

Blocking IL-17R signaling may be a promising therapeutic strategy for the treatment of proliferative and crescentic glomerulonephritis.
Keywords: IL-17, inflammation, kidney
Abstract
In recent years, proinflammatory cytokines in the nephritic kidney appear to contribute to the pathogenesis of AGN. The complex inflammatory cytokine network that drives renal pathology is poorly understood. IL-17, the signature cytokine of Th17 cells, which promotes autoimmune pathology in a variety of settings, is beginning to be identified in acute and chronic kidney diseases as well. However, the role of IL-17-mediated renal damage in the nephritic kidney has not been elucidated. Here, with the use of a murine model of experimental AGN, we showed that IL-17RA signaling is critical for the development of renal pathology. Despite normal systemic autoantibody response and glomerular immune-complex deposition, IL-17RA−/− mice exhibit a diminished influx of inflammatory cells and kidney-specific expression of IL-17 target genes correlating with disease resistance in AGN. IL-17 enhanced the production of proinflammatory cytokines and chemokines from tECs. Finally, we were able to show that neutralization of IL-17A ameliorated renal pathology in WT mice following AGN. These results clearly demonstrated that IL-17RA signaling significantly contributes to renal tissue injury in experimental AGN and suggest that blocking IL-17RA may be a promising therapeutic strategy for the treatment of proliferative and crescentic glomerulonephritis.
Introduction
AGN causes a great degree of mortality and morbidity in patients suffering from chronic kidney diseases [1, 2]. Following kidney injury, infiltration of inflammatory cells into the renal parenchyma leads to disruption of glomerular barrier integrity and development of fibrosis [3]. Recent evidence suggests a critical role of proinflammatory cytokines in the pathogenesis of AGN [4–6]. IL-17 is the signature cytokine of newly identified Th17 cells, which promote autoimmune pathology in a variety of settings and are beginning to be identified in human and experimental AGN [7–11]. In the presence of IL-6, TGF-β, IL-1, and IL-21, naive CD4+ T cells differentiate into Th17 cells in mice and humans [7, 8]. IL-23, a heterodimeric cytokine consisting of a unique p19 subunit and p40 subunit, is important for survival and pathogenic function of Th17 cells [7, 8]. Accordingly, IL-12p40−/− mice (lacking IL-23 and Th1, promoting cytokine IL-12) and IL-12p19−/− (lacking IL-23 with intact IL-12), but not IL-12p35−/− (lacking IL-12 with intact IL-23), were resistant to the development of experimental AGN [12]. However, the role of IL-17 in driving AGN in chronic kidney diseases is poorly understood.
IL-17A is a member of the IL-17 family that includes: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F [13]. IL-17A, the most well-characterized member of the IL-17 family, is produced by Th17 cells [13]. In addition, γδ T, lymphoid tissue inducer, CD8, NK, NKT, and plasma cells also produce IL-17A [14]. In inflammatory conditions, Th17 cells release not only IL-17A but also IL-17F [15]. These cytokines can form IL-17A and IL-17F homodimers, as well as IL-17A/IL-17F heterodimers, and bind to IL-17R complexes, composed of IL-17RA paired with IL-17RC [16]. Following ligand binding, IL-17RA and IL-17RC associate with each other and activate a number of different downstream effector signaling pathways in target cells. In addition to IL-17A and IL-17F, IL-17RA acts as a receptor for IL-17E (IL-25) [15].
A number of mouse model studies have implicated the essential role of the IL-23/Th17 axis in promoting renal injury in human and experimental AGN [17–19]. These studies took advantage of genetically deficient mice lacking Th17 cells (retinoic acid receptor-related organ receptor 1γt−/− and IL-23p19−/− mice) or transfer of in vitro-differentiated Th17 cells in recombination-activating gene 1−/− recipients with planted antigens in the kidney glomeruli [17–19]. Although these reports have provided useful information on the involvement of “classic” Th17 cells as a source of a IL-17 family of cytokines in AGN, these studies are limited by the fact that Th17 cells contribute a fraction of the total IL-17 family of cytokines produced in the nephritic kidney. Moreover, in addition to IL-17A and IL-17F, Th17 cells produce multiple cytokines, including GM-CSF, IL-22, and IL-21 [7, 8], making it difficult to assess the role of IL-17A and IL-17F in the pathogenesis of AGN. To address this issue, recently, Pisitkun et al. [20] demonstrated that mice deficient in Act1 (an adaptor protein absolutely necessary for the IL-17R signaling in the target cells) are protected from developing renal pathology following experimental AGN. Lack of Act1 was shown to protect lupus-prone FcγRIIb−/− mice from development of lethal glomerulonephritis without affecting the systemic autoimmune response and production of anti-nuclear antibodies [20]. These results clearly indicate a previously unappreciated kidney-specific, pathogenic role of IL-17 family cytokines in renal inflammation. However, there are discrepancies regarding the role of Act1 in autoimmunity. Act1 has been shown to be involved in the negative regulation of B cell-activating factor receptor and CD40 signaling in B cells by a separate group [21, 22]. Consequently, deficiency of Act1 resulted in hyperplasia and spontaneous activation of B cells leading to loss of self-tolerance, production of autoantibodies, and development of systemic autoimmune conditions [23]. To overcome these major problems, we decided to revisit the role of IL-17R signaling in the development of AGN in IL-17RA−/− signaling mice. To this end, we took advantage of a prototypic mouse model of AGN, where glomerular injury is induced by generating an autoimmune response against rabbit anti-mouse GBM serum, faithfully recapitulating many essential features of autoimmune kidney diseases [24].
In this report, we showed that IL-17A is up-regulated in the nephritic kidney of AGN mice, resulting in increased expression of IL-17-responsive genes in the late stages of AGN. Consequently, IL-17RA−/− signaling significantly ameliorated glomerular injury and tubulointerstitial inflammation following induction of AGN. Interestingly, IL-17RA−/− mice showed diminished infiltration of innate effector cells in the kidney, despite a normal systemic immune response and glomerular immune complex deposition, correlating with disease resistance. Finally, we were able to show that administration of anti-IL-17A antibody protected mice from the development of AGN, indicating that neutralization of IL-17A may be an attractive, therapeutic approach to control chronic renal inflammation associated with AGN, which may have ramifications in other autoinflammatory chronic kidney disorders.
MATERIALS AND METHODS
Mice
C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). IL-17RA−/− mice were obtained from Amgen (San Francisco, CA, USA) and bred in-house. All mice were housed and bred under specific pathogen-free conditions, and age- and sex-matched mice were used for all the experiments. All animal protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and adhered to the guidelines in the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health.
Induction of experimental AGN
The protocol for inducing AGN has been described before [20]. Briefly, mixed rabbit IgG, at a final concentration of 0.1 mg/ml in CFA (2.5 mg/ml; Sigma, St. Louis, MO, USA), was i.p.-injected on Day −3, and then heat-inactivated rabbit mouse anti-GBM serum (Lampire Biological Laboratories, Pipersville, PA, USA) was i.v.-injected (100 μl) via tail vein on Day 0. Controls were injected with an equal volume of CFA. Mice were killed to collect tissues for analysis on Day 14. For functional studies, blood samples were collected at the time of sacrifice. BUN levels were measured by a commercially available kit (Bioo Scientific, Austin, TX, USA). Mouse anti-IL-17A antibody and isotype control antibody were obtained from Janssen Pharmaceuticals (Titusville, NJ, USA).
Histological analyses and immunofluorescence staining
Kidneys were fixed in 10% neutral-buffered formalin and embedded in paraffin. Tissue sections were stained with H&E or PAS, and histological scores were evaluated blindly as described before [12].
Frozen sections (5 μm thickness) were fixed in acetone and blocked with 1% BSA in PBS. Immune complex deposition was evaluated by staining with FITC-conjugated mouse anti-IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Slides were mounted with Vectashield without 4′,6′-diamidino-2-phenylindole (Vector Labs, Burlingame, CA, USA) and visualized using a Leica AF6000LX fluorescence microscope.
Isolation of single cells from the kidney
Kidneys were perfused with 15 ml prewarmed PBS, while the mice were anesthetized with Avertin. The perfused kidneys were digested with collagenase B (0.230 U/ml; Roche Applied Science, Indianapolis, IN, USA) in 10% FCS in RPMI for 30 min (37°C) with occasional shaking. RBC lysis was performed, and cells were resuspended in 10 ml RPMI media. The resuspended cells were slowly layered over 5 ml Percoll (Sigma). The tubes were spun for 2500 rpm for 30 min at room temperature. The cell layer at the interface of the media and Percoll was collected and washed twice with PBS and used for FACS staining.
Real-time qPCR analyses
Total RNA was isolated from the cells using the RNeasy Micro kit (Qiagen, Valencia, CA, USA), and cDNA was prepared by SuperScript II RT (Invitrogen, Carlsbad, CA, USA). Expression of the gene of interest was measured by real-time qPCR using a PerfeCTa SYBR Green FastMix ROX (Quanta BioSciences, Gaithersburg, MD, USA) on a 7300 real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). The primers for mouse IL-17A, IL-17F, IL-6, CXCL1, CXCL2, and CCL20 were purchased from Qiagen (QuantiTect Primer Assays). The expression of each gene was normalized to the expression of mouse GAPDH.
Isolation of CD4+ T cells from spleen and lymph nodes
Total CD4+ T cells from spleen and lymph nodes of WT and IL-17RA−/− mice were isolated using the CD4 T cell isolation kit (Miltenyi Biotec, Cambridge, MA, USA). Purity of isolated CD4+ T cells was >95% in all the experiments, as determined by FACS analysis.
Single-cell suspension for FACS analyses
RBC-lysed, single-cell suspensions from spleen were surface-stained with fluorochrome-conjugated B220 (RA3-6B2), CD138 (281-2), CD4 (L3T4), CD62L (Ly-22), CD44 (IM7), Ly6G (IA8), Ly6C (AL-21), CD11b (M1/70), and F4/80 (BM8; eBioscience, San Diego, CA, USA) and GL-7 (Ly-77) and CD95 (Jo2) antibodies (BD Biosciences, San Jose, CA, USA) and analyzed by FACS. Intracellular staining of Foxp3 was performed with the mouse Foxp3 staining kit (eBioscience), per the manufacturer's instructions. Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
For intracellular cytokine staining, splenocytes or renal cells were ex vivo-stimulated with PMA and ionomycin (Calbiochem, La Jolla, CA, USA) in the presence of Monensin (BD PharMingen, San Diego, CA, USA) for 4 h. Cells were intracellularly stained for IL-17A and IFN-γ using the BD intracellular staining kit (BD PharMingen), followed by flow cytometry.
Absolute neutrophil count of peripheral blood smears
Blood was collected by saphenous venous puncture or cardiac puncture and collected in EDTA-containing tubes (Microvette tubes; Sarstedt, Newton, NC, USA, and Microtainer tubes; Becton Dickinson, Franklin Lakes, NJ, USA). Total WBC counts were determined by the trypan blue exclusion method. The percentage of neutrophils was determined by enumeration of a blood smear stained with Giemsa (Fisher Scientific, Fair Lawn, NJ, USA). Two hundred cells were counted/sample, and the absolute neutrophil count was determined by multiplying the percentage of neutrophils (mature and band forms) by total WBC.
ELISA
Cell culture supernatants were assayed for cytokine production by murine ELISA kits, IL-17A, IFN-γ, and IL-4 (eBioscience). Mouse anti-rabbit IgG antibody titers were measured by ELISA using sera collected 14 days after induction of nephritis. In brief, ELISA microtiter plates were coated with 100 μl 100 μg/ml rabbit IgG (Sigma) in carbonate–bicarbonate buffer overnight at 4°C. After being blocked with 1% BSA in TBS (Sigma), the plates were incubated with serial dilutions of mouse serum (1:100–1:12,500) for 1 h at room temperature. Bound mouse IgG was detected using peroxidase-conjugated goat anti-mouse IgG (SouthernBiotech, Birmingham, AL, USA) at 1:1000, 3,3′,5,5′-tetramethylbenzidine peroxidase substrate and absorbance readings (at 450 nm) on a spectrophotometer. Ig isotypes (IgG1 and IgG2a) were measured using ELISA. Mouse serum BUN levels were determined using BUN ELISA kit (Bioo Scientific), according to the manufacturer's instructions.
In vitro stimulation of primary mouse tECs
Primary mouse tECs were cultured, according to the manufacturer's instructions (Cell Biologics, Chicago, IL, USA). Cells were left unstimulated or stimulated with IL-17A (50 ng/ml and 200 ng/ml; PeproTech, Rocky Hill, NJ, USA), TNF-α (5 ng/ml; PeproTech), or IL-17 (200 ng/ml) + TNF-α (5 ng/ml) for 24 h.
Transwell chamber migration assays
Neutrophils were isolated from bone marrow of 8- to 10-week-old C57BL/6 mice using the neutrophil isolation kit (Miltenyi Biotec). Purity of isolated neutrophils was >95% in all of the experiments, as determined by FACS analysis. Migration of neutrophils was assayed using a 5.0-μm pore-size transwell chamber (Corning, Corning, NY, USA) in the presence of supernatant from tEC cultures, left untreated or treated with IL-17 and/or TNF-α.
Statistical analyses
Results are expressed as mean ± sd. Differences between groups were calculated for statistical significance using two-tailed paired Student's t-tests. P < 0.05 was considered significant.
RESULTS
IL-17A and IL-17A-responsive, inflammatory genes were up-regulated in the kidney of AGN mice
First, we evaluated whether cortical expression of IL-17 family cytokines, such as IL-17A and IL-17F, were up-regulated in the early and late stages of AGN development. To achieve this, C57BL/6 (WT) mice were preimmunized with rabbit IgG and CFA, 3 days before the administration of NRS. Control mice received CFA only. Seven and 14 days post-NRS administration, mice were killed, and kidney cortex was evaluated for IL-17A and IL-17F transcript expression by qPCR. IL-17A mRNA was significantly up-regulated at early (Day 7) and late (Day 14) stages of AGN development (Fig. 1A). However, there was no statistical significant difference in IL-17F transcript level between nephritic and control mice (Fig. 1B). At Day 14, intracellular staining for IL-17A revealed increased frequency of IL-17A-producing cells within the CD3+CD4+ and CD3+CD4− compartments (Fig. 1C). These results clearly indicated Th17 and innate IL-17-producing cells as cellular sources of IL-17 in the nephritic kidney.
Figure 1. Increased level of IL-17A in the nephritic kidney of AGN mice.
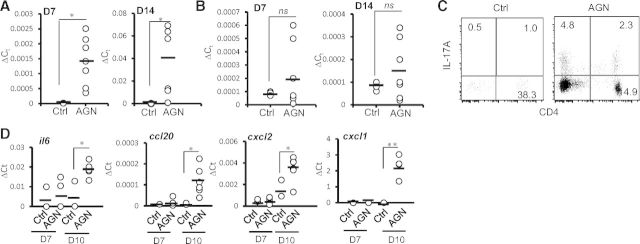
C57BL/6 mice were subjected to AGN, as described in Materials and Methods. Mice were killed at Days (D) 7 [n=7 for AGN, and n=3 for control (Ctrl)] and 14 (n=7 for AGN, and n=3 for control), and kidney cortex is dissected out carefully. (A) IL-17A and (B) IL-17F mRNA expression was evaluated by qPCR. ΔCt, relative expression. (C) At Day 14, leukocytes isolated from the perfused kidneys by Percoll gradient centrifugation were stimulated with PMA/ionomycin for 4 h and intracellularly stained for IL-17A, followed by FACS analyses (gated on CD3+ cells). Numbers in the dot plot represent percentage of cells. (D) IL-17-responsive genes (il6, ccl20, cxcl1, and cxcl2) were measured by qPCR. Each dot represents individual mice, and the horizontal bars indicate mean for each group. The scattered plot is the pooled data from two individual experiments. *P < 0.05; **P >0.001; ns, not statistically significant.
To study the impact of IL-17A expression, we next assessed IL-17-dependent cytokine and chemokine gene expression in the kidney cortex of nephritic mice. Whereas there was no difference in the IL-17-responsive genes between the groups at Day 7, transcript expression of Il6, Ccl20, Cxcl1, and Cxcl2 was increased significantly in the nephritic kidney compared with control mice at Day 10 (Fig. 1D). Overall, these results indicated that IL-17A- and IL-17- responsive, inflammatory genes were up-regulated in the late stages of renal inflammation.
IL-17RA−/− ameliorated renal dysfunction in AGN
To test whether IL-17RA signaling contribute to AGN, we induced AGN in WT and IL-17RA−/− mice and evaluated the mice for AGN development. Specific glomerular binding and deposition patterns of the NRS did not differ between WT and IL-17RA−/− mice (data not shown). At Day 14, while WT mice demonstrated significant loss of kidney function, IL-17RA−/− mice were protected from AGN, as evident by significantly reduced blood urea-nitrogen levels (Fig. 2A). Examination of H&E and PAS-stained kidney sections of nephritic WT mice at Day 14 showed severe focal glomerular and tubular damage with destruction of regular tissue structures. Glomerular changes included hypercellularity and formation of cellular crescents, capillary aneurysms, and intraglomerular deposition of PAS-positive material (Fig. 2B). In addition to massive leukocyte infiltrates, the tubulointerstitial compartment showed tubular dilation, necrosis and atrophy, and protein casts. Interestingly, glomerular and tubulointerstitial tissue damage was less severe in nephritic IL-17RA−/− mice, as shown by representative PAS staining (Fig. 2B). To quantify renal tissue damage, PAS-stained kidney sections were blindly evaluated for the presence of crescents, glomerular sclerosis, and tubulointerstitial injury. The frequency of abnormal glomeruli at Day 14 was decreased significantly in nephritic IL-17RA−/− mice compared with WT mice (Fig. 2C). Furthermore, nephritic kidneys of IL-17RA−/− mice showed a significantly diminished number of crescent formation and reduced tubulointerstitial inflammation, as indicated by a significant decrease in the interstitial area (Fig. 2C). Surprisingly, glomeruli of IL-17RA−/− and WT mice demonstrated comparable immune complex deposition, as revealed by immunofluorescence staining for mouse IgG (Fig. 2D). Collectively, these results indicated an essential role for IL-17RA signaling in renal pathology associated with AGN.
Figure 2. IL-17RA−/− mice were protected from AGN.
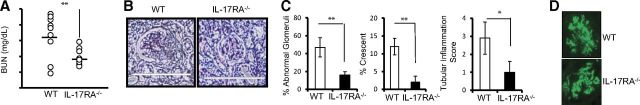
C57BL/6 mice (n=9) and IL-17RA−/− mice (n=10) were subjected to AGN. Renal dysfunction at Day 14 was assessed by determination of the (A) serum BUN level. Each dot represents individual mice, and the horizontal bars indicate mean for each group. (B) Representative photographs of PAS-stained kidney sections of nephritic C57BL/6 and IL-17RA−/− mice at Day 14 (original magnification, 400×). (C) Renal pathology was blindly evaluated and scored for percentage of abnormal glomeruli, percentage crescent formation, and tubular inflammation in C57BL/6 and IL-17RA−/− mice. (D) Representative photographs of mouse IgG deposition in the kidney glomeruli of C57BL/6 and IL-17RA−/− mice at Day 14. Bar diagram represents mean ± SD. Data are pooled from two independent experiments. *P < 0.05; **P >0.001.
Systemic immune response was unaltered in the absence of IL-17RA−/−
Previous studies have suggested a role of IL-17R signaling in B and T cell responses [25, 26]. Therefore, to evaluate whether IL-17RA−/− induces alteration in systemic humoral and cellular immune response against rabbit IgG, we performed detailed phenotypic characterization of splenic B and T cells response in WT and IL-17RA−/− mice. As demonstrated in Fig. 3A, percentages of GC B cells (B220+GL-7+CD95+) were comparable between the WT and IL-17RA−/− mice. Additionally, there was no significant difference in the percentage of plasma cells (B220loCD138+) between the groups (Fig. 3A). For a more precise description of antigen-specific humoral immune response, we analyzed the total IgG and isotype pattern of mouse IgG antibody response directed against rabbit IgG in the serum of WT and IL-17RA−/− mice. There was no significant difference in total mouse IgG antibody titers to total rabbit IgG at Day 14 between the groups (Fig. 3B). Additionally, the analysis of IgG isotypes revealed no bias for Th1 (IgG2a) or Th2 (IgG1) antibody production in the absence of IL-17RA signaling (Fig. 3B).
Figure 3. Comparable systemic immune response between C57BL/6 and IL-17RA−/− mice.
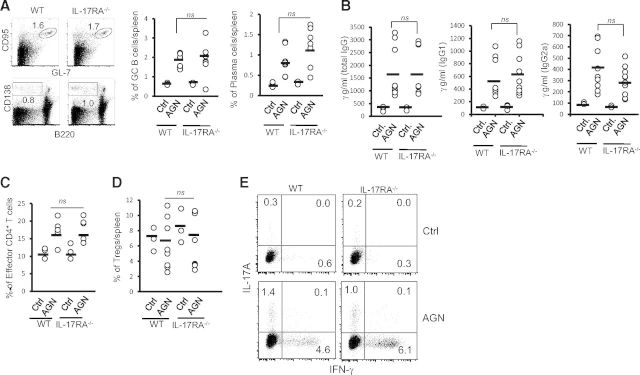
(A) Flow cytometric analysis of splenic cells from C57BL/6 (n=9) and IL-17RA−/− (n=10) was performed at Day 14 to determine the frequency of GC B cells (B220+GL-7+CD95+) and plasma cells (B220loCD138hi). Numbers in the dot plot represent percentage of cells. (B) At Day 14, serum titers of mouse anti-rabbit total IgG, IgG1 isotype, and IgG2a isotype were measured by ELISA in C57BL/6 (n=9) and IL-17RA−/− (n=10) mice. Flow cytometric analysis of splenic cells from C57BL/6 (n=9) and IL-17RA−/− (n=10) was performed at Day 14 to determine the frequency of (C) effector CD4+ T cells (CD4+CD62LloCD44hi) and (D) natural Tregs (CD4+CD25+Foxp3+). Each dot represents individual mice, and the horizontal bars indicate mean for each group. Data are pooled from two independent experiments. (E) At Day 14, splenocytes were stimulated with PMA/ionomycin for 4 h and intracellularly stained for IL-17A and IFN-γ, followed by FACS analyses (gated on CD4+ cells). Numbers in the dot plot represent percentage of cells.
As CD4+ T cells play a major a role in the pathogenesis of AGN, we next characterized the splenic and renal T cell responses in WT and IL-17RA−/− mice. At Day 14, a percentage of effector CD4+ T cells (CD4+CD62LloCD44hi) in the spleen was comparable between the groups (Fig. 3C). Although the number of kidney-infiltrating CD4+ T cells was three- to fourfold less in IL-17RA−/− mice compared with control, 90–95% of these cells have a surface phenotype characteristic of effector cells (CD4+CD62LloCD44hi; data not shown). Additionally, the frequency of natural Tregs (CD4+CD25+Foxp3+) in the spleen was similar between the groups (Fig. 3D).
To determine the effector cytokine production from CD4+ T cells, splenocytes were ex vivo-stimulated with PMA/ionomycin for 4 h and then intracellularly stained for IL-17A and IFN-γ. As shown in Fig. 3E, there was no difference in the percentage of CD4+IL-17A+ and CD4+IFN-γ+ cells in the spleen of WT and IL-17RA−/− following AGN. In addition, purified splenic CD4+ T cells from WT and IL-17RA−/− mice were restimulated with anti-CD3/CD28 for 48 h, and supernatants were evaluated for IFN-γ, IL-17A, and IL-4 production by ELISA. CD4+ T cells from WT and IL-17RA−/− mice produced comparable levels of IL-17A and IL-4 upon restimulation (Supplemental Fig. 1). Although there was a trend toward increased IFN-γ production by IL-17RA−/− CD4+ T cells, it did not achieve statistical significance (Supplemental Fig. 1). Overall, these data indicated that systemic B and T cells responses were largely unaffected in the absence of IL-17RA signaling in AGN mice.
Renal inflammation was severely compromised in the absence of IL-17RA signaling
To evaluate the contribution of IL-17RA signaling in local inflammatory reaction in the kidney cortex, we next determined the frequency of kidney-infiltrating innate effectors at early (Day 7) and late (Day 14) stages of AGN development by FACS analyses. At Day 7, a number of neutrophils (Ly6G+Ly6C−) were significantly diminished in the IL-17RA−/− kidney compared with WT mice (Fig. 4A). At this time-point, the frequency of kidney-infiltrating inflammatory monocytes (Ly6G−Ly6C+CD11b+F4/80lo) and macrophages (Ly6G−Ly6C+CD11b+F4/80hi) was comparable between the groups (Fig. 4A). However, the number of kidney-infiltrating neutrophils and inflammatory monocytes was reduced significantly in the absence of IL-17RA signaling at later stages (Day 14) of nephritis development. Although the frequency of macrophages at Day 14 was also reduced in IL-17RA−/− kidney compared with WT mice, it did not achieve statistical significance. Furthermore, to determine whether IL-17RA−/− mice are neutropenic, we enumerated the absolute number of neutrophils in the blood of WT and IL-17RA−/− mice following Giemsa stain. As shown in Fig. 4B, there was no significant difference in the number of neutrophils between WT and IL-17RA−/− mice. These results clearly indicated that whereas the infiltration of neutrophils at early and late stages of AGN development is IL-17RA-dependent, monocytes and macrophages require IL-17RA signaling to infiltrate the kidney, albeit at a later stage.
Figure 4. Diminished renal inflammation in the absence of IL-17RA.
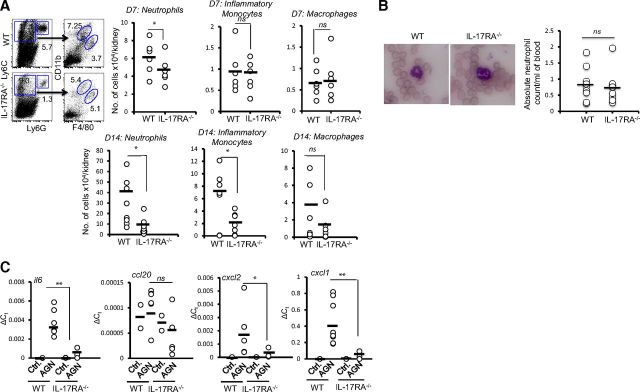
(A) Flow cytometric analysis of isolated renal leukocytes from C57BL/6 and IL-17RA−/− mice were performed at Days 7 (n=6) and 14 (n=9–10) to determine the number of neutrophils (Ly6G+Ly6C−), inflammatory monocytes (Ly6G−Ly6C+CD11b+F4/80lo), and macrophages (Ly6G−Ly6C+CD11b+F4/80lo). Numbers in the dot plot represent percentage of cells. The scattered plots represent absolute number of cells/kidney. (B) Peripheral blood smears of WT (n=11) and IL-17RA−/− (n=9) mice were stained with Giemsa stain and microscopically evaluated for the number of neutrophils/ml blood. (C) qPCR analysis of renal cytokine and chemokine mRNA expression in C57BL/6 (n=3 for control; n=9 for AGN) and IL-17RA−/− (n=3 for control; n=10 for AGN) mice at Day 10. Each dot represents individual mice, and the horizontal bars indicate mean for each group. The scattered plot is the pooled data from two individual experiments. *P < 0.05; **P <0.01.
One of the major mechanisms by which IL-17 drives infiltration of innate effector cells in the inflamed organs is via production of cytokines and chemokines from target cells [27]. To determine whether a diminished number of infiltrating innate effectors correlate with reduced kidney-specific expression of inflammatory mediators, we measured the transcript expression of IL-17-responsive cytokine and chemokine genes in the kidney of WT and IL-17RA−/− mice by qPCR. At Day 10, kidney-specific expression of Il6, Cxcl2, and Cxcl1 mRNA was reduced significantly in IL-17RA−/− mice compared with WT mice. However, there was no difference in the Ccl20 mRNA expression between the groups (Fig. 4C). These results clearly suggested that IL-17-driven chemotactic signals in the nephritic kidney are required for infiltration of innate effector cells in the pathogenesis of AGN.
IL-17 induces proinflammatory cytokine/chemokine expression in primary mouse tECs
Previous studies in mouse models of acute and chronic kidney injury showed that interaction between inflammatory cytokines and tECs activates cell-signaling pathways that precede induction of local inflammatory events in the kidney. The tECs express functional IL-17R and constitute ∼70% of the total cells in the kidney cortex [19]. Therefore, to study the potential role of IL-17 in renal inflammation, we analyzed its regulatory effect on the expression of the cytokines and chemokines in mouse primary tECs. To investigate this issue, we stimulated mouse primary tECs with IL-17 in the presence or absence of TNF-α (with which IL-17 exhibits profound synergy) [27]. As shown in Fig. 5A, mRNA expression of Il6, Cxcl1, Cxcl2, and Ccl20 transcripts was induced significantly by synergistic effects of IL-17 and TNF-α, whereas IL-17 or TNF-α alone had minimal effect in tECs.
Figure 5. IL-17A induces cytokine and chemokine gene expression in tECs.
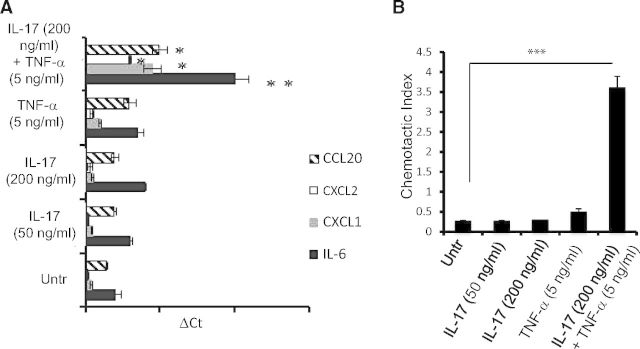
(A) Mouse tECs were left untreated (Untr) or treated with IL-17 (50 ng/ml and 200 ng/ml), TNF-α (5 ng/ml), or IL-17 (200 ng/ml) + TNF-α (5 ng/ml). After 24 h of stimulation, cells were evaluated for cytokine and chemokine gene expression by qPCR. (B) Following 24 h of stimulation, cell culture supernatant from tECs was used to perform in vitro transwell membrane migration assays using bone marrow-derived neutrophils as responding cells. The bar diagram represent mean ± sd. The data represent one of three independent experiments with similar results. *P < 0.05; **P < 0.01; ***P < 0.0001.
To assess whether cytokines and chemokines, produced by tECs in response to IL-17 alone or in synergy with TNF-α, can induce chemotactic signals enough to facilitate migration of innate effectors, we performed transwell chamber migration assays with supernatant from treated or untreated tECs. As our in vivo data suggest that migration of neutrophils is maximally affected in the absence of IL-17RA signaling, we decided to use neutrophils as a prototypic cell type for these assays. Neutrophils purified from the bone marrow of C57BL/6 mice demonstrated a significant increase in chemotactic migration in the presence of tEC supernatant stimulated with IL-17 and TNF-α compared with the untreated group (Fig. 5B). Consistent with the mRNA expression patterns of cytokine and chemokine genes in IL-17-stimulated tECs, we found no difference between the migration of neutrophils in the presence of supernatant from tECs treated with IL-17 or TNF-α alone and untreated groups (Fig. 5B).
Neutralization of IL-17A ameliorated AGN in WT mice
We showed that the absence of IL-17RA diminished the renal pathology and inflammatory response following induction of AGN. Based on this finding, we hypothesized that neutralization of IL-17A may have beneficial impact on the development of AGN. To this end, three groups of C57BL/6 mice were treated with a lower dose (500 μg/week) or higher dose (500 μg, three times/week) of anti-IL-17A or isotype control (500 μg, three times/week) antibodies following induction of AGN. After 14 days, mice were evaluated for the development of AGN. Consistent with the disease-protective phenotype in IL-17RA−/− mice, we observed significant reduction in the serum BUN levels in mice treated with a higher dose of anti-IL-17A antibody compared with control groups (Fig. 6A). However, there was no difference between the group of mice treated with the lower dose of anti-IL-17A antibody and control mice (Fig. 6A). In support of these findings, PAS-stained kidney sections of WT mice treated with a higher dose of anti-IL-17A antibody demonstrated a significantly reduced percentage of abnormal glomeruli and glomeruli with crescent formation compared with the isotype-treated group (Fig. 6B). The tubular inflammation score was also significantly reduced in groups of mice treated with a higher dose of anti-IL-17 antibody compared with control mice. Interestingly, we observed a dose-dependent effect of IL-17A neutralization on the glomerular and tubular inflammatory changes, indicating that a lower dose of anti-IL-17A antibody was insufficient to neutralize IL-17A in the nephritic kidney. Next, we determined whether the resistant phenotype-observed mice treated with anti-IL-17A antibody correlated with diminished innate cell influx in the nephritic kidney, as shown before in IL-17RA−/− mice. As shown Fig. 6C, mice treated with anti-IL-17A antibody (1500 μg/week/mice) demonstrated a significant decrease in the number of kidney-infiltrating neutrophils at Day 14. Although the frequency of inflammatory monocytes and macrophages was also reduced in anti-IL-17A-treated mice compared with the control group, it did not achieve statistical significance. Overall, these results clearly suggested that prophylactic administration of IL-17A has a beneficial impact on the development of renal pathology associated with AGN.
Figure 6. The neutralization of IL-17A attenuated AGN development in WT mice.

Three groups of C57BL/6 mice (n=10) were treated with a lower dose (500 μg/week) or higher dose (1500 μg/week) of anti-IL-17A or isotype (Iso.) control (500 μg, three times/week) antibody following induction of AGN. At Day 14, mice were evaluated for the development of AGN. Renal dysfunction at Day 14 was assessed by determination of the (A) serum BUN level. Each dot represents individual mice, and the horizontal bars indicate mean for each group. (B) Renal pathology was blindly evaluated and scored for percentage of abnormal glomeruli, percentage crescent formation, and tubular inflammation in C57BL/6 and IL-17RA−/− mice. Bar diagram represent mean ± sd. Data are pooled from two independent experiments. (C) Flow cytometric analysis of isolated renal leukocytes from anti-IL-17A antibody and isotype control antibody-treated C57BL/6 mice was performed at Day 14 (n=5) to determine the number of neutrophils (Ly6G+Ly6C−), inflammatory monocytes (Ly6G−Ly6C+CD11b+F4/80lo), and macrophages (Ly6G−Ly6C+CD11b+F4/80lo) following AGN. The scattered plots represent absolute number of cells/kidney. The horizontal bars represents mean for each group. *P < 0.05; **P < 0.01.
DISCUSSION
Th cells infiltrate the kidney in human and experimental glomerulonephritis, and several lines of evidence indicate that Th cells that mediated tissue damage play an important role in the pathogenesis of inflammatory diseases. However, the function of different Th cell subsets, particularly the recently identified Th17 cells and their signature cytokine IL-17, is poorly defined in AGN. With the use of a prototypic mouse model of AGN, we showed that IL-17RA signaling is critical for the development of renal pathology. Consequently, IL-17RA−/− signaling mice exhibited diminished renal inflammation and infiltration of the innate effector cells in the kidney, correlating with the reduced expression of IL-17-responsive cytokine and chemokine genes. In vitro, IL-17 enhanced the production of proinflammatory cytokines and chemokines from tECs, which are implicated in the recruitment of innate effector cells in the kidney. Finally, neutralization of IL-17A ameliorated renal pathology in WT mice following AGN induction. These results clearly demonstrated that IL-17RA signaling contributes to renal tissue injury and suggested that blocking IL-17RA signaling may be a promising therapeutic strategy for the treatment of AGN associated with multiple autoinflammatory kidney disorders.
To date, nearly all documented IL-17R signaling occurs in cells of nonhematopoietic origin, particularly epithelial and mesenchymal cells [27]. However, recent reports indicate a role for IL-17 in hematopoietic compartments [25–29]. IL-17 promotes granulopoiesis and neutrophil survival by stimulating the expression of G-CSF and neutrophil-attracting chemokines in the inflamed organs [28]. In macrophages, IL-17 indirectly induces the expression of TNF-α and IL-1 [29]. Whereas the role of IL-17 on innate cells is well-established, some, but not all, studies suggest a direct role of IL-17 on B cells. IL-17 promotes GC formation and autoantibody production from B cells [20, 26]. Nevertheless, with the argument against the direct role of IL-17 on B cells, a recent report showed that the survival of lupus AGN-prone mice was improved significantly in the absence of IL-17, whereas an abnormal B cell response continued unabated. Subsequently, loss of IL-17 resulted in reduced, IL-17-responsive gene expression and infiltration of inflammatory cells in the nephritic kidney, correlating with disease resistance [20]. These data point to a critical and likely direct role for IL-17 family cytokines on cells of nonhematopoietic origin in mediating kidney pathology. Supporting this line, we also found that IL-17RA−/− mice were protected completely from AGN, as assessed by the development of overt renal pathology and inflammatory changes. Interestingly, IL-17RA−/− exhibit diminished renal inflammation, despite an abnormal systemic immune response, correlating with disease resistance. These data are indicative of the fact that IL-17R signaling in kidney-resident cells plays a central role in driving renal inflammation in AGN. The identification of the IL-17-responsive cells and subsequently, the blockage of IL-17R signaling in these cells may prove beneficial in developing a better-targeted therapeutic approach in controlling AGN.
The two major immune compartments that contribute to AGN are (1) kidney-infiltrating cellular and humoral effectors and (2) kidney-resident cells. Whereas the injurious role of kidney-infiltrating immune cells has been emphasized, the contribution of kidney-resident cells has proved difficult to define. Studies in chronic kidney injury models showed that interaction between inflammatory cytokines and kidney resident cells activates cell-signaling pathways that precede induction of local inflammatory events in the kidney and initiates renal injury and fibrotic events. The major kidney-resident cells implicated in the AGN are podocytes, mesangial cells, and proximal and distal tECs [30–32]. To date, most of the IL-17R signaling outcome studies involve in vitro approaches using fibroblast and epithelial cell lines, and information regarding the pathological consequence of IL-17R signaling in vivo in the nephritic kidney is currently lacking. To investigate the potential contribution of IL-17 in inducing inflammatory mediators from kidney-resident cells, we showed that IL-17, in synergy with TNF-α, induced the production of multiple cytokine and chemokine genes from tECs. Overall, these findings suggest a general proinflammatory effect of IL-17 on resident kidney cells. As cell types interpret IL-17 signals differently, with overlapping but nonidentical patterns of gene expression, the contribution of IL-17R signaling in the other kidney-resident cells types, such as mesangial cells, podocytes, and glomerular endothelial cells, in the pathogenesis of AGN remains to be defined.
Whereas it is becoming ever more apparent that IL-17 plays a central role in the pathogenesis of AGN, therapeutic neutralization of IL-17 to diminish the severity of AGN has never been tested in mouse models of autoinflammatory kidney diseases. Lupus-prone mice treated with anti-IL-6 antibody (IL-6 is required for Th17 differentiation) or IL-17RFc (IL-21 is produced by Th17 cells) resulted in diminished AGN [33, 34]. However, given the pleiotropic effect of IL-6 and IL-21 on other immune cell types, such as B cells and myeloid cells, it is difficult to assess the therapeutic efficacy of anti-IL-6 and anti-IL-21 therapies in diminishing the Th17 response in these settings. On the other hand, administration of IL-17RFc in autoimmune-prone BXD2 mice resulted in diminished GC reaction and autoantibody production. But, the clinical consequence of IL-17R blocking in terms of kidney pathology development is largely unknown [26]. Here, we show for the first time that administration of anti-IL-17A antibody attenuated renal pathology and inflammation and significantly improved renal function associated with AGN.
Multiple clinical trials targeting the IL-17 pathway and signaling mechanisms are ongoing, and positive results have been obtained in psoriasis, rheumatoid arthritis, and ankylosing spondylitis [35]. Thus, our results provide a strong rationale for future clinical trials to test the therapeutic effectiveness of anti-IL-17R antibody in patients suffering from AGN associated with autoinflammatory kidney disorders. One advantage of IL-17 depletion therapy over anti-TNF drugs is that the risk of tuberculosis appears to be reduced. On the other hand, depletion of IL-17 may lead to neutropenia, and the patients may succumb to extracellular bacterial and fungal infections. However, at the current stage of drug development, this has not been a major concern. Although few cases of neutropenia and Candida infection have been noted, the direct association of IL-17 depletion in these patients has not been proven.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Children's Hospital of Pittsburgh of UPMC Health System (to A.R.H.), a Pediatric Infectious Disease Society Fellowship Award (funded by the Stanley A. Plotkin Sanofi Pasteur Fellowship Award to A.R.H.), U.S. National Institutes of Health (Grant DE022550 to S.L.G.), and the Division of Rheumatology and Clinical Immunology, UPMC (to P.S.B.).
We thank Drs. Youhua Liu and Mandy McGeachy for helpful suggestions and discussions.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- −/−
- deficient
- AGN
- autoimmune glomerulonephritis
- BUN
- blood urea nitrogen
- CD62L
- CD62 ligand
- Foxp3
- forkhead box p3
- GBM
- glomerular basement membrane
- GC
- germinal center
- NRS
- nephrotoxic rabbit serum
- PAS
- periodic-acid Schiff
- qPCR
- quantitative PCR
- RBC
- red blood cell
- tEC
- tubular epithelial cell
- Treg
- regulatory T cell
- UPMC
- University of Pittsburgh Medical Center
- WBC
- white blood cell
- WT
- wild-type
AUTHORSHIP
K.R., S.P., K.M., and A.R.H. designed, executed, and analyzed data from the experiments. S.L.G. designed and analyzed the data. P.S.B. designed and analyzed the data and wrote the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1. Kambham N. (2012) Crescentic glomerulonephritis: an update on pauci-immune and anti-GBM diseases. Adv. Anat. Pathol. 19, 111–124. [DOI] [PubMed] [Google Scholar]
- 2. Kulkarni O. P., Anders H. J. (2012) Lupus nephritis. How latest insights into its pathogenesis promote novel therapies. Curr. Opin. Rheumatol. 24, 457–465. [DOI] [PubMed] [Google Scholar]
- 3. Srivastava A., Rao G. K., Segal P. E., Shah M., Geetha D. (2013) Characteristics and outcome of crescentic glomerulonephritis in patients with both antineutrophil cytoplasmic antibody and anti-glomerular basement membrane antibody. Clin. Rheumatol. 32, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 4. Kewalramani R., Singh A. K. (2002) Immunopathogenesis of lupus and lupus nephritis: recent insights. Curr. Opin. Nephrol. Hypertens. 11, 273–277. [DOI] [PubMed] [Google Scholar]
- 5. Tipping P. G., Kitching A. R., Cunningham M. A., Holdsworth S. R. (1999) Immunopathogenesis of crescentic glomerulonephritis. Curr. Opin. Nephrol. Hypertens. 8, 281–286. [DOI] [PubMed] [Google Scholar]
- 6. Makker S. P. (1993) Mediators of immune glomerular injury. Am. J. Nephrol. 13, 324–336. [DOI] [PubMed] [Google Scholar]
- 7. Awasthi A., Kuchroo V. K. (2009) Th17 cells: from precursors to players in inflammation and infection. Int. Immunol. 21, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Veldhoen M. (2009) The role of T helper subsets in autoimmunity and allergy. Curr. Opin. Immunol. 21, 606–611. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Z., Kyttaris V. C., Tsokos G. C. (2009) The role of IL-23/IL-17 axis in lupus nephritis. J. Immunol. 183, 3160–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turner J. E., Paust H. J., Steinmetz O. M., Panzer U. (2010) The Th17 immune response in renal inflammation. Kidney Int. 77, 1070–1075. [DOI] [PubMed] [Google Scholar]
- 11. Gan P. Y., Steinmetz O. M., Tan D. S., O'Sullivan K. M., Ooi J. D., Iwakura Y., Kitching A. R., Holdsworth S. R. (2010) Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J. Am. Soc. Nephrol. 21, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ooi J. D., Phoon R. K., Holdsworth S. R., Kitching A. R. (2009) IL-23, not IL-12, directs autoimmunity to the Goodpasture antigen. J. Am. Soc. Nephrol. 20, 980–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaffen S. L. (2009) Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cua D. J., Tato C. M. (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10, 479–489. [DOI] [PubMed] [Google Scholar]
- 15. Gaffen S. L. (2011) Recent advances in the IL-17 cytokine family. Curr. Opin. Immunol. 23, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaffen S. L. (2008) An overview of IL-17 function and signaling. Cytokine 43, 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitching A. R., Holdsworth S. R. (2011) The emergence of TH17 cells as effectors of renal injury. J. Am. Soc. Nephrol. 22, 235–238. [DOI] [PubMed] [Google Scholar]
- 18. Nalbandian A., Crispin J. C., Tsokos G. C. (2009) Interleukin-17 and systemic lupus erythematosus: current concepts. Clin. Exp. Immunol. 157, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paust H. J., Turner J. E., Steinmetz O. M., Peters A., Heymann F., Holscher C., Wolf G., Kurts C., Mittrucker H. W., Stahl R. A., Panzer U. (2009) The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J. Am. Soc. Nephrol. 20, 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pisitkun P., Ha H. L., Wang H., Claudio E., Tivy C. C., Zhou H., Mayadas T. N., Illei G. G., Siebenlist U. (2012) Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 37, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian Y., Qin J., Cui G., Naramura M., Snow E. C., Ware C. F., Fairchild R. L., Omori S. A., Rickert R. C., Scott M., Kotzin B. L., Li X. (2004) Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity 21, 575–587. [DOI] [PubMed] [Google Scholar]
- 22. Giltiay N. V., Lu Y., Cullen J. L., Jorgensen T. N., Shlomchik M. J., Li X. (2013) Spontaneous loss of tolerance of autoreactive B cells in Act1-deficient rheumatoid factor transgenic mice. J. Immunol. 191, 2155–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qian Y., Giltiay N., Xiao J., Wang Y., Tian J., Han S., Scott M., Carter R., Jorgensen T. N., Li X. (2008) Deficiency of Act1, a critical modulator of B cell function, leads to development of Sjogren's syndrome. Eur. J. Immunol. 38, 2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fu Y., Du Y., Mohan C. (2007) Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin. Immunol. 124, 109–118. [DOI] [PubMed] [Google Scholar]
- 25. O'Connor W., Jr., Kamanaka M., Booth C. J., Town T., Nakae S., Iwakura Y., Kolls J. K., Flavell R. A. (2009) A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10, 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsu H. C., Yang P., Wang J., Wu Q., Myers R., Chen J., Yi J., Guentert T., Tousson A., Stanus A. L., Le T. V., Lorenz R. G., Xu H., Kolls J. K., Carter R. H., Chaplin D. D., Williams R. W., Mountz J. D. (2008) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 9, 166–175. [DOI] [PubMed] [Google Scholar]
- 27. Onishi R. M., Gaffen S. L. (2010) Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forlow S. B., Schurr J. R., Kolls J. K., Bagby G. J., Schwarzenberger P. O., Ley K. (2001) Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 98, 3309–3314. [DOI] [PubMed] [Google Scholar]
- 29. Liu B., Tan W., Barsoum A., Gu X., Chen K., Huang W., Ramsay A., Kolls J. K., Schwarzenberger P. (2010) IL-17 is a potent synergistic factor with GM-CSF in mice in stimulating myelopoiesis, dendritic cell expansion, proliferation, and functional enhancement. Exp. Hematol. 38, 877.e1–884.e1. [DOI] [PubMed] [Google Scholar]
- 30. Muller G. A., Markovic-Lipkovski J., Frank J., Rodemann H. P. (1992) The role of interstitial cells in the progression of renal diseases. J. Am. Soc. Nephrol. 2, S198–S205. [DOI] [PubMed] [Google Scholar]
- 31. Radeke H. H., Resch K. (1992) The inflammatory function of renal glomerular mesangial cells and their interaction with the cellular immune system. Clin. Investig. 70, 825–842. [DOI] [PubMed] [Google Scholar]
- 32. Kelley V. R., Singer G. G. (1993) The antigen presentation function of renal tubular epithelial cells. Exp. Nephrol. 1, 102–111. [PubMed] [Google Scholar]
- 33. Herber D., Brown T. P., Liang S., Young D. A., Collins M., Dunussi-Joannopoulos K. (2007) IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R. Fc reduces disease progression. J. Immunol. 178, 3822–3830. [DOI] [PubMed] [Google Scholar]
- 34. Liang B., Gardner D. B., Griswold D. E., Bugelski P. J., Song X. Y. (2006) Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 119, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miossec P., Kolls J. K. (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 11, 763–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.