

Abstract
Single-cell sequencing is emerging as an important tool for studies of genomic heterogeneity. Whole genome amplification (WGA) is a key step in single-cell sequencing workflows and a multitude of methods have been introduced. Here, we compare three state-of-the-art methods on both bulk and single-cell samples of E. coli DNA: Multiple Displacement Amplification (MDA), Multiple Annealing and Looping Based Amplification Cycles (MALBAC), and the PicoPLEX single-cell WGA kit (NEB-WGA). We considered the effects of reaction gain on coverage uniformity, error rates and the level of background contamination. We compared the suitability of the different WGA methods for the detection of copy-number variations, for the detection of single-nucleotide polymorphisms and for de-novo genome assembly. No single method performed best across all criteria and significant differences in characteristics were observed; the choice of which amplifier to use will depend strongly on the details of the type of question being asked in any given experiment.
Introduction
The recent development of techniques to perform single-cell genome analysis enables direct interrogation of the genetic heterogeneity of cellular populations. Examples of biological phenomena that are accessible for investigation with single-cell sequencing include the clonal diversity within cancer [1]–[3], the role of genetic mosaicism in the biology of multicellular organisms [4]–[6], the genomic variation in gamete cells and embryos [7], [8], and the metabolism of as-yet unculturable microbes [9] [10]–[15].
Whole genome amplification (WGA) is used in order to obtain sufficient material for genetic analyses of DNA isolated from single cells: Illumina and PacBio-based sequencing workflows typically require 1 ng and 500 ng of input material respectively, and a single bacterial or human cell contains on the order of 1 fg or 1 pg of genomic material only. Genome amplification with a factor of 103 to 109 is thus required, depending on the sequencing strategy. WGA can be broadly separated into two categories: temperature-cycled (i.e. PCR-based) methods, and isothermal amplification methods [15]. PCR-based methods rely on ligation of a common primer sequence to sheared DNA, or the use of degenerate oligo-nucleotides for priming. Best-in-class performance for PCR-based methods is achieved with protocols that include a limited MDA pre-amplification phase preceding PCR. The PicoPLEX single-cell WGA kit (NEB-WGA) and the recently described Multiple Annealing and Looping Based Amplification Cycles chemistry (MALBAC) are in this category. The constant region of the primers used in MALBAC is designed such that the products of the initial reaction can form loops, thereby potentially excluding these products as templates for further DNA synthesis [1]. It is unclear to what extent cycling of MDA or loop formation contributes to potential reduction of amplification bias and the performance of MALBAC and NEB-WGA has not been compared systematically.
Isothermal WGA methods, including multiple displacement amplification (MDA) [16], utilize polymerases with high processivity and strand-displacement activity that extend from randomly primed sites. The simplicity of the MDA chemistry makes it relatively straightforward to implement MDA on microfluidic platforms. Improved genomic coverage was reported for MDA in small volumes, but it is unclear what factors contribute to this effect [15], [17], [18].
Although datasets from different WGA methods have been compared [19] in a limited sense, systematic evaluation of the strengths and limitations of each approach on the same samples has been lacking. The present study focuses on single-cell whole genome amplification using MDA, MALBAC and NEB-WGA. We compared numerous metrics of interest, including the specificity (as measured by read mappability), the uniformity of genome coverage, de-novo genome assembly quality and the performance of each method for the identification of single-nucleotide variants (SNVs) and the detection of copy number variants (CNVs).
We chose E. coli as a target organism because of the relatively low cost of deep sequencing the E. coli genome and because amplifications of a bacterial genome from single cells allowed us to study the performance of the different available chemistries in a challenging scenario. Recognizing that the total gain achieved in the amplification reaction is an important parameter, we compared the influence of the gain on the characteristics of interest for the different available methods, where gain is defined as the ratio of DNA output mass over DNA input mass.
Results
Design of experiments
We carried out 41 different reactions based on 8 different experimental designs, distinguished by the DNA template (single E. coli cells or E. coli bulk DNA), the WGA method (MDA, MALBAC or NEB-WGA), and the volume of the amplification chamber (Fig. 1). To obtain single E. coli cells, we sorted individual cells into separate chambers of a microfluidic chip using optical tweezers [20], [21]. Single-cell lysis and WGA reactions were performed on-chip or off-chip as desired. Sequencing of the amplification products was carried out using the Illumina MiSeq 2×250 platform (average depth 158x, minimum 8x, maximum 678x).
Figure 1. Design of experiments.

Overview of experiments, where n denotes the number of experiments of a given type. In the “E. coli single cells” column, the box that straddles both the “Microfluidic” and the “Tube” fields corresponds to the method of carrying out a first round of amplification in a microfluidic chamber and then a second round of amplification in a test tube. This method will be denoted by “microfluidic+tube” in subsequent figure captions.
Specificity
We investigated specificity by computing the fraction of mapped, unmapped and discordantly mapped read pairs resulting from the different WGA reactions (Fig. 2A). Here, a read pair was flagged as unmapped when both reads in the read pair could not be aligned to the reference genome. A paired-end alignment, i.e. a read pair for which both mates aligned to the reference, was flagged as concordant if the mates were in the expected forward–reverse orientation and had an end-to-end separation ≤2000 base pairs, or as discordant otherwise. The discordant alignments are likely due to chimera formation during the amplification process [17] or during the library preparation.
Figure 2. Sequence read classification.

(A) Breakdown of read pairs in each experiment according to type of mapping achieved. (B) Breakdown of unmapped reads by organism of origin, expressed as a fraction of the total number of reads.
We found that the fraction of correctly mapped reads is greater (1) for reactions performed in a smaller volume and/or with lower total reaction gain, and (2) for reactions that required fewer hands-on steps (Fig. 2A). We determined the nature of unmapped reads by comparing them against the NCBI database of known nucleotide sequences with the Basic Local Alignment Search Tool, BLAST [22]. On average, 63% of the unmapped reads were assigned to the human genome (36%) or to other known genomes (27%) (see legend Fig. 2B). The remaining unassigned sequences are likely due to the primer-dimer formation during the preparation of sequencing libraries. We found that microfluidic MDA reactions are relatively insensitive to failure due to contamination: the fraction of unmapped reads was very low, even for reactions performed on single E. coli cells (mean fraction of unmapped reads 0.035%, n = 5). We attribute this to the small and isolated volume in which the reaction is performed [15]. WGA reactions performed in a tube were more sensitive to background contamination, in particular when the amount of starting genomic material, mDNA, was low: MDA, MALBAC and NEB-WGA perform well for mDNA≥15 pg, but are unreliable for mDNA≤1.5 pg. We found that the PCR-based chemistries, which require many hands-on steps, are particularly sensitive to failure by contamination (fraction of unmapped reads for single-cell NEB-WGA>0.98, n = 3; fraction of unmapped reads for single-cell MALBAC>0.93, n = 5).
Amplification bias and uniformity
We also analyzed the bias in amplification that results from the different chemistries. On average 7% (and up to 45%) of mapped read pairs from a sequencing run were identified as PCR or optical duplicates arising from library construction or repeated reading and were removed using Picard tools [23]. Figures 3A and 3B show examples of the local genome coverage density (normalized to a mean of 1) measured for single-cell MDA, single-cell MALBAC and single-cell NEB-WGA. Consistent with previous results [1], we find that power spectra of the noise in mapping density were distinctly different for MDA and the PCR-based methods, with more low frequency noise contributions for MDA and more high frequency noise contributions for MALBAC and NEB-WGA (see Fig. S1).
Figure 3. Amplification bias and uniformity.

(A) Local mapping density from properly mapped reads (at fixed 5x sampling depth), normalized to average 1, as a function of position along the reference sequence, for single-cell MDA (microfluidic in red, microfluidic+tube in orange). (B) Same as panel A, but for single-cell MALBAC and bulk NEB-WGA. (C) Fractional genome coverage from properly mapped read pairs, plotted as a function of gain. Here, each set of properly mapped read pairs was randomly down-sampled to 5x depth. Experiments that did not generate this many properly mapped reads were not included in the figure. (D) Fraction of the genome covered by mapped read pairs when the set of raw read pairs was down-sampled to a fixed depth of 20x, plotted as a function of gain. Filled symbols signify bulk experiments, open symbols single-cell experiments.
To obtain insight into the origin of the amplification bias in MDA, we performed a secondary MDA reaction on the product of a first MDA reaction, and compared the bias before and after the additional round of MDA (Fig. 3A). We found that the bias in amplification that results from the initial reaction is exaggerated by the secondary reaction as regions that were overrepresented in the first reaction generate amplicons at a greater rate in the second reaction. The amplification bias in MDA thus progressively worsens with greater fold amplification [24]. An analysis of the coverage distribution as measured by the Gini index revealed the same dependence of the amplification non-uniformity on gain (see Fig. S2). We next examined the dependence of the genome coverage of reads that mapped to the reference genome as a function of the reaction gain (fixed sampling depth: 5x mapped read pairs; Fig. 3C). We indeed found that the genome coverage is a strong function of the reaction gain in MDA, with greater gain leading to a lower fractional coverage. MALBAC and NEB-WGA however were relatively insensitive to the gain and MALBAC consistently resulted in higher fractional genome coverage than MDA for reactions with a gain greater than 106. Interestingly, we found that isothermal and PCR-based methods performed similarly for reactions with a gain under 106, relevant e.g. for the genome analysis of single human cells (mean fractional coverage 0.82±0.07 for MDA, 0.78±0.07 for MALBAC, 0.69±0.04 for NEB-WGA). We next analyzed the fractional genome coverage achieved given a fixed total sequencing depth (Fig. 3D, 20x sequencing depth), thereby both taking into account reads that mapped and reads that did not map to the reference genome. (The dependence of genome coverage on total sequencing depth is illustrated by the rarefaction curves in Fig. S3.) Remarkably, when considering the fractional coverage at a fixed sequencing depth, we found that the different chemistries perform similarly over the range of reaction gains investigated: the greater inherent uniformity achieved by MALBAC and NEB-WGA (see Fig. 3C) was offset by the larger proportion of unmappable sequences that resulted from these chemistries (Fig. 2A).
Identification of copy number variants
Biases introduced in whole genome amplification make the robust identification of copy number variants in single-cell sequencing challenging. To obtain insight into the performance of the different chemistries in identifying CNVs, we analyzed the minimal resolvable length of a gene duplication. As a proxy for the minimal resolvable duplication length, we considered the minimum width of a sliding window average filter, W, that gives rise to a relative genome mapping density smaller than 2 across all positions in the genome. Consistent with the above observations, we found that the performance of MDA in the detection of CNVs is strongly dependent on the reaction gain. MALBAC and NEB-WGA are more robust to the effects of gain and outperform MDA for reactions with gain exceeding 2.5•103 (Fig. 4). MALBAC and NEB-WGA performed remarkably similarly in the gain range from 102 to 107, relevant for the analysis of eukaryotic genomes (W = (3.28±1.17)•104 for MALBAC, W = (3.30±1.12)•104 for NEB-WGA). Note that in the case of diploid genomes, using the allele fraction in heterozygous sites may help in detecting CNVs; however, it is unclear whether the effectiveness of such an approach will be compromised in the presence of amplification related allele dropout.
Figure 4. CNV resolution.

Size of resolvable duplications (W, minimum width of a sliding window average filter that gives rise to a relative genome mapping density smaller than 2 across all positions in the genome) versus gain. Filled symbols signify bulk experiments, open symbols single-cell experiments.
Error rates
The rate of single-nucleotide errors introduced in the amplification reaction is another important parameter to consider, in particular for applications where single-nucleotide variants (SNVs) are targeted. For all WGA chemistries considered here, for gains ≤5•107, we found that the combined error rate, D, defined as the fraction of measured bases discrepant from the reference genome, increases logarithmically with the total reaction gain and linearly with the effective number of reaction cycles, computed as 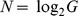 . Given a per-base, per-cycle replication error rate, ε, D is expected to scale with N as
. Given a per-base, per-cycle replication error rate, ε, D is expected to scale with N as 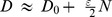 (equation (1), see Appendix S1), where D0 is an offset influenced for example by sequencing errors and inaccuracies in the reference. Fitting this model to the data in Fig. 5 for 1≤ gain ≤5•107 allowed us to extract the effective per-cycle error rates for the different WGA chemistries. The offset D0 was observed to be roughly of order 10−3, consistent with the quality cutoff imposed during quality trimming (Q = 30, corresponding to a per-base error probability of 0.001). We found that the per-base per-cycle error rates for MALBAC and NEB-WGA are similar,
(equation (1), see Appendix S1), where D0 is an offset influenced for example by sequencing errors and inaccuracies in the reference. Fitting this model to the data in Fig. 5 for 1≤ gain ≤5•107 allowed us to extract the effective per-cycle error rates for the different WGA chemistries. The offset D0 was observed to be roughly of order 10−3, consistent with the quality cutoff imposed during quality trimming (Q = 30, corresponding to a per-base error probability of 0.001). We found that the per-base per-cycle error rates for MALBAC and NEB-WGA are similar, 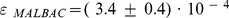 respectively
respectively 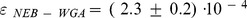 .
.
Figure 5. Combined single-nucleotide error rates.

Main panel: Experimental error rates D versus gain G, for low gains. Here D is the fraction of bases differing from the reference in the mapped reads. Linear fits for D as a function of 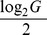 are also shown: their slope approximately indicates the per-base per-cycle replication error rate. Inset: D versus G over the entire gain range. Filled symbols signify bulk experiments, open symbols single-cell experiments.
are also shown: their slope approximately indicates the per-base per-cycle replication error rate. Inset: D versus G over the entire gain range. Filled symbols signify bulk experiments, open symbols single-cell experiments.
The per-base error rate for MDA was found to be at least one order of magnitude lower, 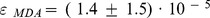 . In order to obtain a more precise estimate of the error rate in MDA, we performed error-corrected sequencing using a random barcoding strategy, as described previously [25]. Briefly, DNA molecules were labeled with uniquely identifiable molecular barcodes. Barcoded molecules were subsequently PCR-amplified to generate groups of molecules that carry the same barcode and multiple molecules from each barcode group were sequenced. Consensus calling was then performed to discriminate true variants from sequencing errors. Using this strategy, we achieved a detection limit as low as (9.7±0.4)•10−7 per base (determined from unamplified genomic DNA). The measured error rate for MDA at gain ∼3000 was (1.93±0.02)•10−5. Estimating the effective number of amplification cycles as log2(3000) and using equation (1), we extract the per-cycle per-base error rate to be
. In order to obtain a more precise estimate of the error rate in MDA, we performed error-corrected sequencing using a random barcoding strategy, as described previously [25]. Briefly, DNA molecules were labeled with uniquely identifiable molecular barcodes. Barcoded molecules were subsequently PCR-amplified to generate groups of molecules that carry the same barcode and multiple molecules from each barcode group were sequenced. Consensus calling was then performed to discriminate true variants from sequencing errors. Using this strategy, we achieved a detection limit as low as (9.7±0.4)•10−7 per base (determined from unamplified genomic DNA). The measured error rate for MDA at gain ∼3000 was (1.93±0.02)•10−5. Estimating the effective number of amplification cycles as log2(3000) and using equation (1), we extract the per-cycle per-base error rate to be 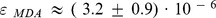 , in agreement with reported error rates for the polymerase used in MDA (of order 10−7–10−6) [16]. The use of a polymerase with proofreading activity and corresponding low error rate is a major advantage of MDA for studies of SNVs.
, in agreement with reported error rates for the polymerase used in MDA (of order 10−7–10−6) [16]. The use of a polymerase with proofreading activity and corresponding low error rate is a major advantage of MDA for studies of SNVs.
Assemblies
De-novo sequence assembly is an important application of single-cell sequencing, for example in studies of novel genomic diversity. We used the SPAdes Genome Assembler 2.5.1 [26] and the quality assessment tool QUAST 2.2 [27] to evaluate the compatibility of the WGA methods with genome assembly. To this end, we investigated LG50, the minimal number of assembled contigs (≥500 bp) required to cover 50% of the E. coli reference genome (fixed sequencing depth 30x, Fig. 6). For reactions with gain ≤5•106, isothermal and PCR-based methods performed similarly (mean LG50 416±188 for MDA, 513±262 for MALBAC, 969±223 for NEB-WGA), indicating reconstruction of extended contigs. At higher gains, the assembly quality deteriorated for all three methods. The assemblies resulting from a subset of reactions did not cover 50% of the genome and were marked as “failed” in Fig. 6.
Figure 6. De-novo assemblies.

LG50, the minimal number of assembled contigs (≥500 bp) needed to cover 50% of the E. coli reference genome, versus reaction gain (at fixed raw sequencing depth 30x). Assemblies that failed to cover 50% of the reference sequence were symbolically assigned the maximum value that LG50 can take in this scenario ((50%•genome length/500) = 4686). Filled symbols signify bulk experiments, open symbols single-cell experiments.
Discussion
Single-cell sequencing offers a powerful approach for the study of the genomic heterogeneity of cellular populations [2], [28] and for the study of the genetics of microbes that are as-of-yet unculturable [11], [29]. The finite capture efficiency of current sequencing workflows necessitates significant amplification of the target genome. In this work, we have conducted a quantitative performance comparison of the different methods that are available for whole genome amplification. These data measure the differences in performance of temperature-cycled (PCR-based, NEB-WGA, MALBAC) and isothermal amplification (MDA) chemistries, and provide a framework for choosing a WGA chemistry based on the characteristics of interest.
We compared the influence of the gain achieved in WGA on the characteristics of interest. Overall, we found that the performance of WGA chemistries deteriorates with gain, but that not every characteristic and every chemistry is affected to the same extent. The amplification bias in MDA is a direct function of the overall reaction gain, with greater gain leading to greater bias. This observation underlines the importance of tailoring the gain of the amplification reaction to yield the amount of DNA required in a subsequent sequencing workflow. The overall gain can be set through a choice of the volume of the reaction chamber, e.g. by performing the reaction in a small microfluidic reaction chamber, or by limiting the reaction time or the concentration of reagents. It should be noted that we were unable to reproduce the results of a recent report which claimed extraordinarily low bias in single cell amplifications by combining low volume MDA with a subsequent detangling reaction [30]. In contrast to MDA, we found that the inherent bias in amplification in MALBAC and NEB-WGA is not strongly affected by gain. In agreement with previous observations [1], [19], we find that the PCR-based methods lead to a lower long-range variability in read mapping, a property that makes these methods particularly well suited for the detection of CNVs. Within the class of PCR-based amplification chemistries, we found MALBAC and NEB-WGA to compare similarly.
Single-cell WGA reactions are notoriously sensitive to DNA contamination. Contamination is in particular problematic for single-cell studies of small microbial genomes. In general, we found that reactions that entail fewer hands-on steps and that are performed in a smaller reaction volume are more robust against contamination. The relative robustness of amplification reactions in small and enclosed microfluidic environments is an important benefit of microfluidic MDA.
The fractional coverage of the genome, rather than the uniformity and noise spectrum of the mapping density are important for analyses of single-nucleotide variants (SNVs). Our experiments indicated that the inherently low bias in amplification offered by MALBAC and NEB-WGA was offset by the higher sensitivity of these methods to background contamination, leading to a smaller proportion of reads that map to the target genome.
The rate of WGA-induced single-nucleotide errors is another important factor to consider in the context of the detection of SNVs. MALBAC and NEB-WGA displayed an amplification-induced error rate approximately 10 times higher than MDA, which is a consequence of the use of a polymerase with an inherently higher error rate. We conclude that the high coverage at fixed gain as well as the inherently low error rate makes MDA best suited for studies of single-nucleotide variants. While our work here does not investigate the performance of the different amplification chemistries in SNP analyses of diploid genomes, the fundamental error rates we have measured here make strong predictions about the limits of performance for many key metrics in the diploid case.
The assembly of short-read sequence data into a long contiguous sequence is often an important goal in single-cell sequencing projects. We compared the compatibility of different WGA chemistries with genome assembly. We found that MDA, MALBAC and NEB-WGA yielded assemblies of comparable quality (as measured by LG50) for reaction gains ≤5•106. Assembly performance metrics tended to deteriorate at very high gain, consistent with our observation of greater sensitivity to background contamination and lower fractional genome coverage for high-gain reactions.
In conclusion, we have conducted a quantitative assessment of the performance of several widely adopted genome amplification chemistries. The data will enable researchers to design amplification reactions and methods to the needs of their specific experiments.
Methods
Bacteria strains and DNA isolation
Invitrogen One Shot TOP10 Chemically Competent E. coli were grown overnight in LB broth. These cells were then cleaned in phosphate-buffered saline without CaCl2 and MgCl2 and either used for single-cell experiments or used for genomic DNA isolation using the QIAamp DNA Mini kit. The gain of the different amplification reactions was modulated by changing the amount of the reaction input material.
Single-cell primary and secondary MDA
Single E. coli cells were sorted in small microfluidic chambers using a laser trap (1 W, 976 nm laser), integrated on a phase-contrast microscope as described previously [20]. Microfluidic devices, attached tygon tubing, and all chemistry not included in the Qiagen REPLIG-g Single Cell MDA kit were U.V. treated for 1 hour. Following U.V. treatment, sorting lines in the device were passivated with phosphate-buffered saline pH 7.4 supplemented with 0.01% pluronic F127 and 0.01% Tween-20 for 15 minutes at room temperature. On the order of 104 E. coli cells per µl were loaded into the device and sorted in individual ∼50 nl reaction chambers, partitioned three ways for single-cell MDA chemistry. The first partition, the lysis component, was dead-end filled with 3 nl of a 30 µl volume DLB aliquot supplemented with 3 µl 1 M DTT and 1.4 µl of 10% Tween-20. The entire device was then placed in an incubator (Labnet Mini Incubator, l-5110) at 65°C for 20 minutes, subsequently placed at room temperature for an additional 10 minutes. The second partition, the lysis neutralizing component, was dead-end filled with 3 nl of Stop solution. The last partition, the amplification component, was dead-end filled with ∼43 nl of a 40 µl volume amplification reaction mix aliquot which included 29 µl of single cell REPLI-g reaction buffer, 4 µl of single cell phi29, 2.5 µl of 6.5% Tween-20, and 4.5 µl of 10 mg/ml BSA. The entire device was then placed in the incubator at 31°C for 12 hrs. 20 µl gel pipette tips were then placed in the outlets of the reaction chambers and 5 µl of elution buffer, Tris-EDTA buffer pH 8.0 supplemented with 0.01% Tween-20, was flushed for MDA product retrieval. The single-cell MDA products were then purified with Qiagen MinElute columns and quantified with a Qubit HS DNA kit. Each single-cell reaction generated 40–50 ng of material of which 30 ng was used for Nextera DNA sample library preparation, quantified again with Qubit, and visualized using an Agilent Bioanalyzer 2100. Final libraries were sequenced on the Illumina 2×250 MiSeq platform.
From each single-cell primary MDA reaction, 3 ng was used for a second round of MDA using the same REPLI-g single cell kit. These reactions were performed in a 50 µl volume following the manufacturer’s protocol. Each secondary reaction generated 30–40 µg of total material of which 30 ng was also used for sequence library construction as written above.
MDA on bulk DNA in tubes
E. coli bulk genomic DNA was amplified using the REPLIG-g single-cell kit in 50 µl volume following manufacturer’s protocol (input material 15 ng, 1.5 ng, 0.15 ng, 0.15 ng (bis), 15 pg, 1.5 pg, 0.15 pg). Each reaction generated between 30–40 µg of material. Library construction, material input, and sequencing were carried out as described above.
NEB-WGA on single-cell and bulk DNA in tubes
Using the integrated optical tweezers, single E. coli cells were sorted into separate chambers and retrieved from the microfluidic chip by flushing with elution buffer (described above) in a final volume of 2 µl. The single cells were subsequently amplified with the NEB single cell WGA kit following the manufacturer’s protocol. Single cells on average generated about 1.6 µg of total material. Additionally, six different reactions were performed on bulk genomic E. coli DNA (input amounts 15 ng, 1.5 ng, 0.15 ng, 15 pg, 1.5 pg, 0.15 pg). These reactions generated on average 1.5 µg of total material. Final library construction, material input, and sequencing were carried out as described above.
MALBAC on single-cell and bulk DNA in tubes
Single cells were sorted into single tubes in a 2 µl final volume as described above. Cells were lysed with 1.5 µl Qiagen DLB supplemented with 1 M DTT and heated to 65°C for 10 minutes. Following lysis, 1.5 µl of Qiagen Stop solution was added for final total reaction volume of 5 µl. These lysed cells were then amplified by MALBAC as described previously [1]. First 25 µl of a linear preamplifcation mix was added to 5 µl lysed cell containing 18 µl H2O, 3 µl 10x ThermoPol buffer, 1 µl 10 mM dNTP, 1 µl 50 mM MgSO4, and 1 µl of 15 mM of each MALBAC Primer (GTGAGTGATGGTTGAGGTAGTGTGGAGNNNNNGGG and GTGAGTGATGGTTGAGGTAGTGTGGAGNNNNNTTT). The reaction was then placed at 94°C for 3 min and immediately quenched on ice. 1 µl of Bst large fragment (NEB 8 U/µl, diluted in 1∶3 in 10X ThermoPol buffer) and 1 µl of PyroPhage3173 DNA Polymerase exo- (Lucigen, 5 U/µl diluted in 4∶25 10X ThermoPol buffer) were added to the reaction and run at 10°C for 45 sec; 20°C for 45 sec; 30°C for 45 sec; 40°C for 45 sec; 50°C for 45 sec; 65°C for 2 min; 94°C for 20 sec, and immediately quenched on ice. Another 1 µl of Bst large fragment (NEB 8 U/µl, diluted in 1∶3 in 10X ThermoPol buffer) and 1 µl of PyroPhage3173 DNA Polymerase exo- (Lucigen, 5 U/µl diluted in 4∶25 10X ThermoPol buffer) were added to the reaction and run at 10°C for 45 sec; 20°C for 45 sec; 30°C for 45 sec; 40°C for 45 sec; 50°C for 45 sec; 65°C for 2 min; 94°C for 20 sec; 58°C for 20 sec and immediately quenched on ice. This last preamplification step was repeated another 5 times. Preamplification reactions were split and PCR amplified with 5 µl 10X ThermoPolbuffer, 1 µl 10 mM dNTP, 3.35 µl 50 mM MgSO4, 2 µl Deep Vent Enzyme (NEB, 2 U/µl), 3.33 µl 15 uM MALBAC PCR primer (GTGAGTGATGGTTGAGGTAGTGTGGAG) and supplemented with H2O for a final 50 µl reaction volume and run for 17 cycles at 94°C for 20 sec; 59°C for 20 sec; 65°C for 1 min; 72°C for 2 min. Final PCR reactions were then cleaned up using Qiagen MinElute columns and measured using the Qubit HS DNA kit. These single cell reactions generated about 600 ng of material.
Additionally reactions were performed on E. coli genomic DNA (input amounts 15 ng, 1.5 ng, 0.15 ng, 15 pg, 1.5 pg, 0.15 pg). The same procedure as detailed above was used for these reactions generating between 900 ng for 0.15 pg starting material to 2 µg for 15 ng starting material. All single-cell and bulk DNA MALBAC reactions were then sheared on a Covaris S2 instrument using the following program: 2 min, 10% duty cycle, intensity 5, 200 cycles/burst and under frequency sweep. Sequencing libraries were then made using the NEBNext DNA Library preparation kit according to the manufacturer’s protocol. Final libraries were quantified with the Qubit HS DNA kit, visualized on an Agilent Bioanalyzer 2100, and sequenced on the 2×250 MiSeq platform.
Barcoded DNA sequencing
200 ng E. coli genomic DNA or bulk MDA products were sheared to 300–500 bp with a Covaris focused-ultrasonicator following manufacturer’s recommendation. Sheared DNA was ligated with pair-end Illumina sequencing adaptors, with 9-bp random barcodes at the beginning of each end. The libraries were PCR amplified to generate redundant molecules and sequenced on Illumina HiSeq 2000 with 2×100 bp mode. Read pairs with the same barcodes were collapsed to generate consensus sequences. At least three reads were required for consensus building. The final duplex consensus sequences were used for error analysis.
Quality trimming and alignment
MALBAC primers were removed from raw reads with the sequence grooming tool Cutadapt [31] (“cutadapt -n 10 -g GTGAGTGATGGTTGAGGTAGTGTGGAG”, respectively “cutadapt -n 10 -a CTCCACACTACCTCAACCATCACTCAC”). All reads were quality trimmed with Trimmomatic [32], using a 4-base-pair sliding-window algorithm with a quality score cutoff of 30, clipping off ends with at least one occurrence of a quality score below 25, and discarding reads that dropped below a length of 35 base-pairs (“java -jar trimmomatic-0.30.jar PE -phred33 SLIDINGWINDOW:4∶30 LEADING:25 TRAILING:25 MINLEN:35”). Processed reads that were still paired were then aligned to the appropriate E. coli strain K12 substrain DH10B reference genome using Bowtie2 [33] in local alignment mode with maximum proper fragment length 2000 (“bowtie2–local –very-sensitive-local -X 2000”). Using Picard tools [23], we removed optical and PCR duplicates (“java -jar MarkDuplicates.jar REMOVE_DUPLICATES = TRUE”) and realigned locally around indels (“java -Xmx4g -jar GenomeAnalysisTK.jar -T IndelRealigner”) over suspicious intervals determined with RealignerTargetCreator (“java -Xmx2g -jar GenomeAnalysisTK.jar -T RealignerTargetCreator”). Processed alignment files were analyzed using SAMtools [34].
Performance comparison of methods
The nature of unmapped reads was elucidated by comparing them against the NCBI database of known nucleotide sequences with the Basic Local Alignment Search Tool, BLAST [22] (“blastn -db nt -evalue 0.0001 -outfmt ‘6 qseqid sseqid sstart send pident length evalue bitscore sscinames’ -perc_identity 90 -culling_limit 2”). For each read, we quoted the top hit (i.e. the best hit according to e-value) as the organism of origin in Figure 2.
For Figures 3A and 3B, the leftmost mapping positions of reads (considering only the first mate in each pair) were histogrammed in bins of size 250 bp and the resulting mapping density was normalized to a mean of 1 across the genome. For Figures 3C and 3D, the quantity plotted on the vertical axis is the fraction of bases in the genome that were covered at least once by a properly mapped read from the considered number of read pairs.
For Figure 3D, down-sampling to a fixed raw depth 20x was performed by down-sampling the mapped read pairs to F•20x depth (i.e. F•20•(genome length)/(2•250) read pairs), where F≤1 is the fraction of read pairs that were mapped. Similarly, for Figure 6, down-sampling to a fixed raw depth 30x was performed by down-sampling the qualitytrimmed read pairs to T•30x depth, where T is the fraction of read pairs that remained after qualitytrimming. The minimal detectable CNV size W was computed as follows. First, the mapping positions of read pairs were histogrammed in bins of size 250 bp and the mapping density normalized to a mean of 1. Then moving average filters with different window sizes, increasing in steps of 1000 bp, were applied to a circularized (i.e. periodically extended) version of the mapping density. W was the minimum window width such that all windows ≥W gave rise to a relative genome mapping density smaller than 2 across all bins in the genome. The rationale for considering this metric W is that it represents the noise threshold above which a gene duplication could be detected in the smoothed data as a mapping density ≥2.
To compute Gini indices for Figure S2, we first calculated the Lorenz curve (not shown), which is a plot of cumulative share of reads against cumulative share of genome positions covered by those reads, ordered from lowest-covered to highest-covered. [1] The Gini index is then defined as the area between the observed Lorenz curve and the straight Lorenz curve that would result from perfectly uniform coverage. Here, a Gini index of 0 indicates perfect uniformity and a Gini index of 1 indicates maximal non-uniformity.
“Combined single-nucleotide error rates” were calculated by going through each base i in the reference sequence and counting the number of times Mi it was matched by a proper alignment and the number of times Ci it was contradicted by a proper alignment. The discrepancy ratio is then 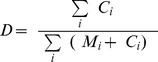 . The base counts necessary for computing Mi and Ci were extracted from the alignment files using bam-readcount [35], [36]. In computing D, we omitted sites i for which
. The base counts necessary for computing Mi and Ci were extracted from the alignment files using bam-readcount [35], [36]. In computing D, we omitted sites i for which 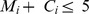 (not enough coverage) and sites i that had
(not enough coverage) and sites i that had 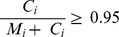 (typically indicative of a faulty reference base at site i).
(typically indicative of a faulty reference base at site i).
De-novo assemblies were created using SPAdes 2.5.1 [26] with the –careful option and LG50-values were computed using QUAST 2.2 [27]. All sequencing data sets were down-sampled to a fixed (pre-qualitytrimming) depth of 30x using seqtk [37] before assembly. For SPAdes assemblies of bulk DNA, we used k-mer lengths 21, 33, 55, 77, 99, 127; for single-cell assemblies, we used the –sc option with k-mer lengths 21, 33, 55. In cases where using SPAdes with the –sc option did not give a result but omitting the –sc option did, or vice versa, we reported the obtained result regardless of whether the sample was a single cell or bulk DNA. The data points lying on the line labeled “failure” in Figure 6 do not correspond to LG50-values calculated by QUAST, as these assemblies did not cover 50% of the reference genome and so do not have a LG50-value; instead, we artificially assigned the maximum possible LG50-value to these assemblies in order to indicate failure.
Error margins indicated in the present paper correspond to 1 standard deviation.
Supporting Information
Power spectra of mapping density. Mapped reads were down-sampled to 4x depth, and power spectra were smoothed using a moving-average filter with window size 2.13•10−6. The MDA, MALBAC and NEB-WGA reactions had gains of the same order of magnitude (2.5•103, 1.5•103 and 1.6•103 respectively). The dashed black lines represent the Lorentzian fits used to extract roll-off frequencies: (4.15±0.06)•10−5 bp−1 for MDA, (7.94±0.08)•10−4 bp−1 for MALBAC, (1.32±0.02)•10−3 bp−1 for NEB-WGA.
(TIF)
Gini indices of coverage distribution. Gini index for the distribution of coverage among sites in the genome, plotted as a function of gain. Here, each set of properly mapped read pairs was randomly down-sampled to 5x depth. Experiments that did not generate this many properly mapped reads (e.g. all single-cell NEB-WGA experiments) were not included in the figure. The Gini index is a measure of non-uniformity: a Gini index of 0 indicates perfect uniformity and a Gini index of 1 indicates maximal non-uniformity. By this metric, we found that the mapping uniformity for MDA decreases with reaction gain, whereas the uniformity of PCR-based methods is only a weak function of gain. The amplification bias in PCR-based reactions was lower than the amplification bias in MDA for reactions that required a gain greater than 106.
(TIF)
Coverage as a function of sequencing depth. (A) Fraction of genome covered versus sampled depth of mapped read pairs. (B) Same as panel A, but with horizontal axis adjusted by the fraction of raw read pairs that were mapped. We show only the curve that yielded the highest coverage at 20x sequencing depth for each listed experimental category.
(TIF)
Modeling discrepancy ratios.
(DOCX)
Acknowledgments
We are grateful to Jeremy A. Dodsworth for providing us with the photograph of a microfluidic flow-cell used in Fig. 1.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. Raw sequencing data from this study are accessible at the NCBI Sequence Read Archive (SRA) under accession numbers SAMN02782248–SAMN02782289.
Funding Statement
This work was funded by the United States Department of Energy Joint Genome Institute (jgi.doe.gov) Emerging Technologies Opportunity Program [supported by the Office of Science of the United States Department of Energy under Contract No. DE-AC02-05CH11231], and by the National Science Foundation (www.nsf.gov) Partnerships for International Research and Education (PIRE) award number 0968421. CFdB is supported by an International Fulbright Science and Technology Award (scienceandtech.fulbrightonline.org) [sponsored by the Bureau of Educational and Cultural Affairs of the United States Department of State] and a Melvin and Joan Lane Stanford Graduate Fellowship (sgf.stanford.edu); CG is supported by the Leukemia and Lymphoma Society (www.lls.org), the American Society of Hematology (www.hematology.org), and the Child Health Research Institute at Stanford (chri.stanford.edu). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zong C, Lu S, Chapman AR, Xie XS (2012) Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338: 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, et al. (2012) Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Science Translational Medicine 4: 149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wells D, Delhanty JD (2000) Comprehensive chromosomal analysis of human preimplantation embryos using whole genome amplification and single cell comparative genomic hybridization. Mol Hum Reprod 6: 1055–1062. [DOI] [PubMed] [Google Scholar]
- 5. McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, et al. (2013) Mosaic Copy Number Variation in Human Neurons. Science 342: 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poduri A, Evrony GD, Cai X, Walsh CA (2013) Somatic Mutation, Genomic Variation, and Neurological Disease. Science 341. [DOI] [PMC free article] [PubMed]
- 7. Wang J, Fan HC, Behr B, Quake Stephen R (2012) Genome-wide Single-Cell Analysis of Recombination Activity and De Novo Mutation Rates in Human Sperm. Cell 150: 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hou Y, Fan W, Yan L, Li R, Lian Y, et al. (2013) Genome Analyses of Single Human Oocytes. Cell 155: 1492–1506. [DOI] [PubMed] [Google Scholar]
- 9. Kvist T, Ahring BK, Lasken RS, Westermann P (2007) Specific single-cell isolation and genomic amplification of uncultured microorganisms. Appl Microbiol Biotechnol 74: 926–935. [DOI] [PubMed] [Google Scholar]
- 10. Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, et al. (2007) Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proceedings of the National Academy of Sciences 104: 11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hongoh Y, Toyoda A (2011) Whole-genome sequencing of unculturable bacterium using whole-genome amplification. Methods Mol Biol 733: 25–33. [DOI] [PubMed] [Google Scholar]
- 12. Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, et al. (2013) Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4: 1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blainey PC, Quake SR (2014) Dissecting genomic diversity, one cell at a time. Nat Meth 11: 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- 15. Blainey PC (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37: 407–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, et al. (2002) Comprehensive human genome amplification using multiple displacement amplification. Proceedings of the National Academy of Sciences 99: 5261–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marcy Y, Ishoey T, Lasken RS, Stockwell TB, Walenz BP, et al. (2007) Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet 3: 1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutchison CA 3rd, Smith HO, Pfannkoch C, Venter JC (2005) Cell-free cloning using phi29 DNA polymerase. Proc Natl Acad Sci U S A 102: 17332–17336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Voet T, Kumar P, Van Loo P, Cooke SL, Marshall J, et al. (2013) Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res 41: 6119–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blainey PC, Mosier AC, Potanina A, Francis CA, Quake SR (2011) Genome of a Low-Salinity Ammonia-Oxidizing Archaeon Determined by Single-Cell and Metagenomic Analysis. PLoS One 6: e16626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Youssef NH, Blainey PC, Quake SR, Elshahed MS (2011) Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Appl Environ Microbiol 77: 7804–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 23.Picard team (2009) Picard. http://picard.sourceforge.net.
- 24. Lasken RS (2009) Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochem Soc Trans 37: 450–453. [DOI] [PubMed] [Google Scholar]
- 25. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, et al. (2012) Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A 109: 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hou Y, Song L, Zhu P, Zhang B, Tao Y, et al. (2012) Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148: 873–885. [DOI] [PubMed] [Google Scholar]
- 29. Lasken RS (2012) Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Micro 10: 631–640. [DOI] [PubMed] [Google Scholar]
- 30. Gole J, Gore A, Richards A, Chiu Y-J, Fung H-L, et al. (2013) Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotech 31: 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads.
- 32. Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, et al. (2012) RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res 40: W622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson D, Abbott T (2009) bam-readcount. https://github.com/genome/bam-readcount: GitHub repository.
- 36.Koboldt DC, Larson DE, Wilson RK (2002) Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Current Protocols in Bioinformatics: John Wiley & Sons, Inc. [DOI] [PMC free article] [PubMed]
- 37.Li H (2012) seqtk. https://github.com/lh3/seqtk: GitHub repository.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Power spectra of mapping density. Mapped reads were down-sampled to 4x depth, and power spectra were smoothed using a moving-average filter with window size 2.13•10−6. The MDA, MALBAC and NEB-WGA reactions had gains of the same order of magnitude (2.5•103, 1.5•103 and 1.6•103 respectively). The dashed black lines represent the Lorentzian fits used to extract roll-off frequencies: (4.15±0.06)•10−5 bp−1 for MDA, (7.94±0.08)•10−4 bp−1 for MALBAC, (1.32±0.02)•10−3 bp−1 for NEB-WGA.
(TIF)
Gini indices of coverage distribution. Gini index for the distribution of coverage among sites in the genome, plotted as a function of gain. Here, each set of properly mapped read pairs was randomly down-sampled to 5x depth. Experiments that did not generate this many properly mapped reads (e.g. all single-cell NEB-WGA experiments) were not included in the figure. The Gini index is a measure of non-uniformity: a Gini index of 0 indicates perfect uniformity and a Gini index of 1 indicates maximal non-uniformity. By this metric, we found that the mapping uniformity for MDA decreases with reaction gain, whereas the uniformity of PCR-based methods is only a weak function of gain. The amplification bias in PCR-based reactions was lower than the amplification bias in MDA for reactions that required a gain greater than 106.
(TIF)
Coverage as a function of sequencing depth. (A) Fraction of genome covered versus sampled depth of mapped read pairs. (B) Same as panel A, but with horizontal axis adjusted by the fraction of raw read pairs that were mapped. We show only the curve that yielded the highest coverage at 20x sequencing depth for each listed experimental category.
(TIF)
Modeling discrepancy ratios.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. Raw sequencing data from this study are accessible at the NCBI Sequence Read Archive (SRA) under accession numbers SAMN02782248–SAMN02782289.