

Abstract
Neuron-glial related cell adhesion molecule (NrCAM) is a regulator of axon growth and repellent guidance, and has been implicated in autism spectrum disorders. Here a novel postsynaptic role for NrCAM in Semaphorin3F (Sema3F)-induced dendritic spine remodeling was identified in pyramidal neurons of the primary visual cortex (V1). NrCAM localized to dendritic spines of star pyramidal cells in postnatal V1, where it was coexpressed with Sema3F. NrCAM deletion in mice resulted in elevated spine densities on apical dendrites of star pyramidal cells at both postnatal and adult stages, and electron microscopy revealed increased numbers of asymmetric synapses in layer 4 of V1. Whole-cell recordings in cortical slices from NrCAM-null mice revealed increased frequency of mEPSCs in star pyramidal neurons. Recombinant Sema3F-Fc protein induced spine retraction on apical dendrites of wild-type, but not NrCAM-null cortical neurons in culture, while re-expression of NrCAM rescued the spine retraction response. NrCAM formed a complex in brain with Sema3F receptor subunits Neuropilin-2 (Npn-2) and PlexinA3 (PlexA3) through an Npn-2-binding sequence (TARNER) in the extracellular Ig1 domain. A trans heterozygous genetic interaction test demonstrated that Sema3F and NrCAM pathways interacted in vivo to regulate spine density in star pyramidal neurons. These findings reveal NrCAM as a novel postnatal regulator of dendritic spine density in cortical pyramidal neurons, and an integral component of the Sema3F receptor complex. The results implicate NrCAM as a contributor to excitatory/inhibitory balance in neocortical circuits.
Keywords: cell adhesion, cortical pyramidal neurons, NrCAM, semaphorin, spine morphogenesis, visual cortex
Introduction
Efficient transmission of information in the brain requires an intricate balance of excitation and inhibition (E/I balance), which is established during development and later refined. Most excitatory synapses in the cerebral cortex are located on dendritic spines and are distributed in such a way that allows correct functioning of cortical networks (Yuste, 2011). During development of the neocortex, spines are initially overproduced then undergo substantial elimination (pruning) in adolescence to form relatively stable adult circuits (Huttenlocher, 1979; Petanjek et al., 2011). Abnormalities in spine pruning may perturb cortical E/I balance in neuropsychiatric diseases such as autism spectrum disorder (ASD; Rubenstein and Merzenich, 2003; Hayashi-Takagi et al., 2011). Cortical pyramidal cells are implicated in ASD and exhibit increased spine densities on apical dendrites, which could arise from defective spine pruning (Parikshak et al., 2013; Willsey et al., 2013), leading to hyperconnectivity and hyperexcitability within affected circuits (Courchesne et al., 2007). However, the mechanisms regulating spine remodeling are incompletely understood.
Neuron-glial related cell adhesion molecule (NrCAM) is an Ig superfamily adhesion molecule (Maness and Schachner, 2007) with an established role in axon growth and repulsion (Falk et al., 2005; Williams et al., 2006; Demyanenko et al., 2011; Sakurai, 2012). NrCAM is one of four L1 family members that reversibly engage the actin cytoskeleton through ankyrin B and ezrin-radixin-moesin adaptors (Maness and Schachner, 2007). In addition NrCAM has a PDZ binding motif with the potential to bind PSD scaffold proteins PSD-95 and SAP102 (Davey et al., 2005; Dirks et al., 2006). Genetic alterations involving the adhesion molecule NrCAM have been associated with vulnerability to ASD (Bonora et al., 2005; Marui et al., 2009; Pinto et al., 2010; Voineagu et al., 2011), intellectual disability (Docherty et al., 2010), and addictive behavior (Sakurai et al., 2006; Ishiguro et al., 2014). NrCAM-null mice display ASD-relevant behaviors including impaired sociability, cognitive inflexibility, and hypersensitivity to sensory stimuli (Moy et al., 2009), and altered visual cortical responses (Demyanenko et al., 2011). In the thalamocortical system, NrCAM deficiency in mice causes misprojection of thalamic axon contingents to the primary visual cortex (V1), as a consequence of impaired repulsion from a gradient of Semaphorin3F (Sema3F) in the embryonic ventral telencephalon (Demyanenko et al., 2011). Interestingly, Sema3F has a distinct postnatal role in constraining dendritic spine density on cortical pyramidal neurons (Tran et al., 2007). During postnatal development NrCAM is strongly downregulated in thalamic neurons but is prominently expressed in the cortex (Demyanenko et al., 2011), raising the possibility that NrCAM might have a dual function in repellent axon guidance and postnatal regulation of dendritic spine remodeling.
Here we report that NrCAM regulates spine density and excitatory synapses, as shown in star pyramidal neurons in V1, by promoting spine remodeling as an essential component of the Sema3F receptor complex. These findings demonstrate for the first time that an Ig-class adhesion molecule couples with repellent signaling receptors to regulate spine and synaptic development in pyramidal neurons, thus potentially impacting E/I balance in cortical circuits.
Materials and Methods
Mice.
NrCAM-null mice (Sakurai et al., 2001; from Dr. Martin Grumet; Rutgers University) were originally on a mixed background of 129S6/EvSvTac and Swiss Webster (CFW) but were subsequently bred for over five generations to C57BL/6 mice. Sema3F-null mutant mice (Sahay et al., 2003) from Dr. Alex Kolodkin (Howard Hughes Medical Institute, HHMI; Johns Hopkins University) were on a C57BL/6 background. E0.5 was defined as the plug date and the day of birth as P0. For the trans-double heterozygous genetic analysis NrCAM+/−;Sema3F+/− mice were produced by intercrossing NrCAM+/− mice with Sema3F+/− mice. All mice were handled according to the University of North Carolina (UNC) Institutional Animal Care and Use Committee policies and in accordance with NIH guidelines.
Immunoreagents.
Monoclonal antibodies used were as follows: Tau, VGluT2 (EMD Millipore), PSD-95 (ThermoFisher Scientific), VGAT (Synaptic Systems), VGluT1 (NeuroMab, University of California Davis/NIH NeuroMab facility, Davis, CA), MAP2, and NCAM clone OB11 (Sigma-Aldrich). Polyclonal antibodies were as follows: GFP, NrCAM, and PlexA3 (Abcam); Npn-2 (R&D Systems); and NR1 (Santa Cruz Biotechnology). Normal goat, rabbit, and mouse IgG and fluorescently labeled (Alexa Fluor 488, TRITC, and Cy5) secondary antibodies were from Jackson ImmunoResearch. Fc fragment of human IgG was from Abcam and Sema3F-Fc fusion protein was from R&D Systems.
In situ hybridization.
DIG-labeled riboprobes for NrCAM were generated by in vitro transcription from a pBlueScript (pBS) plasmid containing mouse NrCAM cDNA (GenBank accession number AJ543321; nucleotides 460–1470), including nine bases from the 5′ untranslated region as described previously (Demyanenko et al., 2011). Riboprobes for Sema3F (Giger et al., 1998; a gift from Dr. David Ginty, HHMI, Johns Hopkins University) were generated from the pBS KS vector. In situ hybridization (ISH) was performed in the histology core facility of the UNC Neuroscience Research Center as described previously (Colbert et al., 1995). Brains from WT P7 and P21 mice were immersion fixed in 4% PFA overnight and cryoprotected in sucrose before sectioning horizontally. Images were captured digitally on an Zeiss Axioplan 2 microscope.
Immunostaining and EGFP expression.
Mice were anesthetized with 1.25% Avertin and transcardially perfused with 0.9% heparin saline, followed by 4% PFA in PBS, and brains were removed at E16.5, P21, P27, or P60. Brains were cryoprotected in a 10, 20, and 30% sucrose series, and sectioned for immunostaining (14–24 μm). Sections were permeabilized with Triton X-100 and labeled overnight with the following antibodies: polyclonal rabbit anti-NrCAM (Abcam #ab24344; 1:100), monoclonal mouse anti PSD-95 (NeuroMab; 1:300), polyclonal guinea pig anti VGLUT2 (Millipore #AB2251; 1:4000), monoclonal mouse anti VGAT (Synaptic System catalog #131011; 1:500), monoclonal mouse anti VGluT1 (NeuroMab #75–066; 1:100), polyclonal guinea pig anti-VGluT1 (Millipore AB-5905; 1:1000), polyclonal goat anti Neuropilin-2 (R&D Systems, #AF567; 1:200), anti-mouse PlexA3 antibody (R&D Systems, catalog #AF4075; 1:100), polyclonal rabbit anti PlexA3 (Abcam #641564; 1:200), chicken anti MAP2 (Millipore Bioscience Research Reagents; 1:1000), polyclonal rabbit anti GFP IgG (Invitrogen; 1:500), and chicken anti-GFP IgG (Abcam #ab13970; 1:1000). Secondary antibodies were added for 1 h include Alexa Fluor488 (1:400), TRITC (1:150), and Cy5 (1:150).
In utero electroporation of pCAGG-IRES-mEGFP plasmid into the embryos of pregnant females (E14.5) was used to label neurons with EGFP to visual spines, as previously described (Demyanenko et al., 2010). Injected mice at P27 were perfused transcardially, brains sectioned, and processed for GFP immunostaining to enhance spine visualization.
Subcellular fractionation.
WT and NrCAM-null mice (P21; n = 3/genotype) were anesthetized, decapitated, and cortex and fractionated as described previously (Cho et al., 1992). Briefly, cortex was dissected from the brains and homogenized in HEPES-buffered sucrose (0.32 m sucrose, 4 mm HEPES, pH 7.4), containing the protease/phosphatase inhibitors: 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 0.11 TIU/ml aprotinin, 200 μm Na-orthovanadate, and 10 mm NaF, and centrifuged at 800–1000 × g at 4°C to remove the pelleted nuclear fraction yielding a supernatant (S1), which represented the clarified homogenate. S1 was centrifuged at 10,000 × g for 15 min to yield the crude synaptosomal pellet (P2) and supernatant (S2), which contained cytosol and light membranes. The P2 pellet was lysed by hypo-osmotic shock, and centrifuged at 25,000 × g for 20 min to yield the S3 supernatant (crude synaptic vesicle fraction) and P3 pellet (lysed synaptosomal membranes). The P3 pellet was resuspended in HEPES-buffered sucrose and centrifuged in a discontinuous gradient containing 0.8–1.0 to 1.2 m sucrose at ∼150,000 × g for 2 h. Crude synaptic plasma membranes fractionated between 1.0 and 1.2 m sucrose, and were extracted twice in 50 mm HEPES, pH7.4, 2 mm EDTA, and 0.5% Triton X-100 then centrifuged at 32,000 × g for 20 min to obtain the PSD-2T pellet. All solutions contained protease and phosphatase inhibitors. Protein concentrations were determined using the bicinchoninic acid assay.
Spine density and Sholl analysis of Golgi-impregnated neurons.
Mice (P21 and P60) were anesthetized with isoflurane and brains were removed and processed for Golgi staining using the FD Rapid GolgiStain Kit (FD NeuroTechnologies). Brains were vibratome sectioned (150 μm) and mounted on gelatin-coated microscope slides after staining. Golgi-labeled neurons were analyzed in coronal sections of mouse brain using a Zeiss Axioplan microscope with AxioVision LE digital image capture. Spines and dendritic branching (Sholl analysis) were measured on digital bright-field images captured using a 40× oil objective using Neurolucida software (MBF Bioscience). For apical dendrites, spines were scored along the primary segment 100–150 μm distal to the soma, as there were few spines proximal to the soma. For basal dendrites, spines were scored along 35–50 μm segments on 3–5 secondary branches. Mean spine number per dendritic length (density) was calculated from measurements on 2D digital images, and thus represents an underestimate. Distinct spine morphologies were manually scored as thin, stubby, or mushroom types on apical dendrite segments from WT and NrCAM-null littermates (P21), as defined previously (Peters and Kaiserman-Abramof, 1970). For Sholl analysis Golgi-labeled image stacks were captured using a 20× objective. Each neuron was reconstructed using Adobe Photoshop CS4 and Neurolucida software. The center of concentric circles was defined as the center of the cell body at a soma detector sensitivity of 1.5 μm, and then automatic tracing mode was used to seed and trace dendritic arbors. Images in DAT format were subjected to Sholl analysis using Neurolucida Explorer with a starting radius of 10 μm and radius increments of 10 μm ending at 300 μm.
For monocular deprivation (MD) mice (P24) were anesthetized using Avertin (0.025 ml/g, i.p.), and eyelids trimmed and sutured for 3 d as described previously (McCurry et al., 2010). At P27 brains were removed and fixed then processed for Golgi labeling as above. Dendritic spine density was analyzed on layer 2/3 pyramidal neurons and layer 4 star pyramidal cells in the binocular zone of V1 within hemispheres contralateral and ipsilateral to the sutured, deprived eye. Binocular V1 was located by stereotaxic coordinates (Franklin and Paxinos, 1997) and by defined morphological criteria as described previously (Antonini et al., 1999; Coleman et al., 2009).
EM.
WT and NrCAM-null littermates (P21) were deeply anesthetized and perfused transcardially with phosphate buffer (0.15 m sodium phosphate, pH 7.4) followed by 2% PFA and 2.5% glutaraldehyde in phosphate buffer. The brains were postfixed in the same fixative for 2 d, and 100-μm-thick coronal vibratome sections were cut using Leica VT1200S vibratome. The vibratome sections were postfixed in 1% osmium tetroxide with 1.25% potassium ferrocyanide in phosphate buffer for 20 min, dehydrated in series of ethanol, and flat embedded in epoxy resin. Semithin sections (1 μm) were cut, stained with toluidine blue, and used for orientation. Ultrathin (70 nm) sections of cortex were cut using a Leica Ultracut UCT microtome (Leica Microsystems) and mounted on 200 mesh copper grids (Electron Microscopy Sciences). Ultrathin sections were contrasted with uranyl acetate and lead citrate, and imaged on an LEO EM 910 transmission electron microscope (Carl Zeiss SMT) at the UNC Microscopy Facility (Dr. Robert Bagnell, Director, Department of Pathology, UNC School of Medicine). The same magnification and size of electron micrograph was used to compare synaptic profiles in WT and NrCAM mutant V1, layer 4. Excitatory synaptic profiles on spines were identified by the presence of asymmetric PSDs aligned with a presynaptic terminal membrane containing clustered synaptic vesicles, as described previously (Coleman et al., 2009). In each micrograph, numerous spines were observed in cross section, and those with asymmetric synaptic profiles were classified as containing one or multiple PSDs. In each micrograph, the numbers of synaptic profiles with PSDs (single or multiple PSDs), terminal profile areas, and PSD lengths were counted on all spines. The number of PSDs on spines within a unit cross-sectional area of cortex was calculated from the magnification and image size, and expressed as mean number of PSDs per 100 μm cortical area ± SEM. PSD length and spine cross-sectional areas were measured using ImageJ software. Means were compared between genotypes by a two-tailed t test assuming unequal variances for statistical significance (p < 0.05). Statistical analyses were performed in Prism (v5.0).
Electrophysiology
Whole-cell patch-clamp recordings.
Mice were anesthetized with ketamine/xylazine (80/8 mg/kg) and decapitated. Coronal slices (350 μm) of the visual cortex were prepared from P27–P32 WT and NrCAM-null mutant mice in ice-cold, oxygenated high-sucrose artificial CSF (ACSF; 240 mm sucrose, 26 mm NaHCO3, 2.5 mm KCl, 1 mm CaCl2, 4 mm MgSO4, 1.25 mm NaH2PO4, and 10 mm d-glucose, pH 7.4, gassed with 95%O2/5% CO2). The slices were then incubated in extracellular recording solution containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgSO4 20 d-glucose, 1.25 NaH2PO4, and 26 NaHCO3; pH 7.4, gassed with 95% O2/5% CO2 for 30 min at 34°C, followed by at least 30 min at room temperature. mEPSCs were recorded from star pyramidal cells in layer 4, in whole-cell voltage clamp with a recording pipette containing 68 mm CsMetSO3, 72 mm CsCl, 5 mm EGTA, 10 mm HEPES, 4 mm ATP, 0.3 mm GTP, 10 mm creatine phosphate, and 3 mm QX-314 chloride. Alexa Fluor 488 (0.1%, w/v; Invitrogen) was added to the internal solution to provide morphological identification of all recorded cells, and only results from star pyramidal neurons are reported. The extracellular solution was supplemented with 1 μm TTX to block action potentials and 10 μm SR95531 to block GABAergic transmission. No series resistance compensation was used. All chemicals were obtained from Sigma-Aldrich, except SR95531(Tocris Bioscience). Recordings were made with an AxoClamp 700B Amplifier (Molecular Devices), and acquired with a custom MATLAB program (R2011a; The MathWorks). All recordings were made at 34°C. mEPSC frequency, 10–90% rise time, and decay time constants were measured in Mini Analysis (Synaptosoft) from 60 to 180 s long traces exported from the MATLAB acquisition program. mEPSCs were recorded from nine neurons per genotype, using littermates. The rise time and decay time constants were measured from the averaged mEPSCs. Statistical analyses were performed in Prism (v5.0).
Site-directed mutagenesis and subcloning.
Mouse NrCAM splice variant 1 (GenBank accession number AJ543321) cDNA in pcDNA3 was provided by Dirk Montag (Leibniz Institute for Neurobiology, Magdeburg, Germany). Mutations in the Npn-2 binding site of the first Ig domain of NrCAM were generated using the QuikChange Site Directed Mutagenesis Kit (Agilent Technologies), according to manufacturer's instructions using the following mutagenic primers: TARΔNER: 5′-CCAGTGCACTGCAAGGGGAGCTGCCGTCTCC-3′; TARNEV: 5′-CCAGTGCACTGCAAGGAACGAGGTTGGAGCTGCCGTCTCC-3′; TARAAA: 5′-CCAGTGCACTGCAAGGGCCGCGGCTGGAGCTGCCGTCTCC-3′.
WT or mutant NrCAM sequences were introduced into pCAGG-IRES-mEGFP at the FseI and AscI restriction sites. FseI was introduced at the 5′ end and AscI at the 3′ end of NrCAM through PCR using Phusion DNA polymerase. The primers were as follows: FseFwd: 5′-CGATTGGCCGGCCCCTTACATGCCGAAGAAGAAGCACTTATCTGC-3′ and AscRev: 5′-GCTAAGGCGCGCCTTATTATTAAACAAAGGAGTTCATTGCG-3′.
PCR products and pCAGG-IRES-mEGFP were digested with FseI and AscI. The restriction-digested pCAGG-IRES-mEGFP vector was treated with calf intestinal alkaline phosphatase to remove terminal phosphate residues at the restriction sites. The FseI/AscI-digested NrCAM cDNA was ligated to pCAGG-IRES-mEGFP using the Quick Ligation kit from New England BioLabs. Ligations were transformed into SoloPack Gold Ultracompetent bacteria (Agilent Technologies), constructs screened, and plasmids were amplified, purified, and sequenced. All enzymes were from New England BioLabs.
HEK293T cell culture and transient transfection.
HEK293T cells were grown in DMEM-H supplemented with gentamicin/kanamycin (Life Technologies) and 5% FBS (Atlanta Biologicals) in a humidified incubator with 5% CO2. On the day before transfection, cells were seeded at 700,000 cells/60 mm dish. Plasmids containing WT or mutant NrCAM (8 μg) were transfected into cells with Lipofectamine 2000 (20 μl; Life Technologies). Media was changed on the day following transfection, and cells harvested 24 h later. Protein concentrations were determined using BCA.
Immunoprecipitation.
Homogenates of mouse forebrain (E15.5 and P21) and cell lysates were prepared in RIPA buffer (20 mm Tris, pH 7.0, 0.15 m NaCl, 5 mm EDTA, 1 mm EGTA, 1% NP-40, 1% deoxycholate, 0.1% SDS, 200 μm Na3VO4, and 10 mm NaF, 1× protease inhibitors; Sigma-Aldrich) as described previously (Demyanenko et al., 2011). Lysates (0.5–1 mg) were precleared for 30 min at 4°C using Protein A/G Sepharose. Precleared lysates were incubated with NrCAM, Npn-2, or nonimmune IgG (nIg; rabbit or goat) antibodies for 2 h at 4°C. Protein A/G Sepharose beads were added for an additional 30 min at 4°C before washing. Immunoprecipitated proteins were released from the beads by boiling in SDS-PAGE sample buffer.
Western blotting.
Lysates (75 μg), protein fractions, or immunoprecipitations were subjected to SDS-PAGE and transferred to nitrocellulose. Blots were blocked in TBS-Tween containing 5% milk and incubated overnight in the following antibodies: anti-NrCAM (1:500), anti-Npn-2 (1:500), anti-NR1 (1:500), anti-PlexA3 (1:500), anti-NCAM (1:1000), anti-PSD-95 (1:2000), and anti-actin (1:5000). Blots were washed and incubated in secondary antibodies (1:7500) for 1 h before being developed using enhanced chemiluminescence (ThermoFisher Scientific) and exposed to film. Films were scanned and analyzed for Npn-2 association using ImageJ software (NIH Image).
Cortical neuron cultures.
Cortical neurons were prepared from brains of WT or NrCAM-null embryos (E14.5–E16.5) and electroporated with pCAGG-IRES-mEGFP plasmid according to the method described previously (Tran et al., 2009). Media was changed on the cultures every 2 d until 18 DIV. At 18 DIV, neurons were treated with Sema3F-Fc or Fc control protein (3 nm) for 2 h before fixation and staining with anti-GFP antibodies (1:200) overnight. Alexa Fluor488 anti-chicken antibodies (1:400) were incubated for 1 h. Images were captured using a 60× oil-immersion lens and 2× optical zoom on an Olympus FV500 microscope at the UNC Microscopy Services Laboratory (Dr. Robert Bagnell, Director). For rescue experiments, NrCAM−/− neurons were transfected with pCAGG-NrCAM-IRES-mEGFP plasmid or vector alone. Spines of labeled neurons were measured in confocal images along 20–30 μm segments of apical dendrites and primary or secondary branches of basal dendrites using Neurolucida software. Spine densities were measured on dendrites of at least 10 neurons (30–148 total spines) in each of four replicate cultures per condition. Mean spine densities ± SEM were determined for each condition and compared by the two-tailed t test (p < 0.05). For colocalization, cortical neuron cultures (18 DIV) were fixed and labeled overnight with rabbit anti-NrCAM (1:100), anti-Npn-2 (1:50), and anti-MAP2 antibodies, processed for double immunofluorescence staining, and visualized by confocal imaging.
Results
Postsynaptic localization of NrCAM in visual cortex
To investigate a potential role for NrCAM in postnatal maturation of pyramidal neurons, we examined the cortical expression of NrCAM in V1 by ISH during early postnatal development (P7–P21) before the onset of the critical period. NrCAM transcripts were expressed during postnatal development (P7 and P21) in V1, where they were enriched in layers 4 and 6 (Fig. 1A). The distribution of NrCAM transcripts at these stages overlapped that of Sema3F, which were also enriched in the middle and deep layers of V1. NrCAM and Sema3F transcripts were also present in primary motor and somatosensory cortical areas and medial prefrontal cortex, where they were observed in a nongraded pattern along the rostrocaudal axis (data not shown). These findings were consistent with previous results showing that NrCAM is expressed in developing postnatal mouse forebrain (P5–P20) and localizes prominently in V1, layers 4 and 6 (Demyanenko et al., 2011).
Figure 1.
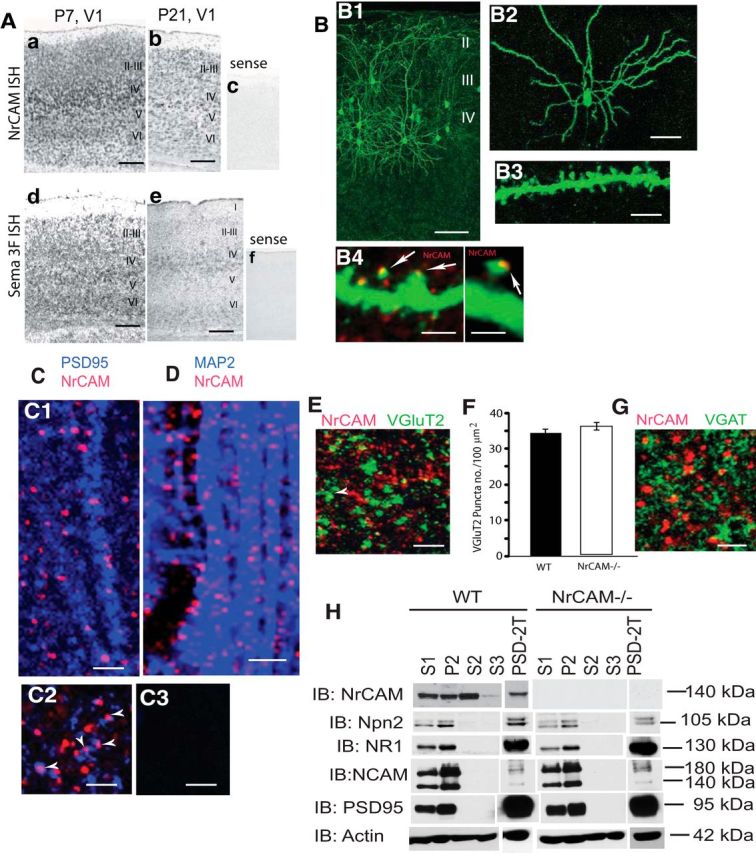
NrCAM expression during postnatal development in mouse visual cortex. A, a, b, ISH showed NrCAM mRNA expression overlaps with (d, e) Sema3F mRNA in developing V1 (P7 and P21). c, f, Sense control. B, B1, Pyramidal neurons in V1, layers 3 and 4 of WT mice (P21) were labeled by in utero electroporation of pCAGG-IRES-mEGFP plasmid at E14.5. B2, B3, Star pyramidal neurons labeled with EGFP in layer 4, V1 by in utero electroporation displayed strong labeling of processes and dendritic spines. B4, Immunostaining for NrCAM (red) showed discrete localization on spine heads of EGFP-labeled, star pyramidal neurons in layer (arrows). C, Punctate immunofluorescent staining of NrCAM on dendritic spines in V1, layer 4 of WT mice (P21). Immunostaining for NrCAM (red) colocalized with PSD-95 (blue) on vertically oriented apical dendrites in coronal (C1) and horizontal sections (C2). Control showing lack of staining of NrCAM-null mutant (P21) sections of V1, layer 4 with NrCAM antibodies (C3). D, Punctate immunofluorescent colocalization of NrCAM (red) with MAP2 (blue) at dendritic spines in V1 at P21 in coronal sections. E, NrCAM immunofluorescence (red) did not colocalize with a presynaptic marker of thalamocortical terminal VGluT2 (green) at P21 in V1, layer 4. F, Quantification of the density of VGluT2-positive puncta in confocal images corresponding to a unit area of 100 μm2 layer 4 of V1 showed no significant difference between WT and NrCAM-null mutant mice at P27. N = 3 mice of each genotype; WT, n = 81 unit area; NrCAM-null, n = 46 unit area (t test; p < 0.05). G, NrCAM immunofluorescence (red) did not colocalize with the presynaptic marker of GABAergic inhibitory neurons VGAT (green) at P21 in V1, layer 4. H, Fractionation of P21 mouse brain showed that NrCAM distributed within PSD fraction (PSD-2T) identified by enrichment in postsynaptic markers, NMDA receptor NR1 subunit, and PSD-95. Loss of NrCAM did not alter the distribution or expression of Npn-2, NR1, or PSD95. S1, clarified homogenate; P2, crude synaptosomes; S2, cytosol and light membranes; S3, synaptic vesicles; PSD-2T, 2 × Triton X-100 washed PSD fraction. Scale bars: A–E, 100 μm; B1, 100 μm; B2, 50 μm; B3, B4, 5 μm; C–F, 5 μm. IB, immunoblot.
To localize NrCAM at the subcellular level, cortical pyramidal neurons were labeled with EGFP by in utero electroporation of pCAGG-IRES-mEGFP plasmid at E14.5 and immunostained for NrCAM postnatally at P21 and P60 (Fig. 1B). Star pyramidal neurons are the major excitatory neuron in layer 4 of V1 and principal target of thalamocortical inputs (Maffei et al., 2006). EGFP-labeled star pyramidal neurons in layer 4 of V1 displayed a typically short apical dendrite lacking an apical tuft, laterally oriented basal dendrites, and numerous dendritic spines (Fig. 1B1–B3) as described previously (Jiang et al., 2010). At P21, when spine maturation is rapid (Li et al., 2010) and critical period plasticity begins (Espinosa and Stryker, 2012), NrCAM in layer 4 was discretely localized to EGFP-labeled spine heads along dendrites of star pyramidal cells (Fig. 1B4). The PDZ binding motif at the C terminus of NrCAM has been reported to bind PSD-95 (Davey et al., 2005; Dirks et al., 2006). NrCAM displayed a striking punctate distribution on dendritic processes identified by colocalization with PSD-95 and the somatodendritic marker MAP2 (Fig. 1C,D).
At embryonic stages NrCAM transcripts are expressed highly in specific domains of the dorsal thalamus but decline to very low levels by P5, while increasing postnatally in the cortical plate (Demyanenko et al., 2011). Consistent with a lack of NrCAM at thalamocortical terminals, NrCAM did not significantly colocalize with VGluT2, a selective marker of thalamocortical synapses in V1 (Nahmani and Erisir, 2005; Coleman et al., 2010) in brain sections within V1, layer 4 of WT mice (P21) (Fig. 1E). Deletion of NrCAM has been shown to cause mistargeting of subpopulations of thalamic axons to V1 (Demyanenko et al., 2011). In accord with the small number of mistargeting axons, the density of VGluT2-positive puncta in V1, layer 4 of WT and NrCAM-null mice (P27) was equivalent (Fig. 1F). NrCAM did not colocalize with the presynaptic GABAergic inhibitory neuronal marker VGAT (Fig. 1G), suggesting that NrCAM may be primarily associated with excitatory synapses. Subcellular fractionation of P21 mouse brain showed that NrCAM was present in the clarified homogenate (S1), the crude synaptosomal pellet (P2), light membranes, and cytosol (S2), with little in the synaptic vesicle fraction (S3). A portion of NrCAM distributed within Triton X-100-resistant PSD fraction (PSD-2T) was identified by an enrichment in postsynaptic markers NR1 and PSD-95 (Fig. 1H). The PSD fraction also contained Npn-2 but only a marginal level of a different NCAM. Fractionation of P21 mouse brain from NrCAM-null mice showed that NrCAM loss did not significantly alter the distribution or expression of Npn-2, NR1, or PSD-95 (Fig. 1H). The postsynaptic localization of NrCAM suggests that in addition to its embryonic role in axon guidance, NrCAM has a potential postnatal role in cortical pyramidal cells.
Increased dendritic spine density in NrCAM-null pyramidal neurons
To evaluate a role for NrCAM in dendritic spine development and synaptogenesis, Golgi labeling was performed on P21 and adult (P60) brains of WT and NrCAM-null mutant mice. Increased spine density was observed on apical dendrites of star pyramidal neurons in V1, layer 4 of NrCAM−/− mice at both P21 and adult (P60) ages (Fig. 2A–G). Spine density was assessed at 100–150 μm from the cell soma, as few spines were observed in soma or proximal dendrites. NrCAM loss specifically resulted in increased spine density on apical dendrites but not on basal dendrites (Fig. 2I). Increased dendrite spine density was also evident on pyramidal neurons of NrCAM mutants in other layers of V1. Analysis of Golgi-impregnated neurons revealed increased spine density in layers 2/3 and 5 of V1 in NrCAM-null mice (P21; Fig. 2J). Increased spine density on the dendritic tree of pyramidal cells in these layers was seen on both apical and basal dendrites of NrCAM mutants. Little is understood about the functional differences involving spine synapses on apical versus basal dendrites of pyramidal neurons in V1; however, in somatosensory cortex (S1) apical and basal dendrites have different connections as well as forms of plasticity (Gordon et al., 2006). Elevated spine density on apical but not basal dendrites was also reported for Sema3F−/− and Npn-2−/− pyramidal neurons in layer 5 of mouse S1 (Tran et al., 2009). These results point to a developmental mechanism in which NrCAM cooperates with Npn-2 and Sema3F in limiting spine number on pyramidal neurons in V1.
Figure 2.
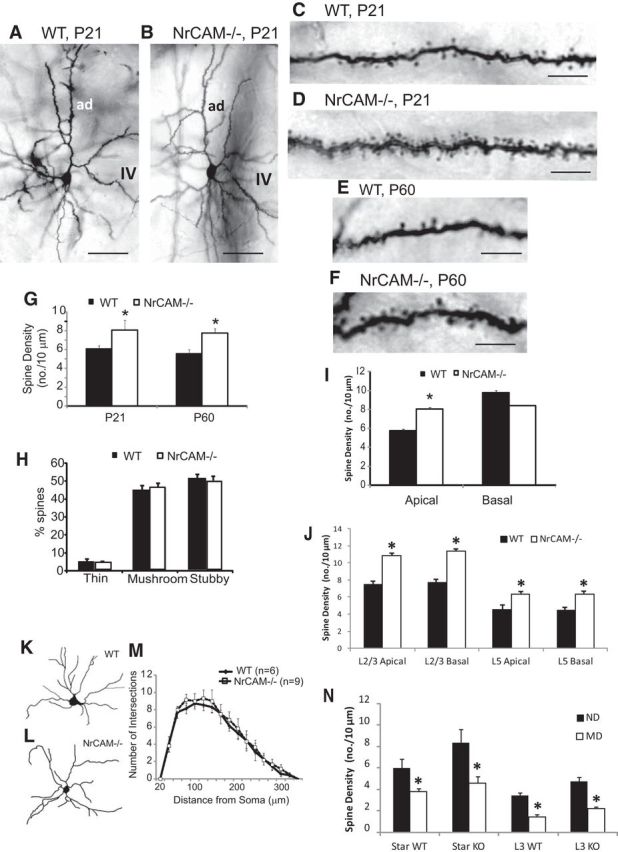
NrCAM regulates dendritic spine density on pyramidal neurons in mouse visual cortex. A–F, Representative Golgi-labeled, star pyramidal neurons in V1, layer 4 of WT and NrCAM-null mice at P21 (A–D) and P60 (E, F). ad, apical dendrite. G, Quantification of spine density on layer 4 apical dendrites of star pyramidal neurons in the binocular zone of V1 showed a significant increase in spine density on dendritic segments located 100–150 μm from the cell body in NrCAM-null compared with WT mice at both P21 and P60 (*p < 0.05). Number of mice: 3 per genotype. Number of neurons scored: WT, n = 51 (P21), n = 57 (P60); NrCAM-null, n = 57 (P21), n = 20 (P60). H, No differences were observed in the percentage of Golgi-labeled spines with thin, mushroom, or stubby morphology on apical dendrites of star pyramidal neurons in V1, layer 4 of three NrCAM-null (n = 26 neurons, 419 spines) and four WT mice (n = 21 neurons, 166 spines; P21; p < 0.05). I, Quantification of spine density showed a significant increase in apical but not basal dendrites of star pyramidal neurons in V1, layer 4 of NrCAM-null compared with WT mice at P21 (*p < 0.05). J, Quantification of spine density on pyramidal neurons in V1, layers 2–3 and 5 showed a significant increase on both apical and basal dendrites in NrCAM-null compared with WT mice (*p < 0.05). Three mice per genotype, n = 22–23 neurons scored for apical or basal dendrites in each genotype. K–M, Representative high-contrast images of Golgi-labeled, star pyramidal neurons in V1, layer 4 of WT (K) and NrCAM-null (L) mice (P21; 3 mice per genotype). Number of neurons scored: layer 2/3 apical WT, n = 160; NrCAM-null, n = 187; basal WT, n = 121; NrCAM-null, n = 127; layer 5 apical WT, n = 50; NrCAM-null, n = 50; basal WT, n = 26; NrCAM-null, n = 33. M, Sholl analysis indicated no difference in branching of dendritic arbors within a 20–300 μm radius from the star pyramidal cell body of WT (6 mice, n = 40 neurons) and NrCAM-null mice (9 mice, n = 38 neurons). N, Brief MD at P24 for 3 d decreased spine density at P27 on Golgi-labeled apical dendrites of layer 2/3 and star pyramidal neurons in binocular V1, layer 4 compared with ND neurons in both NrCAM KO and WT mice (3 mice per genotype; n = 30 neurons scored per condition; *p < 0.05). Scale bars: A, B, 100 μm; C, F, 5 μm.
Dendritic spines are morphologically diverse and are typically divided into three types based on the size of the spine head and neck: thin (filopodial), mushroom (large head, narrow neck), and stubby (uniform head and neck; Holtmaat and Svoboda, 2009). NrCAM deletion had no effect on the proportion of thin, mushroom, or stubby spines on apical dendrites of star pyramidal cells (P21; Fig. 2H). In addition, Sholl analysis of Golgi-labeled star pyramidal neurons in WT and NrCAM−/− V1 (P21) showed that loss of NrCAM did not significantly alter the extent of dendritic arborization (Fig. 2K–M).
The finding that spine density on apical dendrites of star pyramidal cells was increased in NrCAM mutant mice at both postnatal (P21) and adult (P60) stages indicated that the phenotype appeared by P21 and was stable in adulthood. Hence, NrCAM deficiency likely impairs a normal developmental process that regulates spine density before P21 rather than plasticity. The binocular response of visual cortical neurons is altered by MD during a critical period of postnatal development (Gordon and Stryker, 1996). Ocular dominance plasticity induced by brief (3–4 d) MD during the postnatal critical period in mice (P21–P28) is accompanied by decreased spine density on apical dendrites of pyramidal neurons in layers 2/3 of the contralateral binocular zone of V1 (Mataga et al., 2004), as well as by structural remodeling of thalamocortical synapses (Antonini et al., 1999; Coleman et al., 2009). To directly test for involvement of NrCAM in spine alteration following brief MD, we performed 3 d MD in WT and NrCAM-null littermates (P24–P27). We then analyzed spine density on Golgi-labeled star pyramidal cells and layer 2/3 pyramidal neurons in both the contralateral (MD) and ipsilateral (nondeprived, ND) binocular zone of V1. In WT mice MD decreased spine density on apical dendrites of star pyramidal cells, as well as on layer 2/3 pyramidal cells (Fig. 2N), similar to that previously reported (Mataga et al., 2004). Spine density decreased to a similar extent on both cell types in NrCAM-null mice, suggesting that this form of plasticity is unaffected by NrCAM loss. The observation that spine density in the NrCAM mutant normalized to WT levels after MD, despite being somewhat higher initially, raised the possibility that synapse stability might be decreased by NrCAM loss, resulting in enhanced pruning by an NrCAM-independent mechanism.
NrCAM deficiency increases excitatory spine synapse number and neurotransmission
To evaluate whether elevated spine density in the NrCAM-null cortex corresponded to increased numbers of excitatory spine synapses, ultrastructural analysis was performed in V1, layer 4 of NrCAM-null mice and WT littermates at P21. Excitatory synapses on spines were identified in electron micrographs as asymmetric synaptic profiles with a PSD juxtaposed to a presynaptic terminal containing synaptic vesicles (Fig. 3A,B). The numbers of synaptic profiles on spines with single or multiple PSDs were scored within a unit cross-sectional area corresponding to 100 μm2 of cortical area. The mean number of spine synapses with single PSDs per 100 μm2 was significantly increased (two-tailed t test, p < 0.05) in NrCAM mutants compared with WT (Fig. 3C). There was also a small increase in the number of spine synapses with multiple PSDs per unit area, possibly reflecting perforated synapses. Loss of NrCAM did not affect mean spine area or PSD length (Fig. 3D,E). This analysis suggests that elevated spine density on pyramidal neurons of NrCAM mutant mice is associated with increased numbers of excitatory spine synapses.
Figure 3.

NrCAM deficiency increases excitatory spine synapses and neurotransmission in layer 4 of visual cortex. A, B, Electron micrographs showed postsynaptic specializations on spines (*) opposed to presynaptic terminals containing synaptic vesicles, which are characteristic of excitatory synaptic profiles in layer 4 of NrCAM−/− and WT binocular V1 at P21. Scale bar, 1 μm. C, The density of excitatory synaptic profiles with single PSDs or multiple PSDs on spines within a unit area of V1, layer 4 was increased in NrCAM−/− mutants compared with WT mice (two-tailed t test; p ≤ 0.05); n = total number of spine PSDs analyzed in three WT and three NrCAM-null mice (P21). D, Spine cross-sectional area (μm2 ± SEM) calculated from electron micrographs was equivalent in WT and NrCAM-null mice (P21; n = 3 mice/genotype); n = number of spines scored per condition (two-tailed t test; p < 0.05). E, Mean PSD length of spine synaptic profiles (±SEM) was equivalent in WT and NrCAM-null mice (P21; 3 mice per genotype); n = number of PSDs scored per condition (two-tailed t test; p < 0.05).
The increased level of dendritic spines and excitatory synapses in V1, layer 4 of NrCAM-null mice may contribute to an increase in glutamatergic neurotransmission. To test this, whole-cell voltage-clamp recordings were made from star pyramidal cells in layer 4 of V1 in acute brain slice preparations from WT and NrCAM-null mutant littermates (P27–P32; Fig. 4A). The mEPSC frequency, amplitude, 10–90% rise time, and decay time constants were measured. In NrCAM mutants, variability of mEPSC frequency was much larger in layer 4 star pyramidal cells than in cells from WT mice (F(8,8) = 15.90, p = 0.0007; Fig. 4B). The mEPSC frequency was, on average, also significantly higher in mutant mice than WT mice (unpaired t test with Welch's correction for unequal variances, t(9) = 3.867, p = 0.0038; Fig. 4B). However, there was no significant difference in mEPSC amplitude (unpaired t test, t(16) = 1.547, p = 0.14; Fig. 4C), rise (t(16) = 0.873, p = 0.40; Fig. 4D), or decay time constants (t(16) = 0.127, p = 0.90; Fig. 4E) between WT and NrCAM mutants. Plots of the relationship between rise versus decay time constants were also not different between the two genotypes, indicating that the detected synapses were not differentially distributed along the dendritic tree between the two genotypes.
Figure 4.

NrCAM mice have higher rates of spontaneous mEPSCs. A, Example segments of traces of mEPSCs recorded in the presence of 1 μm TTX and 10 μm SR95531 from star pyramidal cells in slices from a WT (left) and a NrCAM-null (right) mouse. Scale bars indicate the same values for both traces. Traces have been low-pass filtered at 2 kHz for display. B, mEPSC event frequency was higher in cells from NrCAM-null mice than WT littermates; **p = 0.038, unpaired t test assuming unequal variances. C, There was no difference in mEPSC amplitude between NrCAM-null and WT littermates. D, mEPSC 10–90% rise time was similar between NrCAM-null and WT littermates. E, The mEPSC decay time constant did not depend on rise time in either genotype. Graphs B–E show geometric mean with 95% confidence intervals; each point is an individual cell. N = 9 cells for each genotype. WT (n = 9 mice), NrCAM-null (n = 9 mice).
The increase in mEPSC frequency in NrCAM-null star pyramidal cells is consistent with increased dendritic spine densities on apical dendrites of these neurons and with increased numbers of excitatory synapses in layer 4 of V1. Similarly, layer 5 pyramidal neurons in primary somatosensory cortex of Sema3F-null mice, which also have an increased number of morphological synapses, displayed increased mEPSC frequencies (Tran et al., 2009). Thus, NrCAM likely functions by limiting the number of dendritic spines and excitatory synapses in developing star pyramidal cells.
Interaction of NrCAM with the Sema3F receptor
Because mice deficient in Npn-2, Sema3F, and PlexA3 display increased spine density on apical but not basal dendrites of cortical pyramidal neurons in the primary somatosensory cortex (Tran et al., 2009), NrCAM may engage Npn-2 as an obligate component of the Sema3F receptor complex (Npn-2 and PlexA3) to promote Sema3F-induced dendritic spine retraction in postnatal cortical neurons, as depicted in Figure 5A. NrCAM has been shown to coimmunoprecipitate with Npn-2 from brain lysates from mouse embryos (Falk et al., 2005; Demyanenko et al., 2011); however, it is not known if NrCAM associates with Npn-2 in brain of postnatal mice or if binding is direct. We found that NrCAM coimmunoprecipitated with Npn-2 not only from lysates of embryonic brain (E15.5) but also from postnatal forebrain at P21 (Fig. 5B). NrCAM also associated with PlexA3 from brain lysates at both stages (Fig. 5B), but not with Npn-1, PlexA1, or PlexA4 (data not shown). A direct interaction was suggested by coimmunoprecipitation of NrCAM and Npn-2 from transfected HEK293T cells (Fig. 5B). The Ig domains of L1 family adhesion molecules mediate homophilic and heterophilic binding to multiple ligands and are targeted for mutation in human mental retardation syndromes (Maness and Schachner, 2007). The Ig1 domain of NrCAM contains a sequence (TARNER) that shares homology with Npn-1 binding sites in the Ig1 domains of L1 (FASNKL120; Castellani, 2002) and Close Homolog of L1 (FASNRL115; Wright et al., 2007). These sequences have been shown to be essential for Sema3A-induced growth cone collapse, and affect a surface reside in the L1 motif (L →V120), which is also a human pathological mutation (Bateman et al., 1996). Site-directed mutagenesis was used to mutate the NrCAM sequence by deleting the residues N121ER (TARΔNER), which is predicted to change its polar character. The TARΔNER deletion effectively disrupted NrCAM binding to Npn-2, as shown by coimmunoprecipitation from transfected COS-7 cells (Fig. 5C). The single-point mutation TARNEV or triple mutation TARAAA in NrCAM were less effective than TARΔNER in reducing binding to Npn-2, as shown by the relative levels of Npn-2 in NrCAM immunoprecipitates (Fig. 5C). All NrCAM mutants and WT NrCAM were expressed at equivalent levels at the cells surface, which was verified by immunofluorescence staining in COS-7 cells (data not shown). Double immunofluorescence staining showed colocalization of NrCAM and Npn-2 at numerous puncta in confocal images of mouse brain (P21) in sections of V1, layer 4, consistent with association studies in COS-7 cells (Fig. 5D). It was reported that Npn-2 colocalizes with PSD-95. In Figure 1C, NrCAM staining of puncta in V1, layer 4 (P21) overlapped with the postsynaptic marker PSD-95, while pyramidal cells labeled with EGFP by in utero electroporation showed punctate labeling at the tip of spines for both NrCAM and Npn2 (Fig. 5E).
Figure 5.

NrCAM associates with Npn-2 through the TARNER sequence in Ig1. A, Postulated mechanism of NrCAM/Npn-2/PlexA3 as a receptor complex for Sema3F in inducing dendritic spine retraction in cortical pyramidal neurons. The sequence TARNER within the Ig1 domain of NrCAM binds Npn-2, which together with PlexA3, comprises a receptor complex for Sema3F in mediating spine retraction. NrCAM binds ezrin-radixin-moesin (ERM), ankyrin, and PDZ domains through specific motifs in the cytoplasmic domain. B, NrCAM (140 kDa) coimmunoprecipitated (IP) with Npn-2 (130 kDa) and PlexA3 (220 kDa) from E15.5 (1 mg) and P21 (1 mg) brain, and with Npn-2 in transfected HEK293T cells, as shown by immunoblotting (IB). Lysate lanes represent 75 μg. Blots were stripped and reprobed for NrCAM (lower row). Lysate lane for E15.5 brain was from the same gel as nonimmune Ig (nIg) IP and NrCAM IP, but the lane was separated, as indicated. C, Mutations in the TARNER sequence of the NrCAM Ig1 domain abrogated binding to Npn-2 (TARΔNER>TARAAA>TARNEV) in transfected COS-7 cells. The level of Npn-2 relative to NrCAM in NrCAM immunoprecipitates was estimated from densitometric scanning of immunoblots and shown below. D, Double immunostaining showed colocalization of NrCAM (green) and Npn-2 (red) at numerous puncta (arrows) in confocal images of V1, layer 4 (P21). Scale bar, 5 μm. E, Pyramidal cells in V1, layer 4 (P21) were labeled with EGFP by in utero electroporation and EGFP-positive, dendritic spine puncta (E1) were labeled for both NrCAM and Npn2 (E2, arrow).
Regulation of Sema3F-induced spine retraction by NrCAM
To investigate whether NrCAM was required for Sema3F-induced spine retraction, we used an assay in which Sema3F-Fc fusion protein was added to cultures of primary cortical neurons from WT and NrCAM-null mice to induce spine retraction, as described previously (Tran et al., 2009). Neuronal cultures were prepared from embryos at E14.5-E16.5, electroporated with pCAGG-IRES-mEGFP to visualize spines, and propagated for 18 DIV. Cells were treated with Sema3F-Fc or control Fc protein (3 nm) for 2 h, fixed, and immunostained for EGFP. Spine density was scored on dendrites of neurons with pyramidal morphology in confocal images using Neurolucida software. Spine density on apical dendrites of cortical neurons from WT and NrCAM−/− mice did not differ between genotypes (Fig. 6A,B). However, treatment with Sema3F-Fc induced a significant reduction in spine density (∼75%) on apical dendrites of WT neurons but not on apical dendrites of NrCAM−/− neurons (Fig. 6A,B). In some Sema3F-Fc-treated NrCAM−/− cells a number of spines acquired a more filopodial morphology, but this was not observed throughout the culture and was likely a response restricted to a subset of neurons rather than an additional overall phenotype. To determine whether re-expression of WT NrCAM could rescue Sema3F-induced spine retraction in NrCAM mutant neurons, cultures from NrCAM-null mice were transfected with pCAGG-NrCAM-IRES-mEGFP or empty vector together with the mEGFP plasmid and treated at 18 DIV with Sema3F-Fc or Fc proteins for 2 h. Spine density on apical dendrites of NrCAM−/− neurons treated with Sema3F-Fc protein or Fc control were unchanged; however, re-expression of WT NrCAM restored the ability of Sema3F-Fc to decrease spine density (Fig. 6C). Spine density on basal dendrites of NrCAM−/− neurons in culture was unaffected by Sema3F-Fc treatment, regardless of re-expression of WT NrCAM (Fig. 6D). The ability of NrCAM to promote Sema3F-induced spine retraction on apical but not basal dendrites was not due to restricted localization, as NrCAM was localized to all dendritic processes and their branches, identified by the somatodendritic marker MAP2, as well as to the axon (arrow) and soma of cultured neurons (Fig. 6E). The selective effect of NrCAM on spine remodeling in apical dendrites likely is due to its interaction with Npn-2, which has a restricted localization on apical and not basal dendrites in cortical neuron cultures (Tran et al., 2009). These results support a postsynaptic function for NrCAM in mediating Sema3F-induced spine retraction on apical dendrites of cortical pyramidal neurons.
Figure 6.
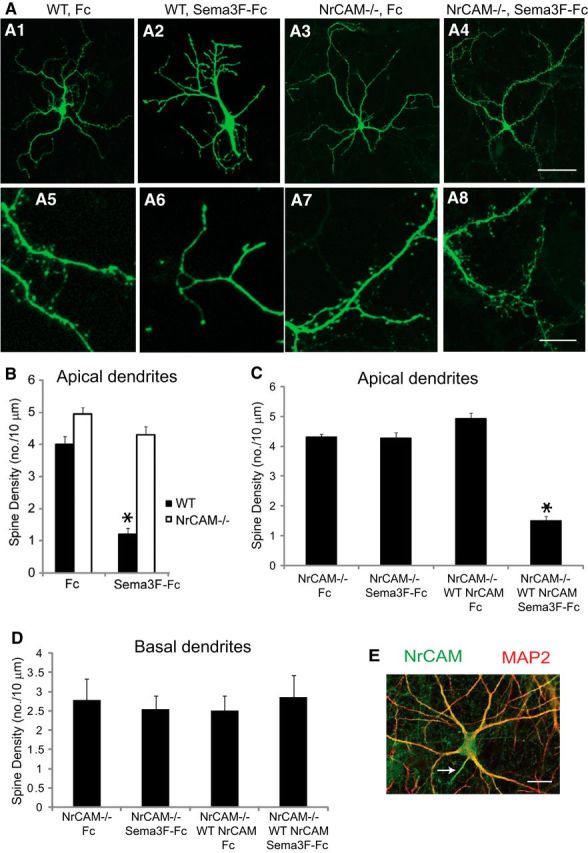
NrCAM regulates Sema3F-induced spine retraction of cortical neurons. A1–A8, Primary cortical neuronal cultures from WT or NrCAM−/− embryos (E14.5–E16.5) were electroporated with pCAGG-IRES-mEGFP, then treated at 18 DIV with 3 nm Fc or Sema3F-Fc protein for 2 h, followed by immunostaining for GFP and quantification of spine density in confocal images. Scale bars: A1–A4, 100 μm; A5–A8, 5 μm. B, Quantification of spine density on apical dendrites of cortical neurons from WT or NrCAM-null mice treated with Fc or Sema3F-Fc protein and means ± SEM compared with significant differences (two-tailed t test; *p < 0.05). C, D, Re-expression of WT NrCAM from pCAGG-NrCAM-IRES-mEGFP electroporated into cortical neuron cultures from NrCAM−/− embryos restored the ability of Sema3F-Fc to decrease spine density on apical but not basal dendrites (two-tailed t test; *p < 0.05). E, Double immunofluorescence staining of cortical neuron cultures showed colocalization of NrCAM (green) and MAP2 (red) in dendritic processes and branches. Scale bar, 25 μm. Spine densities were measured on dendrites of at least 10 neurons (30–148 total spines) in each of four replicate, independent cultures per condition.
Sema3F and NrCAM genetically interact to regulate spine density
To investigate a genetic interaction between Sema3F and NrCAM in Sema3F-mediated spine remodeling in vivo, we conducted a trans-heterozygote genetic test. This test has been used to evaluate dominant interactions between pairs of genes, for example, Sema3A and Fyn, in regulating spine density (Morita et al., 2006). Sema3F+/−; NrCAM+/− double heterozygous mice were generated from intercrosses of single heterozygotes. The resulting double and single heterozygotes and WT littermates were analyzed at P21 for spine density on apical or basal dendrites of star pyramidal neurons in layer 4, V1 after Golgi impregnation (Fig. 7A). Spine densities on apical dendrites of star pyramidal cells in Sema3F+/− or NrCAM+/− heterozygous mice were similar to WT, but density increased strikingly in Sema3F+/−; NrCAM+/− double heterozygotes attaining the same levels as in NrCAM−/− and Sema3F−/− homozygotes (Fig. 7B). In contrast, the basal dendrites of star pyramidal cells were equivalent in all genotypes (Fig. 7C). This genetic test indicates that NrCAM shows strong interaction with Sema3F in regulating the density of excitatory synapses on apical dendrites of star pyramidal cells in vivo. The most likely interpretation of these results is that Sema3F/Npn-2 and NrCAM induce spine retraction within the same molecular pathway at postsynaptic sites in apical dendrites during postnatal stages of cortical development.
Figure 7.
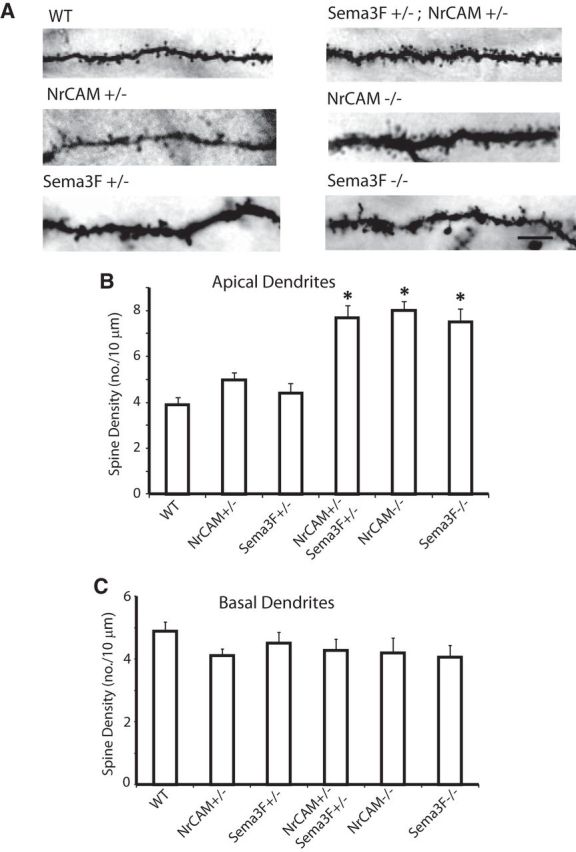
Sema3F and NrCAM genetically interact to regulate spine density. In a double heterozygous interaction test, dendritic spine density was analyzed in Golgi-labeled, star pyramidal neurons in binocular V1, layer 4 of WT (n = 3), NrCAM+/− (n = 3), or Sema3F+/− (n = 8) heterozygotes, NrCAM−/− homozygotes (n = 3), Sema3F−/− homozygotes (n = 3), and NrCAM+/−; Sema3F+/− (n = 3) double heterozygotes at P21; n = 20–23 neurons per mouse. A, Bright-field images of representative apical dendrites of Golgi-labeled, star pyramidal neurons for each genotype. Scale bar, 5 μm. B, Quantification showed increased spine density on apical dendrites of Sema3F+/−; NrCAM+/− double heterozygotes, equivalent to that of NrCAM−/− or Sema3F−/− homozygotes, and significantly greater than that of WT or single heterozygotes (*p < 0.05). C, Quantification showed no difference in spine density on basal dendrites in all genotypes. (*p < 0.05).
Discussion
Here we identify NrCAM as a novel regulator of dendritic spines and excitatory synapses in cortical pyramidal neurons with the potential to modulate E/I balance in cortical circuits. We focused on the visual cortex where development of intrinsic and extrinsic circuitry has been most extensively studied (Sur et al., 2013), and in particular on star pyramidal neurons. Several lines of evidence support the conclusion that NrCAM mediates spine elimination as an integral component of the Sema3F receptor complex in postnatally developing pyramidal neurons. (1) Loss of NrCAM in mutant mice resulted in an elevated spine density on apical dendrites of star pyramidal neurons and an excess of functional excitatory synapses, as indicated by increased mEPSC frequencies in V1, layer 4 pyramidal cells. (2) NrCAM was required for Sema3F-induced spine retraction in cultured cortical neurons. (3) NrCAM associated with the Sema3F receptor subunit Npn-2 at an identified binding site in the NrCAM Ig1 domain, and coimmunoprecipitated with the Sema3F receptor signaling subunit PlexA3. (4) A trans heterozygous genetic test demonstrated that Sema3F and NrCAM pathways functionally interacted to constrain spine density on apical dendrites of star pyramidal cells in vivo. This novel role for NrCAM is not restricted to star pyramidal cells, as NrCAM functioned broadly to limit spine density in the cortex, as shown by increased dendritic spine density in NrCAM-deficient pyramidal neurons of layers 2/3 and 5 of V1, as well as in somatosensory and prefrontal areas (data not shown).
Our studies demonstrate for the first time that a cell adhesion molecule of the Ig superfamily functions cooperatively with repellent signaling molecules to constrain the number of dendritic spines and excitatory synapses in cortical pyramidal neurons. An essential role for NrCAM in the mechanism of Sema3F-induced spine retraction in cortical neurons is analogous to its ability to promote axon repulsion and growth cone collapse to Sema3F through Npn-2 (Falk et al., 2005; Demyanenko et al., 2011), although it remains to be determined whether spine retraction and growth cone collapse are achieved through different signaling pathways impinging on the cytoskeleton. A distinct Ig-class adhesion molecule, SynCAM1, has been shown to have an opposing effect in stimulating spine growth and excitatory synapse formation in the hippocampus (Robbins et al., 2010). NrCAM might be a bidirectional regulator of spine development and/or plasticity as it physically associates with EphB2, a tyrosine kinase receptor that stabilizes dendritic spines (Shi et al., 2009) and modulates ankyrin B binding to NrCAM by phosphorylating a critical tyrosine residue in the NrCAM cytoplasmic domain (Dai et al., 2013).
The cortical expression pattern and localization of NrCAM on dendritic spines of star pyramidal cells, coupled with the ability of NrCAM to rescue Sema3F-induced spine collapse when re-expressed in NrCAM-minus cortical neurons support a postsynaptic role for NrCAM in spine remodeling/elimination. During postnatal development (P7–P21) NrCAM transcripts were enriched in V1, layers 4 and 6, mirroring Sema3F expression, whereas NrCAM expression in thalamic neurons was in steep decline as early as P5 (Demyanenko et al., 2011). These findings are in accord with immunogold labeling in rat hippocampus showing that NrCAM is associated with the PSD during synaptic maturation (Petralia et al., 2005). The subset of postsynaptic specializations that were immunopositive for NrCAM (∼1/3) was consistent with the magnitude of increase in spine density on apical dendrites of NrCAM-minus star pyramidal cells in vivo. In cultured cortical neurons treated with Sema3F-Fc the decrease in spine density was greater, possibly due to a larger proportion of Sema3F-sensitive cells in vitro. Neuropilins have been shown to cross-brace PlexinA subunits and lock secreted semaphorins into a dimerized complex that is signaling ready (Janssen et al., 2012). Through its ability to bind the scaffold proteins SAP102 or PSD-95 (Davey et al., 2005; Dirks et al., 2006), NrCAM may facilitate localization and clustering of Npn-2, which shares a PDZ binding motif (Tran et al., 2009), together with associated PlexA3 subunits at the early developing PSD to activate signaling. Other binding motifs in the cytoplasmic domain of NrCAM might provide additional coupling to the actin cytoskeleton through ezrin-radixin-moesin (Schlatter et al., 2008) and ankyrin (Dai et al., 2013). Adhesive interactions of the L1 family of cell adhesion molecules are mediated through their Ig domains (Sakurai, 2012). Ig1–4 domains are involved in homophilic binding, which promotes neurite outgrowth (Gouveia et al., 2008; Wei and Ryu, 2012), while the Ig1 domain is known to engage the proteoglycans neurocan and phosphacan (Oleszewski et al., 2000; Sakurai, 2012). Other NrCAM partners include Contactin-1/F11/F3, Contactin-2/Axonin1/Tag-1, Neurofascin (Sakurai, 2012), PlexinA1, and Sema6D (Kuwajima et al., 2012). An interesting possibility is that binding of the NrCAM Ig1 domain to Npn-2 competes with or regulates its association with other partners, which may promote Sema3F-induced spine elimination.
The origin of afferent inputs to apical versus basal dendrites of pyramidal neurons in V1 is poorly understood (Karube and Kisvárday, 2011), but the differences we have observed in NrCAM-null mice suggest that cortical circuitry will be altered. The elevated density of excitatory synaptic profiles and increased mEPSCs of star pyramidal cells in NrCAM-null mutants indicate that the observed excess of dendritic spines leads to functional excitatory connections. The majority of excitatory inputs to star pyramidal cells originates in layers 2/3 and 6, which may comprise a recurrent circuit necessary for signal amplification, orientation tuning, or receptive field modulation (Stratford et al., 1996; Binzegger et al., 2004; Douglas and Martin, 2007; Karube and Kisvárday, 2011; Olsen et al., 2012). Altered intracortical circuitry in layer 4 of V1 may contribute to the impairment in visual acuity and visual cortical processing in NrCAM-null mice, as they display normal retinogeniculate and geniculocortical axon targeting (Demyanenko et al., 2011). Mistargeted thalamocortical synapses are unlikely to account for increased dendritic spine density, as the number of VGluT2-positive, thalamocortical synaptic puncta in the NrCAM-null V1 (layer 4) was equivalent to WT. Moreover, only a small fraction of excitatory inputs to layer 4 of V1 (∼5%) are thalamocortical in origin (Ahmed et al., 1994). Generation of NrCAM conditional mutant mice will be needed to ascertain the postulated postsynaptic function for NrCAM in dendritic spines remodeling in vivo.
Activity-dependent mechanisms are known to drive synaptic elimination in development and plasticity (Alvarez and Sabatini, 2007). However, spine elimination induced by brief MD during the critical period of visual cortical development was unaffected by NrCAM loss on apical dendrites of pyramidal cells in layers 2/3 and 4 of binocular V1. It is unclear if secreted Sema3F is produced by pyramidal cells, interneurons, or glia, and whether its expression is activity dependent. A future challenge is to determine whether activity underlies spine remodeling through Sema3F and NrCAM/Npn-2 at earlier stages of postnatal development when synapse generation and elimination occur at high rates to achieve an appropriate E/I balance. Alternatively, Sema3F-induced spine remodeling through NrCAM/Npn-2 might serve to restrict synaptic connections to specific cortical laminae or subcellular locations, analogous to the roles of class 5 semaphorins and DsCAM adhesion molecules in the retina (Zipursky and Sanes, 2010; Matsuoka et al., 2011).
The significance of this work to neurodevelopmental disease is underscored by autism-related polymorphisms at loci containing NrCAM, Ank B (Willsey et al., 2013), Npn-2 (Wu et al., 2007; Brooks-Kayal, 2010), and Sema5A genes (Melin et al., 2006; Weiss et al., 2009). It is also noteworthy that abnormal visual motion detection and binocular switching have been described in cases of ASD (Farzin et al., 2008; Pellicano, 2008; Simmons et al., 2009; Robertson et al., 2013). Our results suggest that NrCAM deficiency may lead to defective spine elimination in cortical neurons during development of visual and other neocortical circuits thus altering E/I balance and relevant behaviors.
Footnotes
This work was supported by National Institutes of Health (NIH) grants R21MH098138 and R01MH101605 (P.F.M.) and R01DC009809 (P.B.M.), Charles and Johanna Busch Biomedical Award (T.S.T.), and NIH P30NS045892 for University of North Carolina (UNC) Neuroscience Research Center core support. We gratefully acknowledge Dirk Montag (Leibniz Institute for Neurobiology, Magdeburg, Germany) for the NrCAM plasmid, Takeshi Sakurai and Carol Mason (Columbia University) for probes, Alex Kolodkin and David Ginty for Sema3F mice and probes, and Ben Philpot and Thorfinn Riday (UNC Chapel Hill) for advice on monocular deprivation. We are also grateful to Sam George, Eli Darnell, Jasbir Dalal, Sneha Venkatramen, and Erin Moore for experimental assistance.
The authors declare no competing financial interests.
References
- Ahmed B, Anderson JC, Douglas RJ, Martin KA, Nelson JC. Polyneuronal innervation of spiny stellate neurons in cat visual cortex. J Comp Neurol. 1994;341:39–49. doi: 10.1002/cne.903410105. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Antonini A, Fagiolini M, Stryker MP. Anatomical correlates of functional plasticity in mouse visual cortex. J Neurosci. 1999;19:4388–4406. doi: 10.1523/JNEUROSCI.19-11-04388.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Jouet M, MacFarlane J, Du JS, Kenwrick S, Chothia C. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 1996;15:6050–6059. [PMC free article] [PubMed] [Google Scholar]
- Binzegger T, Douglas RJ, Martin KA. A quantitative map of the circuit of cat primary visual cortex. J Neurosci. 2004;24:8441–8453. doi: 10.1523/JNEUROSCI.1400-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora E, Lamb JA, Barnby G, Sykes N, Moberly T, Beyer KS, Klauck SM, Poustka F, Bacchelli E, Blasi F, Maestrini E, Battaglia A, Haracopos D, Pedersen L, Isager T, Eriksen G, Viskum B, Sorensen EU, Brondum-Nielsen K, Cotterill R, et al. Mutation screening and association analysis of six candidate genes for autism on chromosome 7q. Eur J Hum Genet. 2005;13:198–207. doi: 10.1038/sj.ejhg.5201315. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal A. Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 2010;32:731–738. doi: 10.1016/j.braindev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Castellani V. The function of neuropilin/L1 complex. Adv Exp Med Biol. 2002;515:91–102. doi: 10.1007/978-1-4615-0119-0_8. [DOI] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Colbert MC, Rubin WW, Linney E, LaMantia AS. Retinoid signaling and the generation of regional and cellular diversity in the embryonic mouse spinal cord. Dev Dyn. 1995;204:1–12. doi: 10.1002/aja.1002040102. [DOI] [PubMed] [Google Scholar]
- Coleman JE, Law K, Bear MF. Anatomical origins of ocular dominance in mouse primary visual cortex. Neuroscience. 2009;161:561–571. doi: 10.1016/j.neuroscience.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JE, Nahmani M, Gavornik JP, Haslinger R, Heynen AJ, Erisir A, Bear MF. Rapid structural remodeling of thalamocortical synapses parallels experience-dependent functional plasticity in mouse primary visual cortex. J Neurosci. 2010;30:9670–9682. doi: 10.1523/JNEUROSCI.1248-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Dai J, Buhusi M, Demyanenko GP, Brennaman LH, Hruska M, Dalva MB, Maness PF. Neuron glia-related cell adhesion molecule (NrCAM) promotes topographic retinocollicular mapping. PLoS One. 2013;8:e73000. doi: 10.1371/journal.pone.0073000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey F, Hill M, Falk J, Sans N, Gunn-Moore FJ. Synapse associated protein 102 is a novel binding partner to the cytoplasmic terminus of neurone-glial related cell adhesion molecule. J Neurochem. 2005;94:1243–1253. doi: 10.1111/j.1471-4159.2005.03271.x. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Halberstadt AI, Rao RS, Maness PF. CHL1 cooperates with PAK1–3 to regulate morphological differentiation of embryonic cortical neurons. Neuroscience. 2010;165:107–115. doi: 10.1016/j.neuroscience.2009.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Riday TT, Tran TS, Dalal J, Darnell EP, Brennaman LH, Sakurai T, Grumet M, Philpot BD, Maness PF. NrCAM deletion causes topographic mistargeting of thalamocortical axons to the visual cortex and disrupts visual acuity. J Neurosci. 2011;31:1545–1558. doi: 10.1523/JNEUROSCI.4467-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks P, Thomas U, Montag D. The cytoplasmic domain of NrCAM binds to PDZ domains of synapse-associated proteins SAP90/PSD95 and SAP97. Eur J Neurosci. 2006;24:25–31. doi: 10.1111/j.1460-9568.2006.04899.x. [DOI] [PubMed] [Google Scholar]
- Docherty SJ, Davis OS, Kovas Y, Meaburn EL, Dale PS, Petrill SA, Schalkwyk LC, Plomin R. A genome-wide association study identifies multiple loci associated with mathematics ability and disability. Genes Brain Behav. 2010;9:234–247. doi: 10.1111/j.1601-183X.2009.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Recurrent neuronal circuits in the neocortex. Curr Biol. 2007;17:R496–R500. doi: 10.1016/j.cub.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Espinosa JS, Stryker MP. Development and plasticity of the primary visual cortex. Neuron. 2012;75:230–249. doi: 10.1016/j.neuron.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Julien F, Bechara A, Fiore R, Nawabi H, Zhou H, Hoyo-Becerra C, Bozon M, Rougon G, Grumet M, Püschel AW, Sanes JR, Castellani V. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48:63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Farzin F, Whitney D, Hagerman RJ, Rivera SM. Contrast detection in infants with fragile X syndrome. Vision Res. 2008;48:1471–1478. doi: 10.1016/j.visres.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Giger RJ, Urquhart ER, Gillespie SK, Levengood DV, Ginty DD, Kolodkin AL. Neuropilin-2 is a receptor for semaphorin IV: insight into the structural basis of receptor function and specificity. Neuron. 1998;21:1079–1092. doi: 10.1016/S0896-6273(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J Neurosci. 2006;26:12717–12726. doi: 10.1523/JNEUROSCI.3502-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia RM, Gomes CM, Sousa M, Alves PM, Costa J. Kinetic analysis of L1 homophilic interaction: role of the first four immunoglobulin domains and implications on binding mechanism. J Biol Chem. 2008;283:28038–28047. doi: 10.1074/jbc.M804991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Barker PB, Sawa A. Readdressing synaptic pruning theory for schizophrenia: combination of brain imaging and cell biology. Commun Integr Biol. 2011;4:211–212. doi: 10.4161/cib.4.2.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Synaptic density in human frontal cortex-developmental changes and effects of aging. Brain Res. 1979;163:195–205. doi: 10.1016/0006-8993(79)90349-4. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Hall FS, Horiuchi Y, Sakurai T, Hishimoto A, Grumet M, Uhl GR, Onaivi ES, Arinami T. NrCAM-regulating neural systems and addiction-related behaviors. Addict Biol. 2014;19:343–353. doi: 10.1111/j.1369-1600.2012.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BJ, Malinauskas T, Weir GA, Cader MZ, Siebold C, Jones EY. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol. 2012;19:1293–1299. doi: 10.1038/nsmb.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Sohya K, Sarihi A, Yanagawa Y, Tsumoto T. Laminar-specific maturation of GABAergic transmission and susceptibility to visual deprivation are related to endocannabinoid sensitivity in mouse visual cortex. J Neurosci. 2010;30:14261–14272. doi: 10.1523/JNEUROSCI.2979-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube F, Kisvárday ZF. Axon topography of layer IV spiny cells to orientation map in the cat primary visual cortex (area 18) Cereb Cortex. 2011;21:1443–1458. doi: 10.1093/cercor/bhq232. [DOI] [PubMed] [Google Scholar]
- Kuwajima T, Yoshida Y, Takegahara N, Petros TJ, Kumanogoh A, Jessell TM, Sakurai T, Mason C. Optic chiasm presentation of Semaphorin6D in the context of Plexin-A1 and Nr-CAM promotes retinal axon midline crossing. Neuron. 2012;74:676–690. doi: 10.1016/j.neuron.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cui Z, Niu Y, Liu B, Fan W, Yu D, Deng J. Synaptogenesis in the developing mouse visual cortex. Brain Res Bull. 2010;81:107–113. doi: 10.1016/j.brainresbull.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O, Nanba E, Nishida H, Sugiyama T, Kasai K, Watanabe K, Kano Y, Sasaki T, Kato N. Association of the neuronal cell adhesion molecule (NRCAM) gene variants with autism. Int J Neuropsychopharmacol. 2009;12:1–10. doi: 10.1017/S1461145708009127. [DOI] [PubMed] [Google Scholar]
- Mataga N, Mizuguchi Y, Hensch TK. Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron. 2004;44:1031–1041. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Matsuoka RL, Nguyen-Ba-Charvet KT, Parray A, Badea TC, Chédotal A, Kolodkin AL. Transmembrane semaphorin signalling controls laminar stratification in the mammalian retina. Nature. 2011;470:259–263. doi: 10.1038/nature09675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat Neurosci. 2010;13:450–457. doi: 10.1038/nn.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin M, Carlsson B, Anckarsater H, Rastam M, Betancur C, Isaksson A, Gillberg C, Dahl N. Constitutional downregulation of SEMA5A expression in autism. Neuropsychobiology. 2006;54:64–69. doi: 10.1159/000096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita A, Yamashita N, Sasaki Y, Uchida Y, Nakajima O, Nakamura F, Yagi T, Taniguchi M, Usui H, Katoh-Semba R, Takei K, Goshima Y. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J Neurosci. 2006;26:2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nonneman RJ, Young NB, Demyanenko GP, Maness PF. Impaired sociability and cognitive function in Nrcam-null mice. Behav Brain Res. 2009;205:123–131. doi: 10.1016/j.bbr.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahmani M, Erisir A. VGluT2 immunochemistry identifies thalamocortical terminals in layer 4 of adult and developing visual cortex. J Comp Neurol. 2005;484:458–473. doi: 10.1002/cne.20505. [DOI] [PubMed] [Google Scholar]
- Oleszewski M, Gutwein P, von der Lieth W, Rauch U, Altevogt P. Characterization of the L1-neurocan-binding site. Implications for L1–L1 homophilic binding. J Biol Chem. 2000;275:34478–34485. doi: 10.1074/jbc.M004147200. [DOI] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H, Scanziani M. Gain control by layer six in cortical circuits of vision. Nature. 2012;483:47–52. doi: 10.1038/nature10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicano E. Autism: face-processing clues to inheritance. Curr Biol. 2008;18:R748–R750. doi: 10.1016/j.cub.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Petanjek Z, Judăs M, S̆imic G, Rasin MR, Uylings HB, Rakic P, Kostovic I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins EM, Krupp AJ, Perez de Arce K, Ghosh AK, Fogel AI, Boucard A, Südhof TC, Stein V, Biederer T. SynCAM 1 adhesion dynamically regulates synapse number and impacts plasticity and learning. Neuron. 2010;68:894–906. doi: 10.1016/j.neuron.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CE, Kravitz DJ, Freyberg J, Baron-Cohen S, Baker CI. Slower rate of binocular rivalry in autism. J Neurosci. 2013;33:16983–16991. doi: 10.1523/JNEUROSCI.0448-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Molliver ME, Ginty DD, Kolodkin AL. Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci. 2003;23:6671–6680. doi: 10.1523/JNEUROSCI.23-17-06671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T. The role of NrCAM in neural development and disorders–beyond a simple glue in the brain. Mol Cell Neurosci. 2012;49:351–363. doi: 10.1016/j.mcn.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Lustig M, Babiarz J, Furley AJ, Tait S, Brophy PJ, Brown SA, Brown LY, Mason CA, Grumet M. Overlapping functions of the cell adhesion molecules Nr-CAM and L1 in cerebellar granule cell development. J Cell Biol. 2001;154:1259–1273. doi: 10.1083/jcb.200104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Ramoz N, Reichert JG, Corwin TE, Kryzak L, Smith CJ, Silverman JM, Hollander E, Buxbaum JD. Association analysis of the NrCAM gene in autism and in subsets of families with severe obsessive-compulsive or self-stimulatory behaviors. Psychiatr Genet. 2006;16:251–257. doi: 10.1097/01.ypg.0000242196.81891.c9. [DOI] [PubMed] [Google Scholar]
- Schlatter MC, Buhusi M, Wright AG, Maness PF. CHL1 promotes Sema3A-induced growth cone collapse and neurite elaboration through a motif required for recruitment of ERM proteins to the plasma membrane. J Neurochem. 2008;104:731–744. doi: 10.1111/j.1471-4159.2007.05013.x. [DOI] [PubMed] [Google Scholar]
- Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DR, Robertson AE, McKay LS, Toal E, McAleer P, Pollick FE. Vision in autism spectrum disorders. Vision Res. 2009;49:2705–2739. doi: 10.1016/j.visres.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Stratford KJ, Tarczy-Hornoch K, Martin KA, Bannister NJ, Jack JJ. Excitatory synaptic inputs to spiny stellate cells in cat visual cortex. Nature. 1996;382:258–261. doi: 10.1038/382258a0. [DOI] [PubMed] [Google Scholar]
- Sur M, Nagakura I, Chen N, Sugihara H. Mechanisms of plasticity in the developing and adult visual cortex. Prog Brain Res. 2013;207:243–254. doi: 10.1016/B978-0-444-63327-9.00002-3. [DOI] [PubMed] [Google Scholar]
- Tran TS, Kolodkin AL, Bharadwaj R. Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol. 2007;23:263–292. doi: 10.1146/annurev.cellbio.22.010605.093554. [DOI] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature. 2009;462:1065–1069. doi: 10.1038/nature08628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CH, Ryu SE. Homophilic interaction of the L1 family of cell adhesion molecules. Exp Mol Med. 2012;44:413–423. doi: 10.3858/emm.2012.44.7.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SE, Grumet M, Colman DR, Henkemeyer M, Mason CA, Sakurai T. A role for Nr-CAM in the patterning of binocular visual pathways. Neuron. 2006;50:535–547. doi: 10.1016/j.neuron.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, Murtha MT, Bichsel C, Niu W, Cotney J, Ercan-Sencicek AG, Gockley J, Gupta AR, Han W, He X, Hoffman EJ, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AG, Demyanenko GP, Powell A, Schachner M, Enriquez-Barreto L, Tran TS, Polleux F, Maness PF. Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J Neurosci. 2007;27:13667–13679. doi: 10.1523/JNEUROSCI.2888-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Yue W, Jia M, Ruan Y, Lu T, Gong X, Shuang M, Liu J, Yang X, Zhang D. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:492–495. doi: 10.1002/ajmg.b.30495. [DOI] [PubMed] [Google Scholar]
- Yuste R. Dendritic spines and distributed circuits. Neuron. 2011;71:772–781. doi: 10.1016/j.neuron.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Sanes JR. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell. 2010;143:343–353. doi: 10.1016/j.cell.2010.10.009. [DOI] [PubMed] [Google Scholar]