

T-box transcription factor expression enables effector differentiation of self-reactive T cells upon antigen engagement, but fails to prevent their deletion.
Keywords: transcription factors, effector differentiation/immunity, immunotherapy
Abstract
CD8+ T cells must detect foreign antigens and differentiate into effector cells to eliminate infections. But, when self-antigen is recognized instead, mechanisms of peripheral tolerance prevent acquisition of effector function to avoid autoimmunity. These distinct responses are influenced by inflammatory and regulatory clues from the tissue environment, but the mechanism(s) by which naive T cells interpret these signals to generate the appropriate immune response are unclear. The identification of the molecules operative in these cell-fate decisions is crucial for developing new treatment options for patients with cancer or autoimmunity, where manipulation of T cell activity is desired to alter the course of disease. With the use of an in vivo murine model to examine CD8+ T cell responses to healthy self-tissue, we correlated self-tolerance with a failure to induce the T-box transcription factors T-bet and Eomes. However, inflammation associated with acute microbial infection induced T-bet and Eomes expression and promoted effector differentiation of self-reactive T cells under conditions that normally favor tolerance. In the context of a Listeria infection, these functional responses relied on elevated T-bet expression, independent of Eomes. Alternatively, infection with LCMV induced higher Eomes expression, which was sufficient in the absence of T-bet to promote effector cytokine production. Our results place T-box transcription factors at a molecular crossroads between CD8+ T cell anergy and effector function upon recognition of peripheral self-antigen, and suggest that inflammation during T cell priming directs these distinct cellular responses.
Introduction
Most self-reactive T cells are selectively eliminated during thymic development. However, some potentially autoreactive T cells make it out of the thymus and must be controlled by mechanisms of peripheral tolerance to avoid autoimmunity. Peripheral tolerance is a multifaceted process that can involve the deletion of autoreactive T cells or the inhibition of their proliferation and effector function [1–4]. This contrasts with protective immune responses, such as those elicited during acute infections, where reactive T cells expand in number, differentiate to acquire effector functions, and eventually form a stable memory population [5]. Yet, how naive T cells are initially directed toward tolerance versus immunity remains a fundamental question, as the T cell-intrinsic pathways that regulate these divergent cellular programs are still largely undefined.
Under the current paradigm, naive CD8+ T cells that engage antigen in the context of a MHC-I molecule must receive subsequent instructions that guide an appropriate immune response. These instructions come primarily from costimulation and inflammatory signals that accompany infections and tend to promote CD8+ T cell activation and robust immunity. Alternatively, engagement of antigen in the absence of inflammation or other danger signals favors tolerance, as this generally indicates surveillance of healthy tissue. The decision between T cell tolerance or immunity is clearly linked to human disease. For instance, suppression of T cell immunity can render patients susceptible to cancer and infection, whereas failure to maintain T cell tolerance can result in autoimmunity. Furthermore, the same tolerizing mechanisms that protect healthy tissues from autoimmunity also present a barrier to eliciting immune responses against tumors, which are derived from self-tissue and often share many of the same antigens [6–9]. Thus, defining the molecular pathways that influence the fate of responding CD8+ T cells has the potential to impact patient health significantly and could inform efforts to clinically alter T cell responses for therapeutic applications.
Insight into the pathways that determine CD8+ T cell tolerance versus immunity may be gleaned from studies of T cell differentiation—the process that guides essentially nonfunctional, naive T cells to become activated effector cells capable of clearing an infection. The T-box transcription factor T-bet directs CD4+ T cell lineage commitment toward a Th1 phenotype and promotes expression of the Th1-associated cytokine IFN-γ [10]. Despite more than one decade of research, the precise contribution of T-bet in CD8+ T cell effector differentiation and IFN-γ production is less clear and is complicated by expression of the related T-box transcription factor Eomes, which may share in these roles [11]. Indeed, several independent studies have reported that T-bet is not intrinsically required for CD8+ effector differentiation and IFN-γ production in response to pathogens—an effect attributed primarily to redundant functions by Eomes [11–16].
T-bet and Eomes influence expression of several other target genes that encode effector molecules important for CD8+ T cell control of infected or malignant cells, including GzmB, perforin, FasL, and the chemokine receptor CXCR3 [11, 12, 14, 17, 18]. Thus, there is a consensus that appropriate expression of T-bet and Eomes helps guide CD8+ T cell effector responses and contributes to pathogen clearance [12, 15, 19–21]. Conversely, the unregulated expression of these molecules in self-reactive T cells could circumvent tolerance and lead to autoimmunity, as is the case for CD4+ T cells [22–24] and for CD8+ T cells, which require T-bet for autoimmunity in Tg rat insulin promoter models of type 1 diabetes [16]. However, in this later system, self-reactive T cells are often described as ignorant rather than tolerant and require inflammation for antigen recognition and activation, impeding the evaluation of T-bet induction in T cells engaging self-antigen in the steady state [25, 26]. The contribution of T-bet expression has not been assessed in systems where induction of peripheral CD8+ T cell tolerance itself is an active process involving suppression of effector function, apoptosis, and subsequent deletion [27–30]. An intriguing possibility is that a dearth of T-box transcription factors directs CD8+ T cells toward a tolerant rather than an immune fate after self-antigen encounter under homeostatic conditions, whereas T-bet and Eomes expression is required to divert tolerance toward effector T cell differentiation under inflammatory conditions. Whereas there is evidence inversely correlating T-bet and Eomes expression with CD8+ T cell anergy [31], deficiency in these T-box transcription factors has not been implicated directly as a molecular mechanism for the promotion of CD8+ T cell tolerance.
Our study addressed the fundamental immunological question of what intrinsic signals guide naive CD8+ T cells away from an anergic fate and toward effector differentiation upon primary encounter with antigen. We confirmed that T-bet and Eomes failed to be induced in CD8+ T cells responding to tolerizing self-antigen, which proliferated briefly but did not acquire effector functions. However, expression of T-bet could be induced by this same self-antigen within an inflammatory environment during acute infection with Listeria monocytogenes, which has been reported to break T cell tolerance [32, 33]. Under these inflammatory conditions, T-bet was required for autoreactive CD8+ T cells to acquire effector capabilities, independent of Eomes. This requirement for T-bet was specific to Listeria, as inflammation during LCMV infection favored higher Eomes expression, which was also sufficient by itself to induce CD8+ T cell effector cytokine production. Despite this apparent redirection of T cell fate, inflammation and subsequent activation of T-bet and Eomes was inadequate to overcome peripheral deletion of T cells primed by tolerizing self-antigen. These results identify T-box transcription factor deprivation as a mechanism for maintaining the functional unresponsiveness that defines CD8+ T cell tolerance to healthy self-tissues.
MATERIALS AND METHODS
Mice
Alb:Gag and rag1−/− TCRGag Tg mice have been described previously [2, 3, 34]. B6, tbx21−/−, eomesf/f, and Lck-cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). tbx21−/−, eomesf/f, and Lck-cre mice were crossed with rag1−/− TCRGag Tg mice. To generate Gag-specific CD8+ T cells deficient for Eomes, eomesf/f × TCRGag mice were crossed with Lck-cre × TCRGag mice to generate eomesf/f × Lck-cre x TCRGag mice. CD2-T-bet Tg mice were graciously provided by Dr. Satoru Takahashi and have been described previously [35]. These mice were crossed with rag1−/− TCRGag Tg mice to generate Gag-specific T cells with constitutive T-bet expression. All animals were maintained under specific pathogen-free conditions and used in accordance with our animal protocol approved by the Animal Care Committee of the Department of Comparative Medicine, St. Louis University School of Medicine (St. Louis, MO, USA).
Cell lines, peptides, and antibodies
FBL is a murine erythroleukemia cell line of B6 origin that expresses the H-2b-restricted Friend murine leukemia virus-derived Gag glycoprotein as a tumor-associated antigen [36, 37]. Gag peptide (CCLCLTVFL) and Ova (SIINFEKL) or LCMV-GP33 (KAVYNFATM) control peptides were purchased from Pi Proteomics (Huntsville, AL, USA). Cell culture was conducted in complete RPMI 1640 with 10% FBS (Sigma, St. Louis, MO, USA). Fluorochrome-conjugated antibodies to CD8 (53–6.7), CXCR3 (CXCR3-173), PD-1 (RMP1-30), T-bet (4-B10), and Eomes (Dan11mag) were purchased from eBioscience (San Diego, CA, USA). Fluorochrome-conjugated antibodies to CD90.1 (OX-7), IFN-γ (XMG1.2), TNF-α (MP6-XT22), and TCR-Vα3 (RR3-16) were purchased from BD Biosciences (San Jose, Ca, USA). CFSE and eFluor 670 cell dyes were purchased from Invitrogen (Carlsbad, CA, USA).
T cell transfer and infection
Gag-specific T cells were isolated from spleens and lymph nodes of rag1−/− TCRGag, tbx21−/− TCRGag, eomesf/f Lck-cre TCRGag, or T-bet-Tg TCRGag donors. Whole-cell suspensions containing 2 × 106 Vα3-TCR+ CD8+ cells were i.v.-injected into sex- and age (6–8 weeks)-matched recipients. In some experiments, transferred cells were labeled with 2 μg/mL CFSE before infusion. To provide an immunogenic environment, 5 × 106 FBL cells were established i.p. in B6 recipients, 3 days before T cell transfer. Infected recipients received 2 × 106 CFU-attenuated (ΔActA) L. monocytogenes or 2 × 105 PFU LCMV Armstrong i.v., 2 h before T cell transfer.
Flow cytometry
Recipient spleen and peripheral lymph nodes were harvested for analysis at indicated time-points. Tissues were homogenized into single-cell suspensions before cell culture or staining for flow cytometry. Cell suspensions were stained for extracellular markers at 4°C for 30 min. For intracellular staining of the transcription factors, T-bet and Eomes, cells were fixed and permeabilized in Foxp3/Transcription Factor Fixation/Permeabilization solution (eBioscience) and proteins stained in Permeabilization buffer (eBioscience). Ex vivo cytokine production was assessed following overnight stimulation with 4 μg/mL Gag, Ova, or Gp33 peptide in the presence of GolgiPlug (BD Biosciences). For intracellular staining of IFN-γ and TNF-α, cells were fixed and permeabilized in Cytofix/Cytoperm buffer (BD Biosciences) and proteins stained in Perm/Wash buffer (BD Biosciences) for 30 min at 4°C, according to the manufacturer's protocol. For simultaneous staining of intracellular effector cytokines (IFN-γ) and transcription factors (Eomes), 4 h ex vivo restimulation with Gag peptide was performed, and then cells were fixed and permeabilized with the eBioscience Fix/Perm solution before antibody staining. All flow cytometry was conducted using an LSR II or FACSCanto II (BD Biosciences) and resulting data analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Quantitative RT-PCR
Transferred T cells were fixed in 4% formaldehyde and sorted to better than 95% purity using a FACSAria III (BD Biosciences). Total RNA was isolated from sorted cells using the PureLink FFPE RNA Isolation Kit (Invitrogen), and cDNA was synthesized using the Transcriptor First Strand cDNA synthesis kit (Roche, Indianapolis, IN, USA). Real-time PCR was performed with SYBR Select Master Mix (Life Technologies, Carlsbad, CA, USA) on a 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Relative amplification values were calculated by normalizing to amplification of β-actin. The following primers were used: tbx21 sense primer: 5′-CAACAACCCCTTTGCCAAAG-3′, tbx21 antisense primer: 5′-TCCCCCAAGCAGTTGACAGT-3′; cxcr3 sense primer: 5′-GCCAAGCCATGTACCTTGAG-3′, cxcr3 antisense primer: 5′-GTTCAGGCTGAAATCCTGTGG-3′; ifng sense primer: 5′-CACGGCACAGTCATTGAAAGC-3′, ifng antisense primer: 5′-GAGATAATCTGGCTCTGCAGG-3′; eomes sense primer: 5′-GCCTACCAAAACACGGATA-3′, eomes antisense primer: 5′-TCTGTTGGGGTGAGAGGAG-3′; gzmb sense primer: 5′-GCTACTGCTGACCTTGTCTCT-3′, gzmb antisense primer: 5′-TCACAGTGAGCAGCAGTCAG-3′; fasl sense primer: 5′-AACCCCAGTACACCCTCTG-3′, fasl antisense primer: 5′-CGTTGATCACAAGGCCACC-3′; pdcd1 sense primer: 5′-CAGGTACCCTGGTCATTCACT-3′, pdcd1 antisense primer: 5′-GACCAGTTGGACAAGCTGC-3′; tnf sense primer: 5′-CTGAACTTCGGGGTGATCGG-3′, tnf antisense primer: 5′-CTCAGCCACTCCAGCTGCTC-3′; β-actin sense primer: 5′-CCTTCGTTGCCGGTCCACAC-3′, β-actin antisense primer: 5′-ACCTCTCTTGCTCTGGGCCT-3′.
In vivo killing assay
Recipient mice received adoptive T cell transfers and infections, as described above. Three days after T cell transfer, B6 splenocytes (targets) were pulsed with 10 μg/ml Gag or control Ova or Gp33 peptide and differentially labeled with 1 or 5 μM eFluor 670, respectively. Targets were then washed twice in PBS, combined at a 1:1 ratio, and injected into recipient mice i.v. Twenty hours later, the frequency of eFluorhigh versus eFluorlow targets from recipient spleens was assessed by flow cytometry.
Immunotherapy assay
One week before treatment, FBL leukemia was established in Alb:Gag mice by i.v. injection of 1 × 105 viable FBL tumor cells. Seven days after tumor inoculation, tumor-bearing mice were vaccinated with Listeria, 2 h before adoptive transfer of 2 × 106 Gag-reactive CD8+ T cells by i.v. injection. Recipient survival was tracked for a minimum of 40 days with daily health monitoring, and recipients were killed upon moribund appearance.
Statistical analysis
The Kruskal-Wallis test was used for statistical comparison (GraphPad Prism 4) of total cell numbers between different treatment groups. A one-way ANOVA was used for statistical comparison of cell frequencies between multiple treatment groups. Survival data were analyzed with a Mantel-Cox log-rank test. P < 0.05 was considered significant.
RESULTS
To define the operative pathways that dictate whether CD8+ T cells become tolerant or immunized following primary antigen recognition in vivo, we compared responses to a single antigen (Gag) expressed in two distinct contexts: naturally, as an immunogenic tumor-associated antigen or as a tolerizing self-antigen. Specifically, TCRGag were transferred into normal B6 mice (naive), B6 mice with an established Gag-positive FBL tumor (immune), or Alb:Gag mice that express the same Gag antigen via a transgene in healthy hepatocytes (tolerant). Whereas transfer of this large number of potentially self-reactive, naive T cells is not considered physiological, it does provide a straightforward model for rigorously examining how antigen context influences naive T cell fate. Here, recognition of antigen in the immunizing or tolerizing context resulted in robust proliferation of responding CD8+ T cells within 3 days of transfer (Fig. 1A). Despite this burst in proliferation, T cells that encountered Gag self-antigen became tolerant, failed to acquire effector function, and were ultimately deleted from the periphery [4]. The signals that influence the fate of responding T cells come from a multitude of extrinsic environmental clues, such as costimulatory and negative regulatory molecules engaged along with antigen. Here, we sought to identify the molecular pathways operative inside of the T cell that guide differentiation away from tolerance and toward an effector cell phenotype.
Figure 1. CD8+ T cells proliferate but fail to express T-bet or Eomes upon self-antigen encounter.
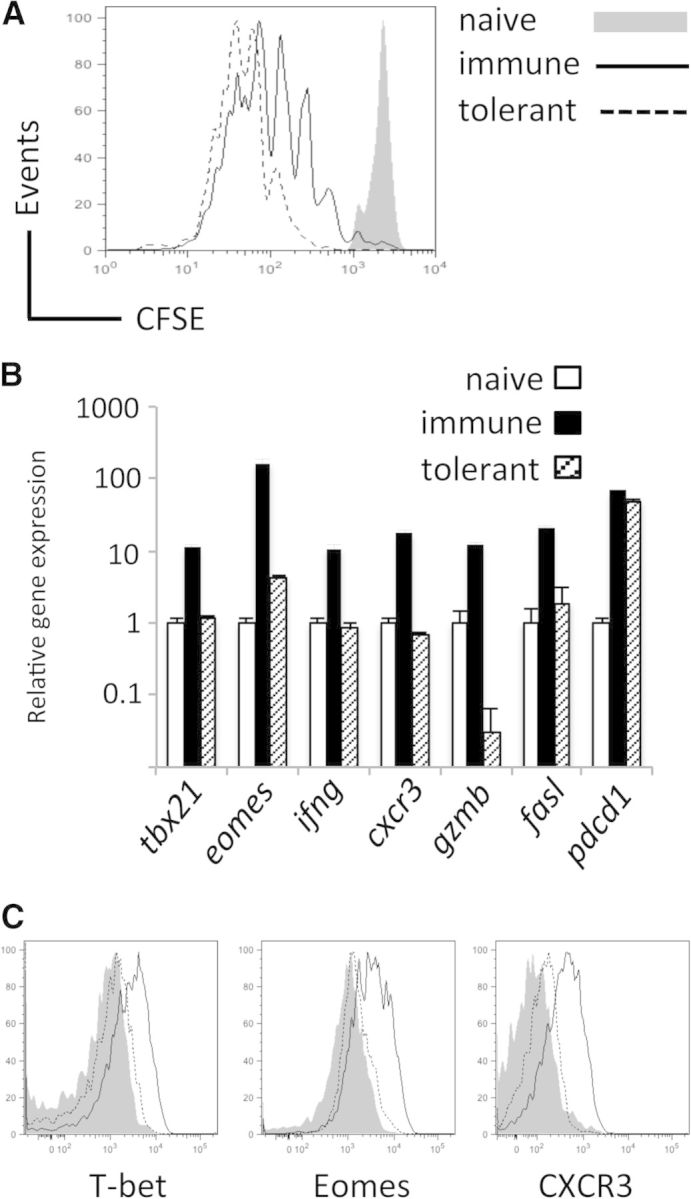
CD8+ CD90.1+ Gag-specific T cells were adoptively transferred into naive (B6), immunizing (B6+FBL), or tolerizing (Alb:Gag) environments. (A) CFSE dilution by transferred T cells was assessed 3 days later and is displayed in overlayed histograms. (B) Real-time PCR analysis of tbx21, eomes, ifng, cxcr3, gzmb, fasl, and pdcd1 mRNA expression in FACS-purified T cells, 3 days after transfer into a naive, immunizing, or tolerizing environment. Data are presented as relative expression versus β-actin control, and error bars are sd. (C) T-bet, Eomes, and CXCR3 protein expression in T cells, 3 days after adoptive transfer, is compared directly by overlaid histograms from naive, immunizing, or tolerizing environments. Data are representative of three experiments, each with three recipients/group.
As the T-box transcription factors T-bet and Eomes have roles in CD8+ T cell differentiation and effector function, we examined whether T-bet, Eomes, and several reported target genes were differentially expressed following encounter with antigen in an immunizing versus a tolerizing context. Compared with naive CD8+ T cells, those stimulated with immunizing Gag tumor antigen induced approximately tenfold more T-bet (encoded by tbx21) and >100-fold more eomes gene expression, 2 days after antigen encounter (Fig. 1B). However, T cells responding to tolerizing Gag self-antigen showed only a modest increase in eomes gene expression and no increase in tbx21. The T-box target genes ifng, cxcr3, gzmb, and fasl associated with T cell effector function were also not induced under tolerizing conditions (Fig. 1B). In contrast, expression of pdcd1, a coinhibitory receptor, up-regulated upon T cell activation [38], was increased by antigen recognition in the immunizing and tolerizing environments (Fig. 1B), supporting the engagement of other conventional activation pathways in T cells within tolerizing hosts. At the protein level, T-bet and Eomes expression was much lower following tolerizing antigen stimulation compared with T cells activated under immunizing conditions, and this pattern of protein expression was also true for the T-box-regulated cell-surface chemokine receptor CXCR3 (Fig. 1C) and the effector cytokine IFN-γ [4]. Thus, despite inducing robust CD8+ T cell proliferation, encounter with tolerizing self-antigen failed to induce substantially T-bet, Eomes, or T-box target genes that encode effector molecules. These data support a role for inadequate expression of T-bet or Eomes in the functional defects characteristic of tolerant CD8+ T cells.
T cell tolerance inversely correlates with T-bet and Eomes expression
If insufficient T-bet or Eomes expression following self-antigen encounter precludes effector CD8+ T cell differentiation and promotes tolerance, then boosting T-bet or Eomes expression should promote T cell differentiation toward an effector phenotype. To achieve this, Gag-specific CD8+ T cells (CD90.1+) were transferred into normal B6 or tolerizing Alb:Gag recipients and left untreated or infected with a live, attenuated ActA-deficient Listeria, which induces inflammation and production of cytokines, such as IL-2, IL-12, type I IFNs, and IFN-γ [39], known to induce T-box transcription factor expression [40–42]. Intracellular T-bet and Eomes, as well as cytokine production, were assessed 3 days later—near the peak of responses in Alb:Gag recipients and before T cell deletion that precludes reliable functional analysis at later times [4, 43]. Under nontolerizing conditions in B6 recipients (no Gag antigen), inflammation associated with Listeria induced a modest increase in T-bet and Eomes compared with unvaccinated hosts but failed to induce T cell expansion or expression of the T-bet target, IFN-γ (Fig. 2A). In the absence of inflammation, CD8+ T cells that encountered a tolerizing self-antigen in Alb:Gag recipients showed minimal expression of T-bet and Eomes and did not produce the effector cytokines IFN-γ or TNF-α upon ex vivo restimulation (Fig. 2B). However, inflammation associated with Listeria infection transformed the T cell encounter with self-antigen from a strictly tolerizing event into one that corresponded with high T-bet expression and production of IFN-γ (Fig. 2B and C). Despite this large increase in T-bet, Eomes expression remained essentially unchanged, suggesting that T-bet may be key to averting tolerance and promoting effector T cell differentiation when self-antigen is encountered under inflammatory conditions.
Figure 2. Induction of T cell effector function corresponds with T-bet expression.

CD8+ CD90.1+ Gag-specific T cells were transferred into (A) B6 or (B) tolerizing Alb:Gag recipients, with or without infection with Listeria. Recipient spleens were analyzed 3 days later for the frequency of transferred T cells by flow cytometry (top). Inset numbers are the percent of total splenocytes within the inscribed square region. Intracellular T-bet and Eomes protein expression was assessed directly ex vivo (middle), and IFN-γ and TNF-α production were determined after overnight restimulation with Gag peptide (bottom). Numbers in each quadrant represent the percent of gated CD8+ CD90.1+ T cells, and data are representative of six separate experiments, each with two to three mice/group. (C) The percent of gated CD8+ CD90.1+ T cells that express T-bet (top) or Eomes (bottom) in differentially treated Alb:Gag recipients is depicted, with each circle representing individual mice pooled from six separate experiments. Horizontal bars represent the average for each group, and P values are indicated.
T-bet is required to divert CD8+ T cell dysfunction toward effector differentiation during Listeria vaccination
To evaluate the contribution of T-bet in directing CD8+ T cells toward an effector cell-differentiation program in response to self-antigen, we examined T cells deficient for the gene that encodes T-bet (tbx21). Here, tolerizing Alb:Gag host mice received a cotransfer of WT and tbx21−/− Gag-specific CD8+ T cells, with or without Listeria, and effector cytokine production was assessed 3 days later. As before, IFN-γ production was not elicited in WT T cells upon encounter with self-antigen alone in Alb:Gag recipients but could be induced following infection with Listeria (Fig. 3A and B). In contrast, despite identical expansion (Fig. 3C), IFN-γ responses were not elicited in tbx21−/− T cells following Listeria infection, suggesting that T-bet is required to promote effector T cell differentiation under these inflammatory conditions. This was not the case for TNF-α, which was produced independently of T-bet within the tolerizing environment (Fig. 3A), echoing prior observations in self-reactive CD4+ Th1 T cells [44].
Figure 3. T-bet is required for effector function within a tolerizing environment.

(A) TCR-Tg, Gag-specific T cells on a WT background (CD90.1+) or tbx21−/− background (CD90.1+/CD90.2+) were cotransferred into tolerizing Alb:Gag recipients, with or without Listeria infection. Three days later, production of IFN-γ and TNF-α by transferred T cells was measured after overnight restimulation with the Gag peptide (right). Numbers in each quadrant represent the percent of gated CD8+ CD90.1+ or CD90.1+/CD90.2+ T cells. (B) The percent of gated CD8+ CD90.1+ T cells that express IFN-γ in differentially vaccinated Alb:Gag recipients from A is depicted, with each circle representing individual mice (n=14) pooled from five separate experiments. (C) Frequency of tbx21−/− or WT of total CD8+ T cells is depicted, 3 days after transfer, with each circle representing individual mice (n=8) pooled from three separate experiments. CD90.1+ Gag-specific CD8+ T cells from (D) WT or (E) tbx21−/− donor mice were transferred separately into tolerizing Alb:Gag recipients (CD90.2+), with or without infection with Listeria. Three days after T cell transfer, recipients were infused with a 1:1 ratio of Gag (eFluor 670low) and control (eFluor 670high) peptide-pulsed target cells. Twenty hours later (Day 4), recipient spleens were harvested, and the frequency of transferred Gag-specific T cells was determined by flow cytometry (upper). Inset numbers are the percent of total splenocytes within the inscribed regions. Target cell frequency is displayed as histograms with the percentage of total eFluor 670-positive cells inset above the indicated regions (lower). (F) Graph displays relative target cell killing (percent eFluor 670high/percent eFluor 670low) pooled from four independent experiments, each with three mice/group (n=12), and error bars represent sd. See also Supplemental Fig. 1.
To characterize further the functional defects induced by T-bet deprivation within the tolerizing environment, the in vivo cytolytic activity of WT or tbx21−/− T cells was assessed after transfer into separate Alb:Gag recipient mice in the presence or absence of Listeria. Consistent with our previous data [4], WT Gag-specific T cells failed to kill Gag-pulsed target cells within the tolerizing environment, but specific cytolytic activity was induced by self-antigen in conjunction with Listeria (Fig. 3D). Despite this robust lytic activity, Alb:Gag recipients did not display symptoms of autoimmunity, as determined by liver enzyme levels in serum (data not shown). T-bet was absolutely required for this effector activity, as tbx21−/− T cells failed to kill target cells even during Listeria infection (Fig. 3E and F). The overall lack of effector function by tbx21−/− T cells was not a result of reduced proliferation, expansion, or persistence, as the frequency of WT and tbx21−/− T cells was comparable between treatment groups at this time (Fig. 3D and E).
Eomes is dispensable for Listeria-mediated CD8+ T cell effector differentiation within the tolerizing environment
Our data indicate a requirement for T-bet in the effector differentiation of CD8+ T cells following engagement of self-antigen in the context of Listeria inflammation and also appear to marginalize any potential contribution by Eomes in this process. However, these data do not rule out completely a role for Eomes during acquisition of CD8+ T cell effector function in the tolerizing environment. To address this directly, TCR Tg mice that lack Eomes in all peripheral T cells through Lck-Cre-mediated gene deletion were generated (eomesf/f Lck-cre TCRGag), and the loss of Eomes expression was confirmed at the mRNA or protein level (Supplemental Fig. 1). In stark contrast to the defects observed in tbx21−/− T cells, IFN-γ production and cytolytic activity by eomes-deficient T cells engaging self-antigen during Listeria infection were similar to WT T cells (Fig. 4A–C). These data demonstrate that Eomes is not required for self-reactive T cells to acquire effector functions during Listeria infection, reinforcing a key role for T-bet in this process.
Figure 4. Eomes is dispensable for Listeria-mediated CD8+ T cell effector differentiation within the tolerizing environment.

(A) TCR-Tg, Gag-specific T cells on a WT background (CD90.1+) or Eomes-deficient (eomesf/f) background (CD90.1+/CD90.2+) were cotransferred into tolerizing Alb:Gag recipients, with or without Listeria infection. Three days later, production of IFN-γ and TNF-α by transferred T cells was measured after overnight restimulation with the Gag peptide (right). Numbers in each quadrant represent the percent of gated CD8+ CD90.1+ or CD90.1+/CD90.2+ T cells. (B) The percent of gated CD8+ CD90.1+ T cells that express IFN-γ in differentially vaccinated Alb:Gag recipients from A is depicted, with each circle representing individual mice (n=9) pooled from three separate experiments. (C) WT or (D) Eomes-deficient CD90.1+ Gag-specific CD8+ T cells were transferred into tolerizing Alb:Gag recipients (CD90.2+), with or without Listeria infection. Three days after T cell transfer, recipients were infused with a 1:1 ratio of Gag (eFluor 670low) and control (eFluor 670high) peptide-pulsed target cells. Twenty hours later (Day 4), recipient spleens were harvested, and the frequency of transferred Gag-specific T cells was determined by flow cytometry (upper). Inset numbers are the percent of total splenocytes within the inscribed regions. Target cell frequency is displayed as histograms with the percentage of total eFluor 670-positive cells inset above the indicated regions (lower). (E) Graph displays relative target cell killing (percent eFluor 670high/percent eFluor 670low) pooled from three independent experiments, and error bars represent sd.
That Eomes was unable to compensate, as least partially, for a loss of T-bet in eliciting CD8+ T cell effector responses under inflammatory conditions was unanticipated, based on previous reports [11–13, 19, 45]. The caveat to this interpretation is that Eomes is not highly expressed by in tbx21−/− T cells in response to Listeria infection (Fig. 2C), likely as a result of repression by IL-12 [40]. This makes it difficult to determine if a lack of effector function by tbx21−/− T cells (Fig. 3) is the result of Eomes failing to compensate or simply not being expressed at sufficient levels. To address this question, self-reactive CD8+ T cells were alternatively studied during LCMV infection, which is dominated by type I IFN rather than IL-12 [46]. As predicted, LCMV infection during self-antigen encounter resulted in higher Eomes expression compared with Listeria (Fig. 5A), and both infections induced similar cytokine responses in WT T cells (Fig. 5A and B). However, LCMV also led to increased Eomes expression in tbx21−/− T cells, which was accompanied by moderate cytokine production, where essentially, none was seen during Listeria infection. Thus, low Eomes expression likely explains why tbx21−/− T cells fail to make IFN-γ in response to Listeria. Collectively, our results imply that effector differentiation of self-reactive T cells during Listeria infection relies more heavily on T-bet than Eomes; however, this reliance is not universal to all pathogens, as Eomes compensates for T-bet under tolerizing conditions when sufficient induction is achieved.
Figure 5. Eomes expression augments IFN-γ responses by T-bet-deficient T cells within the tolerizing environment.

WT (CD90.1+) or tbx21−/− (CD90.1+/CD90.2+) Gag-specific T cells were cotransferred into tolerizing Alb:Gag recipients, with or without infection with Listeria or LCMV. (A) After 3 days, intracellular IFN-γ and Eomes protein expression in transferred T cells was assessed by flow cytometry following ex vivo restimulation. Numbers in each quadrant represent the percent of gated CD8+ CD90.1+ T cells, and data are representative of three experiments. (B) The percent of gated CD8+ CD90.1+ or CD90.1+/CD90.2+ T cells expressing IFN-γ in differentially vaccinated Alb:Gag recipients from A is depicted graphically, with each circle representing individual mice pooled from three separate experiments. Horizontal bars represent the average for each group, and P values for select treatment groups are included.
PD-1 repression during Listeria infection occurs independently of T-bet
The repression of Eomes during Listeria infection provides a unique model to evaluate further the specific role of T-bet in tolerance to self-antigen. One potential mechanism by which T-bet could impact CD8+ T cell effector responses is by repressing transcription of the pdcd1 gene, which encodes the inhibitory receptor, PD-1. It was recently reported that T-bet directly inhibits expression of PD-1, facilitating rescue of exhausted CD8+ T cell function during chronic viral infection [20]. As PD-1 also has a prominent role in CD8+ T cell tolerance in our system [4], down-regulation of PD-1 could contribute to the T-bet-dependent effector T cell responses observed here. Indeed, PD-1 was expressed on Gag-specific CD8+ T cells, 3 days after encounter with self-antigen in Alb:Gag recipients, which was markedly reduced on the surface of these transferred T cells following Listeria infection (Fig. 6A and B) and was mirrored at the level of pdcd1 mRNA (Fig. 6C). However, this was true for WT and tbx21−/− T cells, suggesting that PD-1 repression occurred independently of T-bet and was not likely responsible for Listeria-mediated restoration of effector function, seen only in WT T cells (Fig. 3). Expression of other inhibitory receptor genes, such as ctla4 and lymphocyte-activation gene 3, was also not appreciably different in the absence of T-bet (data not shown).
Figure 6. PD-1 repression during inflammation occurs independently of T-bet.

CD90.1+ Gag-specific CD8+ T cells from WT or tbx21−/− donor mice were transferred separately into tolerizing Alb:Gag recipients (CD90.2+), with or without Listeria infection. (A) Three days later, cell-surface expression of PD-1 on transferred T cells was determined by flow cytometry. (B) Geometric mean fluorescence intensity (geo MFI) of PD-1 expression on WT and tbx21−/− Gag-specific T cells was averaged from three pooled experiments, and error bars represent sd. (C) Real-time PCR analysis of PD-1 (pdcd1) mRNA expression in FACS-purified WT or tbx21−/− cells, 3 days after transfer. Data are relative expression versus β-actin control. Error bars are sd of three replicate samples, and data are representative of two separate experiments.
Constitutive T-bet expression steers self-reactive T cells toward effector differentiation
Our data imply that the level of T-bet expression is a key determinant of whether CD8+ T cells differentiate and gain effector function or become anergic following primary antigen encounter. This hypothesis suggests that aberrant overexpression of T-bet may disrupt the natural induction of T cell tolerance even without inflammatory signals. To address this possibility, we examined Gag-specific T cells that constitutively express T-bet under control of a Tg CD2 promoter, regardless of external stimuli (Fig. 7A) [35]. Here, WT or T-bet-Tg T cells were transferred into normal B6 or tolerizing Alb:Gag recipients or into Alb:Gag recipients infected with Listeria as a positive control for induction of effector function. At 4 days after transfer, WT and T-bet-Tg T cells expanded similarly in response to self-antigen in Alb:Gag recipients and also, in the presence of inflammation (Fig. 7B). As before, WT T cells required inflammation to induce T-bet and differentiate into cytolytic T cells (Fig. 7C). In contrast, T-bet-Tg T cells demonstrated specific target cell killing under tolerizing conditions in Alb:Gag hosts without inflammation, suggesting that elevated T-bet expression had induced CD8+ T cell differentiation despite powerful extrinsic-tolerizing influences. In fact, these Gag-specific T cells even killed Gag-pulsed targets in B6 recipients without prior antigen stimulation (Fig. 7C), suggesting that elevated T-bet expression rendered naive CD8+ T cells poised for cytolytic activity. Constitutive T-bet expression also endowed naive CD8+ T cells with the capacity to produce IFN-γ modestly upon direct ex vivo restimulation (Fig. 7D). Intriguingly, this ability appeared to be actively suppressed upon encounter with a tolerizing self-antigen, unless inflammation was also present (Fig. 7D), regardless of sustained T-bet expression in Alb:Gag recipients (Fig. 7E). To be consistent with our earlier experiments, IFN-γ responses by T-bet-Tg T cells were confirmed at the earlier Day 3 time-point (Supplemental Fig. 2). Therefore, despite inducing cytolytic activity, T-bet expression was not sufficient to induce effector cytokine production under tolerizing conditions. Together, these data illustrate how vital T-bet deprivation is to maintaining anergy in autoreactive CD8+ T cells. Conversely, this provides clear evidence that T-bet expression alone can indeed steer self-reactive CD8+ T cells toward effector cell differentiation. However, it is also apparent that Listeria vaccination provides signals other than just T-bet that further contribute to CD8+ T cell differentiation and acquisition of more potent effector activity within a tolerizing environment.
Figure 7. T-bet expression is sufficient to induce cytolytic activity but not cytokine production within the tolerizing environment.

(A) Peripheral blood lymphocytes from 6-week-old WT and T-bet-Tg, Gag-specific CD8+ T cells were stained for CD90.1 and CD8 surface expression, and CD90.1+ CD8+ cells were assessed for intracellular T-bet protein expression. (B–D) WT or T-bet-Tg T cells were transferred into B6 or Alb:Gag recipients, with or without Listeria vaccination. Three days after T cell transfer, recipients were infused with a 1:1 ratio of Gag (eFluor 670low) and control (eFluor 670high) peptide-pulsed target cells. (B) Twenty hours later (Day 4), recipient spleens were harvested, and the frequency of transferred, Gag-specific T cells was determined by flow cytometry. The percent of CD8+ CD90.1+ T cells among total CD8+ T cells is depicted, with each circle representing individual mice pooled from two separate experiments. (C) Target-cell frequency is displayed in histograms with the percentage of total eFluor 670-positive cells inset above the indicated regions. Graph displays relative target cell killing (percent eFluor 670high/percent eFluor 670low) pooled from three independent experiments, each with three mice/group (n=9), and error bars represent sd. (D) Production of IFN-γ and TNF-α by transferred T cells was measured after overnight restimulation with the Gag peptide. Numbers in each quadrant represent the percent of gated CD8+ CD90.1+ T cells and are representative of two separate experiments, each with three mice/treatment group. (E) Intracellular T-bet expression was assessed in splenic CD8+ CD90.1+ T cells, 4 days after transfer into the indicated environments. See also Supplemental Fig. 2.
T-bet is required for Listeria vaccination to overcome T cell tolerance for cancer immunotherapy
To assess further the strength of T-bet-dependent effector function induced during Listeria infection, the ability to provide effective, adoptive T cell immunotherapy for cancer under tolerizing conditions was examined. Here, a murine FBL leukemia was established in Alb:Gag mice by i.v. injection to promote dissemination, as described previously [4]. One week later, Gag-specific T cells (WT, tbx21−/−, or T-bet-Tg) were infused i.v. and recipients left uninfected or infected with attenuated Listeria as a vaccine. Transfer of WT T cells in the absence of vaccination resulted in tolerance, and these recipients died of a progressive tumor in <16 days (Fig. 8A). Similarly, Listeria infection in the absence of adoptive T cell transfer provided no benefit to tumor-bearing recipients, primarily as Alb:Gag mice lack endogenous Gag-specific T cells as a result of mechanisms of central tolerance [34]. In contrast, the combination of adoptive transfer of WT Gag-specific T cells and Listeria vaccination increased significantly the survival of tumor-bearing Alb:Gag recipients, with 32% of recipients surviving beyond 40 days (Fig. 8A). This anti-tumor immunity was dependent on T-bet, as vaccination of adoptively transferred tbx21−/− T cells failed to provide a substantial survival benefit to mice with leukemia. Surprisingly, despite possessing potent cytolytic capacity (Fig. 7C), T cells with constitutive T-bet expression provided no benefit to tumor-bearing recipients, reinforcing that Listeria vaccination goes beyond just T-bet induction in boosting the activity of tumor/self-reactive CD8+ T cells. Thus, T-bet expression and vaccine-mediated inflammation play integral roles in the generation of effector responses by tumor/self-reactive CD8+ T cells upon antigen encounter.
Figure 8. T-bet is required but not sufficient to overcome functional T cell tolerance during cancer immunotherapy.
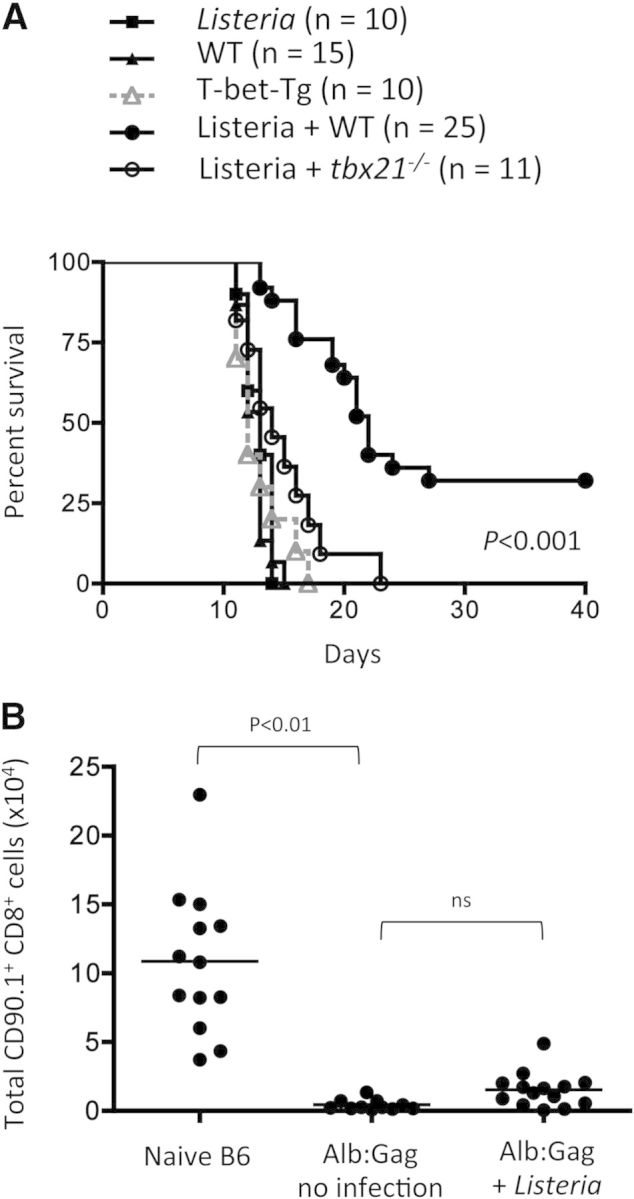
Tolerizing Alb:Gag recipient mice were inoculated i.v. with FBL leukemia. Seven days later, tumor-bearing recipients were left untreated or given adoptive transfers of WT, tbx21−/−, or T-bet-Tg, Gag-specific CD8+ T cells and vaccinated as indicated. (A) Recipient survival was tracked for 40 days, and results pooled from two to three separate experiments are depicted in the graph showing survival (y-axis) over time in days (x-axis), with the number of total mice in each treatment group indicated (n). (B) CD90.1+ Gag-specific CD8+ T cells from WT donor mice were transferred into tolerizing B6 or Alb:Gag recipients (no tumor), with or without Listeria as indicated. The frequency of transferred T cells in recipient spleens was assessed after 8 days. The total number of CD90.1+ CD8+ cells is displayed graphically and shows pooled data from four independent experiments, with each circle representing data from one mouse.
As Listeria immunization induced such vigorous effector function by WT Gag-specific CD8+ T cells within a tolerizing environment (Fig. 3), it was anticipated that adoptive T cell immunotherapy with Listeria vaccination would provide highly protective tumor immunity and even complete tumor eradication in most recipients. Despite achieving significantly increased survival in this treatment group, most recipients still did not survive long-term (Fig. 8A). The failure of adoptively transferred T cells to persist within the tolerizing environment represents one possible explanation for this apparent lack of a durable response. In support of this, elimination of autoreactive effector and memory CD8+ T cells following antigen engagement has been shown in several reports [47–49]. Similarly, while Listeria vaccination readily induced T-bet-dependent effector cytokine production and cytolytic activity here, it was not sufficient to prevent apoptosis and subsequent deletion of these tumor/self-reactive T cells within the tolerizing Alb:Gag environment (Fig. 8B). This confirms that the role of T-bet is limited to regulation of effector function and further shows that inflammatory signals during priming are ultimately insufficient to shield CD8+ T cells from the influences of tolerizing self-antigen. Whether this is a result of the effects of continuous antigen encounter over time (i.e., exhaustion) or the lack of an unidentified survival signal is yet to be determined.
DISCUSSION
The T-box transcription factor T-bet has a well-described role in CD4+ T cell differentiation, but how T-box transcription factors guide CD8+ T cell biology is more controversial. Certainly, T-bet and Eomes can influence IFN-γ responses and regulate other target genes primarily involved in CD8+ T cell effector function. But, the converse of this—that a lack of these transcription factors limits effector cell differentiation and promotes T cell tolerance—has not been demonstrated previously. For this reason, we investigated the roles of T-bet and Eomes in naive CD8+ T cells receiving a primary antigen encounter under immunogenic or tolerogenic conditions. We confirmed that whereas Eomes and T-bet are expressed in response to immunogenic foreign antigen, an encounter with the same protein expressed as a model self-antigen in the absence of inflammation fails to induce T-bet or Eomes. However, induction of these transcription factors when self-antigen was encountered under inflammatory conditions diverted tolerance and promoted effector differentiation of CD8+ T cells.
Our studies using Listeria vaccination showed that T-bet was a key determinant of whether CD8+ T cells differentiate and gain effector function following self-antigen encounter. Studies from other labs have shown that inhibition of T-bet signaling during a CD8+ T cell immune response does not overtly impact effector function, thought largely as a result of the redundancy of Eomes [11, 13, 19, 45]. Thus, we anticipated that Eomes would compensate in the absence of T-bet and that obvious defects would not be observed in tbx21−/− T cells during Listeria vaccination. Instead, IFN-γ production and cytolytic activity were abolished nearly completely in tbx21−/− T cells vaccinated with Listeria in the tolerizing environment, suggesting that Eomes alone was not sufficient to induce effector function of self-antigen-reactive T cells, even under inflammatory conditions. However, for Eomes to compensate for a loss of T-bet, it must be expressed at sufficient levels. Consistent with previous reports, we observed low Eomes induction with Listeria [40], but when induced to higher levels (here during a LCMV infection), Eomes was indeed capable of directing similar effector responses. Thus, reliance on T-bet during Listeria vaccination was not a result of a lack of Eomes activity but rather, of insufficient levels of Eomes protein expression. These findings support the previous idea that T-bet and Eomes expression correlate inversely with CD8+ T cell tolerance [31], but we have now gone further to demonstrate that the level of expression of these transcription factors is a critical determinant of effector programming in self-reactive T cells and that these levels can be regulated by the inflammatory milieu. We provide evidence that T-bet, in particular, plays an integral role under conditions of Listeria vaccination, which is currently being explored clinically as a therapeutic cancer vaccine.
T-box family members have a conserved ability to recruit chromatin-remodeling complexes to affect permissive changes at target promoters and are additionally required for subsequent transactivation events [50]. Thus, the failure to express these transcription factors during tolerance presumably leads to inaccessibility of promoter binding sites for other transcription factors, as well as a loss of target gene transactivation, ultimately resulting in a dearth of the cellular machinery necessary to carry out effector activity. This was supported by experiments using T cells that constitutively express T-bet, which displayed robust cytolytic activity under naive and tolerizing conditions. However, this same dynamic was not reflected in the production of effector cytokines. Whereas naive T-bet-Tg T cells expressed some IFN-γ relative to WT T cells, this was not observed in the tolerizing environment. This illustrates the complex nature of cytokine gene regulation and implies that IFN-γ expression was actively inhibited in T cells that received tolerizing signals, for which enforced T-bet expression was unable to overcome without additional inflammatory factors. The molecular mechanisms that regulate this potential inhibition and how they are reversed under inflammatory conditions remain to be defined.
Exactly how T-box transcription factor expression itself is regulated in CD8+ T cells after primary antigen encounter is also unclear. In our experiments, appreciable T-bet and Eomes expression was not detected in T cells encountering self-antigen in the absence of inflammation. This could be a result of an activation threshold not met under tolerizing conditions, but this seems unlikely, as these T cells proliferate rapidly and even expand in number following self-antigen encounter [4]. It is also possible that induction of T-bet expression requires a signal not received through engagement of self-antigen by steady-state APCs. Consistent with this idea, cytokines, such as IL-12 and type I IFN, which characterize the inflammatory environments during Listeria and LCMV infections [46], have been shown to provide a requisite third signal after antigen and costimulation that is necessary for the development of key effector functions, such as IFN-γ production and cytolytic activity [51]. Indeed, provision of these cytokines to T cells activated by artificial APCs and costimulation in vitro has been correlated with increased T-bet and Eomes transcription [41]. Similarly, preventing ligation of inhibitory coreceptors permits acquisition of effector function [4], but whether these same pathways prevent T-box transcription factor expression remains to be determined. As an alternative to failed induction, it is possible that these molecules are actively repressed under tolerizing conditions by the action of microRNA. For example, microRNA-29 is reported to target and degrade specifically T-bet and Eomes mRNA in CD4+ T cells [52, 53]. However, our unpublished data and work by other labs suggest microRNA-29 may not regulate T-bet and Eomes the same way in CD8+ T cells [31]. Instead, other microRNAs (possibly those yet to be defined) may be involved in control of T-box transcription factor expression and subsequent regulation of tolerance in CD8+ T cells and warrant further investigation.
CD8+ T cell tolerance is not limited to effector cell dysfunction but also involves the deletion of self-reactive T cells through Bim-mediated apoptosis [4, 29, 54]. Here, we observed a disconnect between mechanisms of tolerance associated with a lack of effector functions versus deletion of autoreactive T cells, similar to a previous report on intrahepatic CD8+ T cell priming [55]. As inflammatory stimuli could be combined with self-antigen to induce robust CD8+ T cell effector differentiation, we anticipated that this represented a true rescue from the tolerizing influences of self-antigen, including deletion. This proved to be incorrect, as responding tumor/self-reactive T cells were eliminated nearly completely after 8 days in Alb:Gag hosts, even after differentiation and acquisition of effector function following Listeria vaccination. These data suggest that differentiation alone is inadequate to rescue T cells from peripheral tolerance. The fact that T cells can transiently express effector function or even differentiate into memory cells before tolerance induction has been reported previously [47–49, 56]. It has also been reported that high levels of T-bet expression correlate with commitment to a short-lived effector T cell phenotype over memory precursors, which suggests that relative amounts of lineage-determining transcription factors in activated CD8+ T cells may contribute to their long-term fitness for survival or memory potential [57].
The inability to overcome deletion or promote long-term survival has potential clinical implications, as only a short therapeutic window could be provided to tumor-bearing recipients treated with adoptive immunotherapy and Listeria vaccination, resulting in recipient survival outcomes that were somewhat below expectations. Thus, durable therapeutic responses failed to be achieved in a majority of recipients, presumably because tumor/self-reactive T cells were still deleted from the periphery by apoptosis before all leukemic cells could be eradicated. Previous reports suggest that rescued T cells ultimately succumb to tolerance over time if rescue stimuli are not maintained [2, 8, 31]. However, evidence for long-term rescue within the tolerizing environment has been observed for combination checkpoint-blockade therapy [4], perhaps reflecting that the method by which tolerance is overcome impacts the durability of treatment.
Our study provides novel insight into how naive CD8+ T cells interpret the environment in which antigen is encountered. We conclude that relative expression of T-bet and Eomes intrinsically controls whether stimulation by a self-antigen leads to effector differentiation. The results are fundamental to basic T cell immunology but could have important translational repercussions as well. As the field of immunotherapy approaches an era where customized manipulation of CD8+ T cells and other immune components is almost commonplace [58, 59], questions have shifted from whether immune modulation is possible to determining which alterations will elicit the desired therapeutic outcomes. Listeria-based cancer vaccines are advancing in human clinical trials [60, 61], in part, as a result of the ability of these vaccines to overcome immune tolerance [33]. However, the molecular mechanisms involved have not been defined. Our data place T-box transcription factors in a central role of dictating whether effector T cell differentiation is appropriate upon antigen recognition and identify a role for inflammation in making and reinforcing this decision. Therefore, it may be desirable to invoke these signaling pathways in the setting of adoptive T immunotherapy for cancer, but potential risks to healthy self-tissue should be considered carefully. Conversely, disruption of T-bet and/or Eomes signaling may represent a strategy to alleviate autoimmune diseases or prevent transplant rejection by attenuating effector T cell function.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Disease, U.S. National Institutes of Health (R01AI087764) to R.M.T., by a Cancer Research Institute Investigator Award to R.M.T., and by a National Cancer Institute fellowship, U.S. National Institutes of Health (F30CA180375) to S.R.J.
The authors thank Sherri Koehm and Joy Eslick for technical assistance with flow cytometry and cell sorting.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- ActA
- actin assembly-inducing protein
- B6
- C57BL/6
- Eomes
- eomesodermin
- FasL
- Fas ligand
- FBL
- F-box and leucine-rich repeat protein
- GzmB
- granzyme B
- LCMV
- lymphocytic choriomeningitis virus
- Ova
- ovalbumin
- PD-1
- programmed death 1
- pdcd1
- pyruvate dehydrogenase complex deficiency 1
- rag1−/−
- recombination-activating gene 1-deficient
- T-bet
- T-box expressed in T cells
- tbx21−/−
- T-box protein 21-deficient
- TCRGag
- TCR transgenic Gag-specific CD8+ T cells
- Tg
- transgenic
- WT
- wild-type
AUTHORSHIP
Experiments were conceived and designed by S.R.J. and R.M.T. Methodology was developed by S.R.J., J.Y., M.M.B-E., and C.L.C. Data were acquired by S.R.J., J.Y., M.M.B-E., and C.L.C. Data were analyzed and interpreted by S.R.J., J.Y., M.J.D., and R.M.T. The manuscript was written by S.R.J. and R.M.T. and reviewed by all authors. Administrative and technical support regarding animal husbandry and data/materials organization was provided by J.M.M. and M.J.D. The study was supervised by R.M.T.
DISCLOSURES
The content is solely the interpretation of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health. The authors declare no commercial or financial conflict of interest.
REFERENCES
- 1. Redmond W. L., Sherman L. A. (2005) Peripheral tolerance of CD8 T lymphocytes. Immunity 22, 275–284. [DOI] [PubMed] [Google Scholar]
- 2. Teague R. M., Greenberg P. D., Fowler C., Huang M. Z., Tan X., Morimoto J., Dossett M. L., Huseby E. S., Ohlen C. (2008) Peripheral CD8+ T cell tolerance to self-proteins is regulated proximally at the T cell receptor. Immunity 28, 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ohlen C., Kalos M., Cheng L. E., Shur A. C., Hong D. J., Carson B. D., Kokot N. C., Lerner C. G., Sather B. D., Huseby E. S., Greenberg P. D. (2002) CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J. Exp. Med. 195, 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berrien-Elliott M. M., Jackson S. R., Meyer J. M., Rouskey C. J., Nguyen T. L., Yagita H., Greenberg P., Dipaolo R. J., Teague R. M. (2013) Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T cell tolerance. Cancer Res. 73, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaech S. M., Wherry E. J., Ahmed R. (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2, 251–262. [DOI] [PubMed] [Google Scholar]
- 6. Bai A., Higham E., Eisen H. N., Wittrup K. D., Chen J. (2008) Rapid tolerization of virus-activated tumor-specific CD8+ T cells in prostate tumors of TRAMP mice. Proc. Natl. Acad. Sci. USA 105, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savage P. A., Vosseller K., Kang C., Larimore K., Riedel E., Wojnoonski K., Jungbluth A. A., Allison J. P. (2008) Recognition of a ubiquitous self antigen by prostate cancer-infiltrating CD8+ T lymphocytes. Science 319, 215–220. [DOI] [PubMed] [Google Scholar]
- 8. Teague R. M., Sather B. D., Sacks J. A., Huang M. Z., Dossett M. L., Morimoto J., Tan X., Sutton S. E., Cooke M. P., Ohlen C., Greenberg P. D. (2006) Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat. Med. 12, 335–341. [DOI] [PubMed] [Google Scholar]
- 9. Overwijk W. W., Theoret M. R., Finkelstein S. E., Surman D. R., de Jong L. A., Vyth-Dreese F. A., Dellemijn T. A., Antony P. A., Spiess P. J., Palmer D. C., Heimann D. M., Klebanoff C. A., Yu Z., Hwang L. N., Feigenbaum L., Kruisbeek A. M., Rosenberg S. A., Restifo N. P. (2003) Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 198, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szabo S. J., Kim S. T., Costa G. L., Zhang X., Fathman C. G., Glimcher L. H. (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- 11. Pearce E. L., Mullen A. C., Martins G. A., Krawczyk C. M., Hutchins A. S., Zediak V. P., Banica M., DiCioccio C. B., Gross D. A., Mao C. A., Shen H., Cereb N., Yang S. Y., Lindsten T., Rossant J., Hunter C. A., Reiner S. L. (2003) Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science 302, 1041–1043. [DOI] [PubMed] [Google Scholar]
- 12. Intlekofer A. M., Takemoto N., Wherry E. J., Longworth S. A., Northrup J. T., Palanivel V. R., Mullen A. C., Gasink C. R., Kaech S. M., Miller J. D., Gapin L., Ryan K., Russ A. P., Lindsten T., Orange J. S., Goldrath A. W., Ahmed R., Reiner S. L. (2005) Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244. [DOI] [PubMed] [Google Scholar]
- 13. Szabo S. J., Sullivan B. M., Stemmann C., Satoskar A. R., Sleckman B. P., Glimcher L. H. (2002) Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science 295, 338–342. [DOI] [PubMed] [Google Scholar]
- 14. Zhu Y., Ju S., Chen E., Dai S., Li C., Morel P., Liu L., Zhang X., Lu B. (2010) T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J. Immunol. 185, 3174–3183. [DOI] [PubMed] [Google Scholar]
- 15. Sullivan B. M., Juedes A., Szabo S. J., von Herrath M., Glimcher L. H. (2003) Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA 100, 15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Juedes A. E., Rodrigo E., Togher L., Glimcher L. H., von Herrath M. G. (2004) T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J. Exp. Med. 199, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lord G. M., Rao R. M., Choe H., Sullivan B. M., Lichtman A. H., Luscinskas F. W., Glimcher L. H. (2005) T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood 106, 3432–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eshima K., Chiba S., Suzuki H., Kokubo K., Kobayashi H., Iizuka M., Iwabuchi K., Shinohara N. (2012) Ectopic expression of a T-box transcription factor, eomesodermin, renders CD4(+) Th cells cytotoxic by activating both perforin- and FasL-pathways. Immunol. Lett. 144, 7–15. [DOI] [PubMed] [Google Scholar]
- 19. Intlekofer A. M., Banerjee A., Takemoto N., Gordon S. M., Dejong C. S., Shin H., Hunter C. A., Wherry E. J., Lindsten T., Reiner S. L. (2008) Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kao C., Oestreich K. J., Paley M. A., Crawford A., Angelosanto J. M., Ali M. A., Intlekofer A. M., Boss J. M., Reiner S. L., Weinmann A. S., Wherry E. J. (2011) Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 12, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paley M. A., Kroy D. C., Odorizzi P. M., Johnnidis J. B., Dolfi D. V., Barnett B. E., Bikoff E. K., Robertson E. J., Lauer G. M., Reiner S. L., Wherry E. J. (2012) Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bettelli E., Sullivan B., Szabo S. J., Sobel R. A., Glimcher L. H., Kuchroo V. K. (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 200, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishizaki K., Yamada A., Yoh K., Nakano T., Shimohata H., Maeda A., Fujioka Y., Morito N., Kawachi Y., Shibuya K., Otsuka F., Shibuya A., Takahashi S. (2007) Th1 and type 1 cytotoxic T cells dominate responses in T-bet overexpression transgenic mice that develop contact dermatitis. J. Immunol. 178, 605–612. [DOI] [PubMed] [Google Scholar]
- 24. Neurath M. F., Weigmann B., Finotto S., Glickman J., Nieuwenhuis E., Iijima H., Mizoguchi A., Mizoguchi E., Mudter J., Galle P. R., Bhan A., Autschbach F., Sullivan B. M., Szabo S. J., Glimcher L. H., Blumberg R. S. (2002) The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 195, 1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohashi P. S., Oehen S., Buerki K., Pircher H., Ohashi C. T., Odermatt B., Malissen B., Zinkernagel R. M., Hengartner H. (1991) Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 65, 305–317. [DOI] [PubMed] [Google Scholar]
- 26. Von Herrath M. G., Guerder S., Lewicki H., Flavell R. A., Oldstone M. B. (1995) Coexpression of B7-1 and viral (“self”) transgenes in pancreatic β cells can break peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity 3, 727–738. [DOI] [PubMed] [Google Scholar]
- 27. Belz G. T., Behrens G. M., Smith C. M., Miller J. F., Jones C., Lejon K., Fathman C. G., Mueller S. N., Shortman K., Carbone F. R., Heath W. R. (2002) The CD8α(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 196, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hernandez J., Aung S., Redmond W. L., Sherman L. A. (2001) Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Redmond W. L., Wei C. H., Kreuwel H. T., Sherman L. A. (2008) The apoptotic pathway contributing to the deletion of naive CD8 T cells during the induction of peripheral tolerance to a cross-presented self-antigen. J. Immunol. 180, 5275–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kurts C., Carbone F. R., Barnden M., Blanas E., Allison J., Heath W. R., Miller J. F. (1997) CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 186, 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schietinger A., Delrow J. J., Basom R. S., Blattman J. N., Greenberg P. D. (2012) Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brockstedt D. G., Giedlin M. A., Leong M. L., Bahjat K. S., Gao Y., Luckett W., Liu W., Cook D. N., Portnoy D. A., Dubensky T. W., Jr., (2004) Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc. Natl. Acad. Sci. USA 101, 13832–13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clifton G. T., Peoples G. E. (2009) Overcoming cancer immune tolerance and escape. Clin. Cancer Res. 15, 749–751. [DOI] [PubMed] [Google Scholar]
- 34. Ohlen C., Kalos M., Hong D. J., Shur A. C., Greenberg P. D. (2001) Expression of a tolerizing tumor antigen in peripheral tissue does not preclude recovery of high-affinity CD8+ T cells or CTL immunotherapy of tumors expressing the antigen. J. Immunol. 166, 2863–2870. [DOI] [PubMed] [Google Scholar]
- 35. Kondo Y., Iizuka M., Wakamatsu E., Yao Z., Tahara M., Tsuboi H., Sugihara M., Hayashi T., Yoh K., Takahashi S., Matsumoto I., Sumida T. (2012) Overexpression of T-bet gene regulates murine autoimmune arthritis. Arthritis Rheum. 64, 162–172. [DOI] [PubMed] [Google Scholar]
- 36. Cheever M. A., Thompson J. A., Kern D. E., Greenberg P. D. (1985) Interleukin 2 (IL 2) administered in vivo: influence of IL 2 route and timing on T cell growth. J. Immunol. 134, 3895–3900. [PubMed] [Google Scholar]
- 37. Chen W., Qin H., Chesebro B., Cheever M. A. (1996) Identification of a gag-encoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J. Virol. 70, 7773–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T. (1996) Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772. [DOI] [PubMed] [Google Scholar]
- 39. Unanue E. R. (1997) Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol. Rev. 158, 11–25. [DOI] [PubMed] [Google Scholar]
- 40. Takemoto N., Intlekofer A. M., Northrup J. T., Wherry E. J., Reiner S. L. (2006) Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J. Immunol. 177, 7515–7519. [DOI] [PubMed] [Google Scholar]
- 41. Agarwal P., Raghavan A., Nandiwada S. L., Curtsinger J. M., Bohjanen P. R., Mueller D. L., Mescher M. F. (2009) Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183, 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pipkin M. E., Sacks J. A., Cruz-Guilloty F., Lichtenheld M. G., Bevan M. J., Rao A. (2010) Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morimoto J., Tan X., Teague R. M., Ohlen C., Greenberg P. D. (2007) Induction of tolerance in CD8+ T cells to a transgenic autoantigen expressed in the liver does not require cross-presentation. J. Immunol. 178, 6849–6860. [DOI] [PubMed] [Google Scholar]
- 44. Long M., Slaiby A. M., Hagymasi A. T., Mihalyo M. A., Lichtler A. C., Reiner S. L., Adler A. J. (2006) T-bet down-modulation in tolerized Th1 effector CD4 cells confers a TCR-distal signaling defect that selectively impairs IFN-γ expression. J. Immunol. 176, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Way S. S., Wilson C. B. (2004) Cutting edge: immunity and IFN-γ production during Listeria monocytogenes infection in the absence of T-bet. J. Immunol. 173, 5918–5922. [DOI] [PubMed] [Google Scholar]
- 46. Kolumam G. A., Thomas S., Thompson L. J., Sprent J., Murali-Krishna K. (2005) Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kreuwel H. T., Aung S., Silao C., Sherman L. A. (2002) Memory CD8(+) T cells undergo peripheral tolerance. Immunity 17, 73–81. [DOI] [PubMed] [Google Scholar]
- 48. Bercovici N., Heurtier A., Vizler C., Pardigon N., Cambouris C., Desreumaux P., Liblau R. (2000) Systemic administration of agonist peptide blocks the progression of spontaneous CD8-mediated autoimmune diabetes in transgenic mice without bystander damage. J. Immunol. 165, 202–210. [DOI] [PubMed] [Google Scholar]
- 49. Kyburz D., Aichele P., Speiser D. E., Hengartner H., Zinkernagel R. M., Pircher H. (1993) T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur. J. Immunol. 23, 1956–1962. [DOI] [PubMed] [Google Scholar]
- 50. Lewis M. D., Miller S. A., Miazgowicz M. M., Beima K. M., Weinmann A. S. (2007) T-bet's ability to regulate individual target genes requires the conserved T-box domain to recruit histone methyltransferase activity and a separate family member-specific transactivation domain. Mol. Cell. Biol. 27, 8510–8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Curtsinger J. M., Lins D. C., Mescher M. F. (2003) Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J. Exp. Med. 197, 1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steiner D. F., Thomas M. F., Hu J. K., Yang Z., Babiarz J. E., Allen C. D., Matloubian M., Blelloch R., Ansel K. M. (2011) MicroRNA-29 regulates T-box transcription factors and interferon-γ production in helper T cells. Immunity 35, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith K. M., Guerau-de-Arellano M., Costinean S., Williams J. L., Bottoni A., Mavrikis Cox G., Satoskar A. R., Croce C. M., Racke M. K., Lovett-Racke A. E., Whitacre C. C. (2012) miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol. 189, 1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Davey G. M., Kurts C., Miller J. F., Bouillet P., Strasser A., Brooks A. G., Carbone F. R., Heath W. R. (2002) Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2-inhibitable pathway mediated by Bim. J. Exp. Med. 196, 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bertolino P., Trescol-Biemont M. C., Rabourdin-Combe C. (1998) Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur. J. Immunol. 28, 221–236. [DOI] [PubMed] [Google Scholar]
- 56. Zhou G., Lu Z., McCadden J. D., Levitsky H. I., Marson A. L. (2004) Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. J. Exp. Med. 200, 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Joshi N. S., Cui W., Chandele A., Lee H. K., Urso D. R., Hagman J., Gapin L., Kaech S. M. (2007) Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grupp S. A., Kalos M., Barrett D., Aplenc R., Porter D. L., Rheingold S. R., Teachey D. T., Chew A., Hauck B., Wright J. F., Milone M. C., Levine B. L., June C. H. (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morgan R. A., Dudley M. E., Wunderlich J. R., Hughes M. S., Yang J. C., Sherry R. M., Royal R. E., Topalian S. L., Kammula U. S., Restifo N. P., Zheng Z., Nahvi A., de Vries C. R., Rogers-Freezer L. J., Mavroukakis S. A., Rosenberg S. A. (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Le D. T., Brockstedt D. G., Nir-Paz R., Hampl J., Mathur S., Nemunaitis J., Sterman D. H., Hassan R., Lutz E., Moyer B., Giedlin M., Louis J. L., Sugar E. A., Pons A., Cox A. L., Levine J., Murphy A. L., Illei P., Dubensky T. W., Jr., Eiden J. E., Jaffee E. M., Laheru D. A. (2012) A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin. Cancer Res. 18, 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maciag P. C., Radulovic S., Rothman J. (2009) The first clinical use of a live-attenuated Listeria monocytogenes vaccine: a phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 27, 3975–3983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.