

In this paper, the authors describe the regulation of COX-2 expression, which is required for prostaglandin production, by miR-146a in lung cancer cells. This is achieved by miR-146a targeting a 3′ UTR site that is present in COX-2 mRNAs displaying long 3′ UTRs but is absent in short COX-2 3′ UTRs. They report an inverse correlation between miR-146a levels and COX-2 levels in lung cancer cells. This work leads to a model whereby miR-146a down-regulation in lung cancer cells contributes to the overexpression of COX-2 and therefore to increased cell growth through prostaglandin synthesis.
Keywords: COX-2, gene expression, miRNA, regulation
Abstract
Prostaglandins are a class of molecules that mediate cellular inflammatory responses and control cell growth. The oxidative conversion of arachidonic acid to prostaglandin H2 is carried out by two isozymes of cyclooxygenase, COX-1 and COX-2. COX-1 is constitutively expressed, while COX-2 can be transiently induced by external stimuli, such as pro-inflammatory cytokines. Interestingly, COX-2 is overexpressed in numerous cancers, including lung cancer. MicroRNAs (miRNAs) are small RNA molecules that function to regulate gene expression. Previous studies have implicated an important role for miRNAs in human cancer. We demonstrate here that miR-146a expression levels are significantly lower in lung cancer cells as compared with normal lung cells. Conversely, lung cancer cells have higher levels of COX-2 protein and mRNA expression. Introduction of miR-146a can specifically ablate COX-2 protein and the biological activity of COX-2 as measured by prostaglandin production. The regulation of COX-2 by miR-146a is mediated through a single miRNA-binding site present in the 3′ UTR. Therefore, we propose that decreased miR-146a expression contributes to the up-regulation and overexpression of COX-2 in lung cancer cells. Since potential miRNA-mediated regulation is a functional consequence of alternative polyadenylation site choice, understanding the molecular mechanisms that regulate COX-2 mRNA alternative polyadenylation and miRNA targeting will give us key insights into how COX-2 expression is involved in the development of a metastatic condition.
INTRODUCTION
Lung cancer is the most common cause of cancer-related death in both men and women in the United States (Siegel et al. 2013). The predominant type of lung cancer is non-small cell lung cancer (NSCLC). This type is further classified into subgroups, the most prevalent NSCLC-type being adenocarcinoma (Travis et al. 1995). Cancer progression is marked by the acquisition of several properties, including altered cellular growth (Hanahan and Weinberg 2011). One specific group of macromolecules that function in cellular growth and paracrine signaling are prostaglandins. The rate-limiting step of the prostaglandin synthesis pathway is the conversion of arachidonic acid to prostaglandin intermediates, carried out by cyclooxygenase (COX) isoenzymes (also known as PTGS) (Hla et al. 1999). There are two isoforms of COX enzymes responsible for COX function, COX-1 and COX-2, and both mediate the conversion of arachidonic acid to prostaglandin intermediates (Smith et al. 1996). COX-1 (PTGS-1) is responsible for basal levels of prostaglandins, while COX-2 (PTGS-2) expression is induced in response to various pro-inflammatory cytokines, e.g., LPS, TNF-α, IL-1, EGF (Williams et al. 1999). COX-2 is present at sites of inflammation, injury, and disease (Mitchell et al. 1995; Williams et al. 1999). The induction of COX-2 protein expression is usually transient and returns to baseline within 24–48 h post-cytokine stimulation. However, COX-2 is constitutively and sustainably overexpressed in numerous cancers (Dubois et al. 1998; Hida et al. 1998; Wolff et al. 1998; Prescott and Fitzpatrick 2000).
COX-2 not only plays a role in the inflammatory response and is overexpressed in cancer, but also has distinct messenger RNA (mRNA) features. The COX-2 mRNA is comprised of 10 exons and possesses an unusually large 3′ untranslated region (UTR) that is ∼2.5 kb, almost half the length of the processed mRNA molecule (Fig. 1A; Appleby et al. 1994). The 3′ UTR encompasses several sequence elements that function to regulate gene expression, including two polyadenylation signals that are utilized in a tissue-specific manner (Hall-Pogar et al. 2005, 2007). Along with the alternative poly(A) signals, the COX-2 3′ UTR has 22 AU-rich elements, 5′-AUUUA-3′ (Fig. 1A; Appleby et al. 1994; Newton et al. 1997). AU-rich elements (AREs) confer post-transcriptional control of expression through mRNA stability and/or instability and are found in many transiently expressed cytokines or inducible growth factor mRNAs, such as TNF-α and COX-2 (Clark 2000). AREs can regulate the rate of turnover of a transcript by promoting or inhibiting decay, depending on which trans-acting factor binds to the element(s) (Chen and Shyu 1995; Wilusz et al. 2001). Another mode of post-transcriptional regulation is one that is mediated by another class of RNA molecules, microRNAs (miRNAs). The COX-2 3′ UTR contains several putative miRNA-binding sites (Fig. 1A). miRNAs are small, noncoding RNAs that are predicted to regulate expression of thousands of genes. MiRNAs function as translational repressors of mRNAs through an RNA interference mechanism (Bartel et al. 2004; Liu et al. 2008). When associated with the RNA-induced silencing complex (RISC), miRNAs imperfectly base-pair with the 3′ UTR of target mRNAs, leading to either target mRNA degradation or translational repression. Therefore, aberrant expression of miRNAs could, in turn, contribute to the development and progression of cancers (Calin and Croce 2006; Williams et al. 2008). In lung cancer pathogenesis, miRNA expression often becomes misregulated leading to skewed expression patterns of their target genes (Liu et al. 2009, 2011).
FIGURE 1.

COX-2 is overexpressed in lung cancer cells. (A) Schematic illustration of composition of exonic (blue boxes) and intronic (black lines) regions of the COX-2 gene. This illustration also highlights two polyadenylation signals (red) and potential miRNA-binding sites (green) that were predicted using microRNA.org and TargetScan algorithms. (B, left) Western blot of Beas2B and A549 cell lysates indicated overexpression of COX-2 protein in A549 cells (second lane) and no expression in Beas2B cells (first lane). (B, right) qPCR data confirmed relative abundance of COX-2 mRNA in each cell type. A549 cells exhibited ∼163-fold increase in COX-2 mRNA expression relative to Beas2B cells. COX-2 expression was normalized to U6 RNA. Values were significantly different (two-tailed t-test) between Beas2B and A549 cells. (*) P-value < 0.016, n = 3. (C) qPCR data confirmed relative abundance of COX-2 mRNA in other NSCLC cell lines, H1373, H1299, and H1975 with respect to Beas2B cells. COX-2 expression was normalized to U6 RNA. Values were significantly different (two-tailed t-test) between Beas2B and the other cells. (*) P-value < 0.001, n = 3.
Recent studies have identified several miRNAs that contribute to miRNA-mediated regulation of COX-2 (Strillacci et al. 2009; Su et al. 2009; Yoon et al. 2011; Akhtar and Haqqi 2012). These miRNAs include but are not limited to miR-101, miR-26b, miR-137, miR-16, and miR-146a. One miRNA in particular, miR-146a, is known to negatively regulate inflammatory responses mediated through the NFκB pathway (Perry et al. 2008; Rusca and Monticelli 2011). Other known targets of miR-146a include TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK1), as well as inflammatory cytokines, such as TNF-α, iNOS, and IL-6 in human articular synovial cells (Taganov et al. 2006; Li et al. 2010; Rusca and Monticelli 2011). Interestingly, miR-146a expression is also found to be misregulated in a variety of tumors including but not limited to papillary thyroid carcinoma (Jazdzewski et al. 2008), hormone-refractory prostate cancer (Lin et al. 2008), and cervical cancer (Wang et al. 2008).
Here, we demonstrate an inverse relationship between miR-146a and COX-2 expression in lung cancer cells. In this study, we examined sustained COX-2 protein expression in several lung cancer cell lines, and identified ablated miR-146a expression as a potential contributing factor to this robust protein expression. Synthetic miR-146a introduction through transient transfection caused expression of COX-2 protein to be specifically reduced, as well as a significant and specific decrease in prostaglandin release. We conclude that miR-146a directly and specifically regulates COX-2 mRNA and therefore COX-2 protein expression in lung cancer cells.
RESULTS
COX-2 expression in lung cell lines
COX-2 is overexpressed in several cancers, including but not limited to cancers of the colon, breast, pancreas, skin, and lung (Wolff et al. 1998; Mendes et al. 2009; Young and Dixon 2010). To confirm the relative expression of COX-2 protein in lung adenocarcinoma cells as compared with normal lung epithelial cells, Western blotting was performed on protein extracts from A549 cells (NSCLC) and Beas2B cells (normal immortalized lung cells), as shown in Figure 1B. Immunoblot analysis revealed that COX-2 protein is overexpressed in A549 cells (Fig. 1B, left) compared with the normal lung cells, Beas2B. These data suggest that either the COX-2 mRNA is not made in the normal lung cells, or that there is mRNA regulation at work in the lung cancer cells.
We further investigated the presence and relative abundance of COX-2 mRNA in the A549 lung cancer cells compared with the Beas2B cells by quantitative Real-Time PCR (qPCR). Comparative Threshold Cycle (CT) (ΔΔCT) qPCR data analysis revealed that >100-fold increase in COX-2 mRNA expression was detected in A549 cells as compared with the mRNA from Beas2B cells (Fig. 1B, right). In addition, COX-2 mRNA overexpression was confirmed in three other NSCLC cell lines by qPCR analysis (H1299, H1373, H1975) (Fig. 1C). It has been previously published that transcriptional regulation of COX-2 plays a role in its overexpression; however, transcriptional regulation alone cannot account for its sustained expression (Ristimaki et al. 1994). Therefore, we next examined post-transcriptional mechanisms of regulation of COX-2 expression that can explain these differences.
The 3′ UTR of COX-2 harbors several potential binding sites that contribute to post-transcriptional RNA regulatory mechanisms, including AU-rich elements (AREs), putative miRNA-binding sites, and alternative polyadenylation signals (Fig. 1A; Hall-Pogar et al. 2005, 2007). Our laboratory has previously demonstrated that alternative polyadenylation of COX-2 mRNA has a tissue-specific regulatory capacity (Hall-Pogar et al. 2005, 2007). To determine whether COX-2 protein overexpression in lung cancer cells is in part a result of alternative polyadenylation, we first analyzed expression of the two alternatively polyadenylated COX-2 mRNA transcripts by RNase H-alternative polyadenylation cleavage assay (RHAPA) (Fig. 2A; Cornett and Lutz 2014). RHAPA analysis revealed that both COX-2 polyadenylated transcripts, termed short and long, are expressed in A549 cells (Fig. 2B). Minimal expression of the COX-2 short mRNA was also detected in Beas2B cells, which was intriguing since Beas2B cells did not express COX-2 protein (Fig. 1B). Notably, no COX-2 long mRNA was detected from the Beas2B cells. These data suggest that post-transcriptional regulatory mechanisms might be acting on the longer COX-2 transcript, and may contribute to the overexpression of the COX-2 protein in A549 adenocarcinoma cells. We chose to focus on regulation of the longer COX-2 transcript and will return to possible mechanisms of regulation of the shorter transcript in the Discussion.
FIGURE 2.

Alternative polyadenylation of COX-2. (A) Schematic of COX-2 3′ UTR depicting the relative location of PCR primer sets that are COX-2 mRNA variant specific. Since Oligo(dT) selection was used in the cDNA synthesis reaction, the F1:R1 primer set will specifically amplify the COX-2 short polyadenylated mRNA; the F2:R2 set will specifically amplify the COX-2 long mRNA; the F3:R3 set will ensure that RNase H cleavage reaction took place. Use of the F3:R3 primer set resulted in no amplified PCR product, indicating that RNase H cleavage was complete (data not shown). (B) Quantification of COX-2 alternative polyadenylation (APA) mRNA variant expression as measured by RHAPA analysis revealed expression of both COX-2 mRNA transcripts (short and long) in A549 cells as compared with Beas2B cells. COX-2 expression was normalized to GAPDH mRNA; n = 4.
Microarray analysis and computer predictions of miRNAs targeting the 3′ UTR of COX-2 mRNA
The potential for miRNA-mediated regulation of an mRNA is directly affected by alternative polyadenylation. If a miRNA-binding site is present downstream from a poly(A) signal, then the RNA is cleaved and polyadenylated upstream of that miRNA-binding site and the resulting mRNA loses the potential to be regulated by that particular miRNA. Moreover, the combination of both alternative polyadenylation and miRNA regulation could underlie COX-2 overexpression. In order to evaluate possible regulation of COX-2 expression by miRNAs that target its 3′ UTR, we investigated differences of miRNA expression in our two lung cell types, Beas2B and A549, using a microarray to determine the expression levels of endogenous miRNAs. Our microarray analysis revealed significant variations in several miRNA expression levels between the two cell lines. From those miRNAs that exhibited a significant difference of expression, a class of miRNAs was selected based on their predicted ability to base-pair with the COX-2 3′ UTR (Table 1). These predictions were based upon two algorithmic databases, miRanda (http://www.microRNA.org) and TargetScan (http://www.targetscan.org). From this subclass, miR-146a was selected for further investigation because its level of expression was significantly lower in the A549 cells as compared with the Beas2B cells. In addition, a single miR-146a binding site was predicted in the COX-2 3′ UTR downstream from the first polyadenylation signal, and upstream of the second polyadenylation signal (Fig. 1A). An interesting finding was the high level of miR-146a-binding site conservation within the 8-mer seed sequence in COX-2 orthologs. Although most of the 3′ UTR of the COX-2 gene is highly divergent in evolution, the single miR-146a-binding site present in the COX-2 3′ UTR is highly evolutionarily conserved across 10 mammalian species (Fig. 3). In addition, the miR-146a mature sequence is also almost identical across several species. These findings further support the intriguing nature of this particular miRNA and its potential target COX-2.
TABLE 1.
Microarray data analysis of microRNA expression
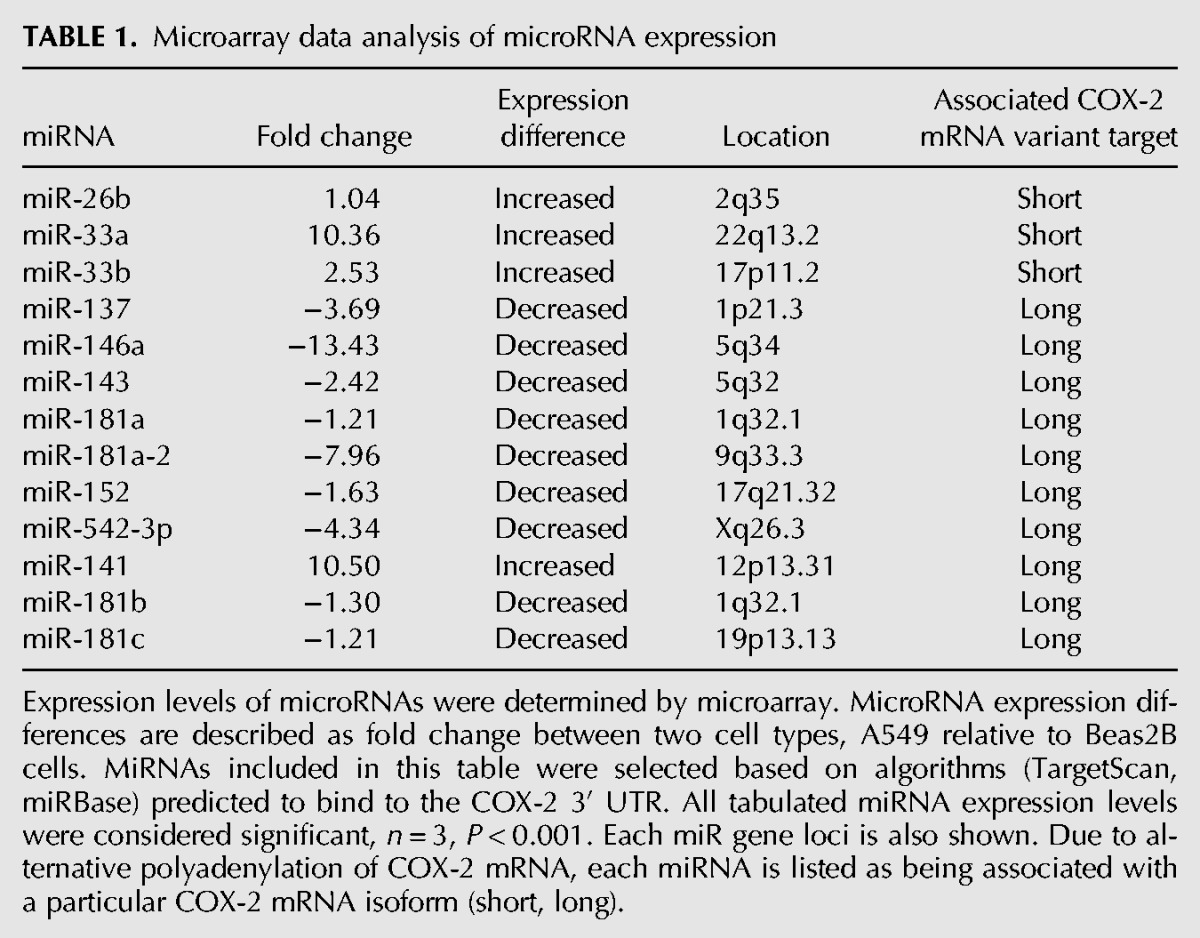
FIGURE 3.

Conservation of miR-146a putative binding site within COX-2 3′ UTR and conservation of mature miR-146a sequence across species. (A) Diagram highlighting the sequence conservation of miR-146a seed region in the COX-2 3′ UTR. (B) Diagram of the mature miR-146a sequence across six species and the chromosomal location of each miR-146a gene.
miR-146a expression in human adenocarcinoma cell lines
Our microarray analysis provided preliminary evidence for the relative expression of miR-146a in Beas2B and A549 cells. To validate miR-146a relative expression, qPCR was utilized. Comparative Threshold Cycle (CT) analysis (ΔΔCT) data confirmed a 20-fold decrease in miR-146a expression in A549 cells, relative to Beas2B cells (Fig. 4). Although miR-146a expression was strikingly lower in the A549 cells, we postulated that it was possible that a miRNA processing defect could potentially cause the significant decrease in mature miR-146a RNA observed in A549 cells. Therefore, precursor-miR-146a expression levels were investigated by qPCR analysis. We observed ∼100-fold less pre-miR-146a expression in A549 cells when compared with expression in Beas2B cells (Supplemental Fig. 1). Therefore, our data suggest that there is not a processing defect that is responsible for the lack of expression of mature miR-146a in the A549 cells. Moreover, we observed low expression levels of miR-146a in H1299 and H1975 cells (Fig. 4). We concluded that an inverse relationship exists between COX-2 expression and miR-146a expression, and this relationship is valid in other NSCLC cancer cell lines (Figs. 1B,C, 4). These data provided the framework to next evaluate the nature of the relationship between miR-146a and COX-2 mRNA, and to determine whether this miRNA directly regulates COX-2 protein expression.
FIGURE 4.

Low miR-146a expression in NSCLC cell lines as measured by qPCR. Real Time qPCR (ΔΔCT) data showed relative expression of miR-146a in Beas2B compared with NSCLC cell lines A549, H1299, and H1975. Expression of miR-146a was normalized to U6 RNA expression. Comparative CT analysis indicated a significantly higher level of miR-146a expression in Beas2B cells when compared with miR-146a expression in NSCLC cell lines, A549, H1299, and H1975. (*) P-value = 0.001, n = 3.
Effect of synthetic miR146a introduction on endogenous COX-2 expression
To examine the effect of exogenous miR-146a on endogenous COX-2 protein expression, A549 cells were transfected with mature synthetic miR-146a at 50 nM and 100 nM concentrations. As a control for nonspecific repression from miRNA transfection, miR-433 was transfected into a subset of A549 cells. Synthetic miR-26b, another miRNA predicted to target COX-2 3′ UTR, was also tested. Cells were harvested 48 h post-miRNA-transfection, and both whole-cell protein lysates and mRNA isolation were analyzed. Western blot analysis revealed that miR-146a transfected samples, at both 50 and 100 nM, showed decreased COX-2 protein expression levels (Fig. 5). Interestingly, we did not observe any effect of miR-26b on COX-2 protein expression. In addition, miR-433, as predicted, had no effect on COX-2 protein levels (Fig. 5). We repeated this experiment in H1975 cells at 50 nm concentration of miR-146a and saw a similar effect on endogenous COX-2 protein expression (Supplemental Fig. 2). These data supported evidence that miR-146a does repress endogenous COX-2 protein. Quantitative-RT-PCR (qPCR) analysis was next performed on mRNA isolated from the transient transfections (Supplemental Fig. 3). We observed that miR-146a caused significant repression of COX-2 mRNA levels, but our data did not conclusively reveal whether the repression of COX-2 protein was due solely to mRNA degradation as opposed to translational repression. Therefore, our data confirm down-regulation of COX-2 protein expression by miR-146a; however, the underlying mechanism of this regulation was not fully determined.
FIGURE 5.

Synthetic miR-146a represses endogenous COX-2 protein in A549 cells. A549 cells were transiently transfected with synthetic miRNAs (x-axis; No miR, miR-26b, miR-146a, miR-433). Cells were lysed and proteins were isolated 48 h post-transfection. (A,B) Western blot analysis was performed on cell lysates. GAPDH served as a loading control. (A) 100 nM miR; (B) 50 nM miR. (C) Densitometric quantification of relative COX-2 protein expression in samples was performed and normalized to GAPDH protein. Each transfection was performed in triplicate in independent experiments.
Effect of miR-146a on reporter protein activity
Our data thus far support an inverse relationship between COX-2 protein and miR-146a expression. In response to introduction of miR-146a, endogenous COX-2 protein expression was significantly reduced. To determine whether miR-146a directly regulates COX-2 through its 3′ UTR, luciferase reporter assays were performed. We purchased Renilla-luciferase reporter constructs (Switch Gear Genomics) that contain an RPL10 promoter, the Renilla luciferase open reading frame (ORF), and either full-length COX-2 3′ UTR (RenSP-COX-2 3′ UTR_WT) or GAPDH 3′ UTR (RenSP-GAPDH 3′ UTR). Beas2B cells were transfected with synthetic miR-146a, miR-433, or miR-26b, or no miRNA; followed by subsequent transfection of the Renilla-luciferase constructs described above (Fig. 6A). We observed ∼50% down-regulation of the relative Renilla-luciferase activity in response to miR-146a (cf. no miR to miR-146a, Fig. 6B). This down-regulation of luciferase activity was not observed in the presence of synthetic miR-26b and miR-433 (Fig. 6B). These data suggest that miR-146a works specifically to regulate COX-2 gene expression through its 3′ UTR.
FIGURE 6.

miR-146a directly regulates Renilla luciferase activity through the COX-2 3′ UTR. (A, top) Schematic diagram of the Renilla luciferase constructs RenSP-COX2 3′ UTR_WT, RenSP-GAPDH 3′ UTR, and RenSP-COX2 3′ UTR_MUT146A. (Bottom) Alignment of the mRNA-miRNA duplex highlighting 8-mer seed sequence (bolded) of COX-2 WT and 5′ end of miR-146a. The alignment also indicates the 4-nt mutation generated in the COX-2 MUT146A construct (TTCT–CCGC). Black lines indicate perfect complementarity and gray lines represent low-affinity T-G matches. (B) Relative Renilla-luciferase activities of RenSP-COX-2 WT constructs were measured in response to synthetic miRNAs (miR-26b, miR-146a, or miR-433) in Beas2B cells. COX-2 WT luciferase activity was normalized to control GAPDH luciferase activity exposed to the same miRNA condition. Luciferase activity was also normalized to total protein concentration for each given sample. Renilla-luciferase activity of COX-2 WT was down-regulated ∼50% in response to miR-146a. This reduction in activity was not observed in response to miR-26b and miR-433 treatments. (*) P-value < 0.03, n = 3. (C) Relative Renilla-luciferase activities of RenSP-COX-2 WT constructs were measured in response to miR-146a alone, anti-miR-146a alone, or both miR-146a and anti-miR-146a treatment in Beas2B cells. Renilla-Luciferase activity was normalized as described above in B. In the presence of both miR-146a and antagomiR, COX-2 WT luciferase activity exhibited no miR-146a down-regulation. Luciferase activity was restored to basal levels. (**) P = 0.006, n = 3. (D) Relative Renilla-luciferase activities of RenSP-COX-2 WT and RenSP-COX MUT146A were measured in response to synthetic miRNAs (no miR, miR-146a, or scrambled miR) in Beas2B cells. Renilla-luciferase activities were normalized as described in B. COX-2 MUT146A luciferase activities were unaffected by miR-146a treatment. Mutation of miR-146a-binding site resulted in no miR-146a regulation of luciferase activity. (*) P < 0.01, n = 3. All experiments were performed in triplicate in independent samples.
To assess the specificity of miR-146a to regulate COX-2 expression through its 3′ UTR, Renilla-luciferase reporter assays were performed in the presence of a miR-146a-specific antagomiR. The antagomiR is the perfect sequence complement of miR-146a and would sequester exogenous miR-146a from binding to the target COX-2 3′ UTR. Beas2B cells were transfected with the synthetic miR-146a in the presence and/or absence of the antagomiR, anti-miR-146a, and sequentially transfected with the Renilla-luciferase constructs (Fig. 6C). In the presence of both miR-146a and anti-miR-146a, relative luciferase activity was restored to basal levels as compared with the measured activity from cells transfected with miR-146a alone (Fig. 6C, cf. miR-146a with miR/anti-146a). Renilla-luciferase activity in the presence of antagomiR alone was also not significantly affected (Fig. 6C, anti-miR-146a). These data suggest that synthetic miR-146a was sequestered from binding the COX-2 3′ UTR luciferase construct by the antagomiR, and that this sequestration prevented miR-146a regulation of luciferase activity. In addition, anti-miR-146a did not cause any off-targeting effect on the reporter protein activity levels. Our data support the importance of a specific interaction between the COX-2 3′ UTR and miR-146a.
Direct interaction of miR-146a with the COX-2 3′ UTR
Repression of endogenous COX-2 protein expression by synthetic miR-146a proposed a relationship between miR-146a and COX-2 (Fig. 5). However, it was unknown whether or not the repressive effect of miR-146a on COX-2 expression was directly linked through miRNA–mRNA sequence interactions.
In order to define the miRNA-binding site responsible for COX-2 3′ UTR luciferase down-regulation, the predicted miR-146a binding site was mutated by site-directed mutagenesis (TTCT to CCGC) (Fig. 6A) (SwitchGear Genomics). Beas2B cells were transfected with either miR-146a or scrambled miR that does not specifically bind any known 3′ UTR sequence in the human genome, followed by transfection of Renilla-luciferase reporter constructs depicted in Figure 6A. We observed a significant down-regulation in relative Renilla-luciferase activity in response to synthetic miR-146a transfection as shown in previous experiments, (Fig. 6B); however, the relative Renilla-luciferase acitivity derived from the COX-2 mutant, COX-2 3′ UTR_MUT146A, exhibited no change in activity in response to miR-146a (Fig. 6D, cf. No miR with miR-146a, black bars). We conclude that the putative miR-146a-binding site is the functioning site responsible for miR-146a regulation of COX-2, and mutation of this miRNA-binding site resulted in loss of miR-146a mediated regulation.
Prostaglandin production, cytotoxicity, and cell viability
COX enzymes are responsible for the conversion of arachidonic acid to prostaglandin intermediates, specifically, PGH2, which is further processed in the cell to make various prostaglandins (Fig. 8, below; Cornett and Lutz 2013). Prostaglandins are a class of molecules that increase cell growth, modulate the immune response, and activate paracrine and autocrine signaling cascades (Tilley et al. 2001; Vancheri et al. 2004). Increases in prostaglandin production are characteristic of cancer cells (Harris et al. 2002; Dannenberg et al. 2005). To examine the downstream effect of miR-146a on the biological function of COX-2 in A549 cells, we performed an enzyme-linked immunosorbent assay (ELISA) specific for prostaglandin E2 in response to transient transfection of synthetic miR-146a. The ELISA revealed a significant decrease in prostaglandin (PGE2) release in response to synthetic miR-146a in A549 cells (Fig. 7A). Indomethacin, a nonspecific COX inhibitor, was used as a positive control for inhibition of PGE2 release (Fig. 7A). The non-COX-2 targeting miRNA, miR-433, showed no effect, and miR-26b revealed little effect (Fig. 7A). These data suggest that introduction of synthetic miR-146a represses COX-2 protein, and thus impairs the biological function of COX-2 to produce prostaglandins.
FIGURE 8.

Model for miR-146a regulation. Schematic depiction of miR-146a regulation of COX-2 expression and how this regulation impacts production of prostaglandins and further downstream, cancer cell viability.
FIGURE 7.

Biological consequences of miR-146a. A549 cells were transiently transfected with synthetic miRNAs (miR-26b, miR-146a, or miR-433) at 50 nM and 100 nM concentrations. (A) Prostaglandin (PGE2) release was measured 48 h post-transfection. As a control for COX activity inhibition, one sample group of cells were exposed to 30 μM indomethacin, a nonselective COX inhibitor, for 3 h prior to supernatant collection. Enzyme-linked immunosorbent assays (ELISA) were performed on cell-free, serum-free supernatants. The bar graph depicts the average of three independent experiments in triplicate. (*) P < 0.0003, n = 3. (B) Lactate dehydrogenase (LDH) cytotoxicity assay was performed on cell-free supernatants from A549 cells treated as described above. (%) Cytotoxicity was determined based on amount of LDH released from treated cells into the supernatant with respect to untreated cells (LDHMIN) and cells exposed to a detergent (LDHMAX). The results show that the higher the concentration of miRNAs, the higher the cytotoxicity. Experiments were performed in independent triplicates. (C) WST-1 cell proliferation assay was performed on A549 cells treated as described above. Optical density (OD) was measured as absorbancy at 450 nm. Percent (%) of viable cells was based on cell proliferation from treated cells with respect to untreated cells (No miR). The results indicate a significant decrease in cell viability in response to miR-146a treatment. This decrease was specific for miR-146a. Experiments were performed in independent triplicates. (*) P < 0.04, n = 3.
The lack of prostaglandin production may be due simply to the introduction of miRNAs, which may lead to increased levels of cellular death, and may not be specific for regulation of COX-2 by miR-146a. In order to investigate this, we carried out a cellular cytotoxicity assay using lactate dehydrogenase (LDH). The LDH cytotoxicity assay is a colorimetric assay that quantifies cell death and lysis by measuring lactate dehydrogenase (LDH) activity in cellular supernatants. Cells were transiently transfected with synthetic miR-146a, miR-26b, and miR-433, at 50- and 100-nm concentrations. Cell-free supernatants were collected 48 h post-miRNA transfections and were subjected to LDH analysis. A control for maximum cell lysis was A549 cells exposed to 1% Triton-X, a cellular detergent. The minimum cell lysis were mock-treated cells that were exposed to transfection reagent, INTERFERin, alone. The introduction of miR-146a and the other miRs resulted in low levels of cytotoxicity at 50 nM, but no significant differences were observed between the miRNAs (Fig. 7B). Higher levels of cytotoxicity were observed at the 100-nM concentration of miRNAs, but there were still no significant differences among the miRNAs (Fig. 7B). These data suggest that decreased prostaglandin production in response to miR-146a is due to miR-146a regulation of COX-2 mRNA, and not because of the synthetic miRNA cytotoxicity.
In carcinogenesis, COX-2 and its downstream products (prostaglandins and thromboxanes) are involved in cellular proliferation, apoptosis, angiogenesis, modulation of the immune response, and tumor invasion (for review, see Brown and Dubois 2004). To assess the effect of miR-146a introduction on A549 cell viability, we performed a WST-1 cellular proliferation analysis on A549 cells in response to synthetic miRNA treatment. In response to miR-146a at 50-nM concentration, which has minimal cytotoxicity (Fig. 7B), A549 cells showed a ∼35% significant reduction in cell viability, whereas the other nontargeting miRNAs showed no significant effect (Fig. 7C). At the higher concentration of the synthetic miRNAs, there was a reduction in cell viability; however, this reduction was observed for each miRNA tested, and may be a consequence of cytotoxicity or miRNA transfection in general (data not shown). Our data support the fact that miR-146a has a significant and specific effect on A549 cell viability and proliferation.
Taken together, our data suggest that miR-146a directly regulates COX-2 mRNA and thus protein expression in lung cancer cells, and that this regulation has biological consequences for prostaglandin production.
DISCUSSION
In this study, we have identified miR-146a as a key regulator of COX-2 expression in lung cancer cells. We observed an inverse correlation of expression: In normal Beas2B lung cells, miR-146a expression was high and COX-2 protein expression was low, while in A549 lung cancer cells, miR-146a expression was low and COX-2 protein expression was high (Figs. 1, 4). Thus, we reasoned that in the absence of miR-146a, COX-2 protein can become highly overexpressed. The restoration of miR-146a in A549 cells resulted in the repression of COX-2 protein expression (Fig. 5). Introduction of a specific miR-146a antagomir ablated this effect (Fig. 6C). Mutation of the miR-146a-binding site in a Renilla-luciferase reporter 3′ UTR construct (RenSP-COX-2_MUT146A) caused introduction of miR-146a to be ineffective in regulation of the luciferase reporter (Fig. 6D). The biological effects of miR-146a introduction resulted in significantly reduced prostaglandin (PGE2) release (Fig. 7A), and reduced cell viability (Fig. 7C). These biological effects are specifically due to regulation of COX-2 by miR-146a and not due to overall cell death caused by the synthetic miR treatment (Fig. 7B). Our data thus provide evidence that miR-146a directly regulates COX-2 gene expression in lung cells.
We propose a model for COX-2 regulation, diagrammed in Figure 8.
In the presence of miR-146a, COX-2 expression is specifically down-regulated, resulting in significant reduction of PGE2 release. This decreased prostaglandin release causes further downstream effect on cancer cell viability. However, COX-1 expression is unaffected by the presence of miR-146a and its enzymatic function is unimpeded. We should note that the low levels of prostaglandins observed upon miR-146a treatment as shown in Figure 7A are likely due to prostaglandins made by COX-1. It is also of note that the COX-1 3′ UTR does not have a binding site for miR-146a. We speculate that this specific COX-2 regulation is kept well in check under normal conditions. In cases of transient inflammation, there is likely miR-146a/COX-2 expression modulation that occurs but this modulation is rapidly restored because of miR-146a. In the case of lung cancer cells, COX-2 expression is constitutive and is not modulated by miR-146a.
It has been noted since 1998 that COX-2 overexpression is found in many cancers and cancer cells (Dubois et al. 1998; Hida et al. 1998; Wolff et al. 1998; Prescott and Fitzpatrick 2000), but the complex relationship of this overexpression with modulated miRNA expression has been less clear. Recently, it was determined that an aberrantly low expression of miR-146a in human non-small cell lung cancer cells exists (Chen et al. 2013). They determined that in five NSCLC cell lines (H358, H1650, H1975, HCC827, and H292) expression of miR-146a by transient transfection led to increased cell death and repressed cellular proliferation (Chen et al. 2013). Although this study did not investigate targets for miR-146a, their findings support our work.
Our data presented here reveal that the low expression levels of miR-146a in A549 cells is not due to a processing defect in the A549 cells (Supplemental Fig. 1). Our data may suggest the notion that precursor-miR-146a is not actively transcribed in A549 cells; however, the mechanism of potential miR-146a transcriptional regulation is not understood. The presence of a G/C single nucleotide polymorphism (SNP) in papillary thyroid carcinoma, rs291064, caused a decrease in mature miR-146a expression and loss-of-function for the miRNA (Jazdzewski et al. 2008). A similar mutation could potentially be responsible for loss of mature miR-146a expression, or affect the transcription of pre-miR-146a resulting in ablation in A549 cells; however, validation in lung cancer cells would need to be performed.
Other mechanisms of COX-2 regulation have also been recognized, including mRNA translation, mRNA stability, and translational regulation (for review, see Cornett and Lutz 2013). COX-2 transcriptional up-regulation is known to play a role in its overexpression in endothelial cells, but enhanced transcription alone cannot account for the sustained induction of COX-2 (Ristimaki et al. 1994). The COX-2 3′ UTR harbors ∼22 AU-rich elements or AREs, known to influence mRNA stability and instability. The COX-2 AREs have been widely investigated, with the proximal ARES appearing to affect mRNA stability and the distal AREs affecting mRNA instability (Gou et al. 1998; Dixon et al. 2000). Additionally, other COX-2 3′ UTR regulatory elements and proteins that bind to them have been identified, including HuR, TIA-1, TIAR, hnRNP U, AUF1, AUF2, FBP, Hsp70, and CUGBP2 (Dixon et al. 2001; Cok et al. 2003; Mukhopadhyay et al. 2003; Sully et al. 2004; Kishor et al. 2013). CUGBP2 not only was reported to stabilize the COX-2 mRNA but also affects the COX-2 mRNA translation (Mukhopadhyay et al. 2003). Translational repression may account for why we observed the shorter COX-2 mRNA from the Beas2B cells in Figure 2B but no COX-2 protein (Fig. 1B). We are currently investigating this finding.
MATERIALS AND METHODS
Mammalian cell culture
Beas2B and A549 cells (ATCC) were grown in DMEM supplemented with 5% fetal bovine serum and 4 mM glutamine at 37°C in a 5% CO2 incubator. H1373 and H1975 cells (ATCC) were grown in Roswell Park Memorial Institute medium (RPMI) supplemented with 10% fetal bovine serum and 4 mM glutamine at 37°C in a 5% CO2 incubator. H1299 cells (ATCC) were grown in Minimal Essential medium (MEM) supplemented with 10% fetal bovine serum and 4 mM glutamine at 37°C in a 5% CO2 incubator. Please note that our strain of Beas2B cells does not cause tumors in nude mice (Jiang et al. 2008).
For prostaglandin E2 (PGE2) release, cells were incubated in serum-free media and stimulated with 4 μM calcium ionophore (A23817, Sigma-Aldrich) 30 min prior to supernatant collection (Gerristen et al. 1987). As a control for COX inhibition, cells were treated with 30 μM indomethacin (Sigma-Aldrich), a known COX-1 and COX-2 inhibitor, 2 h prior to calcium ionophore stimulation.
Microarray analysis
Total RNA (200 ng) was isolated from Beas2B and A549 cells by using the mirVana RNA Isolation kit (Ambion) according to the manufacturer's protocol. The cDNA was hybridized to miRNA-specific oligos and the mixture was bound to streptavidin-conjugated paramagnetic particles to select the cDNA/oligo complexes. An extension step followed by fluorescent labeling of extended products ensued. These labeled assay products correspond to, and are in relative abundance to, specific original miRNAs in the sample. Labeled samples were hybridized to the human MicroRNA expression profiling V2 panel Beadchip (Illumina). The fluorescence intensity was measured by the Illumina BeadArray Reader and was the average of 15–20 beads per oligo. This miRNA panel contained 1146 assays for detecting >97% of the miRNAs described in the miRBase database. GenomeStudio software was used for data analysis.
MicroRNA transfection
Synthetic miRNAs corresponding to miR-146a, miR-26b, and miR-433 were synthesized by Dharmacon. The following constructs were used: hsa-miR-26b mature microRNA sequence: 5′-UUCAAGUAAUUCAGGAUAGGU-3′, hsa-miR-433 mature microRNA sequence: 5′-AUCAUGAUGGGCUCCUCGGUGU; hsa-miR-146a mature microRNA sequence: 5′-UGAGAACUGAAUUCCAUGGGUU. The synthetic miR-433 was used as a negative control because miR-433 was not predicted to bind to COX-2 mRNA. An antagomir specific for miR-146a, anti-hsa-miR-146a-5p, was also used and was purchased from Qiagen. The anti-miR-146a has the sequence: 5′-UGAGAACUGAAUUCCAUGGGUU-3′. A549 and Beas2B cells were transiently transfected with synthetic miRs described above (100 nM; 50 nM) using the INTERFERin transfection reagent (Polyplus-Transfection Inc) according to the manufacturer's instructions. Western blot analysis and quantitative RT-PCR analysis were performed on treated cell samples 48 h post-transfection to examine the effects of the synthetic miRNAs on endogenous COX-2 protein levels and extent of COX-2 mRNA degradation, respectively.
Plasmids
The RenSP-COX-2 3′ UTR_WT and RenSP-GAPDH 3′ UTR Renilla luciferase reporter vectors were purchased from SwitchGear Genomics. The COX-2 and GAPDH 3′ UTR fragments were cloned into the multiple cloning site (MCS) downstream from the Renilla ORF, RenSP. Each construct contained a constitutively active promoter, RPL10. The RenSP-COX-2 3′ UTR_MUT146A, a construct containing the COX-2 3′ UTR fragment that contained a 4-nt mutation (TTCT—CCGC) in the miR-146a seed sequence was also purchased.
Western blot analysis
Beas2B cells and A549 cells were washed with ice-cold PBS and then lysed in RIPA buffer (50 mM Tris at pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1% protease inhibitor). Cell lysis was carried out on ice for 30 min. Protein concentrations were determined by detergent compatible (DC) Bradford Assay analysis (Bio-Rad) according to the manufacturer's protocol. Cleared protein lysates (50 μg) were separated by 10% SDS-PAGE, and were transferred to nitrocellulose membranes by a wet electrophoretic transfer method. Membranes were blocked in 5% nonfat dry milk, 1X PBS, 0.1% Tween-20 (Sigma). Primary antibodies were diluted in blocking solution and were incubated with the blot for 3 h at room temperature. Mouse anti-human COX-2 monoclonal antibody was purchased from Cayman Chemical and diluted 1:1000. Mouse-anti-human GAPDH monoclonal antibody was purchased from ProteinTech Group, Inc. Membranes were washed three times in 1X PBS, 0.1% Tween-20, and then were incubated with goat-anti-mouse horseradish peroxidase-conjugated secondary antibody (HRP) diluted 1:5000 in blocking solution for 1 h at room temperature. Signal detection was accomplished using enhanced chemiluminescence (ECL; Denville Scientific) and autoradiography. For repeated use of the membrane, the membrane was placed in stripping solution, (0.1 M NaOH), for 30 min at room temperature. The membrane was washed three times with deionized distilled H2O and then placed in blocking solution.
RNase-H alternative polyadenylation cleavage assay (RHAPA)
RNA was isolated from A549 and Beas2B cells by the TRIzol method (Invitrogen) according to the manufacturer's protocol. RHAPA Analysis was carried out as described previously (Cornett and Lutz 2014). RNA was incubated with a complementary COX-2-specific DNA oligonucleotide, 5′-GTCATCTTTAAAATGATGTTAAC-3′, at 70°C for 10 min and allowed to cool to room temperature to enable DNA–RNA hybridization. The DNA oligonucleotide will hybridize to the COX-2 mRNA sequence present in the 3′ UTR that resides between the two annotated poly(A) signals (Fig. 2). The RNA mixture was then subjected to RNase H cleavage at 30°C for 1 h. RNase H will cleave DNA–RNA hybrids specifically. Digested RNA was further cleaned using the RNA Clean-Up kit (Qiagen) according to the manufacturer's protocol.
Complementary DNA (cDNA) was synthesized by reverse transcription of RNA using the ThermoScript Reverse Transcriptase according to the manufacturer's protocol (Invitrogen) using an Oligo(dT)20 primer. The Oligo(dT) primer will only amplify RNA molecules that contain a poly(A) tail, and therefore eliminate the possibility that COX-2 mRNA without a poly(A) tail will be reverse transcribed. PCR reactions were carried out using Taq DNA Polymerase (Invitrogen) according to manufacturer's protocol. The PCR cycling conditions were as follows: (1) 95°C for 5 min, (2) followed by 30 cycles of 94°C for 30 sec; 54°C for 1 min; 72°C for 30 sec. PCR primer sets were designed for the COX-2 short mRNA (F1:R1), the COX-2 long mRNA (F2:R2), and a primer set to ensure that the RNase H cleavage reaction occurred (F3:R3) (Fig. 2A). If the RNase H cleavage was incomplete, the F1:R1 primer set would amplify both polydenylated transcripts and no longer be specific for the COX-2 short mRNA. PCR products were analyzed by agarose gel electrophoresis and ethidium bromide staining. Band intensity expressed as relative absorbance units determined relative abundance. The abundance of the COX-2 short mRNA and the COX-2 long mRNA were normalized to the abundance of GAPDH mRNA.
Luciferase assay
Beas2B cells were seeded in a 12-well plate format at a density of 1 × 105 cells/well. Sixteen hours after seeding, cells were transfected with a synthetic miRNA, (miR-26b, miR-146a, miR-433) at 100 or 50 nM as described above (see microRNA transfection). Twenty-four hours post-miR transfection, cells were subsequently transfected with DNA constructs (RenSP-COX-2 3′ UTR_WT; RenSP-GAPDH 3′ UTR; RenSP-COX-2 3′ UTR_MUT146A), using LipoD293 transfection reagent (SignaGen Laboratories) according to manufacturer's protocol. Thirty-hours post-DNA transfection, cells were washed with cold PBS and lysed with 1X Passive Lysis Buffer (Promega). Renilla-luciferase activity (luminescence) was measured using the Renilla-Glo luciferase assay system (Promega) according to the manufacturer's protocol. Briefly, 50 μL of each sample was transferred to black-bottom 96-well dishes and incubated with an equal volume of 1X Renilla-Glo Luciferase Assay Reagent (Promega) for 10 min at room temperature in the dark. Renilla Luminescence was read at 470 nm, with an integration of 5 sec, in a luminometer plate reader, SpectraMax L. Renilla-Luciferase activity from RenSP COX-2 WT or RenSP-COX-MUT146A was normalized activity obtained from samples transfected with RenSP-GAPDH under the same miR-condition. Samples were further normalized to protein concentration of each sample lysate as determined by a Bradford protein determination assay. Assays were conducted in independent triplicates.
Real-time quantitative RT-PCR (qPCR)
Custom miScript Primer Assays were used to detect miR-146a (Assay name: Hs_miR-146a_1), COX-2 (QuantiTect Primer Assay name: Hs_PTGS2_1), precursor-miR-146a (Assay name: Hs_miR-146a_PR_1) expression levels by SYBR Green QuantiTect PCR assay (Qiagen) according to the manufacturer's instructions. Briefly, total RNA was isolated from samples using TRIzol (Invitrogen). RNA was reverse transcribed to cDNA using the miScript RT II kit (Qiagen). PCR was carried out in an Applied Biosystems StepOnePlus Real Time PCR instrument (Applied Biosystems). PCR cycling conditions were according to manufacturer's protocol and as follows: (1) 95°C for 15 min; (2) 40 cycles of 94°C for 15 sec, 57°C for 30 sec, 70°C for 30 sec (collection step); (3) followed by Melt Curve analysis. Samples were normalized to U6 snRNA, (Assay name: Hs_RNU6B_2), which served as an endogenous control. No template (NT) and no Reverse Transcriptase (−RT) controls were also assayed. Quantitative Comparative CT (ΔΔCT) analysis was then conducted using the ABI StepOnePlus v.2.1 software. All assays were performed in triplicate and independently repeated at least twice.
Enzyme-linked immunosorbent assay (ELISA)
Cell-free supernatants from transiently transfected A549 cells were analyzed by an enzyme-linked immunosorbent assay (ELISA) using the Prostaglandin E2 Express EIA Kit (Cayman Chemical). The assay was carried out according to the manufacturer's protocol. Assays were conducted in triplicate and were repeated in three independent experiments.
Lactate dehydrogenase (LDH) cytotoxicity assay
Percent cytotoxicity was assessed with an LDH-cytotoxicity detection kit (Roche Diagnostics), which measures LDH released from cells. A549 cells were seeded in a 24-well format at a density of 0.5 × 105 cells/well. Cells were then transfected with 100 or 50 nM of different miRNAs using INTERFERin as described above. Intracellular LDH release was measured 48 h post-transfection as per the manufacturer's instructions. Briefly, supernatants were transferred to a V-bottom 96-well dish and centrifuged at 250g for 10 min. A total of 100 μL of the cell-free supernatant was transferred to a flat-bottom 96-well dish and was incubated for 30 min with an equal volume of the LDH reaction mixture. The absorbance was read at 490 nm using a plate reader. Cytotoxicity for each treatment was calculated by the following formula: (LDHSAMPLE − LDHMIN)/(LDHMAX − LDHMIN) × 100%, where the LDHMIN was assay medium plus cells not exposed to miRNAs, and the LDHMAX was assay medium plus cells exposed to 1% Triton-X-100, a cellular detergent.
WST-1 cell proliferation assay
Cell viability was assessed with a WST-1 Cell Proliferation Assay kit (Cayman Chemical), which measures the enzymatic conversion of WST-1 to formazan by cell mitochondrial dehydrogenases present in viable cells. A549 cells were seeded in a 96-well format at a density of 5000 cells/well. Cells were then transfected with 100 or 50 nM of different miRNAs using INTERFERin as described above. Cell viability was measured 48 h post-transfection as per the manufacturer's instructions. Briefly, cells were incubated with 10 μL of WST-1 for 30 min—1 h at 37°C, 5% CO2. The absorbance was read at 450 nm using a microplate reader. Optical density (OD) is shown as a percent of viable control cells, No miR. Assays were performed in independent triplicates.
Data analysis
Data are expressed as mean ± SD. Statistical significance was assessed using two-sample two-tailed Student's t-test. P-values <0.05 were considered significant.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melissa Rogers, PhD of NJMS, Rutgers and Annette Lee, PhD of The Feinstein Institute for Medical Research of the North Shore–Long Island Jewish Health System for RNA hybridization and microarray analysis. We also thank Edouard Azzam, PhD, of NJMS for H1299 cells. We thank the New Jersey Commission on Cancer Research (NJCCR) for a Predoctoral Fellowship to A.L.C., and the Lung Cancer Research Foundation and the American Heart Association 12GRNT9120029 for Research Grants to C.S.L.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044149.113.
REFERENCES
- Akhtar N, Haqqi TM 2012. MicroRNA-199a* regulates the expression of cyclooxygenase-2 in human chondrocytes. Ann Rheum Dis 71: 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T 1994. Structure of the human cyclo-oxygenase-2 gene. J Biochem 302: 723–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. Review: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Brown JR, DuBois RN 2004. Cyclooxgenase as a target in lung cancer. Clin Cancer Res 10: 4266s–4269s [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM 2006. MicroRNA signatures in human cancers. Nat Rev 6: 857–866 [DOI] [PubMed] [Google Scholar]
- Chen CA, Shyu A 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465–470 [DOI] [PubMed] [Google Scholar]
- Chen G, Umelo IA, Lv S, Teugels E, Fostier K, Kronenberger P, Dewaele A, Sadones J, Geers C, De Greve J 2013. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One 8: e60317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A 2000. Post-transcriptional regulation of pro-inflammatory gene expression. Arthritis Res 2: 172–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cok SJ, Acton SJ, Morrison AR 2003. The proximal region of the 3′ untranslated region of cyclooxygenase-2 is recognized by a multimeric protein complex containing HuR, TIA-1, TIAR and the heterogenous nuclear ribonucleoprotein U. J Biol Chem 278: 36157–36162 [DOI] [PubMed] [Google Scholar]
- Cornett AL, Lutz CS 2013. Regulation of genes in the arachidonic acid metabolic pathway by RNA processing and RNA-mediated mechanisms. Wiley Interdiscip Rev RNA 4: 593–605 [DOI] [PubMed] [Google Scholar]
- Cornett AL, Lutz CS 2014. RHAPA: a new method to quantify alternative polyadenylation. Methods Mol Biol 1125: 157–167 [DOI] [PubMed] [Google Scholar]
- Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, Dubois RN 2005. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol 23: 254–266 [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM 2000. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem 275: 11750–11757 [DOI] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM 2001. Altered expression of the mRNA stability factor HuR promotes cyclooxgenase-2 expression in colon cancer cells. J Clin Invest 108: 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE 1998. Cyclooxygenase in biology and disease. FASEB J 12: 1063–1073 [PubMed] [Google Scholar]
- Gerristen ME, Nganele DM, Rodrigues AM 1987. Calcium ionophore (A23187)- and arachidonic acid-stimulated prostaglandin release from microvascular endothelial cells: effects of calcium antagonists and calmodulin inhibitors. J Pharmacol Exp Ther 240: 837–846 [PubMed] [Google Scholar]
- Gou Q, Liu CH, Ben-Av P, Hla T 1998. Dissociation of basal turnover and cytokine-induced transcript stabilization of the human cyclooxygenase-2 mRNA by mutagenesis of the 3′ untranslated region. Biochem Biophys Res Commun 242: 508–512 [DOI] [PubMed] [Google Scholar]
- Hall-Pogar T, Zhang H, Tian B, Lutz CS 2005. Alternative polyadenylation of cyclooxygenase-2. Nucleic Acids Res 33: 2565–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Pogar T, Liang S, Hague LK, Lutz CS 2007. Specific trans-acting proteins interact with auxiliary RNA polyadenylation elements in the COX-2 3′-UTR. RNA 13: 1103–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Harris SG, Padilla J, Koumas L, Ray D, Phipps RP 2002. Prostaglandins as modulators of immunity. Trends Immunol 23: 144–150 [DOI] [PubMed] [Google Scholar]
- Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T, Takahashi T 1998. Increased expression of cyclooxgenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res 58: 3761–3764 [PubMed] [Google Scholar]
- Hla T, Bishop-Bailey D, Liu CH, Schaefers HJ, Trifan OC 1999. Cyclooxygenase-1 and -2 isoenzymes. Int J Biochem Cell Biol 31: 551–557 [DOI] [PubMed] [Google Scholar]
- Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A 2008. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci 105: 7269–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Zheng S, Langenfeld L, Lo SC, Rogers MB 2008. Mycoplasma infection transforms normal lung cells and induces bone morphogenetic protein 2 expression by post-transcriptional mechanisms. J Cell Biochem 104: 580–594 [DOI] [PubMed] [Google Scholar]
- Kishor A, Tandukar B, Ly YV, Toth EA, Suarez Y, Brewer G, Wilson GM 2013. HSP70 is a novel posttranscriptional regulator of gene expression that binds and stabilizes selected mRNAs containing AU-rich elements. Mol Cell Biol 33: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vandenboom TG, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH 2010. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res 70: 1486–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Chiang A, Chang D, Ying SY 2008. Loss of mir-146a function in hormone-refractory prostate cancer. RNA 14: 417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Fortin K, Mourelatos Z 2008. MicroRNAs: biogenesis and molecular functions. Brain Pathol 18: 113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sempere LF, Galimberti F, Freemantle SJ, Black C, Dragnev KH, Ma Y, Fiering S, Memoli V, Li H, et al 2009. Uncovering growth-suppressive microRNAs in lung cancer. Clin Cancer Res 15: 1177–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sempere F, Guo Y, Korc M, Kauppinen S, Freemantle SJ, Dmitrovsky E 2011. Involvement of microRNAs in lung cancer biology and therapy. Clin Cancer Res 15: 1177–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes RA, Carvalho JF, Waal I 2009. An overview on the expression of cyclooxygenase-2 in tumors of the head and neck. Oral Oncol 45: e124–e128 [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Larkin S, Williams TJ 1995. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem Pharmacol 50: 1535–1542 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S 2003. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell 11: 113–126 [DOI] [PubMed] [Google Scholar]
- Newton R, Seybold J, Liu SF, Barnes PJ 1997. Alternate COX-2 transcripts are differentially regulated: implications for post-transcriptional control. Biochem Biophys Res Commun 234: 85–89 [DOI] [PubMed] [Google Scholar]
- Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA 2008. Rapid changes in microRNA-146a expression negatively regulate the IL-1β-induced inflammatory responses in human lung alveolar epithelial cells. J Immunol 180: 5689–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SM, Fitzpatrick FA 2000. Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta 1470: M69–M78 [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Garfinkel S, Wessendorf J, Maciag T, Hla T 1994. Induction of cyclooxygenase-2 by interleukin-1 alpha. Evidence for post-transcriptional regulation. J Biol Chem 269: 11769–11775 [PubMed] [Google Scholar]
- Rusca N, Monticelli S 2011. MiR-146a in immunity and disease. Mol Biol Int 10.4061/2011/437301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A 2013. Cancer statistics, 2013. CA Cancer J Clin 63: 11–30 [DOI] [PubMed] [Google Scholar]
- Smith WL, Garavito RM, Dewitt DL 1996. Prostaglandin endoperoxide H syntheases (cyclooxygenase)-1 and -2. J Biol Chem 271: 33157–33160 [DOI] [PubMed] [Google Scholar]
- Strillacci A, Griffoni C, Sansone P, Paterini P, Piazzi G, Lazzarini G, Spisni E, Pantaleo MA, Biasco G, Tomasi V 2009. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res 315: 1439–1447 [DOI] [PubMed] [Google Scholar]
- Su H, Yang JR, Xu T, Huang J, Xu L, Yuan YF, Zhuang SM 2009. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res 69: 1135–1142 [DOI] [PubMed] [Google Scholar]
- Sully G, Dean JLE, Wait R, Rawlinson L, Santalucia T, Saklatvala J, Clark AR 2004. Structural and functional dissection of a conserved destabilizing element of cyclo-oxygenase-2 mRNA: evidence against the involvement of AUF-1, AUF-2, tristetraprolin, HuR, or FBP1. Biochem J 377: 629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D 2006. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci 103: 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley SL, Coffman TM, Koller BH 2001. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest 108: 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis WD, Linder J, Mackay B 1995. Classification, histology, cytology and electron microscopy. In Lung cancer: Principles and practice (ed. Pass HI, et al. ), pp. 361–395 Lippencott Raven, Philadelphia, PA [Google Scholar]
- Vancheri C, Mastruzzo C, Sortino MA, Crimi N 2004. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol 25: 40–46 [DOI] [PubMed] [Google Scholar]
- Wang X, Tang S, Le SY, Lu R, Rader JS, Meyer C, Zheng ZM 2008. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One 3: e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CS, Mann M, DuBois RN 1999. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 18: 7908–7916 [DOI] [PubMed] [Google Scholar]
- Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA 2008. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans 36: 1211–1215 [DOI] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW 2001. The cap-to-tail guide to mRNA turnover. Nat Rev 2: 237–246 [DOI] [PubMed] [Google Scholar]
- Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A 1998. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res 58: 4997–5001 [PubMed] [Google Scholar]
- Yoon S, Choi YC, Lee Y, Jin M, Jeong Y, Yoon J, Baek K 2011. Characterization of microRNAs regulating cyclooxygenase-2 gene expression. Genes & Genomics 33: 673–678 [Google Scholar]
- Young LE, Dixon DA 2010. Posttranscriptional regulation of cyclooxygenase 2 expression in colorectal cancer. Curr Colorectal Cancer Rep 6: 60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.