage display and demonstrated to bind neuropilin-1 (Nrp1) and inhibit VEGF-mediated angiogenesis [30]. The peptide, V1 (ATWLPPR), was remarkably similar to the N-terminal residues in Clone 51, whose full length sequence was ATWLPVPVVGYFMASA. Since V1 did not share exact identity to peptide 51 and the peptides were of differing sequence lengths, preliminary searches for target unrelated peptides did not identify the similarity between V1 and 51.
Examining the role of a homologous V1 sequence in peptide 51
Since peptide 51 shared homology with a vasculature antigen-targeting peptide, it was necessary to determine whether the properties of peptide 51 were mediated by the shared ATWLP sequence. Binding of the full length peptide (51), the N-terminal 7 residues (51N - ATWLPVP) and the V1 peptide (ATWLPPR) was analyzed using fluorescence microscopy for target BT-474 breast cancer cells, 184A.1 breast epithelial cells, and an endothelial cell line demonstrated previously as a target of V1 [31]. Full length 51 bound strongly to BT-474 cells, while neither 51N nor V1 displayed detectable fluorescence to the same cell line, indicating only 51 visibly bound BT-474 cells (Figures 4, 6A). All three peptides did not bind normal breast epithelial cells. Interestingly, both full length 51 and V1 bound strongly to HUVEC endothelial cells, while 51N binding was visibly weaker in fluorescent intensity than 51 or V1.
Figure 6.
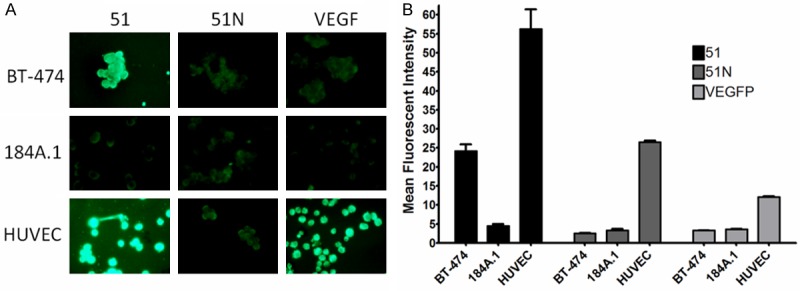
51 and V1 Cell Binding Comparison. A: Biotinylated peptides were examined for BT-474, 184A.1 and HUVEC binding by fluorescent confocal microscopy. Ten µM peptide was incubated with 4% paraformaldehyde fixed cells dried onto microscope slides and peptide binding visualized by anti-biotin Alexafluor-488 conjugated antibody. B: Flow cytometry was performed to quantify peptide binding to cells. Binding is plotted as the mean fluorescent intensity of the total cell population. Bars illustrate a mean of three replicate experiments and error bars correspond to the standard deviation.
Confirmation and quantification of fluorescent peptide binding to each of the cell lines was investigated using flow cytometry. The mean fluorescence intensity (mean FI) was quantified for each cell line incubated with 10 µM of peptide (Figure 6B). Peptide 51 had a significantly higher (P<0.01) BT-474 cell binding (mean FI = 24.2 ± 2.98), 9.5 and 7.4 times higher than 51N (mean FI = 2.53 ± 0.11) and V1 (mean FI = 3.27 ± 0.11), respectively. The mean FI of 51 for 184A.1 cells was 4.47 ± 0.90 in comparison to 3.27 ± 0.73 for 51N and 3.63 ± 0.06 for V1, indicating no binding of any peptide to the breast epithelial cell line. Finally, binding to HUVEC cells was significantly higher (P<0.05) for 51 (mean FI = 56.2 ± 9.05) and V1 (mean FI = 26.5 ± 0.46) than for 51N (mean FI = 12.06 ± 0.46), which was consistent with fluorescent microscopy results.
Since the BT-474 targeting capability of 51 was not mediated by the N-terminal 7 residues, a peptide corresponding to the C-terminal 9 residues (51C - VVGYFMASA) was synthesized to determine if this sequence alone contributed to the cell binding properties. A 96-well, colorimetric cell binding assay was chosen to monitor binding over a number of concentrations for determination of the specificity and affinity of 51, 51N and 51C. Although all peptides bound significantly higher to BT-474 cells than 184A.1 cells (P<0.05) at a concentration of 1 µM, peptide 51 demonstrated higher binding to BT-474 cells than either 51N or 51C. Peptide 51 demonstrated saturable binding and an EC50 = 4.71 ± 0.3 µM. 51N did not reach binding saturation, and 51C bound with a calculated EC50 of approximately 2 mM, 1000 fold worse than full length 51 (Figure 7). These results indicated that the BT-474 targeting properties of 51 were not mediated by the N- or C-terminal sequences by themselves.
Figure 7.
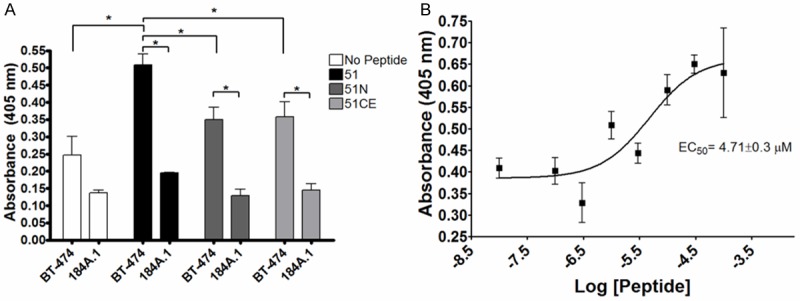
Truncated 51 Binding Analysis. Full length (51), N-terminal (51N) and C-terminal (51C) truncated peptides were assessed for BT-474 and 184A.1 cell binding using a colorimetric 96 well cell binding assay. A: 1 µM biotinylated peptide was incubated with cells and binding was detected by the addition of streptaviding-conjugated horseradish peroxidase followed by addition of 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid). Total bound peptide was measured by absorbance at 405 nm. Bars represent a mean of 3 replicate experiments and error bars denote standard deviation. B: Peptide 51 binding from 10 nM to 100 µM to BT-474 cells is plotted for calculation of EC50.
Discussion
Detection and characterization of breast carcinomas is essential to effectively treating molecularly distinct malignancies. Therapeutic management of breast cancer has benefitted from the introduction of targeted therapies including tamoxifen and trastuzumab [2,3]. Unfortunately, resistance has been demonstrated to occur for both therapies, negating the beneficial effects of targeted therapy in certain patients [5,10]. Development of a targeted imaging agent, such as a radiolabeled peptide, that could detect human breast carcinomas and simultaneously reveal whether the tumor would be susceptible to developing therapeutic resistance would, therefore, be highly beneficial. BT-474 human breast cancer cells express the targets of both tamoxifen and trastuzumab, and have innate resistance to tamoxifen and can develop resistance to trastuzumab [8,32]. Therefore, this cell line may serve as an ideal model for developing targeted peptides for imaging therapy-resistant breast cancer.
In order to develop a peptide with BT-474 imaging capabilities, in vivo phage display was performed with mice bearing BT-474 human breast cancer xenografts. Phages for further characterization were obtained by eliminating target unrelated phages, leaving 68 potential candidates. From these, 14 phages that were identified multiple times in the selection were chosen for cell binding analysis. Binding of the phages was assessed with BT-474 breast cancer target cells and 184A.1 normal breast epithelial cells as a negative control. Clone 51 was chosen for analysis as a synthesized peptide because in addition to minimal non-target binding, the phage bound over 18 times higher than wild-type phage to BT-474 cells. Due to its apparent superior targeting properties, the displayed sequence of clone 51 was synthesized and analyzed for cell binding. Fluorescent microscopy confirmed BT-474 specific binding of the peptide, and flow cytometry was used to quantify a relative EC50 = 2.33 µM of peptide 51 for the target cells. The peptide once again did not bind to 184A.1 cells at any concentration tested. A calculated EC50 that had been previously demonstrated to be sufficient for in vivo imaging, in addition to its specificity for BT-474 cells, lead to synthesis and analysis of the peptide as a radiotracer [15,20].
Radiolabeling of the peptide for in vivo studies was accomplished by conjugating the peptide to the macrocyclic chelator DOTA and incubating with 111In. The radiolabeled peptide was subjected to a peptide binding inhibition assay to confirm specificity and affinity. 111In-DOTA-51 binding inhibition was measured at a relative IC50 = 16.7 nM, confirming retained affinity of the radiolabeled peptide. The difference between the EC50 and IC50 could be the result of enhanced affinity of the radiolabeled peptide, radiolabeling providing a more sensitive means of quantification, or a difference between the concentration of peptide necessary to reach half maximal saturation (EC50) and the concentration necessary to inhibit 50% of submaximal peptide binding (IC50). Further characterization may resolve the difference; however, in vitro cell binding revealed that the peptide affinity had not been diminished by addition of a radiolabeled chelator, which was the goal of the assay. Therefore, 111In-DOTA-51 was subsequently analyzed in BT-474 tumor bearing mice for biodistribution and SPECT imaging capability. Tumor uptake of the peptide was under 1% ID/g, but highly specific. Two measures of specificity, tumor to blood (2.3) and muscle (7.1) ratios, were both greater than 1, indicating tumor uptake was not mediated by blood pooling and was also tissue specific. SPECT imaging of the radiolabeled peptide revealed high uptake in the region surrounding the tumor, consistent with radiolabeled RGD peptide and an anti-VEGF antibody specific for tumor vasculature antigens [28,29]. Vasculature targeting peptides have been selected by in vivo and in vitro phage display previously, and it is known that a sequence as short as 3 amino acids, such as RGD, is sufficient to endow tumor vasculature targeting properties to a peptide [14]. The highly similar uptake pattern between peptide 51 and other tumor vasculature imaging agents suggested that the peptide may have similar targeting properties. Upon a detailed inspection of vasculature targeting peptide sequences in the literature, it was discovered that peptide 51 resembled a Nrp1-targeted peptide selected previously by phage display [30]. The displayed peptide of Clone 51, ATWLPVPVVGYFMASA, was identical in its 5 N-terminal residues to the Nrp-1 targeted V1 peptide, which has the sequence ATWLPPR. Nrp-1 has been demonstrated to be expressed in breast cancer, and its expression has been confirmed in BT-474 cells [33]. It was possible therefore, that the binding of Clone 51 was mediated by the N-terminal residues homologous to V1. In order to test this, 51 and V1 were examined for similar targeting characteristics in vitro and in vivo and several truncated versions of 51 were examined to determine the sequence that mediated BT-474 binding.
In vitro cell binding studies revealed that 51 bound both BT-474 and HUVEC cells, while V1 only targeted HUVEC cells. Although BT-474 cells express Nrp1, it has been shown that they express the receptor at lower levels than HUVEC cells, which may explain the difference in binding of V1 to BT-474 and HUVEC cells [33]. In fact, a monoclonal antibody targeting Nrp1 failed to elicit anti-proliferative effects with BT-474 cells in vitro, consistent with the observed results of V1 binding to BT-474 cells [34]. However, this does not account for the difference in cell binding between 51 and V1. Although it was initially thought that the target of 51 may be Nrp1, the data presented here and previous analysis of V1 indicate this is not likely. In an earlier study examining the critical amino acids necessary for V1 binding, it was determined that the C-terminal LPPR of V1 was the crucial sequence, as demonstrated by binding assays using alanine scanning and truncation variants, and nuclear magnetic resonance spectrometry [35]. Since peptide 51 does not contain the critical LPPR domain, the similarity between the 51 and V1 peptides may only be coincidental, or the ATWLP sequence by itself may not be enough to contribute to the binding properties of the peptide. Finally, truncated peptides were examined to determine the role of shorter sequences, including ATWLP, on the binding of full length 51. It was determined that the full length peptide was required for optimal target affinity and specificity.
In addition to comparison of V1 and 51 in vitro, the in vivo data of this study was compared to previously published V1 in vivo data for pharmacokinetics and imaging comparison. Unfortunately, V1 has only been tested as a 99mTc radiolabeled peptide, and it was used with a different tumor model [36]. Nevertheless, the in vivo analysis of V1 revealed higher tumor uptake (~2% ID/g) than 51, but its tumor to muscle ratio was 0.22, significantly lower than 51. Although blood levels of the peptide were not given, it was reported that blood levels of the V1 peptide prevented detection of tumors by SPECT imaging. Though not directly comparable, the stark differences in pharmacokinetic and SPECT imaging properties suggest that 51 and V1 do have different properties and targets.
Conclusion
The data presented here provide evidence that 111In-DOTA-51 is a potential candidate for imaging BT-474 human breast tumor xenografts. Although the peptide’s properties are not mediated by an Nrp1 targeted sequence, the peptide nonetheless appears to target tumor vasculature, as demonstrated both in vitro and in vivo. In order to progress the peptide further to the clinic, several questions must be addressed. Solid tumor uptake of the peptide is low, however it remains to be seen whether the vasculature uptake of the peptide will be sufficient for imaging in humans. Additionally, although BT-474 cells are a suitable base for developing a peptide targeted at resistance susceptible breast cancer, more in depth models must be used to confirm that the peptide is indeed specific for targeted therapy resistant breast cancer. In that same regard, identification of the target of peptide 51, while not trivial, could provide information for further investigation of proteins that mediate resistance. Regardless, the work here demonstrates that in vivo phage display can be used to select peptides, which target a resistance susceptible breast cancer cell line, and the radiolabeled peptides can be used to identify xenografted tumors in vivo.
Acknowledgements
The authors would like to acknowledge the contributions of Jessica Newton-Northup, Marie T. Dickerson and the VA Biomolecular Imaging Core. This material is based upon work supported by the Department of Veterans Affairs, VeteransHealth Administration, Office of Research and Development, Biomedical Laboratory Research and Development, Clinical Sciences Research and Development including the Cooperative Studies Program, Rehabilitation Research and Development Service, and Health Services Research and Development through a VA Merit Award (I01BX000964). Additional support provided by an NIBIB Training Grant NIBIB 5 T32 EB004822.
References
- 1.Weissleder R. Molecular Imaging in Cancer. Science. 2006;312:1168–1171. doi: 10.1126/science.1125949. [DOI] [PubMed] [Google Scholar]
- 2.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 3.Abe O, Abe R, Enomoto Kt, Kikuchi K, Koyama H, Nomura Y, Sakai K, Sugimachi K, Tominaga T, Uchino J. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 4.Borg Å, Baldetorp B, Fernö M, Killander D, Olsson H, Ryden S, Sigurdsson H. ERBB2 amplification is associated with tamoxifen resistance in steroid-receptor positive breast cancer. Cancer Lett. 1994;81:137–144. doi: 10.1016/0304-3835(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 5.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A Functional Genetic Approach Identifies the PI3K Pathway as a Major Determinant of Trastuzumab Resistance in Breast Cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 6.Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene. 2006;25:3994–4008. doi: 10.1038/sj.onc.1209415. [DOI] [PubMed] [Google Scholar]
- 7.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2005;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y, Wang J, Scollard DA, Mondal H, Holloway C, Kahn HJ, Reilly RM. Imaging of HER2/neu-positive BT-474 human breast cancer xenografts in athymic mice using 111In-trastuzumab (Herceptin) Fab fragments. Nucl Med Biol. 2005;32:51–58. doi: 10.1016/j.nucmedbio.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 9.van Slooten HJ, Bonsing BA, Hiller AJ, Colbern GT, van Dierendonck JH, Cornelisse CJ, Smith HS. Outgrowth of BT-474 human breast cancer cells in immune-deficient mice: a new in vivo model for hormone-dependent breast cancer. Br J Cancer. 1995;72:22–30. doi: 10.1038/bjc.1995.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int J Cancer. 2002;97:306–312. doi: 10.1002/ijc.1614. [DOI] [PubMed] [Google Scholar]
- 11.Deutscher SL. Phage display in molecular imaging and diagnosis of cancer. Chem Rev. 2010;110:3196–3211. doi: 10.1021/cr900317f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 13.Scott J, Smith G. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 14.Ruoslahti E. RGD and Other Recognition Sequences for Integrins RGD. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 15.Karasseva NG, Glinsky VV, Chen NX, Komatireddy R, Quinn TP. Identification and Characterization of Peptides That Bind Human ErbB-2 Selected from a Bacteriophage Display Library. J Protein Chem. 2002;21:287–296. doi: 10.1023/a:1019749504418. [DOI] [PubMed] [Google Scholar]
- 16.Peletskaya E, Glinsky G, Deutscher S, Quinn T. Identification of peptide sequences that bind the Thomsen-Friedenreich cancer-associated glycoantigen from bacteriophage peptide display libraries. Mol Divers. 1996;2:13–18. doi: 10.1007/BF01718695. [DOI] [PubMed] [Google Scholar]
- 17.Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardo-Vila M, Giordano RJ, Mintz PJ, Ardelt PU, Yao VJ, Vidal CI, Chen L, Flamm A, Valtanen H, Weavind LM, Hicks ME, Pollock RE, Botz GH, Bucana CD, Koivunen E, Cahill D, Troncoso P, Baggerly KA, Pentz RD, Do KA, Logothetis CJ, Pasqualini R. Steps toward mapping the human vasculature by phage display. Nat Med. 2002;8:121–127. doi: 10.1038/nm0202-121. [DOI] [PubMed] [Google Scholar]
- 18.Newton JR, Kelly KA, Mahmood U, Weissleder R, Deutscher SL. In vivo selection of phage for the optical imaging of PC-3 human prostate carcinoma in mice. Neoplasia. 2006;8:772–780. doi: 10.1593/neo.06331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung E, Lee NK, Kang SK, Choi SH, Kim D, Park K, Choi K, Choi YJ, Jung DH. Identification of tissue-specific targeting peptide. J Comput Aided Mol Des. 2012;26:1267–1275. doi: 10.1007/s10822-012-9614-6. [DOI] [PubMed] [Google Scholar]
- 20.Larimer BM, Thomas WD, Smith GP, Deutscher SL. Affinity Maturation of an ERBB2-Targeted SPECT Imaging Peptide by In Vivo Phage Display. Mol Imaging Biol. 2014;16:449–458. doi: 10.1007/s11307-014-0724-5. [DOI] [PubMed] [Google Scholar]
- 21.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Ru B, Li S, Lin H, Guo FB. SAROTUP: scanner and reporter of target-unrelated peptides. J Biomed Biotechnol. 2010;2010:101932. doi: 10.1155/2010/101932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rooy I, Cakir-Tascioglu S, Couraud PO, Romero I, Weksler B, Storm G, Hennink W, Schiffelers R, Mastrobattista E. Identification of Peptide Ligands for Targeting to the Blood-Brain Barrier. Pharm Res. 2010;27:673–682. doi: 10.1007/s11095-010-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsubara T, Onishi A, Saito T, Shimada A, Inoue H, Taki T, Nagata K, Okahata Y, Sato T. Sialic Acid-Mimic Peptides As Hemagglutinin Inhibitors for Anti-Influenza Therapy. J Med Chem. 2010;53:4441–4449. doi: 10.1021/jm1002183. [DOI] [PubMed] [Google Scholar]
- 25.Yao ZJ, Kao MC, Chung MC. Epitope identification by polyclonal antibody from phage-displayed random peptide library. J Protein Chem. 1995;14:161–166. doi: 10.1007/BF01980328. [DOI] [PubMed] [Google Scholar]
- 26.Thomas WD, Golomb M, Smith GP. Corruption of phage display libraries by target-unrelated clones: Diagnosis and countermeasures. Anal Biochem. 2010;407:237–240. doi: 10.1016/j.ab.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar SR, Quinn TP, Deutscher SL. Evaluation of an 111In-Radiolabeled Peptide as a Targeting and Imaging Agent for ErbB-2 Receptor-Expressing Breast Carcinomas. Clin Cancer Res. 2007;13:6070–6079. doi: 10.1158/1078-0432.CCR-07-0160. [DOI] [PubMed] [Google Scholar]
- 28.McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nat Med. 2003;9:713–725. doi: 10.1038/nm0603-713. [DOI] [PubMed] [Google Scholar]
- 29.Thakur ML, Aruva MR, Gariepy J, Acton P, Rattan S, Prasad S, Wickstrom E, Alavi A. PET Imaging of Oncogene Overexpression Using 64Cu-Vasoactive Intestinal Peptide (VIP) Analog: Comparison with 99mTc-VIP Analog. J Nucl Med. 2004;45:1381–1389. [PMC free article] [PubMed] [Google Scholar]
- 30.Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, Mazie JC. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000;19:1525–1533. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr M, Byrne A, Duffy A, Condron C, Devocelle M, Harriott P, Bouchier-Hayes D, Harmey J. A peptide corresponding to the neuropilin-1-binding site on VEGF165 induces apoptosis of neuropilin-1-expressing breast tumour cells. Br J Cancer. 2005;92:328–333. doi: 10.1038/sj.bjc.6602308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kute T, Lack CM, Willingham M, Bishwokama B, Williams H, Barrett K, Mitchell T, Vaughn JP. Development of Herceptin resistance in breast cancer cells. Cytometry Part A. 2004;57A:86–93. doi: 10.1002/cyto.a.10095. [DOI] [PubMed] [Google Scholar]
- 33.Glinka Y, Mohammed N, Subramaniam V, Jothy S, Prud’homme GJ. Neuropilin-1 is expressed by breast cancer stem-like cells and is linked to NF-κB activation and tumor sphere formation. Biochem Biophys Res Commun. 2012;425:775–780. doi: 10.1016/j.bbrc.2012.07.151. [DOI] [PubMed] [Google Scholar]
- 34.Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier- Lavigne M, Watts RJ. Blocking Neuropilin-1 Function Has an Additive Effect with Anti-VEGF to Inhibit Tumor Growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Starzec A, Ladam P, Vassy R, Badache S, Bouchemal N, Navaza A, du Penhoat CH, Perret GY. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides. 2007;28:2397–2402. doi: 10.1016/j.peptides.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Perret GY, Starzec A, Hauet N, Vergote J, Le Pecheur M, Vassy R, Léger G, Verbeke KA, Bormans G, Nicolas P, Verbruggen AM, Moretti JL. In vitro evaluation and biodistribution of a 99mTc-labeled anti-VEGF peptide targeting neuropilin-1. Nucl Med Biol. 2004;31:575–581. doi: 10.1016/j.nucmedbio.2004.01.005. [DOI] [PubMed] [Google Scholar]