

This paper examines the role of amino acids connecting the aminoacylation and editing domains of leucyl-tRNA synthetase. Deletion of these amino acids resulted in complete loss of aminoacylation retention of editing activity. These results indicate that the bridge between the aminoacylation domain and editing domain is crucial for the function of the synthetase.
Keywords: Homo sapiens cytoplasm, leucyl-tRNA synthetase, CP1 hairpin, amino acid activation, aminoacylation, tRNA binding
Abstract
Leucyl-tRNA synthetases (LeuRSs) catalyze the linkage of leucine with tRNALeu. LeuRS contains a catalysis domain (aminoacylation) and a CP1 domain (editing). CP1 is inserted 35 Å from the aminoacylation domain. Aminoacylation and editing require CP1 to swing to the coordinated conformation. The neck between the CP1 domain and the aminoacylation domain is defined as the CP1 hairpin. The location of the CP1 hairpin suggests a crucial role in the CP1 swing and domain–domain interaction. Here, the CP1 hairpin of Homo sapiens cytoplasmic LeuRS (hcLeuRS) was deleted or substituted by those from other representative species. Lack of a CP1 hairpin led to complete loss of aminoacylation, amino acid activation, and tRNA binding; however, the mutants retained post-transfer editing. Only the CP1 hairpin from Saccharomyces cerevisiae LeuRS (ScLeuRS) could partly rescue the hcLeuRS functions. Further site-directed mutagenesis indicated that the flexibility of small residues and the charge of polar residues in the CP1 hairpin are crucial for the function of LeuRS.
INTRODUCTION
Aminoacyl-tRNA synthetases (AaRSs) catalyze the esterification of an amino acid to its cognate tRNA (Ibba and Söll 2000; Schimmel and Ribas De Pouplana 2000; Woese et al. 2000; Ling et al. 2009). The products (aa-tRNAs) are the base materials for protein biosynthesis. The reactions catalyzed by AaRSs are the starting points and accuracy-limiting steps in translation (Ibba and Söll 1999). They display an overall error rate (producing noncognate aa-tRNAs) of about 10−3–10−4, which is a very high level for protein translation apparatus (the overall error of DNA replication is 10−8; transcription is 10−4; tRNA selection by the ribosome is 10−3–10−5) (Loftfield and Vanderjagt 1972; Roy and Ibba 2006). The fidelity of AaRSs is crucial for translation.
According to the signature sequence and structural features of the active site, AaRSs are divided into two classes: I and II. Class I synthetases contain a Rossmann fold, which is a dinucleotide-binding domain, with two signature peptides, HIGH and KMSKS. Class II AaRSs contain three antiparallel β-fold motifs (Eriani et al. 1990).
Except for ArgRS, GluRS, and GlnRS, aminoacylation of tRNA is performed by a two-step reaction: The amino acid is initially activated by ATP to form an aminoacyl-adenylate (aa-AMP) intermediate (amino acid activation), and the aminoacyl moiety is then transferred to the 3′ terminus of the cognate tRNA to yield aminoacyl-tRNA (aminoacylation). Several amino acids have similar structures (e.g., Leu, Ile, Met, and Nva) and discrimination between cognate and noncognate amino acids is inefficient for the AaRSs (Martinis and Fox 1997; Ahel et al. 2003; Ling et al. 2009; Ling and Söll 2010; Yadavalli and Ibba 2012). To improve the fidelity of catalysis reactions, proofreading processes evolved to hydrolyze either misactivated aa-AMPs (pretransfer editing) or mischarged tRNAs (post-transfer editing) (Hale et al. 1997; Silvian et al. 1999; Zhu et al. 2007; Martinis and Boniecki 2010; Tan et al. 2010).
Leucyl-tRNA synthetase (LeuRS) belongs to class Ia AaRS. Based on their similar structures, LeuRS, IleRS, and ValRS are collectively known as LIVRS, all of which contain a representative catalytic core consisting of a Rossmann fold. Besides the conservative Rossmann fold, almost all LeuRSs contain a large insertion domain called connective peptide 1 (CP1) within the sequence of the catalytic core (Chen et al. 2000; Li et al. 2011; Palencia et al. 2012). CP1 folds independently in the tertiary structure and is defined as a classic editing domain, in which the aminoacyl bond of mischarged aa-tRNA is hydrolyzed (post-transfer editing) to ensure the fidelity of the catalytic process (Chen et al. 2000; Palencia et al. 2012; Zhou and Wang 2013). The functions of the aminoacylation domain and the editing domain are independent of each other.
During catalysis, enzymes dynamically fit the conformation of their substrates. The function of LeuRS requires the flexible swing of CP1 to the coordinated conformation (Alexander and Schimmel 2001; Zhang and Hou 2005). According to the structure of Pyrococcus horikoshii LeuRS (PhLeuRS), upon aminoacylation-complex (complex A) formation, the editing domain swings by ∼20° relative to the rest of the class Ia core. In the editing intermediate conformation (complex B), the editing domain swings by ∼5° (Supplemental Fig. S1A; Fukunaga and Yokoyama 2005b). The neck between the editing and aminoacylation domains of LeuRS consists of the CP core and the CP1 hairpin. The CP core is fixed on the aminoacylation domain (Ibba and Söll 1999; Fukunaga and Yokoyama 2005a). The CP1 hairpin is regarded as an attachment of CP1. The critical location of the CP1 hairpin suggests that it plays crucial roles in the CP1 swing and appropriate conformation formation, thus affecting the function of LeuRS.
The CP1 hairpin region is present in all archaeal, eukaryotic, and prokaryotic LeuRS, IleRS, and ValRS proteins. The tertiary structures of the CP1 hairpin of archaeal, eukaryotic, and prokaryotic LeuRS, ValRS, and IleRS comprise three β turns (Fukunaga and Yokoyama 2005b). However, the architectural details of the CP1 hairpins of archaeal and eukaryotic LeuRS are unique and different from ValRS, IleRS, and prokaryotic LeuRS. First, the CP1 hairpin of ValRS, IleRS, and prokaryotic LeuRS contains a zinc finger motif and coordinates a single zinc atom. However, the zinc finger motif is missing in archaeal and eukaryotic LeuRS (Supplemental Fig. S1B). Second, the CP1 editing domains are inserted at different positions within the CP1 hairpin. In archaeal and eukaryotic LeuRS, CP1 is inserted within the CP1 hairpin, and thus, the primary sequence of the CP1 hairpin may be separated into two fragments: CP1 hairpin 1 and 2. In prokaryotic LeuRS, CP1 is inserted outside the CP1 hairpin, and the primary sequence of the CP1 hairpin is not separated (Supplemental Fig. S1C). Third, in prokaryotic LeuRS, the CP1 hairpin may interact with a special insertion region of CP1; mutations in the insertion may disrupt the interaction and alter the correct domain orientation, thus lowering the post-transfer editing activity. However, in the archaeal and eukaryotic CP1 hairpin, the position of the insertion region shifts, and the interaction may disappear (Supplemental Fig. S1D). The functions of the CP1 hairpin of archaeal and eukaryotic LeuRS, thus, remain undetermined (Cusack et al. 2000; Fukunaga and Yokoyama 2005a,b).
In the present study, we reported the critical effect of the human cytoplasmic leucyl-tRNA synthetase (hcLeuRS) CP1 hairpin on the activities of amino acid activation, aminoacylation, and tRNA binding. Deletion of the CP1 hairpin of hcLeuRS destroyed the activity of hcLeuRS, and only the CP1 hairpin from yeast partially rescues the function of hcLeuRS, suggesting that this small motif is highly species-specific. Mutation of the conservative small residues and charged residues had a major impact on the activity of the enzyme, suggesting that these residues may be involved in the function of the CP hairpin.
RESULTS
The CP1 hairpin domain is crucial for activities of leucine, leucylation of tRNALeu, and tRNA binding of hcLeuRS
Based on the identification of the CP1 hairpin, as revealed by the structure of PhLeuRS (PDB-1WKB), those of hcLeuRS range from residue R236 to G256 (designated as CP1 hairpin 1, CH1) and from residue V514 to Y534 (designated as CP1 hairpin 2, CH2) (Fig. 1).
FIGURE 1.

Primary sequence and tertiary structure of the CP1 hairpin of LeuRS. (A) Primary sequence alignment of the CP1 hairpins from representative eukaryotic, archaeal, and prokaryotic LeuRSs, or from hcIRS and hcVRS. Conserved and semiconserved residues in all species are highlighted with a red background and red letter; residues conserved only in eukaryotes and archaea are highlighted with a blue background. The amino acids of hcLeuRS on which site-directed mutagenesis was performed in this study are indicated by numbers. (LRS) Leucyl-tRNA synthetase, (IRS) isoleucyl-tRNA synthetase, (VRS) valyl-tRNA synthetase, (hc) human cytoplasm, (Sc) Saccharomyces cerevisiae, (Ce) Caenorhabditis elegans, (Ph) Pyrococcus horikoshii, (Tt) Thermus thermophiles, (Ec) Escherichia coli, (Bs) Bacillus subtilis. (B) Crystal structures of complex of PhLeuRS and PhtRNALeu in the aminoacylation conformation. The CP1 hairpin is colored in magenta; the Rossmann fold is colored in yellow; the CP1 is colored in cyan; the other parts of PhLeuRS are in green. PhtRNALeu is shown as a blue ribbon.
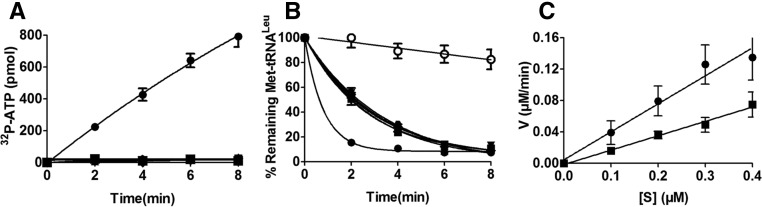
To understand the effect of the CP1 hairpin on various activities of hcLeuRS, we constructed three deletion mutants: hcLeuRS-ΔCH1, hcLeuRS-ΔCH2, and hcLeuRS-ΔCH, in which CH1, CH2, and both were replaced, respectively, by a linker with an Ala tripeptide that maintains the approximate distance between the catalysis and editing domains (Zhou et al. 2008). All deletion mutants were properly folded and displayed little change in their secondary structures compared with hcLeuRS, as revealed by their CD spectrograms (Supplemental Fig. S2). The amino acid activation and aminoacylation assays showed that these mutants had lost both activities (Fig. 2A; Table 1).
FIGURE 2.

Enzymatic activity of wild-type hcLeuRS and the CP1 hairpin domain deletion mutant. (A) The ATP-PPi exchange reaction was catalyzed by 30 nM hcLeuRS (•) and the mutants of hcLeuRS-ΔCH1 (▴), hcLeuRS-ΔCH2 (▾), and hcLeuRS-ΔCH (▪). The upward- and downward-pointing triangles are overlapping with the squares. (B) The post-transfer editing assay was performed using 5 nM of the enzymes described above. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control. The downward-pointing triangle is overlapping with the square. (C) The first-order reaction of deacylation was performed using 2 nM hcLeuRS (•) and the mutants of hcLeuRS-ΔCH (▪). (D) Relative growth rate of yeast knockout strains ScΔleus harboring a WT or mutated hcLeuRS was measured in liquid medium. The error bars are standard deviations from three independent measurements.
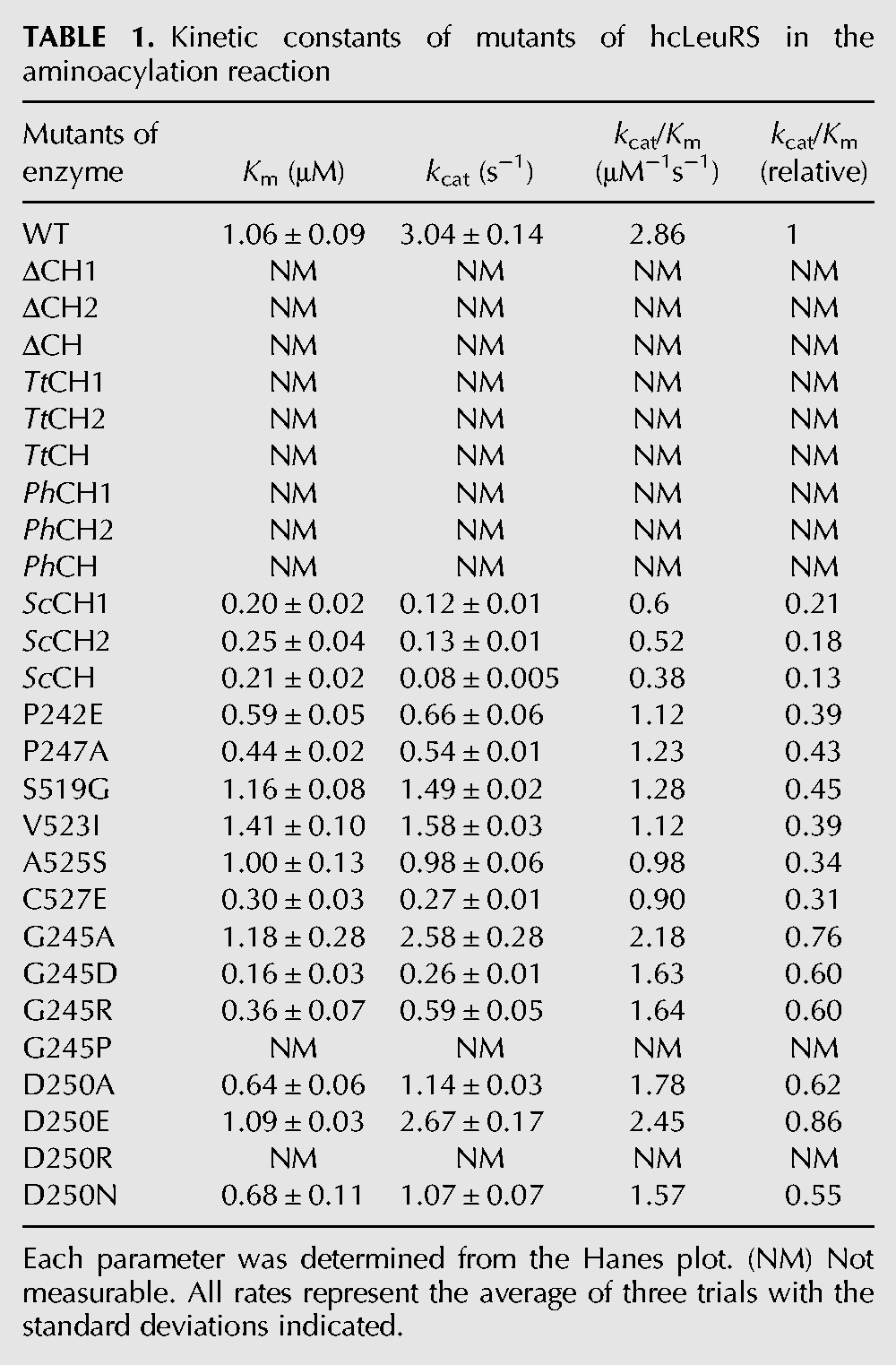
TABLE 1.
Kinetic constants of mutants of hcLeuRS in the aminoacylation reaction
The binding affinity for hctRNALeu was measured by intrinsic tryptophan equilibrium fluorescence. The kd values of hcLeuRS and hcLeuRS-ΔCH were 0.57 μM and 0.83 μM, respectively (Table 2; Supplemental Fig. S3), indicating that the hcLeuRS-ΔCH had a decreased binding affinity with tRNALeu and that the CP1 hairpin is required for optimal tRNALeu binding.
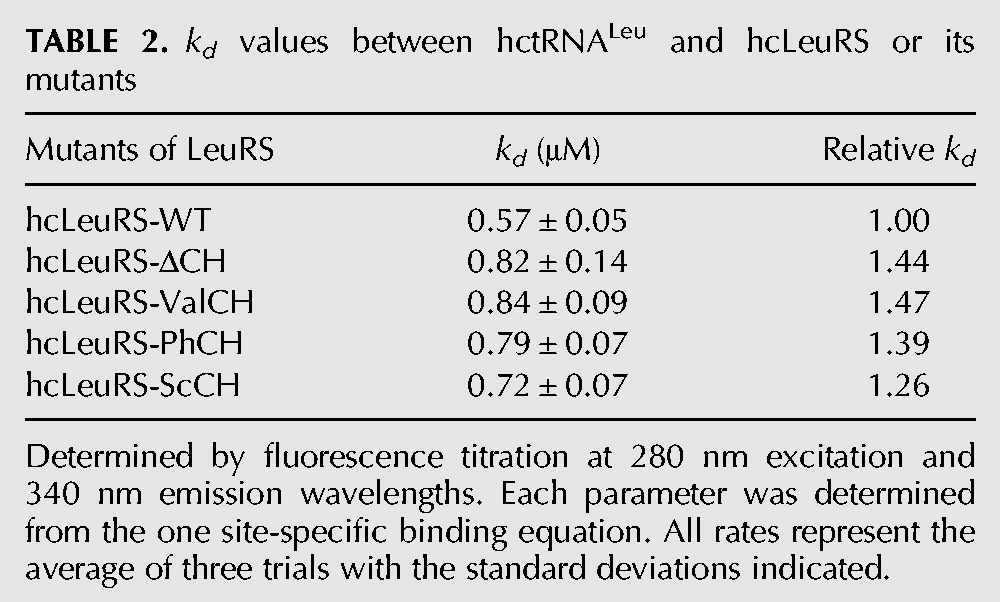
TABLE 2.
kd values between hctRNALeu and hcLeuRS or its mutants
HcLeuRS hydrolyzed a large amount of Met-tRNALeu in 2 min; however, the three deletion variants deacylated less Met-tRNALeu (Fig. 2B). We performed first-order reactions using a low tRNA concentration to measure the precise hydrolysis rate. The rate of deacylation by hcLeuRS-ΔCH declined by ∼43% compared to hcLeuRS-WT (Fig. 2C). Compared to the decreases in the amino acid activation and aminoacylation reactions, the deacylation activities only decreased slightly. We hypothesize that the active site of post-transfer editing within the CP1 domain in each deletion variants is intact. The reduction in the deacylation of Met-tRNALeu might be caused by the decrease in the tRNA binding affinity.
We then measured the kinetic constant for hydrolytic editing of mischarged Met-tRNALeu to further study the function of the CP1 hairpin in editing. By cloning the genes, we obtained the CP1 domain of hcLeuRS spanning from residue G256 to V514, named CPGV, and the CP1 domain plus the CP1 hairpin from residue R236 to Y534, named CHRY. Because of the shorter sequence and simpler structure, the post-transfer editing of CPGV and CHRY declined a lot compared to that of the full-length enzyme. CHRY has a little larger kcat value (2.5 × 10−2 S−1 and 2.1 × 10−2 S−1, respectively), and a much lower Km value compared to CPGV (25 μM and 35 μM, respectively). The data further indicated that the CP1 hairpin contributes a stronger tRNA binding affinity with hcLeuRS (Table 3).
TABLE 3.
Apparent kinetic parameters for hydrolytic editing of mischarged Met-tRNALeu

Previously, we created a leuS (encoding LeuRS) knockout Saccharomyces cerevisiae strain, ScΔleus, and hcLeuRS could cross-recognize yctRNALeu and rescue the ScΔleus lethality in vivo (Yao et al. 2008). We transformed the plasmids of TEF414 containing the gene encoding hcLeuRS-ΔCH1, hcLeuRS-ΔCH2, and hcLeuRS-ΔCH into ScΔleus separately. None of the transformants survived (Fig. 2D), suggesting that these mutants could not leucylate yctRNALeu in vivo.
The CP1 hairpin domains of LIVRS are differentiated
Although their sequences are diverse, the CP1 hairpin structure of archaeal and eukaryotic LeuRS is more similar to ValRS and IleRS than to that of prokaryotic LeuRS. First, the CP1s of ValRS, IleRS, and archaeal and eukaryotic LeuRS are all inserted at point i within the CP1 hairpin; however, the CP1s of prokaryotic LeuRSs are inserted at point ii outside the CP1 hairpin (Supplemental Fig. S1C; Fukunaga and Yokoyama 2005b). Second, the phylogenetic tree constructed using the sequence of diverse CP1 hairpins of LIVRSs indicated that the CP1 hairpin of archaeal and eukaryotic LeuRSs is closer to ValRSs rather than to prokaryotic LeuRSs (Supplemental Fig. S4B).
We constructed several chimeric mutants: hcLeuRS-ValCH1, hcLeuRS-ValCH2, and hcLeuRS-ValCH, in which CH1, CH2, and the entire CH of hcLeuRS were substituted with the corresponding regions of hcValRS, respectively; and hcLeuRS-IleCH1, hcLeuRS-IleCH2, and hcLeuRS-IleCH, in which CH1, CH2, and the entire CH of hcLeuRS were substituted with those of hcIleRS, respectively. All six mutants showed no amino acid activation and aminoacylation activities (Fig. 3A; Table 1). The post-transfer editing activity declined. The hydrolysis of hcLeuRS-IleCH for Met-tRNALeu decreased 49% according to the first-order reaction (Fig. 3B,C). One representative chimeric enzyme, hcLeuRS-ValCH, was chosen to measure the tRNALeu binding affinity. The kd of hcLeuRS-ValCH was 0.84 μM, which was increased by ∼47% compared to that of hcLeuRS-WT (Table 2; Supplemental Fig. S3), indicating weaker binding of tRNALeu to the chimeric enzyme. The activities of these chimeric enzymes in amino acid activation, aminoacylation, post-transfer editing, and their binding affinity to tRNALeu were very similar to those of the hcLeuRS mutants with deleted CP1 hairpins (Fig. 2; Table 1). These results indicated that the CP1 hairpin of hcValRS and hcIleRS could not compensate for the function of the CP1 hairpin of hcLeuRS.
FIGURE 3.
The enzymatic activities of chimeric mutants with the CP1 hairpin of hcLeuRS substituted for that of hcIleRS or hcValRS. (A) The ATP-PPi exchange reaction was catalyzed by 30 nM hcLeuRS (•) and the mutants of hcLeuRS-IleCH1 (▴), hcLeuRS-IleCH2 (▾) hcLeuRS-IleCH (▪), hcLeuRS-ValCH1 (⧫), hcLeuRS-ValCH2 ( ), and hcLeuRS-ValCH (
), and hcLeuRS-ValCH ( ). Except for the solid circles, all other symbols are overlapping. (B) The post-transfer editing assay was performed using 5 nM of the enzymes described above. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control. Except for the solid and open circles, all other symbols are overlapping. (C) The first-order reaction of deacylation was performed using 2 nM hcLeuRS (•) and the mutants of hcLeuRS-ΔIleCH (▪).
). Except for the solid circles, all other symbols are overlapping. (B) The post-transfer editing assay was performed using 5 nM of the enzymes described above. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control. Except for the solid and open circles, all other symbols are overlapping. (C) The first-order reaction of deacylation was performed using 2 nM hcLeuRS (•) and the mutants of hcLeuRS-ΔIleCH (▪).
Only a CP1 hairpin from a eukaryotic LeuRS could compensate for the loss of CP1 from hcLeuRS
We then determined if the CP1 hairpin of LeuRSs from prokaryotes, archaea, and eukaryotes could compensate for the function of the CP1 hairpin of hcLeuRS. The primary sequences of the LeuRSs of prokaryotes, archaea, and eukaryotes are conserved. We constructed nine chimeric mutants, containing hcLeuRS-TtCH1, hcLeuRS-TtCH2, hcLeuRS-TtCH, hcLeuRS-PhCH1, hcLeuRS-PhCH2, hcLeuRS-PhCH, hcLeuRS-ScCH1, hcLeuRS-ScCH2, and hcLeuRS-ScCH, in which the CH1, CH2, and the whole CH of hcLeuRS were replaced by those of Thermus thermophilus, P. horikoshii, or S. cerevisiae, respectively.
The six variants in which the CP1 hairpin of hcLeuRS was substituted for those from an archaeon (hcLeuRS-PhCH1, -PhCH2, and -PhCH) and a prokaryote (hcLeuRS-TtCH1, -TtCH2, -TtCH), respectively, displayed the same enzyme activities as the deletion variants. Their amino acid activation and aminoacylation activities were destroyed completely (Fig. 4A; Table 1). The post-transfer editing activity declined. The hydrolysis of hcLeuRS-TtCH for mischarged Met-tRNALeu decreased 44% according to the first-order reaction (Fig. 4B,C). Representatively, the kd of hcLeuRS-PhCH to hctRNALeu increased by ∼50%, indicating that the binding affinity of the chimeric enzymes to hctRNALeu was weaker (Table 2; Supplemental Fig. S3).
FIGURE 4.

The enzyme activities of chimeric mutants with the CP1 hairpin of hcLeuRS substituted with the homologous regions of LeuRSs from Thermus thermophilus, Pyrococcus horikoshii, and Saccharomyces cerevisiae. (A) The ATP-PPi exchange reaction was catalyzed by 30 nM hcLeuRS (•) and the mutants of hcLeuRS-TtCH1 (▴), hcLeuRS-TtCH2 (▾), hcLeuRS-TtCH (▪), hcLeuRS-PhCH1 (⧫), hcLeuRS-PhCH2 (), and hcLeuRS-PhCH (). Except for the solid circles, all other symbols are overlapping. (B) The post-transfer editing assay was performed using 5 nM of the enzymes described in A. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control. Except for the solid and open circles, all other symbols are overlapping. (C) The first-order reaction of deacylation was performed using 2 nM hcLeuRS (•) and the mutants of hcLeuRS-TtCH (▪). (D) The ATP-PPi exchange reaction was catalyzed by 30 nM hcLeuRS (•) and the mutants of hcLeuRS-ScCH1 (▵), hcLeuRS-ScCH2 (∇), and hcLeuRS-ScCH (□). Except for the solid circles, all other symbols are overlapping. (E) The post-transfer editing assay was performed using 5 nM of the enzymes described in D. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control. Except for the solid and open circles, all other symbols are overlapping. (F) The first-order reaction of deacylation was performed using 2 nM hcLeuRS (•) and the mutants of hcLeuRS-ScCH (□). (G) TLC analysis of [32P]-AMP formation was catalyzed by 1 μM hcLeuRS WT (with or without hctRNALeu), hcleuRS-ScCH1, and hcLeuRS-ScCH2 (with hctRNALeu). (H) The graph represents the quantification of the AMP formation with tRNALeu(UAG)-WT (•), -ScCH1 (▴), -ScCH2 (▾), and without tRNA (○), respectively; the upward- and downward-pointing triangles and the open circles are overlapping. The kobs values of AMP formations were calculated and reported in Table 4.
The chimeric variants of hcLeuRS with the CP1 hairpin from the yeast LeuRS (hcLeuRS-ScCH1, -ScCH2, and -ScCH) maintained partial enzymatic activities. The amino acid activation and aminoacylation activities of the three variants decreased 85%, having 15% of the activities of hcLeuRS (Fig. 4D; Table 1). The deacylating activity of hcLeuRS-ScCH against mischarged tRNALeu (Met-tRNALeu) decreased by 24% according to first-order reactions (Fig. 4E,F). The kd of hcLeuRS-ScCH to hctRNALeu was 0.72 μM (Table 2; Supplemental Fig. S3), indicating that the binding affinity of hcLeuRS-ScCH for tRNALeu was reduced by 26%. The enzymatic activities were much larger than in the deletion mutants and other chimeric mutants of hcLeuRS.
In the presence of hctRNALeu and Nva, TLC was used to assay the observed rate constants (kobs) of the AMP formations of hcLeuRS, hcLeuRS-ScCH1, and hcLeuRS-ScCH2. Compared to that of hcLeuRS-WT (1.58 s−1), the kobs of these mutants were lower (0.14 s−1 and 0.12 s−1). However, the formation of Nva-AMPs was undetectable and less than the Nva-AMP formation without tRNA, indicating no accumulation of misaminoacylation product (Fig. 4G,H; Table 4, below). We hypothesized that the editing ability of the two mutants is maintained. However, norvaline activation by the two mutants is weak; thus, the formation of AMP and Nva-AMP both decreased in the TLC assay.
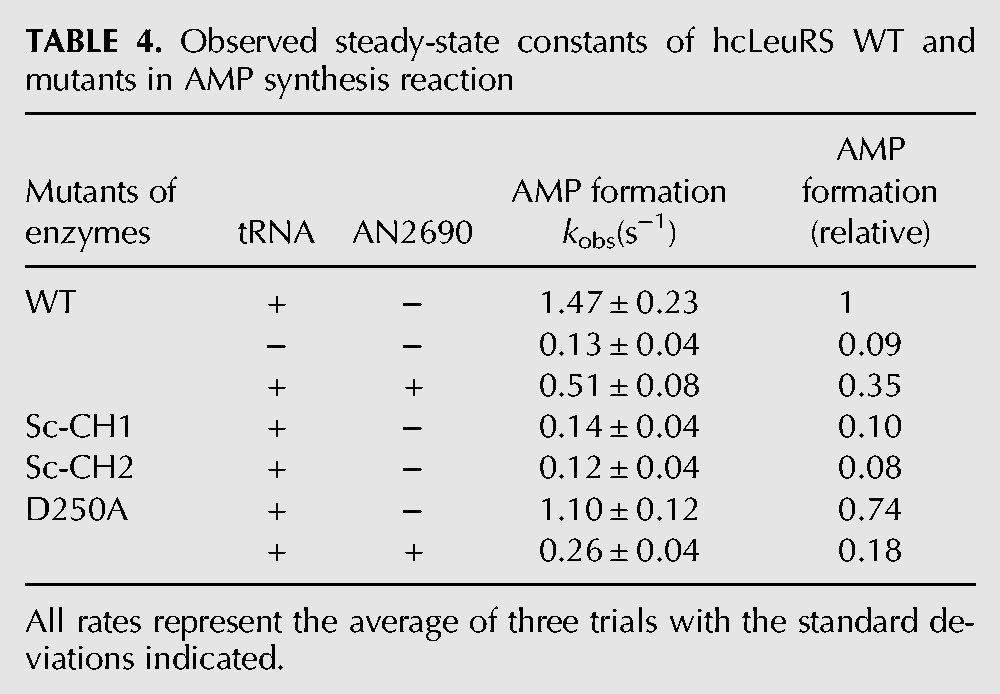
TABLE 4.
Observed steady-state constants of hcLeuRS WT and mutants in AMP synthesis reaction
These results suggested that the CP1 hairpin of LeuRS evolved differently between archaea, eukaryote, and prokaryote. Only the CP1 hairpins from eukaryotic LeuRS could partially rescue the function of the CP1 hairpin of hcLeuRS.
Different residues between hcLeuRS and ScLeuRS are essential for the function of CP1 hairpin
By sequence alignment of CP1 hairpin 1 (CH1) of hcLeuRS and ScLeuRS, only two residues among 21 amino acid residues are different (Fig. 1A). In hcLeuRS, residues 242 and 247 are both proline, while in ScLeuRS, the corresponding positions are aspartic acid and alanine (E256 and A261). Surprisingly, hcLeuRS-ScCH1, which was only mutated at these two residues (P242E and P247A), showed strikingly reduced enzymatic activities (Fig. 4D; Table 1). To study the function of P242 or P247 in hcLeuRS, we constructed the single point mutants hcLeuRS-P242E and hcLeuRS-P247A in which residues at the related positions of hcLeuRS were substituted with those from ScLeuRS. These two mutants had lower amino acid activation and aminoacylation activities than those of hcLeuRS, which were comparable to those of hcLeuRS-ScCH1 (Fig. 5A; Table 1). These results showed that the two proline residues are essential for the function of the CP1 hairpin. Considering that the CP1 hairpin must be rigid to maintain the tertiary structure, we hypothesized that P242 and P247 confer such rigidity on the structure of CP1 hairpin and are thus necessary for its function.
FIGURE 5.

Single different residues between the CP1 hairpins of hcLeuRS and ScLeuRS affect the function of the enzyme. (A,C) The ATP-PPi exchange reaction of mutants with a single different residue between hcLeuRS and ScLeuRS in CP1 hairpin1 (A) and CP1 hairpin2 (C). In each figure, from the top to the bottom, 30 nM hcLeuRS-WT (•), and -P242E (▴), -P247A (▾), -ScCH1 (△) in CP1 hairpin1, -V523I (⧫), -S519G (▪), -A525S (), -C527E (□), and -ScCH2 (∇) in CP1 hairpin2 were used. The hexagon symbol is between the diamond and the square. (B,D) The post-transfer editing assay in CP1 hairpin 1 (B) and CP1 hairpin 2 (D) were performed using 5 nM of the enzymes described above. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control.
Five residues among the 21 amino acid residues of CP1 hairpin 2 (CH2) of hcLeuRS differ from those of ScLeuRS. Thus, based on the sequence of CH2 of ScLeuRS, we constructed four single-site mutants: hcLeuRS-S519G, -V523I, -A525S, and -C527E. All mutants showed slightly decreased amino acid activation and aminoacylation activities. Among them, hcLeuRS-C527E had the lowest activities: 30% aminoacylation activity and 15% amino acid activation activity compared to the wild-type hcLeuRS (Fig. 5C; Table 1). The cysteine of hcLeuRS is a residue without a charge, and glutamic acid is a residue with a negative charge; this replacement introduced a negative charge into CH2 of hcLeuRS. However, the other four substitutions barely changed the polarity of CH2. These results suggested that the polarity of the residues might also be necessary for the function of CH2 of hcLeuRS.
Although the deacylating activity toward mischarged tRNALeu of hcLeuRS-ScCH1 and -ScCH2 decreased by 15% compared to that of hcLeuRS (Figs. 4E, 5B,D), that of all single-site mutants did not change significantly (Fig. 5B,D). The results indicated that these mutations in the CP1 hairpin of hcLeuRS did not affect its post-transfer editing function. However, multisite mutation at once may affect the editing activity of hcLeuRS severely, suggesting that the function of the crucial residues may be dose-dependent.
Identification of significant residues within the CP1 hairpin of hcLeuRS by amino acid scanning
The primary sequence alignment of LeuRSs from diverse species showed that the CP1 hairpin is highly conserved. Most of the conserved residues consist of polar amino acid residues and small amino acid residues. In the above experiment, we hypothesized that these two types of residues are necessary for the function of hcLeuRS. To identify the critical residues in the CP1 hairpin for the function of hcLeuRS, 15 conserved or semiconserved residues were mutated according to their properties (Fig. 1A). The flexible or small residues were mutated to proline to obtain four mutants—hcLeuRS-G245P, -G256P, -S516P, and -L532P; the charged residues were mutated to the residues with opposite charge to form six mutants—hcLeuRS-R236D, -D250R, -H251D, -D252R, -R517D, and -D528R; and the aromatic residues were changed to alanine to form other five mutants—hcLeuRS-Y240A, -Q529A, -W530A, -Y531A, and -Y534A.
All the mutants showed strikingly decreased activities. Seven mutants, hcLeuRS-G245P, -G256P, -S516P, -L532P, -D250R, -H251D, and D252R, lost their amino acid activation and aminoacylation activities completely; eight mutants, hcLeuRS-R236D, -Y240A, -Q529A, -W530A, -Y531A, -Y534A, -R517D, and -D528R, displayed 70%, 50%, 30%, 50%, 50%, 40%, 10%, and 15% of their amino acid activation activity compared to the wild-type, respectively. The decreases in the aminoacylation activities of these mutants were similar to those of amino acid activation (Fig. 6). However, the post-transfer editing activities of these mutants were almost the same as that of wild-type hcLeuRS (Fig. 6). The results indicated further that the CP1 hairpin domain affects the activity of the first step reaction of aminoacylation (amino acid activation) and then that of aminoacylation, but not the post-transfer editing.
FIGURE 6.

Activities of mutants obtained by amino acid scanning at conserved residues in the CP1 hairpin of hcLeuRS. Relative activity of amino acid activation (gray bar), aminoacylation (black bar), and deacylation (white bar) of various mutants obtained by amino acid scanning at conserved residues in the CP1 hairpin of hcLeuRS. The activity of hcLeuRS was assigned as 100%. The activities of mutants were compared with hcLeuRS, and the ratios were calculated.
Functional analysis of pivotal residues by site-directed mutagenesis
To determine the mechanism of the critical residues in the CP1 hairpin, further site-directed mutagenesis was performed. Two representative residues within the CP1 hairpin of hcLeuRS were selected, and multiple mutagenesis experiments were carried out. G245 is a flexible residue and is conserved in LeuRSs from both archaea, eukaryotes, and prokaryotes (Fig. 1A). The amino acid activation and aminoacylation activities of hcLeuRS-G245A were unchanged compared to hcLeuRS. The other two variants, hcLeuRS-G245D and hcLeuRS-G245R, in which G245 was replaced by the longer side-chain residues with negative and positive charges, decreased the activities to 50% of those of hcLeuRS. However, the replacement of G by the rigid residue proline completely eliminated both activities (Fig. 7A; Table 1). The post-transfer editing activity of the four mutants, hcLeuRS-G245A, -G245D, -G245R, and -G245P, did not change (Fig. 7B). The amino acid activation and aminoacylation activities of the mutants at position 245 of hcLeuRS seem to be independent of the charge of the amino acid but rely on flexibility of the amino acid at this position to maintain the tertiary structure of the CP1 hairpin and to allow a suitable swing of the CP1 hairpin.
FIGURE 7.

Activities of mutants at G245 and D250. (A,C) The ATP-PPi exchange reaction of mutants at G245 (A) and D250 (C). In each figure, from the top to the bottom, symbols were hcLeuRS (•), or -G245A (▪), -G245D (▴), -G245R (▾), -G245P (⧫) in (A); in (C), hcLeuRS-D250E (▴), -D250A (▪), -D250N (⧫), and -D250R (▾) were used. (B,D) The post-transfer editing assays of mutants at G245 (B) and D250 (D) were performed using 5 nM of the enzymes described above. A reaction without tRNA (○) was performed as a spontaneous hydrolysis control.
The highly conserved negative residue D250 within the CP1 hairpin was selected to further determine the effect of amino acid polarity on activity of hcLeuRS. HcLeuRS-D250E, in which D250 was replaced by a longer side-chain residue, E, with the same charge, had the same amino acid activation and aminoacylation activities as hcLeuRS. Both activities of hcLeuRS-D250A (D replaced by a small residue, A) and those of hcLeuRS-D250N (D replaced by a neutral polar residue, N) declined to ∼50% of those of hcLeuRS. In contrast, the activities of hcLeuRS-D250R (D replaced by a positive charge residue, R) were completely abolished; possibly because R has the opposite charge of D (Fig. 7C; Table 1). However, hcLeuRS-D250R could hydrolyze the mischarged tRNALeu, just like the wild-type hcLeuRS (Fig. 7D). These results further indicated that the conserved charged amino acid residues within the CP1 hairpin are essential for aminoacylation but do not affect the post-transfer editing activity of hcLeuRS.
The CP1 hairpin may affect the pretransfer editing pathway
We measured the editing activity of hcLeuRS using the AMP formation assay in the presence of tRNALeu and Nva. AN2690 is a benzoxaborole compound and could block the post-transfer editing of hcLeuRS by forming a covalent adduct with the tRNALeu, while pretransfer editing is kept intact. In the AMP formation assay with tRNALeu and Nva, the rate of AMP formation represents the total editing activity of hcLeuRS; however, when in the presence of AN2690, this represents the pretransfer editing activity of hcLeuRS (Chen et al. 2011). We compared the pretransfer editing pathway of hcLeuRS and its mutants by calculating the ratio of pretransfer editing to total editing activities.
Most of the mutations in the CP1 hairpin decreased the activity of amino acid activation. We selected hcLeuRS-D250A to analyze the pretransfer editing because the amino acid activation of this variant decreases mildly. The kobs of hcLeuRS-WT with and without 100 μM AN2690 was 0.51 S−1 and 1.47 S−1, respectively, indicating that the pretransfer editing pathway of hcLeuRS was ∼35% of total editing. However, the pretransfer editing of hcLeuRS-D250A accounted for ∼24% (0.26 S−1 with AN2690 vs. 1.10 S−1 without AN2690) of total editing (Fig. 8; Table 4). Compared to the wild-type hcLeuRS, hcLeuRS-D250A had almost unchanged post-transfer editing activity; however, pretransfer editing decreased obviously, indicating that the pretransfer editing would be affected when the CP1 hairpin was mutated.
FIGURE 8.

Separation of pretransfer and post-transfer pathways of hcLeuRS-WT and -D250A. (A) TLC analysis of [32P]-AMP formation was catalyzed by 1 μM hcLeuRS WT and hcLeuRS-D250A with or without AN2690. (B) Histogram summarizing the relative contributions of each editing pathway in AMP formation. Percentages were calculated from kobs values of AMP formation reported in Table 4.
DISCUSSION
The CP1 hairpin coordinated with the main body of LeuRS during evolution
The CP1 hairpin of LeuRS determines the insertion site and the rotational state of the CP1 domain. The insertion site and the rotational state of the CP1 domain of LeuRSs from eukaryotes and archaea are different from those of other species because of the unique structure of the CP1 hairpin (Supplemental Fig. S1). According to the phylogenetic tree constructed with full-length LeuRS, IleRS, and ValRS from various species, LeuRSs from archaea, eukaryotes, and prokaryotes are similar to each other and separated from ancient ValRS and IleRS at a very early evolutionary stage (Supplemental Fig. S4A). However, if the phylogenetic tree was solely constructed using the sequences of the CP1 hairpin region of these LIVRS, which belong to class Ia AaRS, surprisingly, the CP1 hairpin of prokaryotic LeuRS are similar to those of IleRS and ValRS but not to that of LeuRSs from archaea and eukaryotes (Supplemental Fig. S4B). This result indicated that the CP1 hairpins of ValRS, IleRS, and prokaryotic LeuRS are conserved and retained the ancient form during evolution. However, in archaea and eukaryotes, the CP1 hairpin varies significantly to adapt to the alteration of the CP1 domain, separating them from other LIVRS.
The CP1 hairpin plays important roles in the amino acid activation and aminoacylation activities of hcLeuRS
The CP1 hairpin of hcLeuRS is separated into two fragments because of the insertion of the CP1 domain. Deletion of either of these two fragments, or substitution of either of these two fragments with those from other species, almost completely eliminates the amino acid activation and aminoacylation activities (Figs. 2–4). Our results showed that these two fragments are both crucial for the function of hcLeuRS. They act together to achieve the catalytic function of amino acid activation and aminoacylation of hcLeuRS.
The CP1 hairpin domain is located in a crucial position of hcLeuRS (Fig. 1; Fukunaga and Yokoyama 2005b). It links the synthetic-active domain and the editing-active CP1 domain, serving as a joint. The conformation of LeuRS is dynamic, and the CP1 domain must swing by large degrees during catalysis (Fukunaga and Yokoyama 2005a,b). The joint may be critical for the conformational change of the CP1 domain during catalysis (Mascarenhas and Martinis 2008). The conformation change causes hcLeuRS to adopt an appropriate structure with optimal function.
The interaction between the CP1 hairpin and the catalysis core may be involved in aminoacylation
The CP1 hairpin is close to the synthetic-active domain. According to the structure of PhLeuRS, the side chains of the PhLeuRS residue W465 (corresponding to W530 in the CP1 hairpin of hcLeuRS) are directed toward conserved residue D521 (D586 in the Rossmann fold of hcLeuRS), which are within 2.70 Å distance based on the tertiary structure of PhLeuRS (Supplemental Fig. S5A). Similarly, the distance between Y469 (Y534 in the CP1 hairpin of hcLeuRS) and N624 (N688 in the Rossmann fold of hcLeuRS) are 2.69 Å apart (Supplemental Fig. S5B). Deletion the CP1 hairpin or mutations of the crucial residues in the CP1 hairpin may attenuate the interaction and disturb the structure of the Rossmann fold, thus affecting the function of hcLeuRS. Additionally, PhLeuRS-H181 corresponds to hcLeuRS-R236 and is close to C74 of tRNALeu, at a distance of 2.72 Å (Supplemental Fig. S5C). Thus, R236 may influence the binding and swing of the 3′ end of hctRNALeu. Further mutagenesis studies will be performed to determine if the CP1 hairpin interacts with the active site of the hcLeuRS in the Rossmann fold and if the region interacts with the tRNALeu.
The CP1 hairpin domain is not involved in the post-transfer editing activity of hcLeuRS
According to the structure of PhLeuRS, the CP1 hairpin may be essential for post-transfer editing (Fukunaga and Yokoyama 2005a). The authors reported a special insertion in CP1 that interacts with the CP1 hairpin. Mutations of the residues located in the insertion region within the editing CP1 domain probably disrupt the interaction, interfere with domain orientation, and then decrease the post-transfer editing activity (Supplemental Fig. S1D; Fukunaga and Yokoyama 2005b).
However, the present results showed that, in hcLeuRS, most variants in the CP1 hairpin barely affected the editing activity of hcLeuRS. Deletion of the entire CP1 hairpin of hcLeuRS reduced the editing activity by no more than 50%. We compared the sequence and structure of CP1 of hcLeuRS and PhLeuRS (Seiradake et al. 2009). The primary sequences of these two special insertions in CP1 are not conserved (Supplemental Fig. S6). In addition, although most of the structures of the CP1 domain between hcLeuRS and PhLeuRS are similar, the sites of the “special insertion” are different between hcLeuRS and PhLeuRS (Supplemental Fig. S6). We observed that, in hcLeuRS, the special insertion was not as close to the CP1 hairpin as it is in PhLeuRS; thus, in hcLeuRS, mutations of the CP1 hairpin may not affect the structure of the editing active site in CP1.
However, the crystal structure of the complex of full-length hcLeuRS and tRNALeu remains unresolved. If the structure is solved, the function and mechanism of the CP1 hairpin of hcLeuRS would be understood more clearly.
MATERIALS AND METHODS
Materials
Leucine, ATP, and DTT were purchased from Sigma. Restriction endonucleases and T4 DNA ligase were obtained from Thermo Fisher. The KOD plus DNA polymerase and dNTPs were purchased from TOYOBO. [3H] L-leucine, [3H] L-methionine, tetrasodium [γ-32P] PPi, and 5′-[α-32P] triphosphate were obtained from PerkinElmer Life Sciences. GF/C filters and 3# filters were from GE Healthcare-Whatman. PEI Cellulose F plates for thin-layer chromatography (TLC) were purchased from Merck. Ni2+-NTA superflow columns were purchased from Qiagen.
Gene cloning and site-directed mutagenesis
The plasmids pET16-hcleus containing the gene encoding hcLeuRS was constructed in our laboratory (Chen et al. 2011). The single-site mutants of hcLeuRS were constructed by PCR according to the protocol provided by the KOD-Plus-Mutagenesis Kit (Zhou et al. 2008), using mutation-containing primers and pET16-hcleus as a template.
Preparation of proteins and tRNALeus
hcLeuRS and its mutants were purified as described previously (Chen et al. 2011). The Escherichia coli strain BL21(DE3) was transformed with the plasmids PET16-hcleuS or its mutants. Growth of transformants and the purification of hcLeuRS and its mutants by Ni-NTA Superflow column were performed as previously described. These enzymes were analyzed by SDS-PAGE and appeared as a single band. The preparations were stored in 10 mM potassium phosphate buffer (pH 7.5) containing 50% glycerol.
hctRNALeu was isolated from an E. coli overproduction strain containing the plasmid pTrc99b-hctRNALeu constructed in our laboratory. hctRNALeu was purified by DEAE-Sepharose CL-6B column chromatography and C4 reversed-phase HPLC. The plateau value of the purified hctRNALeu reached 1600 pmole/A260.
ATP-PPi exchange assay
The leucine activation reaction was determined at 37°C in a reaction mixture containing 100 mM HEPES-KOH (pH 7.6), 2 mM KF, 6 mM MgCl2, 2 mM [32P]-pyrophosphate (PPi), 2 mM ATP, and 20 µM leucine, and initiated upon addition of 30 nM LeuRS.
Aminoacylation and deacylation assay
The aminoacylation assays for hcLeuRS or its mutants were determined at 37°C in a reaction mixture containing 1 mM spermine, 50 mM HEPES-KOH (pH 7.6), 25 mM KCl, 6 mM MgCl2, 20 µM [3H]-Leu, 4 mM ATP, and 10 µM tRNALeu, and initiated by adding 15 nM LeuRS.
The kinetic constants of hcLeuRS and its mutants in the aminoacylation reaction were determined in the mixture mentioned above, in the presence of various concentrations of the hctRNALeu (from 0.01–8 μM).
[3H]-Met-tRNALeu was obtained by mischarging hctRNALeu with [3H]-Met catalyzed by hcLeuRS-D399A, which is a mutant defective in post-transfer editing (Chen et al. 2011). The editing activities of hcLeuRS or its mutants toward mischarged [3H]-Met-tRNALeu were measured at 37°C in reactions identical to the aminoacylation condition, except that the leucine and tRNALeu were replaced by 1 µM [3H]-Met-tRNALeu. The reactions were initiated upon addition of 5 nM hcLeuRS or its mutants.
AMP formation assays
The editing reaction consumes ATP; therefore, it is typically measured using the formation of AMP (breakdown of ATP) in the presence of noncognate amino acids (Chen et al. 2011). AMP formation was measured in reaction mixtures containing 1 mM spermine, 50 mM HEPES-KOH (pH 7.6), 25 mM KCl, 6 mM MgCl2, 5 mM DTT, 3 mM ATP, 20 nM [α-32P]-ATP and 10 mM Nva, 5 units/mL pyrophosphatase, in the presence or absence of 5 mM tRNA. The reactions were incubated at 37°C and initiated by adding 1 mM hcLeuRS or the corresponding mutants. Aliquots (1.5 μL) were quenched in 6 μL of 200 mM sodium acetate (pH 5.0). Quenched aliquots (1.5 μL) were dripped onto a TLC plate. Nva-[32P]-AMP, [32P]-AMP, and [32P]-ATP were separated in 0.1 M ammonium acetate and 5% acetic acid. The plates were visualized by phosphor imaging, and the data was further analyzed.
Determination of kd by tryptophan fluorescence quenching
Fluorescence titrations were performed at room temperature with 0.1 μM of hcLeuRS or its mutants in 60 mM Tris-HCl (pH 8.2), 10 mM MgCl2, and 2 mM DTT. Tryptophan fluorescence was excited at 280 nm. An emission wavelength of 340 nm was used to quantify binding after correction for dilution and for the inner filter effect. Bovine serum albumin was used as a negative control to reflect that there was no fluorescence response to tRNA. The kd values were calculated by “one special binding equation” according to fitting fluorescence intensity change data vs. tRNA concentration, using Graphpad Prism software (Hu et al. 2013).
Complementation assays of LeuRS knockout yeast strains
The yeast LeuRS knockout strain ScΔleus was constructed and stored in our laboratory (Yao et al. 2008). hcLeuRS cross-recognized the yeast cytoplasmic tRNALeu and rescued ScΔleuS lethality in vivo.
The genes encoding hcLeuRS or its mutants were PCR-amplified from pET16-hcleus, digested by NdeI and SalI, and inserted into the gap between the NdeI and SalI sites of pTEF414 (yeast expression plasmid). Plasmids (pTEF414) carrying mutated genes of hcLeuRS were transformed into the strain ScΔleus separately.
Transformants were grown at 30°C on plates of synthetic-defined (SD) medium minus tryptophan (SD-Trp−) for 2∼3 d. Transformants were then replicated onto SD-Trp− plates containing 5-FOA (5-Fluoroorotic acid) and uracil to allow shuffling of the maintenance plasmid (containing the URA3 gene, whose product converts 5-FOA to toxic 5-fluorouridine monophosphate [5-FUMP]). The growth of the yeast cells was then observed, and the growth rates were measured at A600 (Yao et al. 2008).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Gilbert Eriani of Université de Strasbourg for his suggestions and Dr. Mo-Fang Liu and Dr. Lan-Tao Gou of the Shanghai Institute of Biochemistry and Cell Biology for their kind technical support. We also thank Prof. Tom Kellie of the University of the Chinese Academy of Sciences for editorial assistance. This work was supported by the Natural Science Foundation of China (Nos. 30930022 and 31130064), and the National Key Basic Research Foundation of China (No. 2012CB911000), and the China Postdoctoral Science Foundation (Nos. 2013M541562 and 2014T70438).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044404.114.
REFERENCES
- Ahel I, Korencic D, Ibba M, Söll D 2003. Trans-editing of mischarged tRNAs. Proc Natl Acad Sci 100: 15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander RW, Schimmel P 2001. Domain-domain communication in aminoacyl-tRNA synthetases. Prog Nucleic Acid Res Mol Biol 69: 317–349 [DOI] [PubMed] [Google Scholar]
- Chen JF, Guo NN, Li T, Wang ED, Wang YL 2000. CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry 39: 6726–6731 [DOI] [PubMed] [Google Scholar]
- Chen X, Ma JJ, Tan M, Yao P, Hu QH, Eriani G, Wang ED 2011. Modular pathways for editing non-cognate amino acids by human cytoplasmic leucyl-tRNA synthetase. Nucleic Acids Res 39: 235–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack S, Yaremchuk A, Tukalo M 2000. The 2 Å crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J 19: 2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriani G, Delarue M, Poch O, Gangloff J, Moras D 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347: 203–206 [DOI] [PubMed] [Google Scholar]
- Fukunaga R, Yokoyama S 2005a. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition. Nat Struct Mol Biol 12: 915–922 [DOI] [PubMed] [Google Scholar]
- Fukunaga R, Yokoyama S 2005b. Crystal structure of leucyl-tRNA synthetase from the archaeon Pyrococcus horikoshii reveals a novel editing domain orientation. J Mol Biol 346: 57–71 [DOI] [PubMed] [Google Scholar]
- Hale SP, Auld DS, Schmidt E, Schimmel P 1997. Discrete determinants in transfer RNA for editing and aminoacylation. Science 276: 1250–1252 [DOI] [PubMed] [Google Scholar]
- Hu QH, Huang Q, Wang ED 2013. Crucial role of the C-terminal domain of Mycobacterium tuberculosis leucyl-tRNA synthetase in aminoacylation and editing. Nucleic Acids Res 41: 1859–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Söll D 1999. Quality control mechanisms during translation. Science 286: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Ibba M, Söll D 2000. Aminoacyl-tRNA synthesis. Annu Rev Biochem 69: 617–650 [DOI] [PubMed] [Google Scholar]
- Li L, Boniecki MT, Jaffe JD, Imai BS, Yau PM, Luthey-Schulten ZA, Martinis SA 2011. Naturally occurring aminoacyl-tRNA synthetases editing-domain mutations that cause mistranslation in Mycoplasma parasites. Proc Natl Acad Sci 108: 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Söll D 2010. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc Natl Acad Sci 107: 4028–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Reynolds N, Ibba M 2009. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol 63: 61–78 [DOI] [PubMed] [Google Scholar]
- Loftfield RB, Vanderjagt D 1972. The frequency of errors in protein biosynthesis. Biochem J 128: 1353–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinis SA, Boniecki MT 2010. The balance between pre- and post-transfer editing in tRNA synthetases. FEBS Lett 584: 455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinis SA, Fox GE 1997. Non-standard amino acid recognition by Escherichia coli leucyl-tRNA synthetase. Nucleic Acids Symp Ser 36: 125–128 [PubMed] [Google Scholar]
- Mascarenhas AP, Martinis SA 2008. Functional segregation of a predicted “hinge” site within the β-strand linkers of Escherichia coli leucyl-tRNA synthetase. Biochemistry 47: 4808–4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia A, Crépin T, Vu MT, Lincecum TL Jr, Martinis SA, Cusack S 2012. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat Struct Mol Biol 19: 677–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H, Ibba M 2006. Molecular biology: sticky end in protein synthesis. Nature 443: 41–42 [DOI] [PubMed] [Google Scholar]
- Schimmel P, Ribas De Pouplana L 2000. Footprints of aminoacyl-tRNA synthetases are everywhere. Trends Biochem Sci 25: 207–209 [DOI] [PubMed] [Google Scholar]
- Seiradake E, Mao W, Hernandez V, Baker SJ, Plattner JJ, Alley MR, Cusack S 2009. Crystal structures of the human and fungal cytosolic Leucyl-tRNA synthetase editing domains: a structural basis for the rational design of antifungal benzoxaboroles. J Mol Biol 390: 196–207 [DOI] [PubMed] [Google Scholar]
- Silvian LF, Wang J, Steitz TA 1999. Insights into editing from an Ile-tRNA synthetase structure with tRNAIle and mupirocin. Science 285: 1074–1077 [PubMed] [Google Scholar]
- Tan M, Zhu B, Zhou XL, He R, Chen X, Eriani G, Wang ED 2010. tRNA-dependent pre-transfer editing by prokaryotic leucyl-tRNA synthetase. J Biol Chem 285: 3235–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Olsen GJ, Ibba M, Söll D 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev 64: 202–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadavalli SS, Ibba M 2012. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv Protein Chem Struct Biol 86: 1–43 [DOI] [PubMed] [Google Scholar]
- Yao P, Zhou XL, He R, Xue MQ, Zheng YG, Wang YF, Wang ED 2008. Unique residues crucial for optimal editing in yeast cytoplasmic Leucyl-tRNA synthetase are revealed by using a novel knockout yeast strain. J Biol Chem 283: 22591–22600 [DOI] [PubMed] [Google Scholar]
- Zhang CM, Hou YM 2005. Domain–domain communication for tRNA aminoacylation: the importance of covalent connectivity. Biochemistry 44: 7240–7249 [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang E 2013. Transfer RNA: a dancer between charging and mis-charging for protein biosynthesis. Sci China Life Sci 56: 921–932 [DOI] [PubMed] [Google Scholar]
- Zhou XL, Zhu B, Wang ED 2008. The CP2 domain of leucyl-tRNA synthetase is crucial for amino acid activation and post-transfer editing. J Biol Chem 283: 36608–36616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Zhao MW, Eriani G, Wang ED 2007. A present-day aminoacyl-tRNA synthetase with ancestral editing properties. RNA 13: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.