

Abstract
The adipocyte-derived hormone leptin modulates neural systems appropriately for the status of body energy stores. Leptin inhibits lateral hypothalamic area (LHA) orexin (OX; also known as hypocretin)-producing neurons, which control feeding, activity, and energy expenditure, among other parameters. Our previous results suggest that GABAergic LHA leptin receptor (LepRb)-containing and neurotensin (Nts)-containing (LepRbNts) neurons lie in close apposition with OX neurons and control Ox mRNA expression. Here, we show that, similar to leptin, activation of LHA Nts neurons by the excitatory hM3Dq DREADD (designer receptor exclusively activated by designer drugs) hyperpolarizes membrane potential and suppresses action potential firing in OX neurons in mouse hypothalamic slices. Furthermore, ablation of LepRb from Nts neurons abrogated the leptin-mediated inhibition, demonstrating that LepRbNts neurons mediate the inhibition of OX neurons by leptin. Leptin did not significantly enhance GABAA-mediated inhibitory synaptic transmission, and GABA receptor antagonists did not block leptin-mediated inhibition of OX neuron activity. Rather, leptin diminished the frequency of spontaneous EPSCs onto OX neurons. Furthermore, leptin indirectly activated an ATP-sensitive potassium (KATP) channel in OX neurons, which was required for the hyperpolarization of OX neurons by leptin. Although Nts did not alter OX activity, galanin, which is coexpressed in LepRbNts neurons, inhibited OX neurons, whereas the galanin receptor antagonist M40 (galanin-(1–12)-Pro3-(Ala-Leu)2-Ala amide) prevented the leptin-induced hyperpolarization of OX cells. These findings demonstrate that leptin indirectly inhibits OX neurons by acting on LHA LepRbNts neurons to mediate two distinct GABA-independent mechanisms of inhibition: the presynaptic inhibition of excitatory neurotransmission and the opening of KATP channels.
Keywords: EPSC, GABA, leptin, LHA, neurotensin, orexin
Introduction
The lateral hypothalamic area (LHA) receives and integrates many signals, including metabolic stimuli, to modulate feeding, activity, and attention/alertness as appropriate for current nutritional and environmental conditions (Morrison et al., 1958). The LHA contains several groups of neurons that contribute to these processes, including glutamatergic orexin (OX; also known as hypocretin)-containing cells that are activated by signals of energy deficit. OX neurons project widely throughout the brain, including to monoaminergic targets in which OX-derived neuropeptides act at G-protein-coupled OX receptors 1 and 2 to inhibit sleep and to promote food seeking and vigilance (Toshinai et al., 2003; Yamanaka et al., 2003; Tsujino and Sakurai, 2009). Acute activation of the OX system, as by OX injection into the brain, triggers arousal and induces feeding (Lubkin and Stricker-Krongrad, 1998; Sakurai et al., 1998; Sweet et al., 1999; Jones et al., 2001; Perez-Leighton et al., 2012).
The hormone leptin, which is produced by adipocytes to signal the sufficiency of energy reserves, represents a crucial modulator of OX function. Leptin inhibits the firing of OX neurons (Yamanaka et al., 2003), and the drop in leptin levels during fasting activates OX neurons (Diano et al., 2003; Leinninger et al., 2011), increasing food seeking and alertness. Although the leptin-mediated inhibition of OX neurons is central to the appropriate control of these neurons and their behavioral outputs (as well as to the physiologic and behavioral response to fasting), the mechanisms by which leptin modulates the activity of OX neurons has remained unclear.
Intermingled with OX neurons in the LHA is a distinct population of neurons that express the leptin receptor (LepRb) and that therefore respond to direct leptin action. Essentially all of these LHA LepRb neurons contain vesicular GABA transporter (vGAT) and GAD1 (Leinninger et al., 2009; Vong et al., 2011), suggesting that they may act in part by releasing GABA; many (∼60%) also contain the neuropeptide neurotensin (Nts) (Leinninger et al., 2011). We demonstrated previously that LHA LepRb neurons and LHA Nts neurons lie in close contact with OX cells and that LHA leptin action modulates Ox mRNA expression, suggesting that leptin may control OX neuron activity via Nts-containing LHA LepRb (LepRbNts neurons; Leinninger et al., 2011). However, the cellular mechanisms that underlie leptin-dependent regulation of OX neurons remain unclear. Here, we use the electrophysiological analysis of OX neuron function to directly demonstrate the importance of LepRbNts neurons for the control of OX cells and to elucidate the mechanisms by which leptin inhibits OX neurons.
Materials and Methods
Animals.
All procedures were approved by the University of Michigan University Committee on the Use and Care of Animals in accordance with Association for Assessment and Accreditation of Laboratory Animal Care and National Institutes of Health guidelines. Animals were bred at the University of Michigan and maintained in a 12 h light/dark cycle with ad libitum access to food and water. Transgenic mice expressing enhanced green fluorescent protein (EGFP) under the control of the human prepro-orexin promoter (OX–EGFP mice; Sakurai et al., 1999; Li et al., 2002) were a generous gift from Yuchio Yanagawa (Gunma University, Gunma, Japan). Ntscre and LeprNtsKO mice (as described previously; Leinninger et al., 2011) were bred onto the OX–EGFP background for electrophysiology experiments.
DREADD activation of LHA Nts neurons.
AAV–Flex–hM3Dq–mCherry virus (Alexander et al., 2009) was purchased from the UNC Vector Core (University of North Carolina, Chapel Hill, NC). Virus was stereotaxically injected bilaterally into the LHA of Ntscre; OX–EGFP mice at 5–6 weeks of age. Mice were administered presurgical analgesic, anesthetized using isoflurane, and placed in a stereotaxic frame. After exposing the skull, a guide cannula with a stylet (Plastics One) was lowered into the target regions. Coordinates to the LHA (from bregma) were as follows: anteroposterior, −1.34 mm; mediolateral, −1.12 mm; and dorsoventral, −5.20 mm, in accordance with the atlas of Paxinos and Franklin (2001). The stylet was removed and replaced by an injector, and 250 nl of virus was injected to the LHA using a 500 nl Hamilton syringe at a rate of 50 nl/30 s. After 5 min, the injector and cannula were removed from the skull, and the incision was sutured.
After 1 week recovery, animals were killed for electrophysiological analysis of OX neuron function. Stereotaxic injection sites were confirmed by visualization of mCherry fluorescence in the LHA of slices used for electrophysiological recording. Mice were only included for study if DREADD–mCherry-expressing cell bodies were confined to the LHA.
Electrophysiological recordings.
Four- to 7-week-old mice of either sex were killed by decapitation, and 250 μm coronal slices were prepared using a VT 1200S vibratome (Leica) in oxygenated ice-cold sucrose solution containing the following (in mm): 220 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 5 glucose, 6 MgCl2, and 1 CaCl2. Slices were allowed to recover for at least 1 h in a holding chamber containing oxygenated artificial CSF (aCSF) solution containing the following (in mm): 130 NaCl, 3 KCl, 1.25 NaH2PO4, NaHCO3, 5 glucose, 1 MgCl2, and 2.5 CaCl2, pH 7.4, before electrophysiological analysis. Electrophysiological parameters were measured from individual OX–EGFP neurons in the LHA visualized using an Olympus BX51WI upright microscope equipped with infrared differential interference contrast optics (Olympus). Patch electrodes made from borosilicate glass capillaries (Warner Instruments) were pulled to a tip resistance of 3–7 MΩ using a Brown/Flaming P-97 micropipette puller (Sutter Instruments) and filled with a solution containing the following (in mm): 130 K-gluconate, 10 KCl, 1 EGTA, 10 HEPES, 0.6 NaGTP, 2 MgATP, and 8 phosphocreatine, pH 7.2. In a subset of experiments, intracellular ATP was increased to 5 mm by the addition of 3 mm K2ATP. Results obtained from neurons dialyzed with 5 mm intracellular ATP did not differ from those filled with 2 mm ATP; therefore, both groups were pooled for analysis. Slices were superfused with aCSF solution warmed to 34°C using a TC-344 temperature controller and preheater (Warner Instruments) and continuously bubbled with 5% CO2 and 95% O2. Drugs were applied via bath perfusion. Data shown are not adjusted for a liquid junction potential of 14.7 mV. Whole-cell currents and membrane potentials were measured using tight-seal whole-cell voltage- or current-clamp (Hamill et al., 1981) using an Axopatch 200B amplifier (Molecular Devices). Currents were filtered at 2 kHz, digitized at 5 kHz, and analyzed offline. Neurons selected for analysis had stable series resistances <25 MΩ that were not compensated. Spontaneous EPSCs (sEPSCs) and sIPSCs were measured from neurons voltage clamped to −50 mV using standard intracellular and aCSF solutions. At this voltage, sEPSCs are inward currents and sIPSCs are outward currents. sIPSCs were also recorded from neurons voltage clamped to −65 mV in the presence of the glutamate receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) and d-(−)-2-amino-5-phosphonopentanoic acid (APV; 20 μm) and using an intracellular solution containing the following (in mm): 140 KCl, 1 EGTA, 10 HEPES, 0.6 NaGTP, 2 MgATP, and 8 phosphocreatine. Current–voltage (I–V) curves were generated using a voltage ramp command (−120 to +40 mV) and whole-cell conductance calculated from the slope of the current elicited from −100 to −50 mV. Leptin-elicited difference currents were plotted versus clamp voltage.
Data analysis and statistics.
sEPSC and sIPSC events were analyzed using a commercial software package (Synaptosoft). Individual events were detected using an amplitude threshold value set at 10 pA and confirmed visually. Event amplitude, interevent interval, 10–90% rise time, and half-width were determined before and after drug treatments. Statistical comparisons of cumulative probability distributions were made with MATLAB using the nonparametric Kolmogorov–Smirnov test, and significance was defined as p < 0.001. All other statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software), and statistical significance was determined using Student's t test for comparison of two groups or ANOVA and Tukey's multiple comparison post hoc test for comparison of multiple groups. Statistical differences were defined as p < 0.05. Error bars shown depict mean ± SEM.
Chemicals and reagents.
Leptin was a generous gift from Amylin Pharmaceuticals. Galanin (Gal) and Nts were purchased from American Peptide. CNQX, APV, M40 (galanin-(1–12)-Pro3-(Ala-Leu)2-Ala amide), and CGP52432 (3-[[(3,4-dichlorophenyl)-methyl]amino]propyl](diethoxymethyl)phosphinic acid) were purchased from R&D Systems. Bicuculline methiodide (BMI) and tolbutamide were purchased from Sigma. Clozapine-N-oxide (CNO) was a generous gift from Dr. Bryan Roth (University of North Carolina, Chapel Hill, NC). All drugs were made as stock solutions (tolbutamide and CGP52432 in DMSO) and freshly diluted in aCSF for recording. Drug treatments were compared with vehicle control.
Results
Leptin inhibits the activity of OX neurons via LepRbNts neurons
To examine the mechanisms by which leptin modulates OX neurons, we used transgenic mice expressing EGFP under the control of the human prepro-orexin promoter (Sakurai et al., 1999), permitting the visualization of OX–EGFP neurons in acute slice preparations for electrophysiological recordings. We assessed the electrical activity of OX–EGFP neurons in the LHA of hypothalamic slices using whole-cell patch-clamp recordings in current-clamp mode to measure membrane potential and action potential firing. As shown previously, acute leptin (10 nm) hyperpolarized OX neuron membrane potential (Fig. 1A,B; control, −51.0 ± 1.1 mV; leptin, −58.6 ± 1.7 mV; p < 0.0001, t(30) = 5.16) and reduced action potential firing (Fig. 1C; control, 5.4 ± 0.7 Hz; leptin, 1.3 ± 0.4 Hz; p < 0.0001, t(19) = 5.34). Leptin action progressed over several minutes with the initial response and maximal hyperpolarization occurring 77 ± 10 and 265 ± 34 s after the start of leptin treatment, respectively. Leptin did not significantly alter action potential amplitude, 10–90% rise time, half-width, or firing threshold (data not shown).
Figure 1.

Leptin inhibits OX neuron activity. A, Current-clamp recording of membrane potential and action potential firing from an individual OX–EGFP neuron in the LHA of an acute brain slice preparation in response to leptin. Leptin significantly hyperpolarized the mean membrane potential (B; n = 31, ***p < 0.001) and reduced the mean action potential firing frequency of LHA OX neurons (C; n = 21, ***p < 0.001). D, Leptin-induced membrane hyperpolarization was reversed in a subset of responders, with mean membrane potential restored near control values after the washout of leptin (n = 17, ***p < 0.001 control vs leptin, *p < 0.05 wash vs leptin, p > 0.05 wash vs control). E, Mean firing frequency remained depressed after the washout of leptin (n = 17, ***p < 0.001 leptin vs control and wash vs control).
Leptin hyperpolarized membrane potential by ≥5 mV in 68% (21 of 31) of the cells examined, and the effect was reversible in 53% of responders (Fig. 1D; control, −51.3 ± 1.9 mV; leptin, −62.4 ± 2.3 mV; wash, −56.9 ± 2.8 mV; p < 0.0001, F(2,32) = 14.77). However, the reduction in spike frequency only partially reversed in 29% of neurons during a wash period of 10–30 min (Fig. 1E; control, 5.9 ± 0.8 Hz; leptin, 1.3 ± 0.5 Hz; wash, 1.8 ± 0.6 Hz; p < 0.0001, F(2,32) = 24.18), consistent with the long-term effects of leptin and previous observations in other systems. It should be noted that these percentages are based on recordings from GFP-identified cells in a transgenic model that labels only a subset of OX neurons (Sakurai et al., 1999).
Although OX neurons themselves do not express LepRb (Leinninger et al., 2009; Louis et al., 2010; Laque et al., 2013), our previous studies reveal that LHA LepRb and LHA Nts neurons lie in close contact with OX neurons (Louis et al., 2010; Leinninger et al., 2011). We thus hypothesized that leptin indirectly modulates OX neuron function via action at presynaptic LHA LepRbNts neurons (neurons that coexpress LepRb and Nts are restricted to the LHA). Indeed, deletion of LepRb from LepRbNts neurons in LeprNtsKO mice prevents the accumulation of c-Fos in OX neurons during fasting, as well as the modulation of Ox mRNA by leptin (Leinninger et al., 2011). To examine the functional connectivity between LHA Nts neurons and OX neurons, we used a recombinant adeno-associated virus (AAV) that mediates the cre-dependent expression of DREADDs (designer receptors exclusively activated by designer drugs; expressed as DREADD–mCherry fusion proteins), which are genetically engineered muscarinic receptor variants that are insensitive to endogenous ligands but are activated by the otherwise biologically inert agonist CNO. CNO activates neurons containing the Gq-coupled DREADD, hM3Dq (Armbruster et al., 2007; Alexander et al., 2009; Krashes et al., 2011).
Thus, the injection of the cre-inducible DREADD–mCherry AAV into the LHA of Ntscre mice will promote the expression of hM3Dq specifically in LHA Nts neurons.
We injected the cre-inducible hM3Dq virus into the LHA of Ntscre animals on the OX–EGFP background to permit the electrophysiological examination of OX neurons during the selective activation of LHA Nts cells. In this system, acute CNO (5 μm) application to hypothalamic slices from animals expressing hM3Dq DREADDs in LHA Nts neurons hyperpolarized membrane potential in 50% (6 of 12) of OX neurons (Fig. 2A,B; control, −49.2 ± 1.8, CNO, −56.7 ± 2.5; p = 0.003, t(11) = 3.80) and decreased their action potential firing (Fig. 2C; control, 4.0 ± 1.4 Hz; CNO, 1.0 ± 0.5 Hz; p = 0.04, t(8) = 1.98), similar to the results observed with acute leptin treatment. This suggests that LHA Nts neurons are functionally afferent to OX neurons, and the activation of LHA Nts neurons inhibits OX neurons. Note that, although we could not confine DREADD expression to LepRbNts neurons specifically, DREADD expression in LHA Nts neurons includes LepRbNts neurons (as well as non-LepRb Nts cells in the LHA), suggesting that LepRbNts neurons may mediate the inhibition of OX neurons by leptin. Importantly, because leptin (like the hM3Dq DREADD) presumably activates LepRbNts cells (Leinninger et al., 2011), the inhibition of OX neurons by activation of LHA Nts cells is consistent with the potential role for leptin action on LepRbNts cells in the suppression of OX neuron activity.
Figure 2.
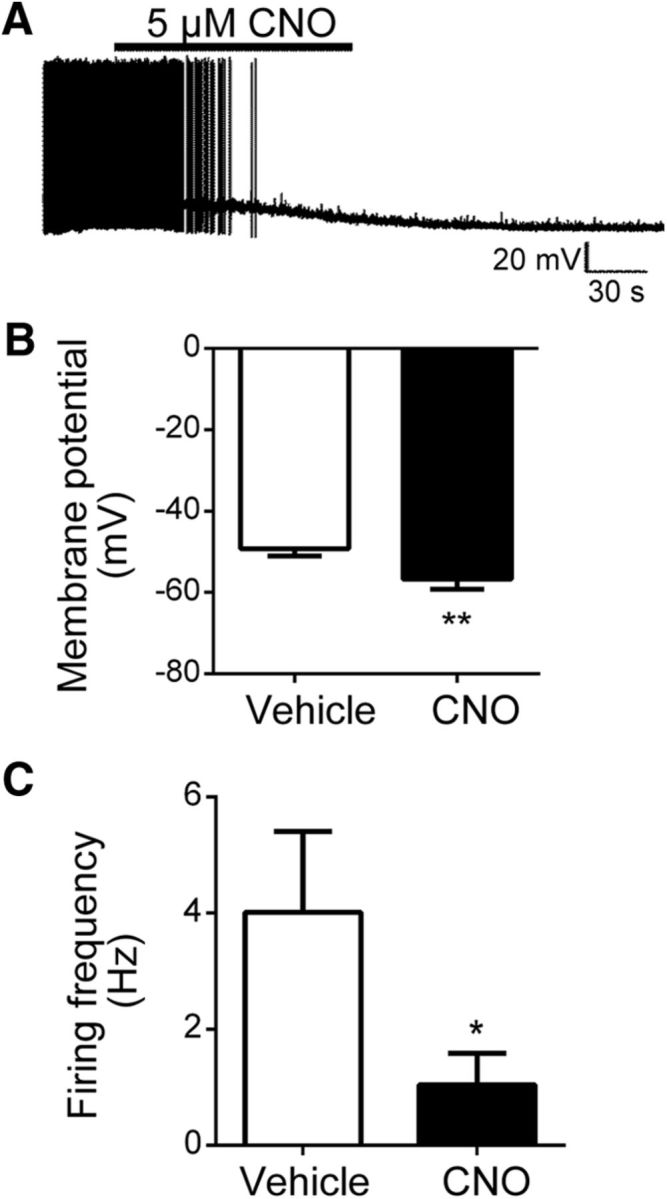
Selective stimulation of LHA Nts neurons via activation of the excitatory DREADD, hM3Dq, inhibits OX neuron activity. A, Current-clamp recording depicting CNO-induced membrane hyperpolarization in an individual OX neuron from a mouse expressing hM3Dq in LHA Nts neurons. Acute CNO application significantly reduced mean OX neuron membrane potential (B; n = 12, **p < 0.01) and suppressed mean action potential firing in OX neurons (C; n = 12, *p < 0.05).
To directly examine leptin regulation of OX neurons by LepRbNts neurons, we bred Ntscre;Leprflox/flox (LeprNtsKO) mice onto the OX–EGFP background to identify and record from OX neurons in hypothalamic slices of animals lacking leptin receptors in LHA Nts neurons. The effects of leptin that we observed in control animals was attenuated in LeprNtsKO mice. Although the basal membrane potential and action potential frequency of OX neurons did not differ between LeprNtsKO mice and littermate controls, leptin failed to modulate activity in most OX neurons of LeprNtsKO mice (Fig. 3A). Although leptin hyperpolarized (≥5 mV) 78% of OX neurons (seven of nine) from control mice (Fig. 3B; control, −51.6 ± 1.1 mV; leptin, −61.1 ± 3.0 mV; p = 0.008, t(8) = 3.50), leptin action was significantly diminished in LeprNtsKO mice because only 20% (2 of 10) of OX neurons exhibited leptin-induced hyperpolarization, with no significant decrease in mean membrane potential (control, −53.4 ± 1.5 mV; leptin, −54.8 ± 2.2 mV; p = 0.45, t(9) = 0.79). Similarly, leptin reduced action potential firing in OX neurons from control mice (Fig. 3C; control, 4.4 ± 1.0 Hz; leptin, 1.2 ± 0.8 Hz; p = 0.0007, t(8) = 5.39) but had no significant effect on the firing of OX neurons from LeprNtsKO mice (control, 5.0 ± 1.6 Hz; leptin, 4.2 ± 2.0 Hz; p = 0.32, t(9) = 1.05). Thus, LHA-restricted LepRbNts neurons play an important role in the modulation of OX neuron activity by leptin, consistent with the blunted activation of OX neurons observed during fasting in LeprNtsKO mice (Leinninger et al., 2011).
Figure 3.

LepRb deletion from LHA Nts neurons attenuates leptin-induced suppression of OX neuron activity. A, Current-clamp recordings of membrane potential from an individual OX neuron in a brain slice preparation from Nts LepRb KO mice show reduced responsiveness to acute leptin. Acute leptin treatment of brain slices from Nts LepRb KO mice did not significantly alter mean OX neuron membrane potential (B) or mean action potential firing (C; n = 9, *p < 0.05 for control and n = 10, p > 0.05 for KO).
GABA-independent inhibition of OX neurons by leptin
LHA LepRb neurons express GAD1 and vGat, consistent with the GABAergic nature of these cells (Leinninger et al., 2009; Vong et al., 2011) and suggesting that leptin might inhibit OX neurons by the direct release of GABA onto OX cells. Indeed, application of the GABAA receptor agonist muscimol or the GABAB receptor agonist baclofen hyperpolarizes OX neurons (Xie et al., 2006; Matsuki et al., 2009), demonstrating that GABA signaling can inhibit OX neurons.
Therefore, we assessed the effect of leptin on GABA-mediated inhibitory synaptic transmission by measuring sIPSCs from OX neurons voltage clamped to −65 mV in the presence of the glutamate receptor blockers APV (20 μm) and CNQX (10 μm) and using symmetrical intracellular and extracellular Cl−. These conditions produced inward sIPSCs that were completely abolished by the GABAA antagonist BMI (30 μm; data not shown), confirming their mediation by GABAA receptors. Mean sIPSC frequency was 0.57 ± 0.13 Hz (range, 0.09–1.02 Hz). Mean sIPSC amplitude was 53.4 ± 9.4 pA (range, 29.2–99.4 pA). This relatively low frequency of sIPSCs at baseline is consistent with previous observations (Horvath and Gao, 2005). Leptin variably affected the sIPSCs of OX neurons, with a subset of neurons exhibiting enhanced sIPSC frequency (three of seven) and/or amplitude (three of seven) and a subset having decreased sIPSC frequency (four of seven) and/or amplitude (three of seven; Fig. 4). Overall, we observed no significant differences in sIPSC frequency or amplitude in the presence of leptin that could account for leptin-induced inhibition of OX activity that we observed. Thus, leptin neither shifted the cumulative distribution of sIPSC amplitude or interevent interval (Fig. 4D,E) nor altered mean normalized sIPSC frequency (Fig. 4D, inset; 92 ± 18% of control, p = 0.13, t(6) = 1.75) or amplitude (Fig. 4E, inset; 121 ± 17% of control, p = 0.28, t(6) = 1.19). These findings suggest that, although LepRbNts neurons may synthesize and package GABA into vesicles, increased GABA-mediated synaptic transmission onto OX neurons is unlikely to underlie leptin-induced OX neuron inhibition.
Figure 4.

Leptin does not enhance GABA-mediated synaptic input to OX neurons. A, Voltage-clamp recording of sIPSCs (downward deflections) from an individual OX neuron, clamped to −65 mV with symmetrical intracellular/extracellular Cl−, before and after acute leptin treatment. Time course of average sIPSC interevent interval (B) and sIPSC amplitude in response to leptin (C). Although subsets of OX neurons displayed either increased or decreased sIPSC frequency and/or sIPSC amplitude in response to leptin, cumulative probability distributions for control (black line) and leptin (gray line) reveal no overall shift in sIPSC interevent interval (D; inset mean normalized sIPSC frequency) or sIPSC amplitude (E; inset: mean normalized sIPSC amplitude). n = 7, p > 0.05.
Furthermore, leptin-induced OX inhibition persisted in the presence of the selective GABAA or GABAB receptor antagonists BMI (30 μm) and CGP52432 (10 μm), respectively (Fig. 5). Leptin-induced hyperpolarization of OX membrane potential persisted in the presence of BMI (BMI, −50.1 ± 0.5; BMI plus leptin, −55.0 ± 1.3; leptin only, −55.8 ± 1.5 mV; Fig. 5A,B, p = 0.003, F(2,12) = 9.69), with a subset of neurons displaying intermittent bursts of activity during treatment with BMI plus leptin (Fig. 5A), likely attributable to changes in excitatory/inhibitory balance caused by BMI. Similarly, CGP52432, which occludes baclofen-induced (and GABAB-mediated) inhibition of OX activity (Xie et al., 2006), did not prevent leptin-induced hyperpolarization of OX neurons (control, −48.3 ± 3.8; CGP52432 plus leptin, −58.8 ± 4.7; leptin only, −64.0 ± 3.1 mV; Fig. 5C,D, p = 0.006, F(2,4) = 25.01).
Figure 5.

GABA receptor antagonists do not prevent leptin-induced inhibition of OX neurons. A, Current-clamp recording of membrane potential in an individual neuron in response to coapplication of BMI (30 μm) plus leptin (10 nm). B, Leptin significantly hyperpolarized mean resting OX membrane potential in the presence of BMI (n = 7, *p < 0.05 for BMI plus leptin and leptin only vs BMI). C, Current-clamp recording of membrane potential in an individual neuron in response to coapplication of CGP52432 (10 μm) plus leptin (10 nm). D, Leptin significantly hyperpolarized mean resting OX membrane potential in the presence of CGP52432 (n = 3, **p < 0.01 for CGP plus leptin and leptin only vs vehicle control).
Leptin suppresses excitatory synaptic input onto OX neurons
Excitatory input to OX neurons is substantially greater than inhibitory input to OX cells, and the excitatory input is modulated by a variety of stimuli (Li et al., 2002; Acuna-Goycolea and van den Pol, 2004; Acuna-Goycolea et al., 2004; Fu et al., 2004; Horvath and Gao, 2005; Liu and Gao, 2007; Rao et al., 2007, 2013), including short-term (24 h) fasting (Horvath and Gao, 2005), which decreases circulating leptin concentrations. To investigate whether leptin modulates excitatory input to OX neurons, we recorded sEPSCs in OX–EGFP neurons, voltage clamped to −50 mV, before and after the application of leptin (10 nm). At −50 mV, sIPSCs and sEPSCs mediate outward and inward currents, respectively, under our recording conditions (Fig. 6A). sEPSCs (inward currents) were blocked by APV (20 μm) and CNQX (10 μm), confirming their mediation by glutamate receptors (data not shown). Mean sEPSC frequency was 4.2 ± 0.8 Hz (range, 0.9–14.4 Hz), and mean sEPSC amplitude was 22.6 ± 1.6 pA (range, 12.8–42.3 pA). Acute leptin significantly decreased sEPSC frequency (14 of 18 neurons), manifested as a prolongation (rightward shift) of sEPSC interevent interval, but had no effect on the sEPSC amplitude distribution (Fig. 6B–E). Likewise, leptin decreased mean normalized sEPSC frequency (Fig. 6D, inset; 68.3 ± 8.1% of control, p = 0.001, t(17) = 3.92) but did not alter mean normalized sEPSC amplitude (Fig. 6E, inset; 99.8 ± 3.5% of control, p = 0.93, t(17) = 0.09). Leptin did not alter sEPSC rise time, half-width, or charge transfer (data not shown). The specific decrease in sEPSC frequency suggests leptin-induced alterations in presynaptic glutamate release rather than changes in postsynaptic glutamate receptor function.
Figure 6.

Leptin suppresses sEPSC input to OX neurons. A, Voltage-clamp recording of sEPSCs (downward deflections) from an individual OX neuron, clamped to −50 mV before and after acute leptin treatment. Time course of average sEPSC interevent interval (B) and sEPSC amplitude in response to leptin (C). D, Leptin decreased sEPSC frequency, as indicated by a rightward shift of the cumulative probability distribution of sEPSC interevent interval (n = 18, **p < 0.001) and a significant reduction of mean normalized sEPSC frequency (inset: n = 18, p < 0.01). E, Leptin did not significantly alter the distribution of sEPSC event amplitudes or mean normalized sEPSC amplitude (inset; n = 18, p > 0.05). F, Coapplication of CGP52432 did not reverse leptin-induced suppression of sEPSC frequency, indicated by a significant reduction in mean normalized sEPSC frequency in the presence of leptin (n = 6, **p < 0.01) and leptin plus CGP52432 (n = 6, *p < 0.05).
The pharmacologic activation of GABAB receptors can inhibit presynaptic glutamate release onto OX neurons (resulting in reduced sEPSC frequency but not amplitude). Thus, GABA release from LHA LepRb neurons could theoretically act presynaptically on GABAB receptors to decrease sEPSC frequency onto OX neurons. Therefore, we tested the ability for the GABAB antagonist CGP52432 to ameliorate leptin-induced suppression of sEPSC frequency in OX neurons. As shown in Figure 6F, CGP52432 had no significant effect on the leptin-induced decrease in sEPSC frequency (normalized frequency: leptin, 63.8 ± 9.5% of control; leptin plus CGP52432, 74.9 ± 11.9% of control; p = 0.007, F(2,10) = 8.45). This finding is consistent with the failure of CGP52432 to reverse the leptin-induced hyperpolarization of OX neurons (Fig. 5C,D) and with a GABA-independent mechanism of OX regulation by leptin.
Leptin elicits KATP channel activation in OX neurons
In addition to suppressing excitatory transmission, leptin decreased the input resistance of OX neurons measured in current clamp (data not shown). Indeed, leptin elicited an outward current in 68% (13 of 19) of OX neurons voltage clamped to −50 mV, with a mean amplitude of 24.7 ± 9.2 pA (Fig. 7A). To identify the channel(s) underlying this current, we used voltage ramp protocols to generate I–V relationships in the presence and absence of leptin (10 nm) and calculated leptin-elicited difference currents. As shown in Figure 7, B and C, leptin activated a current that exhibited a leftward shift in reversal potential and significantly increased whole-cell conductance compared with controls (Fig. 7D; control, 1.3 ± 0.2 nS; leptin, 2.3 ± 0.3 nS; p = 0.0003, t(16) = 4.58). The mean reversal potential of leptin-activated difference currents, −94.3 ± 3.5 mV (adjusted here for liquid junction potential) was near the calculated K+ equilibrium potential of −101 mV for the recording solutions we used, strongly suggesting activation of a K+ channel. OX neurons possess a multitude of potassium channels capable of producing a similar degree of hyperpolarization to that observed with leptin, including ATP-sensitive potassium (KATP) channels, and the opening of OX KATP channels generates currents having similar rectification, reversal potential, and conductance as observed for leptin-elicited current (Parsons and Hirasawa, 2010; Liu et al., 2011; Parsons et al., 2012a,b; Karnani et al., 2011). Therefore, we tested the sensitivity of the leptin-elicited current to the selective KATP blocker tolbutamide (200 μm). A comparison of I–V curves generated in the presence of leptin with or without tolbutamide show that tolbutamide blocked the leptin-activated current (Fig. 8A) and reversed the leptin-induced increase in mean whole-cell conductance (Fig. 8B; control, 1.4 ± 0.3 nS; leptin, 2.3 ± 0.6 nS; leptin/tolbutamide, 1.3 ± 0.3 nS; p = 0.01, F(2,12) = 6.49). In further support of leptin-induced activation of OX KATP channels, the coapplication of leptin with tolbutamide also reversed the leptin-induced hyperpolarization (Fig. 8C). Mean OX membrane potential during leptin treatment (−53.9 ± 1.2 mV) was significantly hyperpolarized compared with vehicle (−47.1 ± 2.6 mV) and leptin plus tolbutamide (−48.5 ± 2.2, p = 0.03, F(2,6) = 6.32), whereas membrane potential in the presence of leptin with tolbutamide did not differ from control (Fig. 8D).
Figure 7.

Leptin activates an outward current in OX neurons. A, Voltage-clamp recording from an individual OX neuron clamped to −50 mV depicting the activation of an outward current in response to acute leptin. B, Representative whole-cell currents recorded from an individual OX neuron in response to linear voltage ramps from −100 to −40 mV in the presence (gray) and absence (black) of 10 nm leptin. C, Representative leptin-activated whole-cell current (presence of leptin minus control) plotted against membrane potential. D, Leptin significantly increased mean whole-cell conductance measured from −100 to −50 mV (n = 17, ***p < 0.001).
Figure 8.

Leptin induces the activation of KATP channels in OX neurons. A, Representative whole-cell currents recorded from an individual OX neuron in response to linear voltage ramps from −100 to −40 mV in the presence of 10 nm leptin (gray) and 10 nm leptin plus 200 μm tolbutamide (black). B, The coapplication of leptin plus tolbutamide reversed the leptin-induced increase in mean whole-cell conductance (n = 7, *p < 0.05 for control vs leptin; *p < 0.05 for leptin vs leptin plus tolbutamide; p > 0.05 for control vs leptin plus tolbutamide). C, Current-clamp recording of membrane potential from an individual OX neuron in response to the application of leptin followed by leptin plus tolbutamide. D, The coapplication of leptin plus tolbutamide reversed leptin-induced hyperpolarization of OX membrane potential (n = 6, *p < 0.05 for control vs leptin; *p < 0.05 for leptin vs leptin plus tolbutamide; p > 0.05 for control vs leptin plus tolbutamide). E, The coapplication of leptin plus tolbutamide did not reverse the leptin-induced decrease in sEPSC frequency. Graph depicts mean normalized sEPSC frequency (n = 9; **p < 0.01 for control vs leptin; ***p < 0.001 for control vs leptin plus tolbutamide; *p < 0.05 for leptin vs leptin plus tolbutamide).
Because OX neurons are proposed to directly and indirectly excite other LHA OX neurons via glutamatergic transmission and OX peptide signaling (Li et al., 2002; Yamanaka et al., 2010), we examined whether the reduction in sEPSC frequency that we observed with leptin was caused by the opening of KATP channels and consequent suppression of activity in neighboring OX neurons. Thus, we tested whether the leptin-induced inhibition of sEPSCs was blocked by the application of tolbutamide, which restored OX neuronal activity in the continued presence of leptin. However, as shown in Figure 8E, tolbutamide did not reverse the leptin-induced suppression of sEPSC frequency (normalized frequency: leptin, 68% of control; leptin plus tolbutamide, 48% of control; p < 0.0001, F(2,16) = 38.66). This implies that the reduced activity of neighboring OX neurons attributable to leptin activation of KATP channels does not underlie the decrease in excitatory input in response to leptin. Interestingly, sEPSC frequency decreased further in the presence of leptin plus tolbutamide versus leptin alone, suggesting that KATP channel blockade in as yet undefined neurons may suppress excitatory input onto OX neurons.
Gal receptor signaling is required for leptin action
Our findings indicate that leptin does not regulate OX neurons via GABA signaling but that other neurotransmitters and/or neuropeptides are released in response to leptin and mediate OX inhibition. In addition to Nts, LepRbNts neurons express the neuropeptide Gal (Laque et al., 2013). Thus, we examined whether acute application of Nts and/or Gal modulated OX activity in hypothalamic slices. Although we observed no significant effect of Nts (100 nm) on Ox activity overall, Nts tended to stimulate OX activity by depolarizing membrane potential in four of six OX neurons (control, −56.9 ± 3.1 mV; Nts, −53.8 ± 4.0 mV; p = 0.15, t(5) = 1.69) and increasing action potential firing in three of five OX neurons (mean normalized frequency, 156% of control; p = 0.41, t(4) = 0.91, Fig. 9A,B). Conversely, Gal (100 nm) hyperpolarized membrane potential in 55% (6 of 11) of OX neurons (Fig. 9C–E; Vm: control, −53.7 ± 2.3 mV; Gal, −57.6 ± 3.4 mV; p = 0.03, t(10) = 2.47) and decreased action potential firing in seven of eight OX neurons (mean normalized frequency, 44% of control; p = 0.003, t(7) = 4.46). Although these findings demonstrate an ability for Gal to inhibit OX activity, they are inconsistent with a role for Nts in the leptin-induced inhibition of OX neurons. Furthermore, coapplication of the Gal receptor antagonist M40 (Bartfai et al., 1993) prevented leptin-induced hyperpolarization of OX neurons (Figure 10; Vm: control, −48.7 ± 1.2 mV; M40, −48.0 ± 1.1 mV; M40/leptin, −49.2 ± 1.1 mV; leptin only, −53.0 ± 0.3 mV; p = 0.0005, F(3,15) = 10.85), suggesting a role for Gal release and receptor activation in the regulation of orexin neurons by leptin.
Figure 9.

The effect of Nts and Gal on OX activity. Acute Nts (100 nm) treatment did not significantly alter mean OX neuron membrane potential (A) or mean action potential firing (B; n = 5–6, p < 0.05). C, Current-clamp recording of membrane potential and action potential firing from an individual OX–EGFP neuron in response to 100 nm Gal. Gal significantly hyperpolarized the mean membrane potential (D; n = 11, *p < 0.05) and reduced action potential firing of LHA OX neurons (E; n = 8, **p < 0.01).
Figure 10.

Gal receptor antagonism prevents leptin inhibition of OX neurons. A, Current-clamp recording of membrane potential from an OX neuron in response to M40 (100 μm) followed by M40 plus leptin (10 nm) and leptin only. B, Coapplication of M40 plus leptin prevented the hyperpolarization of OX neuron membrane potential versus leptin only (n = 6, **p < 0.01).
Discussion
Fasting activates OX neurons, presumably to increase overall activity, alertness, and food-seeking behavior, thereby increasing feeding in response to negative energy balance. Decreased leptin levels mediate a crucial component of this effect, because leptin hyperpolarizes and inhibits OX neurons in hypothalamic slices and leptin replacement inhibits the activation of OX neurons during fasting. Although it was originally postulated that direct leptin action on OX neurons might mediate this inhibition, OX neurons do not express LepRb (Leinninger et al., 2009; Louis et al., 2010; Laque et al., 2013). However, neighboring LHA LepRb neurons lie in close contact with OX neurons, as do LHA Nts cells, many of which contain LepRb. Furthermore, deletion of LepRb from LepRbNts cells (which are restricted to the LHA) prevents the modulation of Ox mRNA by leptin, as well as blocks the accumulation of c-Fos in OX neurons during fasting, suggesting a role for LepRbNts neurons in the control of OX neurons by leptin (Leinninger et al., 2011). Indeed, our present data reveal that leptin inhibits OX neurons indirectly, by acting on LHA LepRbNts cells. Furthermore, we demonstrate that two separate GABA-independent mechanisms mediate this inhibition: (1) the postsynaptic activation of an OX neuron KATP channel; and (2) presynaptic suppression of excitatory input onto OX neurons.
The stimulation of LHA Nts neurons inhibited the activity of OX neurons, and the ability of leptin to hyperpolarize and decrease the activity of OX neurons was abrogated in LeprNtsKO mice. However, residual leptin responsiveness persisted in a small percentage of the OX neurons of LeprNtsKO animals. Thus, although leptin regulation of OX neurons is predominantly mediated by LHA LepRbNts neurons, other LHA (non-Nts) LepRb neurons may also contribute, albeit to a lesser extent.
LHA LepRb neurons contain the GABA-synthesizing enzyme GAD1, as well as vGAT (which transports GABA into synaptic vesicles for release; Leinninger et al., 2009; Vong et al., 2011), suggesting their ability to release inhibitory GABA onto their synaptic targets. Because the expression of the trans-synaptic tracer wheat germ agglutinin in LHA LepRb or LHA Nts neurons accumulates in OX neurons (Louis et al., 2010; Leinninger et al., 2011), we reasoned that LHA LepRb neurons might mediate the inhibition of OX neuron activity via direct GABA release onto these cells. However, leptin failed to significantly potentiate GABAA-mediated sIPSCs in OX neurons. Furthermore, the inhibition of neither GABAA nor GABAB receptors altered the ability of leptin to hyperpolarize OX neurons. Although there are inherent limitations when using a pharmacological approach to assess the involvement of specific receptor subtypes in physiological action, our data indicate that GABA action at the two main GABA receptor subtypes does not underlie leptin modulation of OX function. Rather, leptin decreased the frequency, but not amplitude, of sEPSCs in OX neurons, consistent with a presynaptic effect. GABAB receptor antagonism also failed to attenuate this leptin-promoted inhibition of sEPSCs in OX neurons. Thus, the inhibition of OX neurons by leptin is not mediated by GABA but is attributable, in part, to the GABA-independent inhibition of excitatory input to these cells. This selective modulation of excitatory synaptic transmission is not surprising given that the majority of synaptic input that we measured in OX neurons was glutamate-dependent excitatory transmission, consistent with the predominantly excitatory synaptic architecture of OX neurons (Horvath and Gao, 2005).
A single 24 h fasting episode in vivo increases the frequency of mEPSCs and the number of excitatory synapses present on OX cell bodies (Horvath and Gao, 2005). These data imply that normal levels of circulating leptin exert a tonic inhibitory effect on OX neurons, partly because of the suppression of excitatory neurotransmission, and that the removal of this restraining influence not only enhances OX activity but may induce more long-lasting plasticity. In this way, leptin levels may set the dynamic range for the excitation of OX neurons and influence the integration of other physiological stimuli that modulate glutamatergic synaptic transmission in OX neurons.
In addition to the effects of leptin on excitatory inputs to OX neurons, leptin activated a KATP channel in these neurons, which contributes to the leptin-mediated hyperpolarization of OX neurons. Leptin-induced KATP activation is consistent with a reduced need for OX neuron activity, and therefore foraging behavior, during times of energy abundance and vice versa. OX KATP channels, which comprise Kir6.1/SUR1 subunits (Parsons and Hirasawa, 2010), do not directly sense glucose (González et al., 2008), but their activity is linked to multiple indicators of nutritional and energy status (Parsons and Hirasawa, 2010; Karnani et al., 2011; Liu et al., 2011). Thus, leptin modulation of OX KATP channels provides an additional mechanism by which OX neurons may integrate signals of energy state.
Afferents to OX neurons originate from diverse neuronal populations that reside both within and outside the LHA (Yoshida et al., 2006). Although the identities of the glutamatergic projections to OX neurons are not fully resolved, some evidence supports their regulation by local glutamatergic cells in the LHA, including other OX neurons (Li et al., 2002; Burt et al., 2011). In fact, OX peptides are autoregulatory and have been shown to stimulate OX neuron activity both directly (Yamanaka et al., 2010) and indirectly by enhancing excitatory input from LHA glutamatergic interneurons (Li et al., 2002). However, a mechanism whereby reduced sEPSC frequency occurs secondary to the silencing of neighboring and afferent OX neurons cannot explain the leptin-mediated inhibition of sEPSCs onto OX neurons, because tolbutamide, which restored OX neuron activity, failed to reverse the decrease in sEPSC frequency in response to leptin. It is possible that leptin inhibits the activity of other (non-OX) glutamatergic neurons that provide excitatory synaptic input to OX neurons. Alternatively, decreased sEPSC frequency may reflect presynaptic inhibition of glutamate release from axon terminals that are in synaptic contact with the OX neurons.
Despite the GABAergic nature of LHA LepRb neurons (Leinninger et al., 2009), leptin-mediated inhibition of OX neurons does not appear to involve the GABA system. Consistently, the disruption of GABA signaling in LepRb neurons minimally increases food intake and body weight, although animals lacking LepRb in GABA neurons are extremely hyperphagic and obese (Vong et al., 2011; Xu et al., 2012). Thus, although GABAergic LepRb neurons are integral to leptin action, GABA does not appear to represent the major neurotransmitter that mediates leptin action. Indeed, our results suggest that leptin regulation of OX neurons requires the involvement of other neurotransmitters and/or neuropeptides. Although LHA LepRbNts neurons contain Nts, as well as GABA, this peptide tended to stimulate OX activity and thus seems an unlikely mediator of leptin-mediated inhibitory signaling. In contrast, a recent publication demonstrates that some LHA LepRb neurons contain the inhibitory peptide Gal, which also colocalizes extensively with Nts in the LHA (Laque et al., 2013). Furthermore, Gal has been shown to activate KATP channels (de Weille et al., 1988; Dunne et al., 1989; Zini et al., 1993) and inhibit synaptic glutamate release (Zini et al., 1993; Kinney et al., 1998). In this study, we further demonstrate that Gal inhibits OX activity whereas Gal receptor blockade prevents the hyperpolarization of OX neurons by leptin, suggesting that Gal secretion from LepRbNts neurons and subsequent Gal receptor activation contributes to leptin regulation of OX function. Here, we demonstrate the ability for LHA LepRb neurons to inhibit OX neurons in the LHA. Our results are consistent with a model whereby leptin exerts a tonic inhibitory effect on OX neurons, and their disinhibition by decreasing leptin levels stimulates OX neuron activity to trigger arousal, locomotor activity, and feeding during periods of acute starvation. Although our studies address the response of OX neurons to acute changes in leptin levels, it will be important for future studies to examine how OX neurons integrate signals, including leptin, over the long term to maintain energy homeostasis.
Footnotes
This work was supported by the Animal Phenotyping Core of the Michigan Diabetes Research Center [National Institutes of Health (NIH) Grant P30 DK020572], the Michigan Nutrition and Obesity Research Center (NIH Grant P30 DK089503), the American Diabetes Association, the American Heart Association, the Marilyn H. Vincent Foundation (M.G.M.), and NIH Grants DK078056 (M.G.M.), DK090101 (G.M.L.), and DK46409 (L.S.S.). We thank Amylin Pharmaceuticals for the generous gift of leptin, Dr. Yuchio Yanagawa for OX-EGFP mice, Dr. Bryan Roth for CNO, and members of the Myers laboratory for helpful discussions.
References
- Acuna-Goycolea C, van den Pol A. Glucagon-like peptide 1 excites hypocretin/orexin neurons by direct and indirect mechanisms: implications for viscera-mediated arousal. J Neurosci. 2004;24:8141–8152. doi: 10.1523/JNEUROSCI.1607-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Goycolea C, Li Y, Van Den Pol AN. Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci. 2004;24:3013–3022. doi: 10.1523/JNEUROSCI.5416-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfai T, Langel U, Bedecs K, Andell S, Land T, Gregersen S, Ahrén B, Girotti P, Consolo S, Corwin R, Crawleys J, Xu X, Wiesenfeld-Hallin Z, Hokfelt T., II Galanin-receptor ligand M40 peptide distinguishes between putative galanin-receptor subtypes. Proc Natl Acad Sci U S A. 1993;90:11287–11291. doi: 10.1073/pnas.90.23.11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt J, Alberto CO, Parsons MP, Hirasawa M. Local network regulation of orexin neurons in the lateral hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2011;301:R572–R580. doi: 10.1152/ajpregu.00674.2010. [DOI] [PubMed] [Google Scholar]
- de Weille J, Schmid-Antomarchi H, Fosset M, Lazdunski M. ATP-sensitive K+ channels that are blocked by hypoglycemia-inducing sulfonylureas in insulin-secreting cells are activated by galanin, a hyperglycemia-inducing hormone. Proc Natl Acad Sci U S A. 1988;85:1312–1316. doi: 10.1073/pnas.85.4.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Horvath B, Urbanski HF, Sotonyi P, Horvath TL. Fasting activates the nonhuman primate hypocretin (orexin) system and its postsynaptic targets. Endocrinology. 2003;144:3774–3778. doi: 10.1210/en.2003-0274. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, Bullett MJ, Li GD, Wollheim CB, Petersen OH. Galanin activates nucleotide-dependent K+ channels in insulin-secreting cells via a pertussis toxin-sensitive G-protein. EMBO J. 1989;8:413–420. doi: 10.1002/j.1460-2075.1989.tb03392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LY, Acuna-Goycolea C, van den Pol AN. Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci. 2004;24:8741–8751. doi: 10.1523/JNEUROSCI.2268-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JA, Jensen LT, Fugger L, Burdakov D. Metabolism-independent sugar sensing in central orexin neurons. Diabetes. 2008;57:2569–2576. doi: 10.2337/db08-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metab. 2005;1:279–286. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Jones DN, Gartlon J, Parker F, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Hatcher JP, Johns A, Porter RA, Hagan JJ, Hunter AJ, Upton N. Effects of centrally administered orexin-B and orexin-A: a role for orexin-1 receptors in orexin-B-induced hyperactivity. Psychopharmacology (Berl) 2001;153:210–218. doi: 10.1007/s002130000551. [DOI] [PubMed] [Google Scholar]
- Karnani MM, Apergis-Schoute J, Adamantidis A, Jensen LT, de Lecea L, Fugger L, Burdakov D. Activation of central orexin/hypocretin neurons by dietary amino acids. Neuron. 2011;72:616–629. doi: 10.1016/j.neuron.2011.08.027. [DOI] [PubMed] [Google Scholar]
- Kinney GA, Emmerson PJ, Miller RJ. Galanin receptor-mediated inhibition of glutamate release in the arcuate nucleus of the hypothalamus. J Neurosci. 1998;18:3489–3500. doi: 10.1523/JNEUROSCI.18-10-03489.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laque A, Zhang Y, Gettys S, Nguyen TA, Bui K, Morrison CD, Münzberg H. Leptin receptor neurons in the mouse hypothalamus are co-localized with the neuropeptide galanin and mediate anorexigenic leptin action. Am J Physiol Endocrinol Metab. 2013;304:E999–E1011. doi: 10.1152/ajpendo.00643.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG., Jr Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Opland DM, Jo YH, Faouzi M, Christensen L, Cappellucci LA, Rhodes CJ, Gnegy ME, Becker JB, Pothos EN, Seasholtz AF, Thompson RC, Myers MG., Jr Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 2011;14:313–323. doi: 10.1016/j.cmet.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/S0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Liu ZW, Gao XB. Adenosine inhibits activity of hypocretin/orexin neurons by the A1 receptor in the lateral hypothalamus: a possible sleep-promoting effect. J Neurophysiol. 2007;97:837–848. doi: 10.1152/jn.00873.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Gan G, Suyama S, Gao XB. Intracellular energy status regulates activity in hypocretin/orexin neurones: a link between energy and behavioural states. J Physiol. 2011;589:4157–4166. doi: 10.1113/jphysiol.2011.212514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis GW, Leinninger GM, Rhodes CJ, Myers MG., Jr Direct innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons. J Neurosci. 2010;30:11278–11287. doi: 10.1523/JNEUROSCI.1340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubkin M, Stricker-Krongrad A. Independent feeding and metabolic actions of orexins in mice. Biochem Biophys Res Commun. 1998;253:241–245. doi: 10.1006/bbrc.1998.9750. [DOI] [PubMed] [Google Scholar]
- Matsuki T, Nomiyama M, Takahira H, Hirashima N, Kunita S, Takahashi S, Yagami K, Kilduff TS, Bettler B, Yanagisawa M, Sakurai T. Selective loss of GABA(B) receptors in orexin-producing neurons results in disrupted sleep/wakefulness architecture. Proc Natl Acad Sci U S A. 2009;106:4459–4464. doi: 10.1073/pnas.0811126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SD, Barrnett RJ, Mayer J. Localization of lesions in the lateral hypothalamus of rats with induced adipsia and aphagia. Am J Physiol. 1958;193:230–234. doi: 10.1152/ajplegacy.1958.193.1.230. [DOI] [PubMed] [Google Scholar]
- Parsons MP, Hirasawa M. ATP-sensitive potassium channel-mediated lactate effect on orexin neurons: implications for brain energetics during arousal. J Neurosci. 2010;30:8061–8070. doi: 10.1523/JNEUROSCI.5741-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Belanger-Willoughby N, Linehan V, Hirasawa M. ATP-sensitive potassium channels mediate the thermosensory response of orexin neurons. J Physiol. 2012a;590:4707–4715. doi: 10.1113/jphysiol.2012.236497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Burt J, Cranford A, Alberto C, Zipperlen K, Hirasawa M. Nociceptin induces hypophagia in the perifornical and lateral hypothalamic area. PLoS One. 2012b;7:e45350. doi: 10.1371/journal.pone.0045350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin B. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Perez-Leighton CE, Boland K, Teske JA, Billington C, Kotz CM. Behavioral responses to orexin, orexin receptor gene expression, and spontaneous physical activity contribute to individual sensitivity to obesity. Am J Physiol Endocrinol Metab. 2012;303:E865–E874. doi: 10.1152/ajpendo.00119.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Liu ZW, Borok E, Rabenstein RL, Shanabrough M, Lu M, Picciotto MR, Horvath TL, Gao XB. Prolonged wakefulness induces experience-dependent synaptic plasticity in mouse hypocretin/orexin neurons. J Clin Invest. 2007;117:4022–4033. doi: 10.1172/JCI32829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Mineur YS, Gan G, Wang AH, Liu ZW, Wu X, Suyama S, de Lecea L, Horvath TL, Picciotto MR, Gao XB. Repeated in vivo exposure of cocaine induces long-lasting synaptic plasticity in hypocretin/orexin neurons in the lateral hypothalamus in mice. J Physiol. 2013;591:1951–1966. doi: 10.1113/jphysiol.2012.246983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richarson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92 doi: 10.1016/S0092-8674(00)80892-2. 1 page following 696. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Moriguchi T, Furuya K, Kajiwara N, Nakamura T, Yanagisawa M, Goto K. Structure and function of human prepro-orexin gene. J Biol Chem. 1999;274:17771–17776. doi: 10.1074/jbc.274.25.17771. [DOI] [PubMed] [Google Scholar]
- Sweet DC, Levine AS, Billington CJ, Kotz CM. Feeding response to central orexins. Brain Res. 1999;821:535–538. doi: 10.1016/S0006-8993(99)01136-1. [DOI] [PubMed] [Google Scholar]
- Toshinai K, Date Y, Murakami N, Shimada M, Mondal MS, Shimbara T, Guan JL, Wang QP, Funahashi H, Sakurai T, Shioda S, Matsukura S, Kangawa K, Nakazato M. Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology. 2003;144:1506–1512. doi: 10.1210/en.2002-220788. [DOI] [PubMed] [Google Scholar]
- Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–176. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Crowder TL, Yamanaka A, Morairty SR, Lewinter RD, Sakurai T, Kilduff TS. GABA(B) receptor-mediated modulation of hypocretin/orexin neurones in mouse hypothalamus. J Physiol. 2006;574:399–414. doi: 10.1113/jphysiol.2006.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, O'Brien WG, 3rd, Lee CC, Myers MG, Jr, Tong Q. Role of GABA release from leptin receptor-expressing neurons in body weight regulation. Endocrinology. 2012;153:2223–2233. doi: 10.1210/en.2011-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami Ki, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/S0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Tabuchi S, Tsunematsu T, Fukazawa Y, Tominaga M. Orexin directly excites orexin neurons through orexin 2 receptor. J Neurosci. 2010;30:12642–12652. doi: 10.1523/JNEUROSCI.2120-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, McCormack S, España RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2006;494:845–861. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zini S, Roisin MP, Langel U, Bartfai T, Ben-Ari Y. Galanin reduces release of endogenous excitatory amino acids in the rat hippocampus. Eur J Pharmacol. 1993;245:1–7. doi: 10.1016/0922-4106(93)90162-3. [DOI] [PubMed] [Google Scholar]