

Abstract
Objective
The NR4A orphan nuclear receptor NOR1 functions as a constitutively active transcription factor regulating cellular inflammation and proliferation. In the present study, we employed bone marrow transplantation to determine the selective contribution of NOR1 expression in hematopoietic stem cells to the development of atherosclerosis.
Methods and Results
Reconstitution of lethally irradiated apoE−/− mice with NOR1-deficient hematopoietic stem cells accelerated atherosclerosis formation and macrophage recruitment following feeding a diet enriched in saturated fat. NOR1 deficiency in hematopoietic stem cells induced splenomegaly and monocytosis, specifically the abundance of inflammatory Ly6C+ monocytes. Bone marrow transplantation studies further confirmed that NOR1 suppresses the proliferation of macrophage and dendritic progenitor (MDP) cells. Expression analysis identified RUNX1, a critical regulator of hematopoietic stem cell expansion, as a target gene suppressed by NOR1 in MDP cells. Finally, in addition to inducing Ly6C+ monocytosis, NOR1 deletion increased the replicative rate of lesional macrophages and induced local foam cell formation within the atherosclerotic plaque.
Conclusion
Collectively, our studies demonstrate that NOR1 deletion in hematopoietic stem cells accelerates atherosclerosis formation by promoting myelopoiesis in the stem cell compartment and by inducing local pro-atherogenic activities in the macrophage, including lesional macrophage proliferation and foam cell formation.
Keywords: nuclear receptor, monocyte, macrophage and dendritic progenitors, atherosclerosis
INTRODUCTION
Recruitment of circulating monocytes, their differentiation into macrophages, uptake of LDL-derived cholesterol, and the ensuing activation of a broad range of inflammatory responses are the major events leading to atherosclerosis initiation and progression[1]. In addition to these cellular functions of the macrophage, increased numbers of circulating monocytes are associated with atherosclerosis and predict future cardiovascular disease[2,3]. Hypercholesterolemia for example is intimately associated with monocytosis and gives rise to proinflammatory Ly-6C+ monocyte subsets, which infiltrate the arterial wall to contribute to atherosclerosis formation[4]. The majority of these circulating monocytes originate from hematopoietic stem cell commitment to myeloid progenitor cells and their proliferation in response to atherogenic cues[5]. However, the mechanisms underlying atherosclerosis-associated proliferation of myeloid progenitor cells in the bone marrow remain to be defined.
Members of the nuclear hormone receptor superfamily constitute a highly conserved group of transcription factors that have emerged as important regulators of gene expression in the process of atherosclerosis formation[6]. Among this superfamily, the orphan nuclear receptors Nur77 (NR4A1)[7], Nurr1 (NR4A2)[8], and NOR1 (NR4A3)[9] function as ligand-independent early response genes to integrate environmental cues into adaptive gene expression programs to control cell proliferation, differentiation, survival, and inflammation[6]. The transcription factor NOR1 is rapidly induced in response to growth factors and inflammatory stimuli[10]. NOR1 is highly expressed in atherosclerotic lesions[11], increases monocyte adhesion[12], regulates the expression of proinflammatory genes[13], and modulates cholesterol uptake by macrophages[14]. In addition to these cellular functions, NOR1 has previously been reported to induce the expansion of hematopoietic stem cells and myeloid progenitors[15,16]. Considering this collective evidence, we investigated in the present study the contribution of NOR1 expression in hematopoietic stem cells to myelopoiesis, monocyte differentiation, and atherosclerosis development.
MATERIALS AND METHODS
Mice
Littermate NOR1+/+ and NOR1−/− mice on a mixed C57BL/6J/129Sv background were used as previously described[17,18]. apoE−/− mice on a C57BL/6J background (N10) were obtained from The Jackson Laboratory (stock no. 002052). Bone marrow transplantation (BMT) studies were performed by repopulating lethally irradiated female apoE−/− mice with bone marrow–derived cells of female NOR1+/+ and NOR1−/− mice as described[19]. Briefly, mice were maintained on water containing antibiotics (sulfamethoxazole/trimethoprim) for 1 week before BMT until 4 weeks after BMT. Recipient mice were irradiated with a total of 900 Rads from a cesium source that was delivered in 2 doses within 3 to 4 hours. Bone marrow–derived cells of female NOR1+/+ and NOR1−/− mice were obtained from the tibias and femurs of donor mice and were injected into the tail vein of 10-week-old irradiated female apoE−/− recipient mice (1×107 bone marrow cells per mouse, n=15 for NOR1+/+ and n=15 for NOR1−/−). After 4 weeks of recovery, a saturated fat-enriched diet was fed for 12 weeks for atherosclerosis analysis (Harlan Teklad TD.88137). All studies were performed with the approval of the University of Kentucky Institutional Animal Care and Use Committee.
Atherosclerosis quantification
En face atherosclerosis was quantified as lesion area on the intimal surface of aortic arches as previously described[20,21]. The data are presented as the percentage of lesion area on the aortic arch.
Histology
Aortic roots were frozen in OCT media (Tissue-Tek, Miles Inc.) and serial 10μm sections were cut as previously described[20]. Accumulation of lipids in lesions was visualized by staining with oil red O. Macrophages were detected using rabbit anti-mouse macrophage antisera (Accurate Chemicals)[22]. Percentage of macrophage and lipid (oil-red-o+) area in aortic root plaques was calculated using computer-assisted image analysis (Image-Pro, Media Cybernetics). Macrophage proliferation in atherosclerotic lesions was identified by immunostaining consecutive aortic root sections with antibodies against macrophages or proliferating cell nuclear antigen (PCNA, Abcam ab2426)[23]. Proliferating macrophages were identified as regions staining positive for both macrophage and PCNA and were quantified using computer-assisted image analysis (Image-Pro, Media Cybernetics). The ratio of proliferating macrophages to total macrophages at the aortic root was calculated by dividing macrophage positive areas by macrophage and PCNA positive areas.
Quantification of total cholesterol concentration and lipoprotein-cholesterol distribution
Total serum cholesterol concentration and lipoprotein-cholesterol distribution were analyzed as previously described[12,21]. Briefly, total serum cholesterol was measured by enzymatic colorimetric method using the Wako Cholesterol E kit (Wako Chemicals USA). Lipoprotein-cholesterol distribution was detected by size exclusion chromatography using a fast performance liquid chromatographic machine (Pharmacia LKB Biotechnology, Uppsala, Sweden).
Macrophage recruitment during peritonitis
Thioglycollate-elicited peritoneal macrophages were isolated from wild-type and NOR1−/− female mice (12 mice/group) as previously described[24]. Macrophages were quantified by counting using a hemocytometer.
Quantitative real-time RT-PCR
RNA was isolated using Trizol (Invitrogen) and reverse transcribed with Superscript II (Invitrogen) per manufacturer protocols. Quantitative real-time polymerase chain reaction analysis of target gene expression was performed using the iCycler and SYBR Green I system (Bio-Rad) as described[12]. Samples were analyzed in triplicate and normalized to expression values of mouse housekeeping gene TFIIB or human housekeeping gene TBP. Data were calculated using the 2-ΔΔCT method[25]. The following primer sequences were used: mouse NOR1 (forward: 5′-AGACGCCGAAACCGATGT-3′ and reverse: 5′-TCGGACAAGGGCATTCA-3′), mouse RUNX1 (forward: 5′-GCAGGCAACGATGAAAACTACT-3′ and reverse: 5′-GCAACTTGTGGCGGATTTGTA-3′), mouse TFIIB (forward: 5′-CTCTCCCAAGAGTCACATGTCC-3′ and reverse: 5′-CAATAACTCGGTCCCCTACAAC-3′), human RUNX1 (forward: 5′-TCTTCACAAACCCACCGCAA-3′ and reverse: 5′- CTGCCGATGTCTTCGAGGTTC-3′), human NOR1 (forward: 5′-GGGCTTTTTCAAGAGAACAGTG-3′ and reverse: 5′-ATCTCTGGGTGTTGAGTCTGTT-3′), human TBP (forward: 5′-GGAGAGTTCTGGGATTGTACCGC-3′ and reverse: 5′-ATATTCGGCGTTTCGGGCAC-3′).
Western blotting
Western blotting was performed as described using antibodies against NOR1 (PP-H7833, R & D Systems), and GAPDH (FL 335, Santa Cruz) [17,18].
Flow cytometry
For phenotypic identification of hematopoietic stem cell (HSC), progenitor cells, and mature hematopoietic cells, cells were stained with cell surface markers and analyzed by flow cytometry. Bone marrow cells were stained with the differentiated lineage cell markers (CD5, CD3, B220, Mac-1, Gr-1, and Ter119 from BD Pharmingen) and stem cell markers (Sca-1 from Invitrogen, c-kit from BD Pharmingen). HSC-enriched population is negative for the lineage markers but positive for Sca-1 and c-kit (Lin−Sca-1+c-kit+). Committed progenitor cells, including common myeloid progenitors (CMP), granulocyte-macrophage precursors (GMP), macrophage and dendritic cell progenitors (MDP), were identified by the markers of CD16/CD32 (eBioscience), CD34 and CD115 (BD Pharmingen). CMP were identified as Lin−c-kit+Sca-1− CD16/CD32low CD34+; GMP were identified as Lin−c-kit+Sca−1− CD16/CD32hi CD34+; MDP were identified as Lin−Sca-1− c-kit+ CD16/CD32+CD115+[5,16,26]. Dead cells were excluded by propidium iodide (PI) selection. Bone marrow cells were analyzed and sorted on a BD FacsAria II flow cytometer (Becton Dickinson). Each experimental group was sorted independently. Flow cytomerty and FACS data were analyzed using FlowJo software (Tree Star) (Supplementary Fig S-1, Fig S-2).
For identification of mature hematopoietic cells in bone marrow, spleen, and blood, T cells were identified asCD3e+; B cells were identified as CD19+; granulocytes were identified as CD3e−CD19−NK1.1−CD11b+Ly6Ghi; natural killer T cells (NKT) were identified as NK1.1+; dendritic cells (DC) were identified as CD11c+; total monocytes were identified as CD3e−CD19-NK1.1− Ly6G− CD11b+ CD115+, and monocyte subsets were identified as Ly6C+ monocytes or Ly6C− monocytes by Ly6C fluorescence intensity gated on total monocytes[5,26]. Antibodies used for staining are as follows: APC mouse NK1.1, APC mouse CD19, APC mouse CD3e, APC-Cy7 mouse CD11c, FITC mouse Ly6C, PE-cy7 mouse Ly6G, Percp-cy5.5 mouse CD11b (BD Biosciences); PE anti-mouse CD115 (Biolegend). Antibodies were used according to the manufacturer’s protocol. Cellular fluorescence was assessed with FACSCalibur (BD Biosciences) and data were analyzed with Cell Quest software (version3.3, Becton Dickinson) (Supplementary Fig S-3).
BrdU incorporation
Mice were injected intraperitoneally with 1mg BrdU (5-bromodeoxyuridine, BD Biosciences); 2 hours post injection mice were sacrificed and bone marrow cells from both femurs and tibias were harvested. Red blood cells were lysed in RBC Lysis Buffer (eBioscience) and cells were stained with MDP cell surface markers (lineage panel, Sca-1, c-Kit, CD16/CD32, CD115) and anti-BrdU antibodies according to manufacturer instructions (BD Biosciences). Cells were analyzed by flow cytometry using a FACsCalibur (BD Biosciences) and data were analyzed with Cell Quest software (version3.3, Becton Dickinson).
Transfection of HEK 293 cells
HEK 293 cells were transiently transfected with a NOR1 or GFP expression vector using Lipofectamine 2000 (Invitrogen) as described[17,27]. RNA was collected with Trizol (Invitrogen) 48 hours after transfection. NOR1 and RUNX1 expression were analyzed by quantitative real-time PCR.
Inflammation and foam cell formation of macrophages
Macrophage inflammatory gene expression was quantified using the NanoString nCounter System [28]. Bone marrow differentiated macrophages (BMM) isolated from NOR1-deficient mice or wild-type littermates (n=6/group) were treated with 100ng/ml LPS for eight hours. RNA was collected from cells with Trizol (Invitrogen). Purified RNA was hybridized against the nCounter GX mouse inflammation kit (NanoString Technologies) and data was analyzed using nSolver™ Analysis Software (NanoString Technologies). The geometric mean of the code counts for the positive control genes was used for normalization.
NOR1-deficient or wild-type BMM were treated with 100μg/ml oxLDL overnight (Intracel Corp) and the capability of lipid uptake was measured by oil red O staining. Representative pictures were acquired and lipid uptake was quantified spectrophotometrically at 510nm.
Statistical Analysis
Results were represented as means or medians depending on the distribution of data. Unpaired Student’s t-test was utilized to compare the means between two independent groups on a single variable. The effect of NOR1 on atherosclerosis was compared using the Kruskal-Wallis test followed by Dunn Test post-hoc analysis. P values < 0.05 were considered to be statistically significant.
RESULTS
NOR1 deficiency in hematopoietic stem cells accelerates atherosclerosis formation and macrophage recruitment in apoE−/− mice
We previously demonstrated that whole body deletion of the NOR1 locus in apoE−/− mice reduces atherosclerosis formation by decreasing VCAM-1 expression in endothelial cells[12]. In these prior studies, selective deletion of NOR1 in resident aortic cells reduced adhesion of monocytes that were wild-type for NOR1. While these studies pointed to a key role of NOR1 expression in resident endothelial cells to atherosclerosis formation, the contribution of NOR1 in the hematopoietic system and monocytes/macrophages to atherosclerosis in apoE−/− mice remains to be investigated. Addressing this question is particularly intriguing considering recent evidence that NOR1 may serve as a tumor suppressor of myeloid leukemogenesis[15,16], and inhibits inflammatory macrophage activation[14]. To determine whether NOR1 deletion in hematopoietic bone marrow cells contributes to the development of atherosclerotic lesions, apoE−/− mice were lethally irradiated and reconstituted with bone marrow cells derived from NOR1-deficient or wild-type littermate mice. As depicted in Fig. 1A, repopulation of apoE−/− mice with NOR1−/− bone marrow cells resulted in increased atherosclerosis after 12 weeks of feeding an atherogenic diet (NOR1+/+→apoE−/−, 13.56% [n=10]; NOR1−/−→apoE−/−, 21.82% [n=10] median atherosclerotic lesion area of aortic arches; P<0.05. Fig.1A). Specific deletion of NOR1 in hematopoietic cells did not affect serum cholesterol concentration or distribution (NOR1+/+→apoE−/−, 543.56 ± 52.22 mg/dl [n=10]; NOR1−/−→apoE−/−, 563.83 ± 27.31 mg/dl [n=10]; P>0.05; Fig. S-4) indicating a cell-intrinsic role of NOR1 in bone marrow cells for atherosclerosis formation. Consistent with the observed increase in lesion formation, both aortic lipid content and macrophage infiltration (Fig. 1B-E) into the intima of aortic root lesions were significantly increased in apoE−/− mice repopulated with NOR1-deficient bone marrow cells. Furthermore, using an alternative model of macrophage recruitment during inflammatory responses, we confirmed a robust increase in the number of thioglycollate-elicited peritoneal macrophages in NOR1−/− mice relative to wild-type controls (Fig. 2). Collectively, these findings indicate that the selective deletion of NOR1 in bone marrow cells enhances atherosclerosis formation and increases macrophage recruitment into the arterial wall.
FIGURE 1.
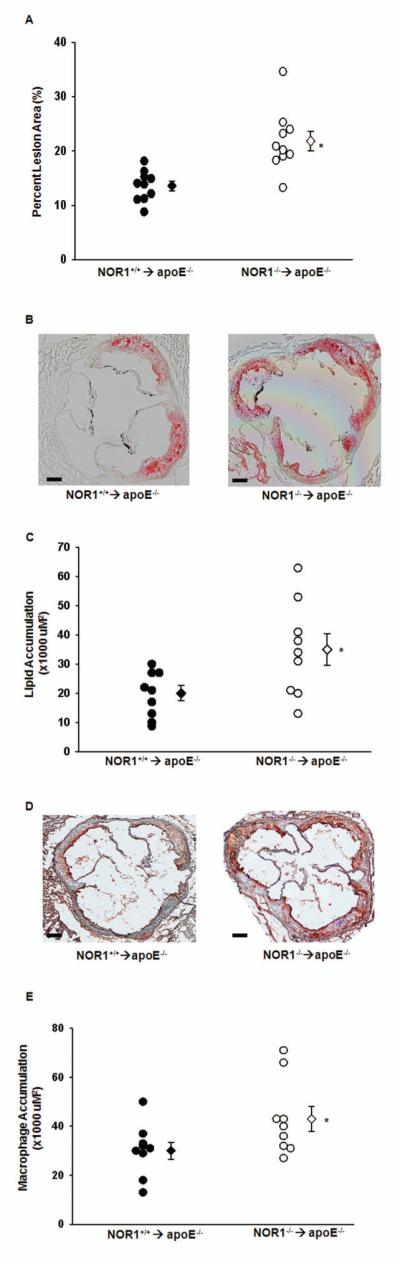
Repopulation of apoE−/− mice with NOR1-deficient bone marrow cells promotes atherosclerosis formation and increases lipid content and macrophage infiltration of the aortic arch. A. Atherosclerotic lesion size was measured on aortic arches from female NOR1+/+→apoE−/− and NOR1−/−→ apoE−/− mice (n=10 per group). Circles represent individual mice; diamonds represent medians (*P<0.05 vs NOR1+/+→apoE−/−). B. Representative oil red O staining of aortic root sections (scale bars 100uM). C. Quantification of lipid content from oil red O staining. Circles represent individual mice; diamonds represent medians (*P<0.05 vs NOR1+/+→apoE−/−). D. Representative immunostaining of aortic root sections with rabbit antiserum for mouse macrophages (scale bars 100uM). Positive cells are stained red. E. Quantification of macrophage content from macrophage immunostaining. Circles represent individual mice; diamonds represent medians (*P<0.05 vs NOR1+/+→apoE−/−).
FIGURE 2.
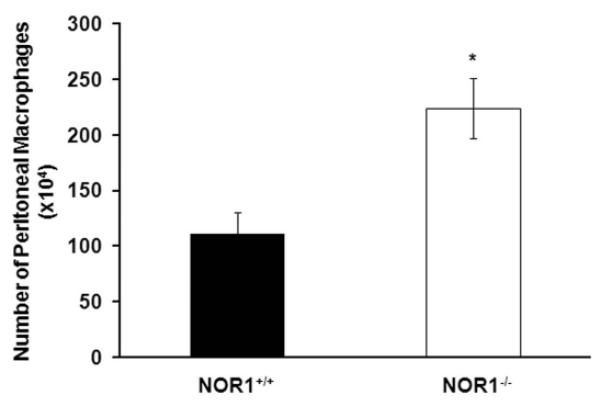
NOR1 deficiency increases peritoneal macrophage recruitment during thioglycollate-induced peritonitis. Quantification of thioglycollate-elicited peritoneal macrophages from NOR1+/+ or NOR1−/− mice by cell counting using a hemocytometer (n=12 per group). Values are mean ± SEM (*P<0.05).
NOR1 deficiency induces splenomegaly and monocytosis
The enhanced macrophage recruitment at two independent sites of inflammation upon NOR1 deletion led us to hypothesize that this phenotype may be due to increased numbers of circulating monocytes. Circulating monocytes originate primarily from differentiation of hematopoietic stem cells and assemble in the red pulp of the spleen[29]. Consistent with our hypothesis that NOR1-deficiency induces monocytosis, we observed splenomegaly in whole body NOR1-deficient mice (Fig. 3A-B). This phenotype was the result of a cell autonomous effect of NOR1 deletion on the hematopoietic stem cell compartment since lethally irradiated apoE−/− mice reconstituted with NOR1−/− hematopoietic cells exhibited similar splenomegaly (Fig. 3C). Quantification of differential white blood cell counts in NOR1−/− mice by flow cytometry analysis (Fig. 3D) further confirmed a two-fold increase in circulating monocyte counts without any obvious changes in T and B-cell numbers, granulocytes, natural killer T-cells, dendritic cells, platelets counts (data not shown), or hematocrit (data not shown).
FIGURE 3.
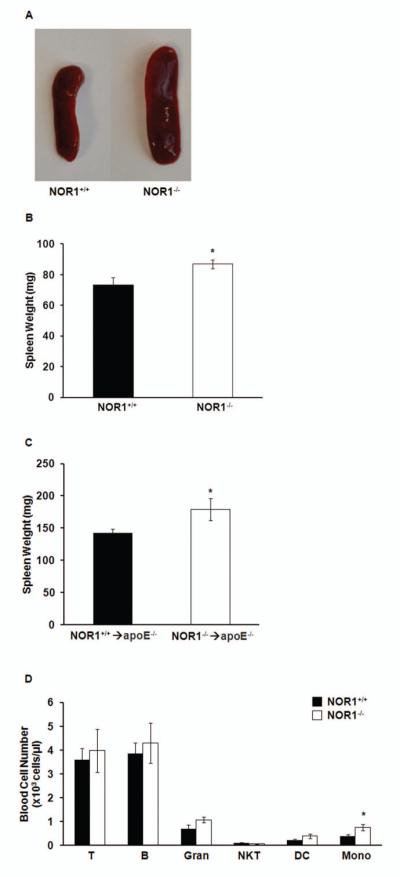
NOR1 deficiency induces splenomegaly and monocytosis in mice. A. Representative images of spleens harvested from NOR1+/+ or NOR1−/− mice. B. Weight of spleens isolated from NOR1+/+ or NOR1−/− mice (n=9 per group). Values are mean ± SEM (*P<0.05). C. Weight of spleens isolated after bone marrow transplantation in NOR1+/+→apoE−/− and NOR1−/−→ apoE−/− mice (n=10 per group). Values are mean ± SEM (*P<0.05). D. Quantification of differential white blood cell populations in NOR1+/+ and NOR1−/− mice (n=6 per group) analyzed by flow cytometry. T, T cells; B, B cells; Gran, granulocytes; NKT, natural killer T cells; DC, dendritic cells; Mono, monocytes. Values are mean ± SEM (*P<0.05).
NOR1 deficiency increases Ly6C+ monocytes in bone marrow, circulation, and spleen
Macrophages recruited to atherosclerotic lesions in response to hypercholesterolemia are predominantly derived from Ly6C+ monocytosis[4], which gives rise to proinflammatory M1 macrophages[30,31]. Considering the increased atherosclerosis, monocytosis, and splenomegaly in apoE−/− mice transplanted with NOR1−/− hematopoietic stem cells, we next quantified Ly6C+ monocytes in blood, bone marrow, and spleen. Despite similar plasma cholesterol levels and distribution, NOR1−/− mice displayed a more than 2-fold increase in blood Ly6C+ monocytes compared to their wild-type littermate controls (Fig. 4A). In contrast, there was only a modest and non-significant increase in Ly6C− monocytes. To further investigate whether a similar monocyte distribution exists in the hematopoietic compartment, we next quantified monocytes and monocyte subset populations in the bone marrow and spleen of NOR1-deficient mice. Consistent with the observations in the peripheral circulation, NOR1-deficient mice exhibited monocytosis in bone marrow and spleen that was primarily the result of increased Ly6C+ monocyte numbers (Fig. 4B-C). To determine whether monocytosis in NOR1-deficient mice is stem cell intrinsic and independent of the background strain, we engrafted irradiated apoE−/− recipient mice with NOR1-deficient bone marrow cells. Following feeding an atherogenic diet for 12 weeks, NOR1−/− → apoE−/− mice revealed similar monocytosis and increased Ly6C+ monocyte numbers in bone marrow, spleen, and blood as compared to the whole body NOR1-deficient mice (Fig. 4D-F). These data confirm a role for NOR1 as a suppressor of myelopoiesis and point to a previously unrecognized role for NOR1 in regulating inflammatory monocyte populations.
FIGURE 4.
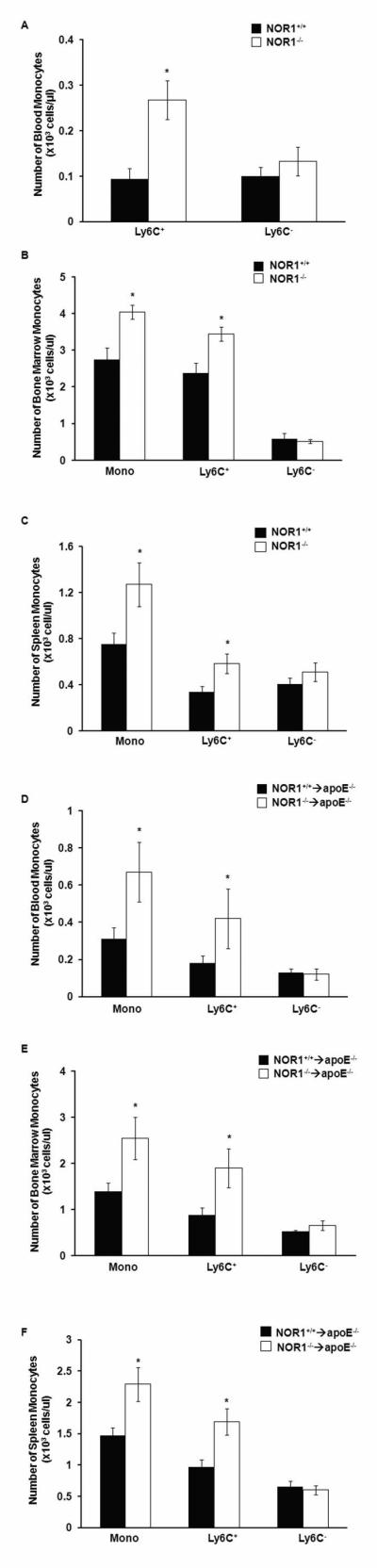
NOR1 deficiency increases total monocytes and Ly6C+ monocytes in the blood, spleen, and bone marrow. A. Quantification of monocyte subsets in the blood of NOR1+/+ and NOR1−/− mice (n=6 per group) analyzed by flow cytometry. Values are mean ± SEM (*P<0.05). B-C. Quantification of total monocytes and monocyte subsets in (B) bone marrow and (C) spleen of NOR1+/+ and NOR1−/− mice (n=6 per group) analyzed by flow cytometry. Values are mean ± SEM (* P<0.05). D-F. Quantification of total monocytes and monocyte subsets in (D) blood, (E) bone marrow, and (F) spleen of NOR1+/+→apoE−/− and NOR1−/− →apoE−/− mice (n=10 per group) analyzed by flow cytometry. Values are mean ± SEM (* P<0.05).
NOR1 represses macrophage and dendritic cell progenitors proliferation
Based on the observation that NOR1 deletion in hematopoietic stem cells selectively induces monocytosis in the bone marrow, we suspected that this phenotype is due to increased proliferation of specifically committed progenitor cells. Circulating blood monocytes arise from hematopoietic stem cells (HSC) via successive commitment of hematopoietic stem cells to common myeloid progenitors (CMP), granulocyte-macrophage precursors (GMP), and finally macrophage and dendritic cell progenitors (MDP)[5]. The latter MDP are bone marrow-resident progenitor cells that ultimately give rise to Ly6C+ monocytes[5]. To explore whether MDP cells constitute the progeny induced by NOR1 deficiency, we first quantified HSC, CMP, GMP, and MDP cells in the bone marrow of NOR1-deficient mice. Consistent with the selective Ly6C+ monocytosis, NOR1 deficiency was associated with increased MDP precursor cells in the bone marrow (Fig. 5A). In contrast, no significant difference between NOR1−/− mice and littermate wild-type mice was observed for HSC, CMP, or GMP populations. These data define a selective increase in MDP cell population as the progeny of bone marrow, blood, and spleen monocytosis in NOR1-deficient mice.
FIGURE 5.
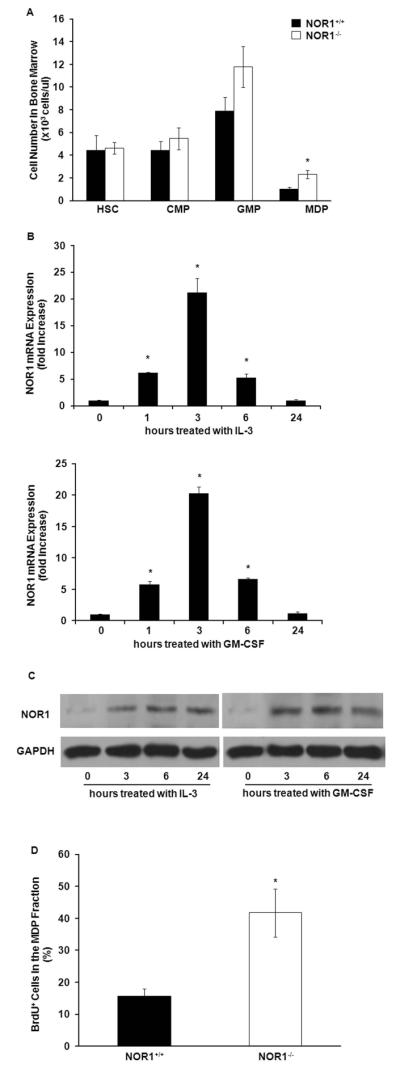
NOR1 is induced in proliferating bone marrow cells and NOR1 deficiency increases the proliferation of MDPs. A. Quantification of hematopoietic cell populations in bone marrow of NOR1+/+ and NOR1−/− mice (n=15 per group). Values are mean ± SEM (* P<0.05). B-C. Wild-type murine bone marrow cells were stimulated with IL-3 (6ng/ml) or GM-CSF (2ng/ml). B. At the indicated time points, NOR1 mRNA expression levels were analyzed by quantitative real-time PCR and normalized to transcript levels of the housekeeping gene TFIIB. Values are mean ± SEM (*P<0.05). C. NOR1 protein expression levels were analyzed at the indicated time points by Western blotting. Cohybridization for GAPDH was performed to control for equal protein loading. The autoradiographs shown are representative of 3 independently performed experiments. D. In vivo proliferation of MDP was analyzed by BrdU incorporation as detected by flow cytometry (n=6 per group). Values are mean ± SEM (*P<0.05).
A key function of NOR1 as an early response gene constitutes mitogenic regulation[6]. Therefore, we next investigated whether NOR1 is expressed during the expansion of progenitor cells. In particular, IL-3 and GM-CSF induce proliferation of HSC and commitment to the myeloid lineage. As depicted in Fig. 5B, time course experiments confirmed increased NOR1 transcript levels following stimulation of bone marrow cells with IL-3 or GM-CSF. Maximal induction of NOR1 mRNA was observed after 3 hours of stimulation, consistent with immediate-early response gene kinetics. The increase in NOR1 mRNA levels was followed by a maximal induction of NOR1 protein expression after 6 hours of stimulation (Fig. 5C). This potent regulation of NOR1 expression may point to an important functional role of NOR1 in progenitor cell proliferation.
To address whether NOR1 deletion specifically increases the proliferation of MDP cells in vivo, we next injected NOR1-deficient mice with BrdU and quantified DNA synthesis in isolated MDP cells. As depicted in Fig. 5D, flow cytometry analysis revealed that deletion of the NOR1 locus increased proliferation of MDP cells, assessed by enhanced DNA synthesis during S phase. Therefore, NOR1 function serves as growth inhibitor in myeloid progenitor cells, which limits monocyte abundance in the circulation.
NOR1 deficiency increases RUNX1 expression in MDP cells
Among the transcriptional programs implicated in leukemogenesis of the myeloid lineage, the RUNX1 (runt-related transcription factor 1) gene mutation is the most frequently mutated locus that induces aberrant myeloid progenitor proliferation[32]. Therefore, we investigated whether NOR1 deficiency increases RUNX1 expression in MDP cells, providing a reasonable mechanism underlying MDP proliferation in NOR1−/− mice. For these studies, bone marrow-derived MDP cells were isolated by FACS (fluorescence-activated cell sorting) and analyzed for RUNX1 transcript levels. Consistent with our hypothesis, NOR1 deletion resulted in a more than 6-fold increase in RUNX1 levels (Fig. 6A). In contrast, other transcription factors implicated in the control of myeloid progenitor cell proliferation, such as PU.1, were not regulated by NOR1 (data not shown). To conversely corroborate an inhibitory effect of NOR1 on RUNX1 expression, we next employed NOR1 overexpression. As depicted in Fig. 6B, NOR1 repressed RUNX1 expression in transiently transfected HEK293 cells. These findings represent the first evidence that expression of RUNX1 in MDP cells, a key mechanism underlying progenitor cell proliferation of the myeloid lineage, is repressed by NOR1.
FIGURE 6.
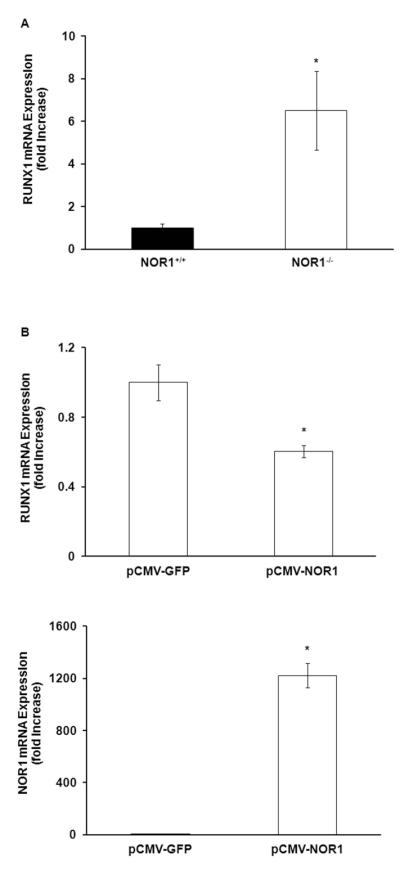
NOR1 represses RUNX1 transcription. A. RUNX1 mRNA expression in MDP sorted from wild-type and NOR1-deficient bone marrow was analyzed by quantitative real-time PCR and normalized to transcript levels of the housekeeping gene TFIIB. Values are mean ± SEM (*P<0.05). B. RUNX1 and NOR1 mRNA expression in HEK 293 cells transfected with NOR1 or GFP overexpression vector were analyzed by quantitative real-time PCR and normalized to transcript levels of the housekeeping gene TBP. Values are mean ± SEM (*P<0.05).
NOR1 deletion promotes local macrophage proliferation and foam cell formation
In addition to the myeloproliferative phenotype and associated monocytosis, NOR1 deletion may elicit pro-atherogenic activities on lesional macrophages. To assess this possibility, we first assessed the proliferation of local macrophages in atherosclerotic lesions. As shown in Fig. 7A and B, NOR1 deletion in the bone marrow increased the replicative rate of lesional macrophages within the aortic root, as assessed by staining for the S phase gene PCNA. Since NOR1 is potently induced by oxidized LDL[33], we next investigated foam cell formation in NOR1-deficient bone marrow-derived macrophages. Consistent with a prior study[14], we documented that NOR1-deletion increased foam cell formation induced by treatment with oxidized-LDL and subsequent oil red O staining of lipid accumulation (Fig. 7C). Finally, the expression of inflammatory genes was examined in activated bone marrow-derived macrophages using Nanostring nCounter Technology. Compared to wildtype macrophages, the deletion of NOR1 in macrophages had no major effect on inflammatory gene expression (Supplementary Fig. S-5). Collectively, these findings indicate that NOR1 deletion increases lesional macrophage proliferation and foam cell formation, without inducing major changes in inflammatory gene expression profiles.
FIGURE 7.
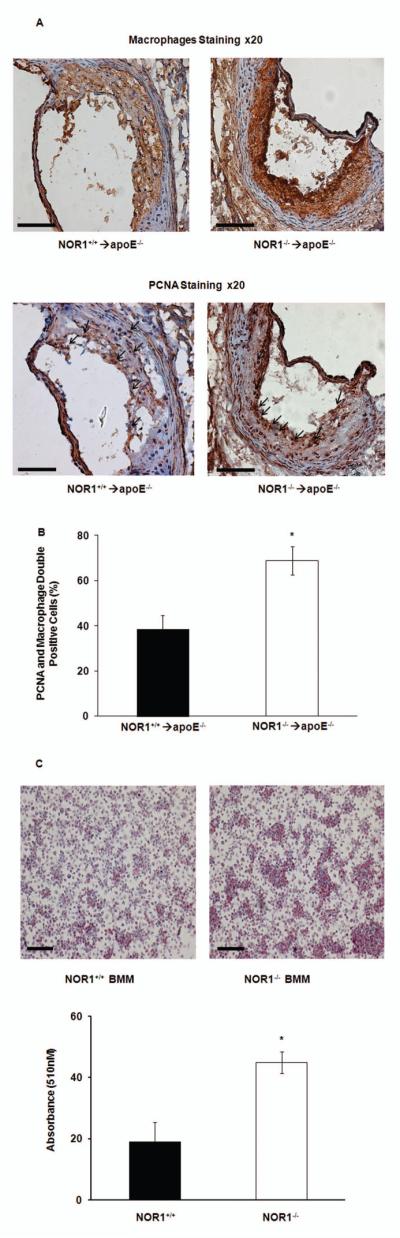
NOR1 deletion promotes macrophage proliferation in atherosclerotic lesions and foam cell formation. A. Representative immunostaining of macrophages and PCNA in consecutive aortic root sections from NOR1+/+→apoE−/− and NOR1−/−→ apoE−/− mice. Positive antibody staining is red. Arrows indicate PCNA positive staining (scale bars 100uM). B. Quantification of PCNA/macrophage double-positive areas at aortic root (*P<0.05 vs NOR1+/+→apoE−/−). C. Top: representative image of oil red O staining of BMM from NOR1-deficient or wild-type mice (scale bars 100uM). Bottom: spectrophotometric quantification of oil red O staining of BMM from NOR1-deficient or wild-type mice. Values are mean ± SEM (*P<0.05).
DISCUSSION
The observation that mice deficient for monocytes are protected from atherosclerosis represents compelling evidence that recruitment of macrophages to atherosclerotic lesions constitutes a key mechanism during atherosclerosis formation[34]. Similarly, in humans monocytosis is associated with the development of cardiovascular disease[2,3]. In the present study, we demonstrate that deletion of NOR1 in hematopoietic stem cells accelerates atherosclerosis formation and increases lesional macrophage content in apoE−/− mice. Bone marrow transplantation studies further confirm a cell intrinsic role of NOR1 to suppress monocytosis and decrease inflammatory Ly6C+ monocyte abundance. Consistent with these observations, NOR1-deficiency induces splenomegaly and a selective expansion of the myeloid lineage by increasing the proliferation of MDP cells in the hematopoietic stem cell compartment. Further in vitro studies confirm that NOR1 expression is induced by IL-3 and GM-CSF stimulation in hematopoietic stem cells. Finally, we identify RUNX1, a key transcriptional activator of myeloid progenitor cell proliferation, as a gene differentially regulated by NOR1. Collectively, these studies characterize the nuclear hormone receptor NOR1 as a previously unrecognized suppressor of monocytosis during atherosclerotic lesion development.
NOR1 functions as an important transcription factor in the control of gene activation during the vascular remodeling processes underlying neointima formation and atherosclerosis[12,18,35,36]. NOR1 is rapidly induced in response to inflammatory and mitogenic signaling in all major cell types participating in vascular disease development, including macrophages, endothelial cells, and smooth muscle cells[17,33,35,36]. Our previous studies demonstrated that whole body deletion of the NOR1 locus decreases neointima and atherosclerosis formation[12,18]. An important finding in our prior atherosclerosis experiments was the observation that selective deletion of NOR1 in resident aortic endothelial and smooth muscle cells decreased adhesion of wild-type monocytes to the endothelium. We inferred from these studies that NOR1 expression in resident aortic cells promotes monocyte adhesion being the major inciting event during atherosclerosis formation. Consistent with this hypothesis, we confirmed that the key adhesion molecules VCAM-1 and ICAM-1 constitute bona fide NOR1 transcriptional target genes induced in response to inflammatory signaling. However, a limitation of this prior research was the inability to define the role of NOR1 expression in macrophages during atherosclerotic lesion formation. Therefore, in the present study we extend these experiments and demonstrate that selective deletion of NOR1 in the hematopoietic lineage using bone marrow transplantation increases atherosclerosis formation in apoE-deficient mice. These findings are indicative of an antagonistic function of NOR1 in resident vascular cells versus monocytes infiltrating the intima from the circulation and hematopoietic system. Under conditions where selective NOR1 deletion in the hematopoietic lineage induces monocytosis, the ensuing increased number of circulating monocytes will promote their recruitment and atherosclerosis formation. While whole body NOR1 deletion induces a similar phenotype of monocytosis (as confirmed in our present study), the deletion of NOR1 in resident endothelial and smooth muscle cells would prevent atherosclerosis to occur due to defective adhesion mechanisms. This hypothesis can be reconciled if accepting that monocyte adhesion and their transendothelial recruitment constitute the critical early events leading to atherosclerosis development[37].
Whether expression of NR4A receptors in the hematopoietic lineage contributes to atherosclerosis formation remains highly controversial. A previous study reported that neither deletion of the NR4A receptor Nur77 nor NOR1 in hematopoietic precursor cells affects atherosclerosis formation[38]. However, with respect to Nur77 these observations were in contrast to two prior studies demonstrating that Nur77 deficiency in bone marrow cells enhances atherosclerosis development in LDL-receptor- and apoE-deficient mice[39,40]. Despite their homology, the three NR4A receptors are thought to mediate transcription through distinct mechanisms and may regulate different but also overlapping target genes[6]. The latter is supported by our observation that the atherosclerosis-prone phenotype in mice with NOR1 bone marrow deletion is consistent with that of Nur77 deletion observed in the two cited studies[39,40], pointing to a potentially common function of Nur77 and NOR1 in hematopoietic cells. However, the discrepancy in atherosclerosis phenotypes upon NOR1 deletion between Chao et al.[38] and our studies may be the result of technical differences, including the employed animal model system. Bone marrow recipients in our experiments were hypercholesterolemic apoE−/− mice whereas the previously published report employed fetal liver cell transplantation into LDL-receptor−/− mice[38]. Therefore, differences in plasma cholesterol levels and lipoprotein size between both strains may affect the effect of NOR1 on atherosclerosis formation[41]. Since the myeloproliferative phenotype was also present in the whole body NOR1−/− mouse, it is unlikely that increased atherosclerosis in apoE−/− mice transplanted with NOR1−/− bone marrow can be ascribed to the previously reported anti-proliferative effect of apoE on hematopoietic stem cells[42]. Consequently, further studies, including in particular cell-specific NOR1 targeting and mechanistic experiments, are required to define the detailed role of NOR1 in atherosclerosis development.
NOR1 has previously been identified as a novel tumor suppressor gene that is repressed in patients with acute myeloid leukemia and inhibits the expansion of myeloid progenitor cells[15,16]. While prior studies employed whole body deletion of both Nur77 and NOR1[16], we extend these observations and confirm that selective deletion of NOR1 in the hematopoietic lineage induces monocytosis and a phenotype in mice reminiscent of myeloid leukemogenesis. Monocytosis is intimately associated with enhanced atherosclerosis formation[34]. Consistent with this notion, our data further characterize NOR1 as a negative regulator of atherosclerosis formation and macrophage lesion content in mice. Using in vivo labeling experiments, we observed that NOR1 deletion induces a selective increase in myeloid progenitor cell proliferation leading specifically to Ly6C+ monocytosis. Ly6C+ monocytes are well established to differentiate into M1 macrophages and secrete various inflammatory cytokines during atherosclerosis formation[4,30,31]. Consequently, the increase in proinflammatory Ly6C+ monocytes associated with NOR1 deletion likely constitutes a major source of monocytes recruited into the lesion and inflammation during atherosclerosis development. In addition to increasing Ly6C+ monocyte number, NOR1 deletion may activate macrophage functions known to increase atherosclerosis development[14]. In the current study, NOR1-deficient macrophages revealed increased local proliferation rates in the intima of aortic root lesions and enhanced foam cell formation. Both these pro-atherogenic activities could mechanistically contribute to the atherosclerosis phenotype associated with NOR1 deletion in the bone marrow. In contrast, the deletion of NOR1 did not result in altered inflammatory cytokine expression in macrophages, consistent with a previous study[38]. Quantification of the extent to which the increased proliferation and foam cell formation of lesional NOR1-deficient macrophages contribute to atherosclerotic plaque formation, will require further investigation. Experimental approaches to differentiate these local pro-atherogenic effects of NOR1 deletion in macrophages from the phenotype of monocytosis due to increased MDP proliferation will depend on the future availability of genetic models allowing inducible deletion of NOR1 selectively in lesional macrophages using Cre-LoxP recombination models.
An important question for future studies will be to define the specific genetic program, which confers the selectivity of NOR1-mediated stem cell proliferation towards the myeloid lineage. Although there is a paucity of studies addressing the transcriptional control of myeloid progenitor cell proliferation, one of the genes most frequently associated with myeloid leukemia is the RUNX1 locus. Also referred to as AML1 (acute myelogenous leukemia-1), RUNX1 is critical for the development of hematopoiesis[43]. While chromosomal translocations and point mutations constitute the key genomic alterations of RUNX1 during leukemogenesis in humans[32], recent evidence has indicated that RUNX1 expression in adult hematopoietic progenitor cells induces proliferation of myeloid progenitors and myeloid leukemogenesis[44]. Consistent with these findings, we confirmed increased RUNX1 expression in highly proliferating NOR1-deficient myeloid progenitor cells. Conversely, overexpression of NOR1 repressed RUNX1 transcription. Since sequence analysis did not detect a functional NBRE target site for NOR1 in the RUNX promoter, the mechanisms underlying this inhibition of RUNX1 expression likely involve indirect repression, as has been described for NOR1[45] and other members of the NR4A family of transcription factors[46]. Therefore, increased RUNX1 expression in NOR1-deficient myeloid progenitor cells may provide a reasonable mechanism underlying increased proliferation of this cell type.
In summary, we demonstrate in the present study that NOR1 deletion in hematopoietic stem cells increases atherosclerosis formation in mice. This phenotype of enhanced atherosclerosis development is associated with an increased abundance of Ly6C+ monocytes, proliferation of macrophages within the intima, and increased foam cell formation. Altered repression of myeloid progenitor cell proliferation in response to NOR1 deletion likely constitutes a key mechanism underlying monocytosis. These findings identify NOR1 as a critical component of monocyte development and macrophage function during atherosclerosis formation.
Supplementary Material
Supplementary figure S-1. Gating strategy for identification of HSC, CMP, GMP and MDP. HSC: Lin−Sca-1+c-kit+; CMP: Lin−c-kit+Sca-1−CD16/CD32lowCD34+; GMP: Lin−c-kit+Sca-1-CD16/CD32hiCD34+; MEP: Lin−c-kit+Sca-1−CD16/CD32lowCD34−; MDP: Lin−c-kit+Sca-1-CD16/CD32+CD115+
Supplementary figure S-2. Representative flow cytometric images for MDP in bone marrow of NOR1+/+ or NOR1−/− mice.
Supplementary figure S-3. Gating strategy for identification of total monocytes and monocyte subsets.
Supplementary figure S-4. Lipoprotein cholesterol distribution analyzed by size exclusion chromatography. Values represent each fraction by mean ± SEM.
Supplementary figure S-5. NanoString nCounter analysis of inflammatory gene expression in LPS-treated NOR1-deficient or wild-type BMM. Data are presented as fold increase relative to untreated cells. Values are mean ± SEM (*P<0.05).
Acknowledgments/Source of Funding
This work was supported in part by National Institutes of Health Grant R01HL084611 and the American Recovery and Reinvestment Act of 2009 Grant R01HL084611-04S1 (to D. B.).
Footnotes
Author Contributions: H.Q.: concept and design, collection and assembly of data, data analysis and interpretation, manuscript writing; Y.L.: collection and assembly of data; Y. Z.: collection and assembly of data; J. A.: collection and assembly of data; K. L. J.: collection and assembly of data; E. B. H.: collection and assembly of data, manuscript writing; D. H.: collection and assembly of data; C. M. B.: collection and assembly of data; A.D.: administrative support; Y.L.: collection and assembly of data; D.B.: concept and design, financial support, collection and assembly of data, data analysis and interpretation, manuscript writing.
Disclosures The authors have no relationships to declare.
REFERENCES
- 1.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnsen SH, Fosse E, Joakimsen O, et al. Monocyte count is a predictor of novel plaque formation: a 7-year follow-up study of 2610 persons without carotid plaque at baseline the Tromso Study. Stroke. 2005;36:715–719. doi: 10.1161/01.STR.0000158909.07634.83. [DOI] [PubMed] [Google Scholar]
- 3.Chapman CM, Beilby JP, McQuillan BM, et al. Monocyte count, but not C-reactive protein or interleukin-6, is an independent risk marker for subclinical carotid atherosclerosis. Stroke. 2004;35:1619–1624. doi: 10.1161/01.STR.0000130857.19423.ad. [DOI] [PubMed] [Google Scholar]
- 4.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geissmann F, Manz MG, Jung S, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Bruemmer D. NR4A orphan nuclear receptors: transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30:1535–1541. doi: 10.1161/ATVBAHA.109.191163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milbrandt J. Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron. 1988;1:183–188. doi: 10.1016/0896-6273(88)90138-9. [DOI] [PubMed] [Google Scholar]
- 8.Law SW, Conneely OM, DeMayo FJ, et al. Identification of a new brain-specific transcription factor, NURR1. Mol Endocrinol. 1992;6:2129–2135. doi: 10.1210/mend.6.12.1491694. [DOI] [PubMed] [Google Scholar]
- 9.Ohkura N, Ito M, Tsukada T, et al. Structure, mapping and expression of a human NOR-1 gene, the third member of the Nur77/NGFI-B family. Biochim Biophy Acta. 1996;1308:205–214. doi: 10.1016/0167-4781(96)00101-7. [DOI] [PubMed] [Google Scholar]
- 10.Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal. 2006;4:e002. doi: 10.1621/nrs.04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkenbout EK, de Waard V, van Bragt M, et al. Protective function of transcription factor TR3 orphan receptor in atherogenesis: decreased lesion formation in carotid artery ligation model in TR3 transgenic mice. Circulation. 2002;106:1530–1535. doi: 10.1161/01.cir.0000028811.03056.bf. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Howatt DA, Gizard F, et al. Deficiency of the NR4A orphan nuclear receptor NOR1 decreases monocyte adhesion and atherosclerosis. Circ Res. 2010;107:501–511. doi: 10.1161/CIRCRESAHA.110.222083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol. 2006;20:786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 14.Bonta PI, van Tiel CM, Vos M, et al. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler Thromb Vasc Biol. 2006;26:2288–2294. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- 15.Ramirez-Herrick AM, Mullican SE, Sheehan AM, et al. Reduced NR4A gene dosage leads to mixed myelodysplastic/myeloproliferative neoplasms in mice. Blood. 2011;117:2681–2690. doi: 10.1182/blood-2010-02-267906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullican SE, Zhang S, Konopleva M, et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13:730–735. doi: 10.1038/nm1579. [DOI] [PubMed] [Google Scholar]
- 17.Nomiyama T, Nakamachi T, Gizard F, et al. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J Biol Chem. 2006;281:33467–33476. doi: 10.1074/jbc.M603436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nomiyama T, Zhao Y, Gizard F, et al. Deficiency of the NR4A neuron-derived orphan receptor-1 attenuates neointima formation after vascular injury. Circulation. 2009;119:577–586. doi: 10.1161/CIRCULATIONAHA.108.822056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cassis LA, Rateri DL, Lu H, et al. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27:380–386. doi: 10.1161/01.ATV.0000254680.71485.92. [DOI] [PubMed] [Google Scholar]
- 20.Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. doi: 10.1385/1-59259-340-2:293. [DOI] [PubMed] [Google Scholar]
- 21.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saraff K, Babamusta F, Cassis LA, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 23.Bravo R. Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res. 1986;163:287–293. doi: 10.1016/0014-4827(86)90059-5. [DOI] [PubMed] [Google Scholar]
- 24.Ranalletta M, Wang N, Han S, et al. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arterioscler Thromb Vasc Biol. 2006;26:2308–2315. doi: 10.1161/01.ATV.0000242275.92915.43. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Hanna RN, Carlin LM, Hubbeling HG, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. 2011;12:778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gizard F, Zhao Y, Findeisen HM, et al. Transcriptional regulation of S phase kinase-associated protein 2 by NR4A orphan nuclear receptor NOR1 in vascular smooth muscle cells. J Biol Chem. 2011;286:35485–35493. doi: 10.1074/jbc.M111.295840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fortina P, Surrey S. Digital mRNA profiling. Nature biotechnology. 2008;26:293–294. doi: 10.1038/nbt0308-293. [DOI] [PubMed] [Google Scholar]
- 29.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci. 2012;17:1120–1139. doi: 10.2741/3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pei L, Castrillo A, Chen M, et al. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem. 2005;280:29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- 34.Smith JD, Trogan E, Ginsberg M, et al. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A. 1995;92:8264–8268. doi: 10.1073/pnas.92.18.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Gonzalez J, Rius J, Castello A, et al. Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ Res. 2003;92:96–103. doi: 10.1161/01.es.0000050921.53008.47. [DOI] [PubMed] [Google Scholar]
- 36.Arkenbout EK, van Bragt M, Eldering E, et al. TR3 orphan receptor is expressed in vascular endothelial cells and mediates cell cycle arrest. Arterioscler Thromb Vasc Biol. 2003;23:1535–1540. doi: 10.1161/01.ATV.0000084639.16462.7A. [DOI] [PubMed] [Google Scholar]
- 37.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 38.Chao LC, Soto E, Hong C, et al. Bone marrow NR4A expression is not a dominant factor in the development of atherosclerosis or macrophage polarization in mice. J Lipid Res. 2013;54:806–815. doi: 10.1194/jlr.M034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamers AA, Vos M, Rassam F, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res. 2012;110:428–438. doi: 10.1161/CIRCRESAHA.111.260760. [DOI] [PubMed] [Google Scholar]
- 40.Hanna RN, Shaked I, Hubbeling HG, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daugherty A, Lu H, Howatt DA, et al. Modes of defining atherosclerosis in mouse models: relative merits and evolving standards. Methods Mol Biol. 2009;573:1–15. doi: 10.1007/978-1-60761-247-6_1. [DOI] [PubMed] [Google Scholar]
- 42.Murphy AJ, Akhtari M, Tolani S, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okuda T, van Deursen J, Hiebert SW, et al. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka T, Tanaka K, Ogawa S, et al. An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 1995;14:341–350. doi: 10.1002/j.1460-2075.1995.tb07008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martens C, Bilodeau S, Maira M, et al. Protein-protein interactions and transcriptional antagonism between the subfamily of NGFI-B/Nur77 orphan nuclear receptors and glucocorticoid receptor. Mol Endocrinol. 2005;19:885–897. doi: 10.1210/me.2004-0333. [DOI] [PubMed] [Google Scholar]
- 46.Saijo K, Winner B, Carson CT, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure S-1. Gating strategy for identification of HSC, CMP, GMP and MDP. HSC: Lin−Sca-1+c-kit+; CMP: Lin−c-kit+Sca-1−CD16/CD32lowCD34+; GMP: Lin−c-kit+Sca-1-CD16/CD32hiCD34+; MEP: Lin−c-kit+Sca-1−CD16/CD32lowCD34−; MDP: Lin−c-kit+Sca-1-CD16/CD32+CD115+
Supplementary figure S-2. Representative flow cytometric images for MDP in bone marrow of NOR1+/+ or NOR1−/− mice.
Supplementary figure S-3. Gating strategy for identification of total monocytes and monocyte subsets.
Supplementary figure S-4. Lipoprotein cholesterol distribution analyzed by size exclusion chromatography. Values represent each fraction by mean ± SEM.
Supplementary figure S-5. NanoString nCounter analysis of inflammatory gene expression in LPS-treated NOR1-deficient or wild-type BMM. Data are presented as fold increase relative to untreated cells. Values are mean ± SEM (*P<0.05).