

Abstract
Antiretroviral therapy extends the lifespan of human immunodeficiency virus (HIV)-infected patients, but many survivors develop premature impairments in cognition. These residual cognitive impairments may involve aberrant deposition of amyloid β-peptides (Aβ). By unknown mechanisms, Aβ accumulates in the lysosomal and autophagic compartments of neurons in the HIV-infected brain. Here we identify the molecular events evoked by the HIV coat protein gp120 that facilitate the intraneuronal accumulation of Aβ. We created a triple transgenic gp120/APP/PS1 mouse that recapitulates intraneuronal deposition of Aβ in a manner reminiscent of the HIV-infected brain. In cultured neurons, we found that the HIV coat protein gp120 increased the transcriptional expression of BACE1 through repression of PPARγ, and increased APP expression by promoting interaction of the translation-activating RBP heterogeneous nuclear ribonucleoprotein C with APP mRNA. APP and BACE1 were colocalized into stabilized membrane microdomains, where the β-cleavage of APP and Aβ formation were enhanced. Aβ-peptides became localized to lysosomes that were engorged with sphingomyelin and calcium. Stimulating calcium efflux from lysosomes with a TRPM1 agonist promoted calcium efflux, luminal acidification, and cleared both sphingomyelin and Aβ from lysosomes. These findings suggest that therapeutics targeted to reduce lysosomal pH in neurodegenerative conditions may protect neurons by facilitating the clearance of accumulated sphingolipids and Aβ-peptides.
Keywords: amyloid, dementia, endosome, HIV, lysosome, neuron
Introduction
The use of combinational antiretroviral therapy (cART) has dramatically increased the survival of people infected with the human immunodeficiency virus (HIV). However, several lines of evidence suggest that accelerated brain aging may accompany this extended lifespan. Nearly half of HIV-infected individuals will develop some form of cognitive impairment. The majority of patients showing deficits in attention, executive functioning, memory and processing speed that are reminiscent of advanced age, and overlap with Alzheimer's disease (AD; Heaton et al., 2010). Functional MRI data suggest that HIV infection is associated with premature failure in neuroadaptive mechanisms that normally compensate for age-related declines of attention by tapping into reserve, and top-down attentional networks (Chang et al., 2013). These neuroprocessing deficits may arise from structural damage that degrades temporal and parietal networks at faster rates than expected during normal aging (Chang et al., 2008; Ances et al., 2012a; Holt et al., 2012; Jahanshad et al., 2012). Structural damage associated with cognitive deficits in HIV-infected patients can be detected early in the course of infection and includes increased permeability of the blood–brain barrier, synaptic and dendritic simplification, white matter damage, glial activation, and alterations in protein processing with subsequent accumulation of proteins that may promote neuronal degeneration including amyloid β-peptides (Aβ; Zhong et al., 2008; Ramirez et al., 2010; Eugenin et al., 2011; Akay et al., 2012; Bachis et al., 2012; Ragin et al., 2012; Stubbe-Drger et al., 2012).
There is considerable evidence that Aβ accumulates earlier than expected by age in the brains of HIV-infected subjects. However, the Aβ deposition pattern in HIV appears to be distinct from AD, in which extracellular senile plaques are a predominant feature. In the HIV-infected brain, Aβ accumulates primarily as diffuse and intraneuronal deposits (Cozzi et al., 1992; Esiri et al., 1998; Andersson et al., 1999; Green et al., 2000, 2005; Izycka-Swieszewska et al., 2000; Gelman and Schuenke, 2004; Brew et al., 2005; Anthony et al., 2006; Ances et al., 2010). These pathological findings are consistent with negative results obtained with middle-aged HIV-infected subjects imaged with the Pittsburg compound B (this probe was designed to bind Aβ plaques; Ances et al., 2010, 2012b). The neuropathogenic mechanisms which produce this unique pattern of Aβ deposition in the HIV-infected brain are not understood. Based on considerable data suggesting that the HIV-1 coat protein gp120 contributes to the neurodegenerative process in HIV, and data that the gp120 transgenic mouse mimics clinical, pathologic, and mechanistic aspects of the HAND26-28 we developed a triple transgenic gp120/APP/PS1 mouse that recapitulates intraneuronal pathology consistent with accelerated aging and intracellular Aβ deposition. Here we describe a signaling network evoked by gp120 that results in intracellular accumulation of Aβ into sphingomyelin and calcium engorged lysosomes. We further demonstrate that activation of the mucolipin transient receptor potential channel 1 (TRPML1) induces release of intraluminal calcium, with consequent reductions in endolysosomal pH, sphingomyelin, and Aβ.
Materials and Methods
Cell culture and experimental treatments.
Primary rat cortical and hippocampal neuronal cultures were prepared as described previously (Xu et al., 2011b). Hippocampal neurons were plated at a density of 150,000 cells/ml and cortical neurons were plated at a density of 500,000 cells/ml in neurobasal medium supplemented with B27 and 1% antibiotic solution (104 U of penicillin g/ml, 10 mg streptomycin/ml, and 25 μg amphotericin B/ml; Invitrogen). Three hours after cell plating the medium was completely replaced and supplemented every 7 d with fresh medium containing B27. Hippocampal cultures are routinely >98% MAP-2+ neurons, with the remainder of cells predominantly GFAP+ astrocytes. Cortical cultures were used between 7 and 10 d, and hippocampal cultures were used between 14 and 21 d in vitro. SHSY5Y cells stably transfected with human APP were cultured in DMEM/F12 nutrient mixture containing 10% heat-inactivated fetal bovine serum and 1% antibiotic solution (104 U of penicillin G/ml, 10 mg streptomycin/ml, and 25 mg amphotericin B/ml; Sigma-Aldrich). Cells were maintained at 37°C in 5% CO2 and the culture medium was changed every 3 or 4 d.
Cell treatments included X4-gp120 (IIIB; Advanced Bioscience Laboratories), R5-gp120 (CM; NIH-AIDS Repository), dual X4/R5 gp120 (MN; Immunodiagnostics), and 4-hydroxynonenal (4HNE; Sigma-Aldrich). Cultured cells were treated in culture maintenance medium, unless otherwise indicated.
Pharmacological agents included inhibitors of CXCR4 (AMD3100 octahydrochloride, 10 μm; Tocris Bioscience), protein kinase C (PKC; chelerythrine chloride, 1 μm; Tocris Bioscience), protein kinase A (PKA; KT5720, 1 μm; Tocris Bioscience), protein kinase B (AKT; API-1, 20 μm; Tocris Bioscience), ceramide synthase (Fumonisin B1, 20 μm,), serine palmitoyl transferase (ISP-1, 10 μm; Tocris Bioscience), and a cholesterol complexing agent (β-cyclodextran, 1.5 mm). The cJun N-terminal kinase (JNK) pathway was inhibited with the JNK inhibitor II (SP600125, 20 μm; EMD Millipore), and the interaction between JNK and c-Jun was inhibited with JNK inhibitor III (10 μm; EMD Millipore). Drug controls included a JNK inhibitor II inactive isomer (20 μm; EMD Millipore), and a JNK inhibitor III inactive isomer (10 μm; EMD Millipore). PPARγ agonists included SR202 (20 μm) and T007907 (10 μm). Antagonists of PPARγ included Pioglitazone (20 μm) and Triglitazone (20 μm).
Lysosomal calcium measurement.
The lysosomal-targeted calcium indicator GCaMP3-TRPML1 was transfected into primary neurons by electroporation using two 5 ms pulses at 120 V (BTX ECM830, Harvard Apparatus) immediately following the isolation of primary fetal neurons. This indicator has recently been shown to accurately and reliably measure juxta-lysosomal calcium increases (Shen et al., 2012). Imaging was performed on mature hippocampal neurons 14–21 d in vitro. Fluorescence (F470) images were acquired with a 40× objective lens using a Zeiss inverted microscope (Observer Z1), equipped with Apotome for optical sectioning, and Axiovision 4.8 software. Lysosomal calcium release was induced with the TRPML1 agonist ML-SA1 (20 μm), as previously reported (Shen et al., 2012).
Transgenic mice.
Two- and 6-month-old C57BL-6 nontransgenic (nTg) mice, C57BL-6 mice transgenic for gp120 (gp120tg), transgenic C57BL-6 mice expressing mutations in human amyloid precursor protein (APPSWE; K670N/M671L) and human presenilin 1 (PS1; dE9), and gp120/APP/PS1 transgenic mice were used in these studies. Gp120 mice were obtained from Dr Lennart Muke (Gladstone Institute, San Fransicso, CA) and express a GFAP promoter-driven HIV-1 env gene (HIV-1LAV; Toggas et al., 1994). HIV-gp120 mice were back-crossed for >10 generations. APP/PS1 mice were obtained from D Alena Savonenko (Johns Hopkins University School of Medicine, Baltimore, MD) and express APP and PS1 under the control of prion promoters (Jankowsky et al., 2003). APP/PS1 mice were maintained as a heterozygote genotype. APP/PS1/gp120 mice were obtained from first generation pairings of gp120tg mice with APP/PS1 mice. Mice were housed in an AAALAC accredited facility on a 12 h light/dark cycle with ad libitum access to food and water. All procedures were conducted in accordance with NIH guidelines for the Use of Animals and Humans in Neuroscience Research and approved by Institutional Animal Care and Use Committee (Johns Hopkins University School of Medicine).
Immunohistochemical analysis.
Mice were anesthetized and perfused transcardially with buffered 4% paraformaldehyde (Sigma-Aldrich). Brains were rapidly removed and fixed for 24 h with 4% paraformaldehyde, and then cryoprotected in 30% sucrose (v/v, Sigma-Aldrich). Microtome sections (40 μm, HM450; Mikron Instruments) were cut and preserved at −20°C with antifreeze solution consisting of 30% sucrose, 30% ethylene glycol, and 0.05 m PBS. Endogenous peroxide activity was quenched with 1% H2O2 in TBS (100 mm Tris-Cl, pH 7.5, 150 mm NaCl), and nonspecific binding sites blocked with 5% goat serum in TBS containing 0.1% Triton X-100 (Sigma-Aldrich). Antibodies directed against Aβ (82E1, 1:200), and Iba-1 (1:200, WAKO Pure Chemical Industries) were applied overnight at 4°C. Sections were washed in TBS and incubated for 2 h with the appropriate secondary antibodies (1:2000; Vector Laboratories). Staining was visualized with diaminobenzidine (Vector Laboratories). Images were captured on a Zeiss upright microscope (AXIO Scope.A1) equipped a with a Qimaging Retiga 2000R camera, and quantified with Openlab 5.0.1 (Improvision, Imaging software) by an observer blinded to the genotype.
Stereological quantifications of Iba-1+ cells, Aβ plaque size and number were performed in a one-in-five series (200 μm apart; 5 sections in total), from the rostral point of bregma −1.22 mm to the caudal point of bregma −2.80 mm) using Openlab 5.0.1 (Improvision, Imaging). Stereological counts of Iba-1+ cells were performed in a similar manner in adjacent sections using the optical dissector technique with 200 × 200 μm2 as the guard height and a dissector frame area of 100 × 100 μm (Stereo investigator 7.50.4, Microbright Field).
Isolation and quantification of human Aβ.
For intracellular Aβ, cell lysates were homogenized in RIPA buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 10 mm EDTA, 2 mm EGTA, 50 mm NaF, 0.5% SDS, 1% NP-40) supplemented with protease inhibitor cocktails (Roche Applied Science). Total protein levels of each sample were measured using Micro BCA Protein Assay Reagent Kit (Thermo Scientific). Cell homogenates were centrifuged at 14,000 × g 4°C for 10 min and the supernatant was collected in a separate tube for analysis. For secreted Aβ, cell culture media were collected, centrifuged at 14,000 × g 4°C for 10 min and supernatant transferred to a new tube. Aβ1–42 was quantitatively detected by enzyme-linked immunosorbent assay (Invitrogen). Absorbance was read at 450 nm using a spectrophotometer (SpectraMax M2, Molecular Devices). Standard curves were linear from 0–1 ng/ml of human Aβ1–42.
Quantitative RT-PCR.
Total RNA was isolated from primary cortical cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized using total RNA, N6 random primers and SuperScriptII Reverse Transcriptase (Invitrogen). cDNA was then mixed with RNase free water, gene-specific primers, primers for actin, and 2× PCR universal master mix (Applied Biosystems), RNA was amplified using an ABI 7500 Real Time PCR system. The gene specific primers used in this study were as follows: APP (Rn00570673-m1; Applied Biosystems), Beta-secretase 1 (BACE1; Rn00569988-m1; Applied Biosystems), and actin (00607939S1, Applied Biosystems). The relative levels of mRNA were calculated using the ΔΔCt method by normalization to the internal control actin.
Western blotting.
Cells were washed in cold PBS and scraped in RIPA buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 10 mm EDTA, 2 mm EGTA, 50 mm NaF, 0.5% SDS, 1% NP-40) with a protease inhibitor cocktail and phosphoSTOP (Roche Applied Science). Cell suspensions were sonicated, and pelleted by centrifugation at 14,000 rpm for 5 min. Protein concentrations were determined using the BCA protein assay reagent kit (Thermo Scientific). A Proteome Profiler Array (RandD) was used to identify kinases phosphorylated following gp120 treatments. Following treatments, cells were lysed with RIPA buffer. Lysates were incubated with membranes overnight at 4°C. Membranes were washed and incubated with detection antibody conjugated to a streptavidin-HRP antibody.
For Western blots the proteins were separated by SDS-PAGE and transferred to immune-Blot PVDF membranes (Bio-Rad). Membranes were preblocked for 1 h in the presence of 5% nonfat milk and incubated overnight at 4°C with primary antibodies to APP (1:1000, Cell Signaling Technology), BACE1 (1:1000, Millipore), JNK (1:1000, Cell Signaling Technology), pJNK (1:1000, Cell Signaling Technology), GSK (1:1000, Cell Signaling Technology) or AKT (1:1000, Cell Signaling Technology), in TBS containing 0.1% Tween 20. Membranes were washed in TBS and exposed to the appropriate horseradish peroxidase-conjugated secondary antibody (1:2000; Cell Signaling Technology). Immunoreactive proteins were visualized by chemiluminescence (Millipore) using a QBOX imaging system (Syngene). Densitometric analysis was performed using Alpha view (Alpha Innotech).
Immunofluorescence and quantitation.
Lipid raft membrane microdomains were identified using a cholera toxin subunit B conjugated to AlexaFluor 555 that binds the ganglioside GM1 (CTB-555; Invitrogen; Wheeler et al., 2009). CTB-555 (1 ng/ml) was incubated with neurons for 10 min at 37°C in a 5% CO2 atmosphere. Media was rapidly removed and cells were fixed with ice-cold 4% paraformaldehyde in TBS. Membranes were permeabilized and nonspecific binding was blocked for 1 h at room temperature in TBS containing 0.1% Triton X-100, 2.5% normal goat serum and 2.5% normal horse serum. Cells were incubated with primary antibodies: APP (1:1000, Cell Signaling Technology), BACE1 (1:1000, Calbiochem), Aβ (82E1, 1:500, IBL), EEA1 (1:500, Calbiochem), LC3 (1:500 Cell Signaling Technology), LAMP1 (1:200, Calbiochem), or Rab11 (1:500, Cell Signaling Technology) overnight at 4°C. Slides were washed with TBS and incubated for 2 h at room temperature with the appropriate secondary antibodies conjugated to AlexaFluor 488, or AlexaFluor 546 (1:1000; Invitrogen). Immunopositive puncta on dendritic branches were imaged with a 100× objective by optical sectioning using structure illumination (Carl Zeiss). Fluorescence quantitation was performed using methods similar to those previously described (Xu et al., 2011b). Colocalization was confirmed by three-dimensional reconfiguration of z-stack images using orthogonal views. All images for quantification were taken with identical settings, and performed on a single plane of focus through the brightest point. Quantifications were performed within 100 μm of the soma, and dendritic areas were calculated for each region of interest. The number of pixels with individual or colocalized fluorescence per square micrometer was determined using Axiovision (4.8.2) imaging software (Carl Zeiss). Each immunopositive signal was normalized to background fluorescence and to area by tracing the outline of the dendrite. A minimum of 21 cells from at least three separate cultures was quantified for each experimental condition.
qPCR and RIP analysis.
Following treatment, cells were lysed in RIPA buffer, disrupted by sonication, and centrifuged at 10,000 × g for 15 min at 4°C. After three washes and digestion with DNase I and proteinase K, supernatants were incubated with protein A-sepharose beads coated using primary antibodies directed against heterogeneous nuclear ribonucleoprotein C (hnRNP C; 1:500; Santa Cruz Biotechnology) or control IgG (Santa Cruz Biotechnology). For ribonucleoprotein immunoprecipitation (RIP) analysis, RNA present in ribonucleoprotein complexes was isolated using Triazol (Invitrogen), reverse-transcribed using random hexamers and SSII reverse transcriptase (Invitrogen), and assayed for abundance of transcripts by real-time, quantitative PCR (qPCR) analysis using SYBR Green PCR master mix (Applied Biosystems). Gene-specific primer sets (forward and reverse primers, respectively) were GCCAAAGAGACATGCAGTGA and AGTCATCCTCCTCCGCATC for APP mRNA, TGCACCACCAACTGCTTAGC and GGCATGGACTGTGGTCATGAG for GAPDH mRNA, and GGACTTCGAGCAAGAGATGG and AGCACTGTGTTGGCGTACAG for ACTB mRNA (encoding β-Actin).
Measurement of sphingolipids.
Total lipids from samples were prepared according to a modified Bligh and Dyer procedure (Haughey et al., 2004). The chloroform layer was removed into a glass storage vial, flushed with nitrogen until dry and stored at −20°C until use. The dried layer was dissolved in 200 μl methanol containing internal standards (ceramide d18:1/C12:0 and sphingomyelin d18:1/C12:0). Lipid analyses were performed on a high-pressure liquid chromatograph coupled electrospray ionization quadrupole tandem mass spectrometer (API3000; AB/Sciex) operated in the positive mode using methods similar to those used in previous studies (Bandaru et al., 2007, 2011).
Results
Intracellular Aβ accumulation is accelerated in gp120/APP/PS1 transgenic mice
To determine the influence of gp120 on Aβ formation we created a triple transgenic line of mice that express gp120 together with mutant forms of human APP and PS1 that cause early onset inherited AD. Aβ deposition was not detected in 2-month-old wild-type (wt) or gp120tg mice, but was apparent in the hilus region of APP/PS1 and gp120/APP/PS1 mice (Fig. 1A–D). Aβ deposition was not seen in 6-month-old wt or gp120tg mice (Fig. 1E,F; because the amino acid sequence of mouse Aβ differs from that of human Aβ, mice do not normally develop Aβ plaques in their brain at any age). However, the number and size of Aβ deposits was increased in 6-month-old triple transgenic gp120/APP/PS1 mice compared with double transgenic APP/PS1 mice (Fig. 1G–J). Diffuse Aβ staining was increased in the cortex and hippocampus of gp120/APP/PS1 mice compared with APP/PS1 mice and, notably, in gp120/APP/PS1 mice diffuse intraneuronal Aβ staining was readily apparent, appearing as granular cytoplasmic deposits in the soma and neurites (Fig. 1K–N). The number of microglial cells in hippocampus and cortex were not different among wt, gp120, APP/PS1, and gp120/APP/PS1 mice, and there was no morphological evidence for global microglial activation. However, activated microglia were commonly found in and around amyloid plaques, and thus microglia with an activated phenotype were more frequent in gp120/APP/PS1 mice compared with APP/PS1 mice due to the increased plaque load (Fig. 2A–G).
Figure 1.
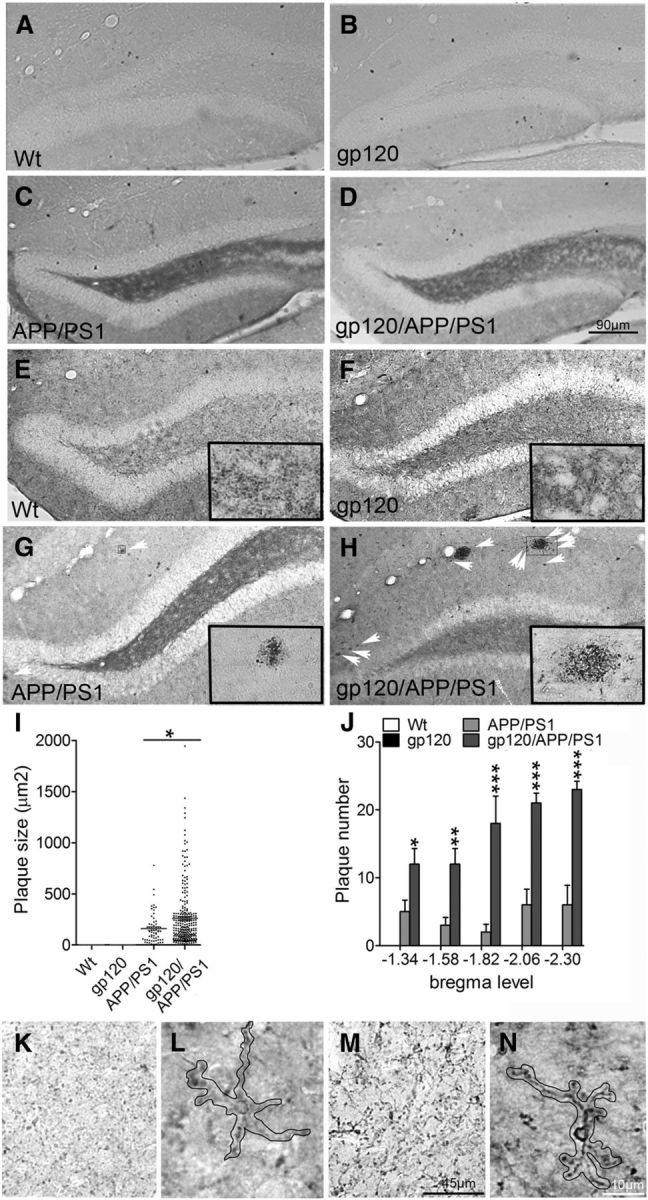
Aβ deposition is accelerated in gp120/APP/PS1 mice compared with APP/PS1 mice. A, B, Two-month-old wt and gp120 mice do not show evidence of Aβ deposition. C, D, Diffuse Aβ appears in the dentate gyrus of 2-month-old APP/PS1, and gp120/APP/PS1 mice. E, F, Six-month-old wt and gp120 mice are negative for Aβ deposition. G, H, In 6-month-old APP/PS1 and gp120/APP/PS1 mice Aβ containing plaques are apparent throughout cortical and subcortical regions. Plaques are more frequent and larger in gp120/APP/PS1 mice compared with APP/PS1 mice. Arrows show examples of Aβ plaques, and insets show magnification of the indicated region. I–L, Sterological counts of plaque size, and number of plaques by genotype and bregma level. In 6-month-old APP/PS1 mice faint staining of diffuse Aβ can been seen in neuropil, and as small intraneuronal deposits in some cells. M, N, In 6-month -old gp120/APP/PS1 mice prominent intraneuronal staining is apparent. A single neuron in each panel is outlined. Data are mean ± SD; n = 3 mice/group. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with control.
Figure 2.
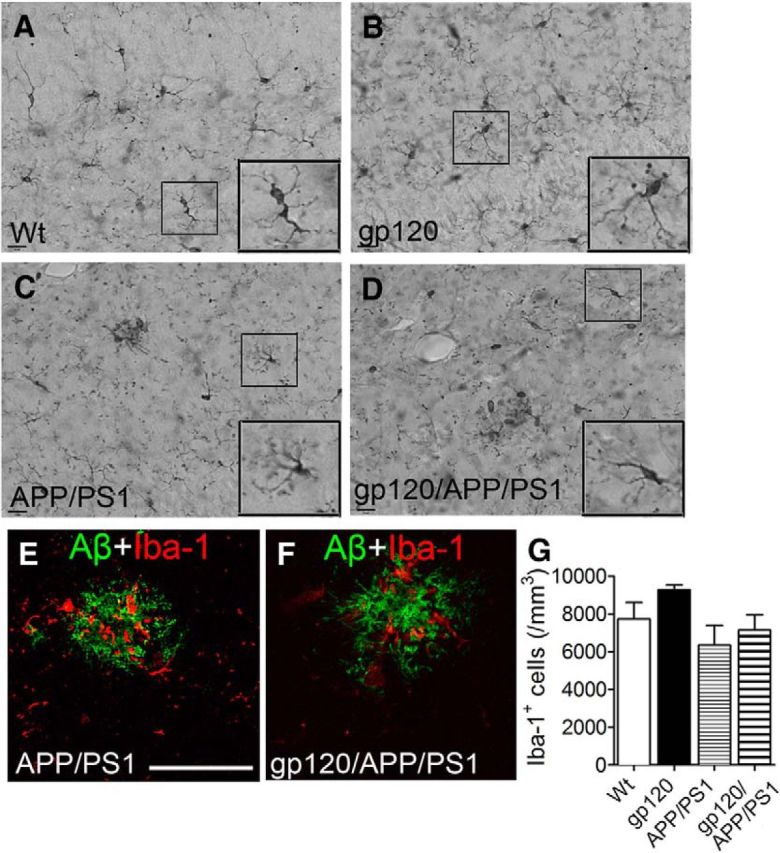
Microglial cells surround amyloid plaques. Iba-1 staining of (A) wt, (B) gp120, (C) APP/PS1, and (D) gp120/APP/PS1 mouse hippocampus. Insets are magnifications of the indicated regions. Immunofluorescent staining showing an Aβ immunopositive plaques (82E1) and microglia (Iba-1) in (E) APP/PS1 and (F) gp120/APP/PS1 mice. G, Sterological quantification of Iba-1-immunopositive cells for the indicated mouse genotypes. Scale bar, 100 μm. ANOVA with Tukey post hoc comparisons.
Sphingolipidomic analyses of cortex showed that multiple ceramides, dihydroceramides, monohexosyl ceramides, and sphingomyelins were increased in gp120/APP/PS1 compared with wt mice. Only a few of these lipid species were increased in gp120 or APP/PS1 compared with wt mice (Fig. 3A,B). Because sphingolipidoses are commonly associated with lipid accumulations in lysosomes, we used scanning electron microscopy to image subcellular compartments. Small electron dense organelles consistent with lysosomes were found in neurons of wt mice. In gp120 mice, electron dense lysosomes were often enlarged and contained lipid inclusions. Although the morphology of lysosomes in APP/PS1 was similar to wt mice, lysosome enlargement with lipid inclusions were frequently found in gp120/APP/PS1 compared with gp120 mice (Fig. 3C). These inclusions are consistent with intracytoplasmic lipofuscin, a cross-linked and oxidized lipoprotein aggregate that is a characteristic feature of neuronal cells in the aged brain (Gray and Woulfe, 2005; Riga et al., 2006). Lipofuscin accumulation interferes with lysosome degradation, and is a site for the intracellular accumulation of Aβ peptides (Brunk and Terman, 2002; Gray and Woulfe, 2005; Gómez-Ramos and Asuncion Moran, 2007). Thus, gp120 and to a greater extent gp120/APP/PS1 mice, show neuropathologies consistent with lysosome dysfunction.
Figure 3.
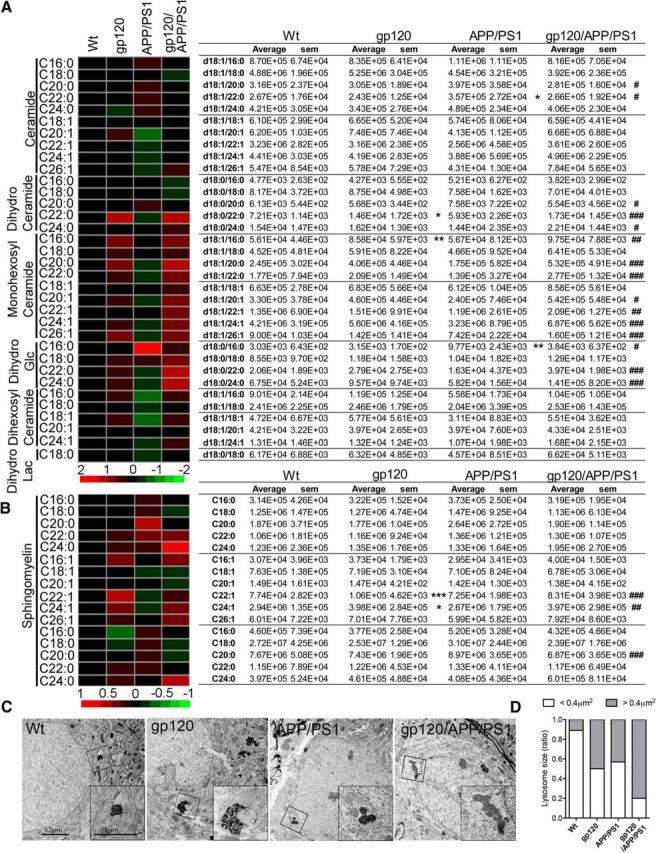
Sphingolipid accumulation and lysosomal pathology in gp120/APP/PS1 mice. A, B, Heat maps and associated counts of the indicated ceramide and sphingomyelin species for wt, gp120, APP/SP1, and gp120/APP/PS1 mice showing that multiple-long and very-long chain ceramides were increased in the cortex of the gp120/APP/PS1 transgenic mice. Data are mean ± SEM; n = 5/group. ANOVA with Tukey post hoc comparisons; *p < 0.05, p < 0.01, ***p < 0.001 compared with control, #p < 0.05, ##p < 0.01, ###p < 0.001 compared with gp120. C, Representative electron microscopy images showing ultrastructural analysis of endolysosomal phenotypes in brain tissues from the indicated genotype. Lysosomes in wt mice show typical electron dense lysosomal inclusions. In gp120 mice, lysosomes are enlarged and contain lipid inclusions known as lipofuscin. Although lysosomes appear to be phenotypically normal in APP/PS1 mice, they are enlarged, partially fused, and contain lipofuscin in gp120/APP/PS1 mice. Insets, are magnifications of the indicated region. Scale bars: 2 μM in main figure and 1 μM in insets. D, Quantitative analysis of lysosomal size expressed as percentage of lysosomes with size <0.4 μm2 (white bars) compared with lysosomes >0.4 μm2 (black bars) for the indicated genotypes. Data are mean ± SEM of 45–60 cells in each of three independent experiments per condition. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with wt.
HIV-gp120 increases Aβ production and reduces its clearance from neurons
Human neuronal (SH-SY5Y) cells expressing APP constitutively produce Aβ, with a predominance of Aβ1-42 (hereafter referred to as Aβ). In a 24 h period we found that ∼1/3 of Aβ1-42 was exported into the media and 2/3 was intraneuronal (Fig. 4A). In these basal conditions 9.58 ± 6.26% of intraneuronal Aβ was localized to EEA1+ early endosomes, 0.93 ± 1.77% to Rab11+ recycling endosomes, 17.48 ± 7.32% to Lamp2+ lysosomes, and 3.27 ± 1.25% to LC3+ autophagosomes (Fig. 4B). Using this human neuronal culture system, we determined whether gp120 perturbed the production and/or trafficking of Aβ. Because CXCR4 (X4), CCR5 (R5), and dual (X4/R5) tropic HIV have each been implicated in the neuropathogenesis of HIV (Gabuzda et al., 1998; Gorry et al., 2001; Gray et al., 2009), we initially screened forms of gp120 that preferentially use X4 (gp120IIIB), R5 (gp120CM), or are dual tropic (gp120MN). SY5Y cells expressing human APP were treated in reduced serum (2%). X4 gp120 produced small increases in the secretion of Aβ at doses ≥100 pm (Fig. 4C), and dose-related intracellular deposition of Aβ (Fig. 4D) that localized primarily to lysosomes and autophagosomes (Fig. 4E). R5 gp120 increased the secretion of Aβ at concentrations ≥250 pm (Fig. 4F), and increased intracellular Aβ deposition in a concentration-dependent manner (Fig. 4G), with early and sustained increases in endosomes, transient increases in lysosomes, and slow accumulation in autophagosomes (Fig. 4H). X4/R5 gp120 at the highest concentration (500 pm) significantly increased Aβ secretion (Fig. 4I), and increased the intracellular deposition of Aβ at concentrations ≥100 pm (Fig. 4J), producing early and sustained increases in endosomes, with slow accumulations in lysosomes (Fig. 4K). The lipid peroxidation product 4HNE, also increased Aβ production, but distinct from the effects of gp120, a majority of this Aβ was secreted into the culture medium (Fig. 4L), in conjunction with intracellular increases (Fig. 4M), and lysosome deposition (Fig. 4N). Representative immunofluorescence images for each of these conditions are shown in Figure 5. Heat-denatured gp120 did not alter the production or distribution of Aβ (Fig. 5). All subsequent experiments used X4-gp120 (hereafter referred to as gp120). Although R5 strains of HIV productively infect microglia and macrophages, X4 strains of HIV play important roles in the pathogenesis of HIV-associated neurocognitive disorders (HAND). X4 and dual trophic strains of HIV have been isolated from brain, and monocytes infected with X4 HIV intermittently traffic into the CNS. This monocyte transmigration is enhanced when the blood brain barrier is compromised during HIV infection, inflammation, drug abuse, or immune reconstitution inflammatory syndrome (Liu et al., 2000; Gorry et al., 2001; Miller et al., 2004; Gray et al., 2005; El-Hage et al., 2006; Dhillon et al., 2008; Fischer-Smith et al., 2008; Ramirez et al., 2009; Yao et al., 2011).
Figure 4.
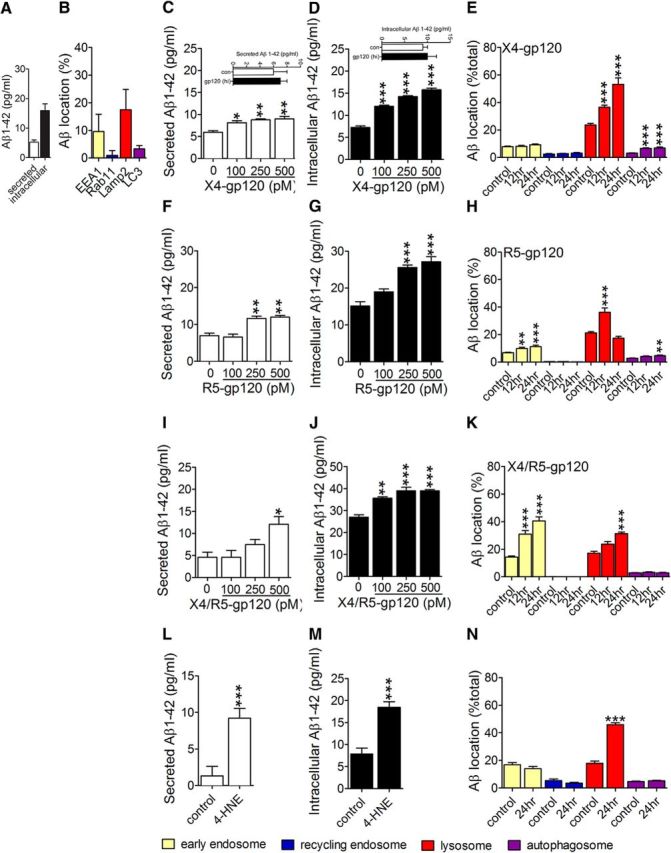
HIV-gp120 induces Aβ formation and perturbs clearance. A, Secreted and intracellular levels of Aβ1–42 in SHSY5Y cells stably expressing human APP were determined by ELISA 24 h after media change. B, Intracellular distribution of Aβ (82E1) in early endosomes (EEA1), recycling endosomes (Rab11), lysosomes (LAMP2), and autophagosomes (LC3) were determined by quantitative immunofluorescence. C, D, Cells were treated for 24 h with the indicated concentrations of X4-gp120. Secreted and intracellular concentrations of Aβ1-42 were determined by ELISA. Insets show results for heat inactivated (hi) X4-gp120 (500 pM). E, Colocalization of Aβ with EEA1, Rab11, LAMP2, and LC3 was quantified 12 and 24 h after exposure to X4-gp120 (250 pm). F, G, Secreted and intracellular Aβ1–42 was quantified 24 h after exposure to the indicated concentrations of R5-gp120 by ELISA. H, Colocalization of Aβ with EEA1, Rab11, LAMP2, and LC3 was quantified 12 and 24 h after exposure to R5-gp120 (250 pm). I, J, Secreted and intracellular levels of Aβ1-42 were determined by ELISA 24 h after exposure to the indicated concentrations of X4/R5-gp120. K, Colocalization of Aβ with Rab11, LAMP2, and LC3 was quantified 12 and 24 h after exposure to X4/R5-gp120 (100 pm). L, M, Aβ1-42 secretion and intraneuronal deposition were quantified by ELISA in neuronal cells treated with 4-HNE for 24 h (100 nM). N, Immunofluorescent quantification of Aβ colocalized with Rab11, LAMP2, and LC3 24 h after exposure to 4-HNE (100 nM). Data are mean ± SEM of at least 21 cells, and three independent experiments per condition. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with corresponding control.
Figure 5.
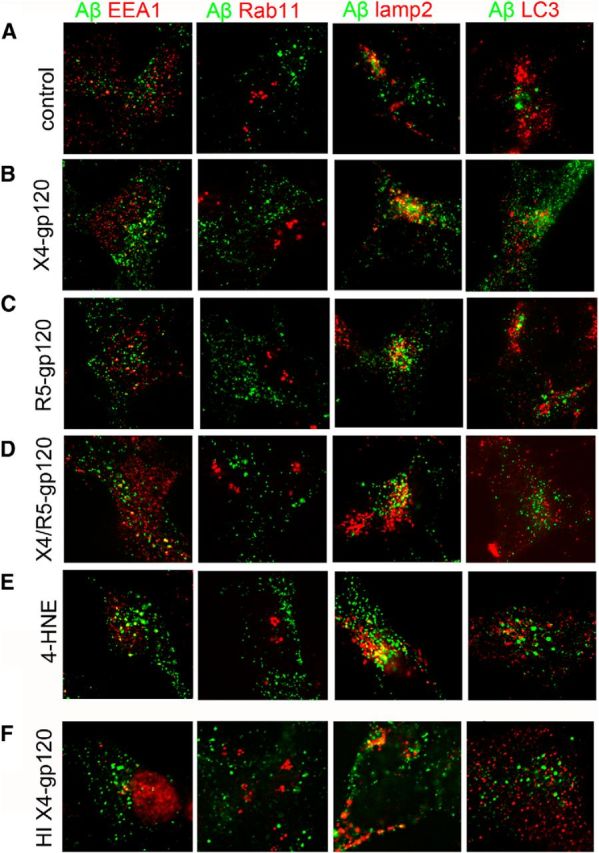
Aβ localizes to lysosomes in neuronal cells exposed to gp120. Representative fluorescent images from SHY5Y cells stably expressing human APP exposed to (A) control, (B) X4-gp120 (250 pm), (C) R5-gp120, (D) X4/R5gp120 (250 pm), (E) 4HNE (10 nm), and (F) heat-denatured X4-gp120 (500 pM) for 24 h. Merged images for Aβ (82E1) with markers for early endosomes (EEA1), recycling endosomes (Rab11), lysosomes (lamp2), and autophagosomes (LC3).
HIV gp120 enhances β- and γ-secretase processing of APP
We next focused on determining if and how gp120 increases amyloidogenic processing of APP. APP is a large type I transmembrane protein that can be differentially processed by either α- (nonamyloidogenic pathway), or β- (BACE) followed by γ-secretase (amyloidogenic pathway that generates Aβ). HIV gp120 did not alter α-secretase activity in cultured primary cortical neurons at any concentration tested (Fig. 6A), but concentration-dependently increased BACE and γ-secretase activities with minimal effective concentrations in the 10–100 pm range (Fig. 6B,C). Because BACE is the rate-limiting enzyme in Aβ production, we focused on identifying the mechanisms by which gp120 modified BACE. BACE1 mRNA (Fig. 6D) and protein levels (Fig. 6E) were increased within 6 h following exposure to gp120 exposure and remained elevated for at least 24 h. A small molecule antagonist of CXCR4 (AMD3100) prevented gp120 from increasing BACE1 activity. However, the endogenous agonist of CXCR4, SDF-1α had no effect on BACE1 protein expression (Fig. 6E). Inhibition of CXCR4 and PKA, but not PKC prevented gp120 from increasing BACE activity (Fig. 6F), and gp120 induced a rapid increase in PKA phosphotransferase activity (Fig. 6G), suggesting that increased BACE1 protein expression was mediated by PKA signaling. This atypical activation of PKA by gp120 signaling through CXCR4 has been previously reported (Masci et al., 2003; Xu et al., 2011a,b).
Figure 6.

Induction of BACE1 transcriptional activation by gp120-mediated suppression of PPARγ. Figures show (A) ∝-secretase (B) β-secretase, and (C) γ-secretase activity in primary neurons treated for 6 h with the indicated dose of gp120 (10–1000 pm). D, qPCR results showing BACE1 mRNA for the indicated time points following control or gp120 exposures. E, Representative Western blot showing BACE1 protein levels in primary neurons treated for 6 h with gp120 (250 pm), or SDF 1α (20 nm), or pretreated for 30 min with the CXCR4 inhibitor AMD 3100 (10 μm) before the addition of gp120. Densitometric analyses of BACE1 protein expression for the indicated conditions are normalized to β-actin. F, β-secretase activity measured in neurons treated with gp120 (250 pm) for 6 h, or pretreated for 30 min with AMD3100 (AMD, 10 μm), the PKA inhibitor KT5720 (KT; 1 μm), or the PKC inhibitor chlerythrine (Chel; 1 μm) before additions of gp120. G, PKA phosphotransferase activity was measured in primary neurons treated with gp120 (250 pm) for 6 h. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding control or time 0; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with gp120.
A phosphokinase array using lysates from control and gp120-treated primary rodent cortical neurons suggested that c-JNK, glycogen synthase kinase B (GSK), and AKT could be effectors downstream of PKA activated by gp120 (Fig. 7A). The phosphorylation of JNK and GSK, but not AKT was confirmed by individual Western blot analyses (Fig. 7B–D). We further confirmed these findings in vivo where increased phosphorylation of JNK and GSK, but not was apparent in gp120, APP/PS1, and APP/PS1/gp120 (Fig. 7E–G). Inhibition of CXCR4 and PKA, but not PKC prevented gp120 from increasing the phosphorylation of JNK (Fig. 8A). Inhibition of JNK activation prevented gp120 from increasing the phosphorylation of JNK (Fig. 8B), and from increasing BACE1 protein levels (Fig. 8C). Although JNK is known to increase BACE1 expression through the immediate early response transcription factor cJun (Guglielmotto et al., 2011), inhibiting the interaction of JNK with cJun did not prevent gp120 from increasing BACE1 mRNA and protein levels, nor did inhibiting AKT (Fig. 8C). These data suggest that gp120 increased BACE1 protein expression through a mechanism that involved PKA signaling to JNK that was independent of the transcriptional influence of cJun.
Figure 7.

HIV gp120 activated a restricted set of kinases. A, Proteome profiler array to identify phosphorylated kinases in primary neurons under control conditions and following gp120 (100 pm) treatment for 30 min. The indicated spots refer to kinases confirmed by the corresponding Western blots. The expression and phosphorylation of (B) c-JNK, (C) GSK, and (D) AKT were confirmed in neurons treated with gp120 (100 pm) for the indicated time points. Protein expression and phosphorylation of (E) JNK, (F) GSK, and (G) AKT were also confirmed in hippocampus from 6-month-old mice of the indicated genotypes. Data are mean ± SD for at-least n = 3 independent experiments per condition. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with gp120.
Figure 8.
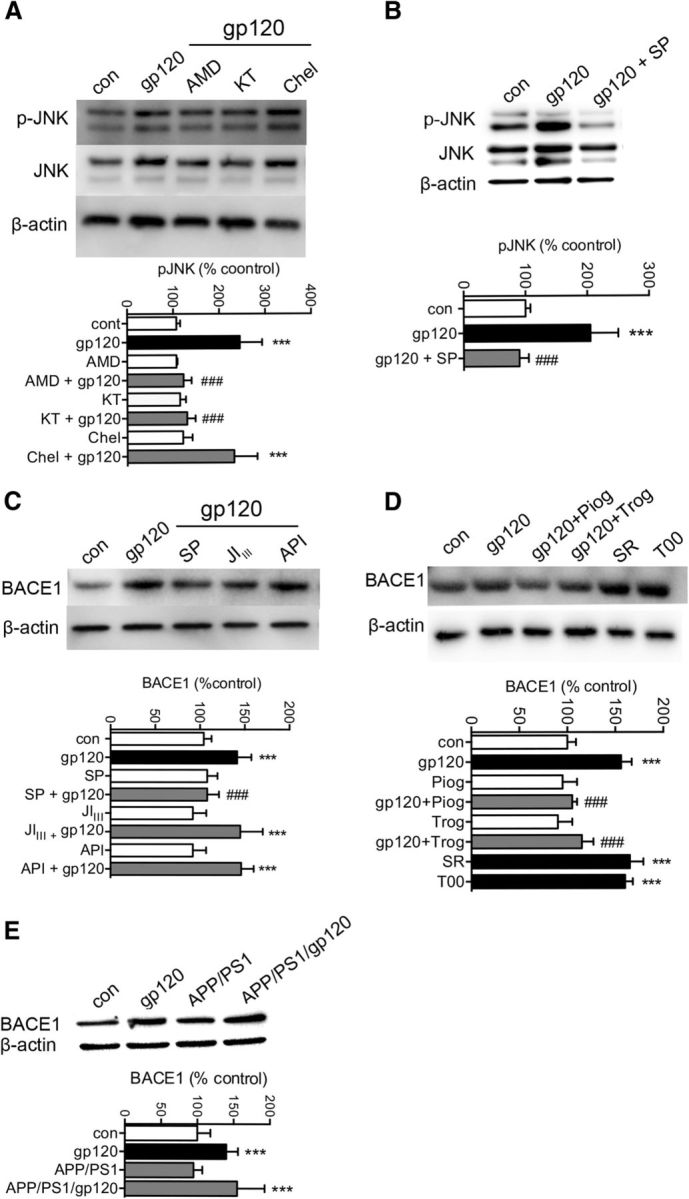
HIV gp120 promotes the release of Bace1 transcriptional repression through induction of the MAP kinase pathway and PPARγ. A, Representative Western blot showing total JNK and pJNK protein expression in cells treated for 30 min with gp120 (250 pm), or pretreated with AMD 3100 (10 μM), KT 5720 (1 μM), or Chelerythrine (1 μM) for 30 min before gp120 treatments. Quantitative densitometric analysis of Western blots showing pJNK for the indicated treatments. B, Representative Western blot and densitometric analysis showing total JNK and pJNK in cells treated for 30 min with gp120 (250 pm) or pretreated with the JNK inhibitor SP600125 (SP, 20 μm) for 30 min before gp120. C, Representative Western blot and densitometric analysis showing BACE1 protein levels in primary neurons treated with gp120 (250 pm) for 6 h, or pretreated with SP600125 (SP; 20 μm), a peptide inhibitor that blocks the interaction of JNK with cJUN (JIIII; 10 μm), or an inhibitor of AKT (API; 20 μm) for 30 min before gp120. D, Representative Western blot and densitometric analysis showing BACE1 protein levels in neurons treated with gp120 (250 pm) for 6 h, or pretreated for 30 min with the agonists of PPARγ Pioglitazone (Piog; 10 μm) and Triglitazone (Trog; 20 μm) before gp120 treatments. Neurons were exposed to antagonists of PPARγ SR202 (SR; 20 μm) and T007907 (T00; 10 μm) for 6 h in the absence of gp120. E, Representative Western blot and densitometric analysis of BACE1 protein levels in hippocampus for the indicated genotypes of 6-month-old mice. Data are mean ± SD for at least n = 3 independent experiments per condition. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with gp120.
The Bace1 gene promoter contains a putative binding site for the peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor protein that functions as a transcription factor. Activation of PPARγ causes repression of Bace1 gene promoter activity, whereas reduction of PPARγ levels leads to increases of BACE1 mRNA (Sastre et al., 2006). Because JNK MAP kinase is known to have an inhibitory effect on PPARγ (Bhatt et al., 2012), we reasoned that gp120 induction of Bace1 transcription could involve JNK inhibition of PPARγ. Two different agonists of PPARγ blocked gp120 from increasing BACE1 mRNA and protein levels in primary rodent cortical neurons (Fig. 8D). The involvement of PPAPRγ was confirmed using two different PPARγ antagonists that each mimicked the effect of gp120 to increase BACE1 mRNA and protein levels (Fig. 8D). These data suggest that gp120 promoted the release of Bace1 transcriptional repression through induction of the MAP kinase pathway that inhibited PPARγ. BACE1 expression was likewise increased in gp120 transgenic mice, in APP/PS1/gp120 mice, but not in APP/PS1 mice, suggesting that increased BACE1 expression was largely driven by gp120 (Fig. 8E).
HIV gp120 increases APP protein production by removal of a translational block
In human SY5Y cells HIV-gp120 did not alter APP mRNA levels at any time following gp120 treatments (Fig. 9A), but increased immature and mature APP protein levels within 3 h following stimulation compared with controls (Fig. 9B). Following gp120 exposures, APP remained increased for up to 6 h following exposure to gp120, then declined (Fig. 9B), presumably due to processing of APP to Aβ. Inhibition of CXCR4, PKA, and JNK, but not CCR5, PKC, or AKT prevented gp120 from increasing APP (Fig. 9C), suggesting that gp120 increases APP protein production via JNK MAP kinase signaling. However, because the effects on APP were independent of transcriptional modulation, we postulated that JNK-regulated APP production at a posttranscriptional stage.
Figure 9.

Increased APP production by gp120 is linked to increased interaction of the translation enhancer hnRNP C with APP mRNA. A, Time course (0–24 h) showing APP mRNA levels in SHSY5Y cells stably expressing human APP exposed to gp120 (250 pm) for the indicated time points. B, Representative Westen blot, and densitometric quantification showing immature (lower band; i) and mature (upper band; m) APP protein levels in cells exposed to gp120 (250 pm) for the indicated treatment times. C, Representative Western blot, and densitometric quantification of neuronal cells exposed to gp120 (250 pm) for 6 h, or pretreated with AMD3100 (AMD, 10 μm), KT5720 (KT, 1 μm), chlerythrine (Chel, 1 μm), SP600125 (SP, 20 μm), API-1 (API, 20 μm), or DAPTA (5 nm) 30 min before gp120 treatments. D, RIP analysis of APP mRNA associated with hnRNP C. At the times indicated following treatment with gp120, hnRNP C was immunoprecipitated using anti-hnRNP C antibody (IgG was used in control parallel immunoprecipitations) and the levels of APP mRNA present in the IP materials were quantified by RT-qPCR analysis. Data were calculated as the levels of APP mRNA relative to GAPDH mRNA in each IP sample. hnRNP IP results were then normalized to IgG IP. Data are the means ± SEM from n > 3 independent experiments per condition. E, Representative Western blot and densitometric quantification of APP from hippocampus of 6-month-old mice with the indicated genotypes. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with gp120.
The near parallel increases in immature and mature forms of APP suggest that gp120 did not promote the maturation of APP (which involves O-linked glycosylations; (Thinakaran and Koo, 2008; Fig. 9B). These data suggest that gp120 regulates APP protein expression at a post-transcriptional stage before maturation. It was recently discovered that the hnRNP C promotes APP translation through displacement of the RNA-binding translational inhibitor Fragile X mental retardation protein (FMRP; Rajagopalan et al., 1998; Lee et al., 2010). Therefore, we measured the interaction of APP mRNA with the translation-activating RBP hnRNP C by using the RIP assay. After immunoprecipitation using either an anti-hnRNP C antibody or control IgG, levels of APP mRNA were measured in the bound material by reverse transcription followed by RTPCR. The association of APP mRNA with hnRNP C was calculated as the enrichment of APP mRNA in hnRNP C IP relative to the IgG IP samples. RIP analysis revealed that gp120 robustly and transiently increased the interaction of hnRNP C with APP mRNA (Fig. 9D), suggesting that gp120 increased APP by promoting the association of hnRNP C with APP mRNA. We confirmed increased expression of APP in vivo. We found that APP protein expression was increased in in mice transgenic for gp120 compared with controls (Fig. 9E). As expected there was a large increase in APP expression in APP/PS1 mice, which was reduced in APP/PS1/gp120 mice (Fig. 9E), presumably reflecting increased processing of APP to Aβ.
The association of BACE1 with APP is stabilized in membrane microdomains
Amyloidogenic processing of APP occurs preferentially in membrane microdomains enriched in cholesterol and ceramides, where APP and APP-processing proteins are concentrated (Lee et al., 1998; Parkin et al., 1999; Wahrle et al., 2002; Ehehalt et al., 2003; Marlow et al., 2003; Kawarabayashi et al., 2004; Watanabe et al., 2004). Several recent reports have demonstrated the importance of membrane microdomains in regulating secretase activities and Aβ formation, and we recently demonstrated that gp120 increases the size and stabilizes the structure of membrane microdomains in neurons (Tamboli et al., 2005, 2011b; Chi et al., 2007; Okada et al., 2008; Mao et al., 2010; Xu et al., 2011b; Ogawa et al., 2011). Thus, we considered whether gp120 could enhance the formation of Aβ by stabilizing the spatial location of BACE1 with APP within membrane microdomains. HIV-gp120 increased the size and stabilized the structure of GM1+ lipid rafts, increased the expression of BACE1 (Fig. 10A–C) and APP in primary rodent neurons (Fig. 10D–F). APP and BACE1 were colocalized in these membrane microdomains (Fig. 10G–I). Preventing the stabilization of membrane microdomains through inhibition of de novo ceramide formation with the serine palmitoyl transferase inhibitor myriocin, or salvage ceramide pathways with Fumonisin B1 prevented gp120 from increasing β-secretase activity (Fig. 10J). Thus, gp120 promotes the formation and stabilization of a membrane microenvironment that favors Aβ production.
Figure 10.
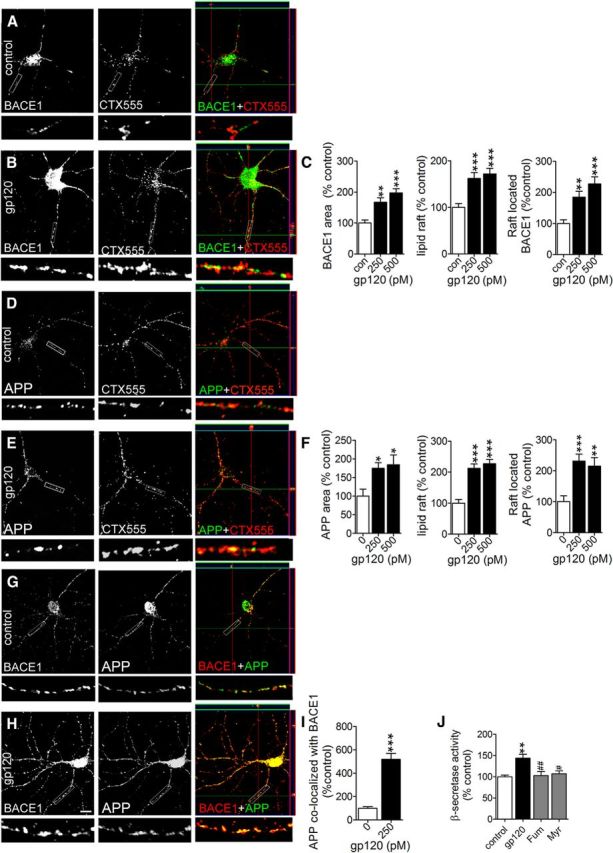
The association of APP with BACE1 is stabilized in lipid raft membrane microdomains following treatment with gp120. Representative images are 100× magnifications of primary neurons. At the bottom of each image are shown enlargements of the indicated neurite. Immunofluorescent images showing BACE1, CTX555 immunopositive membrane microdomains (lipid rafts), merged images, and orthogonal views for (A) control and (B) cultures treated for 6 h with gp120 (250 pm). Colocalized BACE1 and CTX555 appear yellow in merged images. C, Quantitation of fluorescence for BACE1, lipid rafts, and BACE1 colocalized to lipid rafts for the indicated concentrations of gp120. Representative immunofluorescent images show APP, and CTX555 immunopositive lipid rafts for (D) control and (E) cultures treated for 6 h with with gp120 (250 pm). Colocalized APP and CTX555 appear yellow in merged images. F, Quantitation of fluorescence for APP, lipid rafts, and APP colocalized to lipid rafts for the indicated concentrations of gp120. Representative images showing APP, BACE1, and merged images for (G) control and (H) neurons treated for 6 h with gp120 (250 pm). Colocalized APP and BACE1 appear yellow in merged images. I, Quantitation of colocalized APP and BACE1. J, BACE activity in primary neuronal cultures treated for 6 h with gp120 (250 pm), or pretreated with fumonisin β1 (Fum, 10 μm), or myriocin (Myr, 10 μm) followed by gp120. K, Secreted and intracellular Aβ1-42 measured by ELISA in cells treated with gp120 (250 pm) or with gp120 in the presence of β-cyclodextrin (5 mm). Quantitative data are mean ± SEM for at least n = 3 independent experiments per condition. ANOVA with Tukey post hoc comparisons; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the corresponding control.
Activation of the TRPML1 channel clears sphingomyelin and Aβ from lysosomes
In primary neurons, gp120 increases the formation of ceramide and sphingomyelin (Haughey et al., 2004; Xu et al., 2011b). When overproduced, sphingomyelin accumulates in endolysosomal/lysosomal compartments, inhibits TRP channels, and blocks lysosome calcium release (Shen et al., 2012). The accumulation of luminal calcium inhibits hydrogen ion transport and increases lysosome pH. Even small increases in luminal pH can disrupt derivative capacity, as lysosome hydrolases have low pH optima. Thus, we reasoned that the accumulations of Aβ in lysosomes following gp120 treatments may involve the buildup of sphingomyelin, and calcium in lysosomes with a consequent increase in luminal pH. We expressed the lysosomal calcium release indicator GCaMP3-TRPML1 in primary neurons, and stimulated lysosome calcium release with the TRPML1 agonist ML-SA1 in calcium free buffer. The specificity of this probe to measure lysosome calcium release was recently published (Shen et al., 2012). ML-SA1 induced lysosome calcium release was doubled following 6 h treatments with gp120 compared with untreated control neurons (Fig. 11A–G). HIV-gp120 had no effect on ionomycin-induced increases in GCaMP3-TRPML1 fluorescence, demonstrating that the observed experimental differences were not due to variances in transfection efficiency or performance of the calcium probe (Fig. 11C,F,G). These findings suggest that lysosmal calcium content was higher in gp120-treated cells. The ML-SA1 induced release of lysosome calcium rapidly induced endolysosomal acidification in both control and gp120 treated conditions (Fig. 11H,I). Next, we determined whether activation of TRPML1 could facilitate the clearance of lysosome sphingomyelin and Aβ deposits. HIV-gp120-induced deposition of sphingomyelin (Fig. 12A–F), and Aβ (Fig. 12G–M) were both cleared following treatment with ML-SA1. These results demonstrate that under pathogenic conditions which mimic interneuronal Aβ deposition in the HIV-infected brain, stimulating lysosome calcium release with a TRPML1 agonist promoted luminal acidification and the clearance of intraneuronal sphingomyelin and Aβ.
Figure 11.
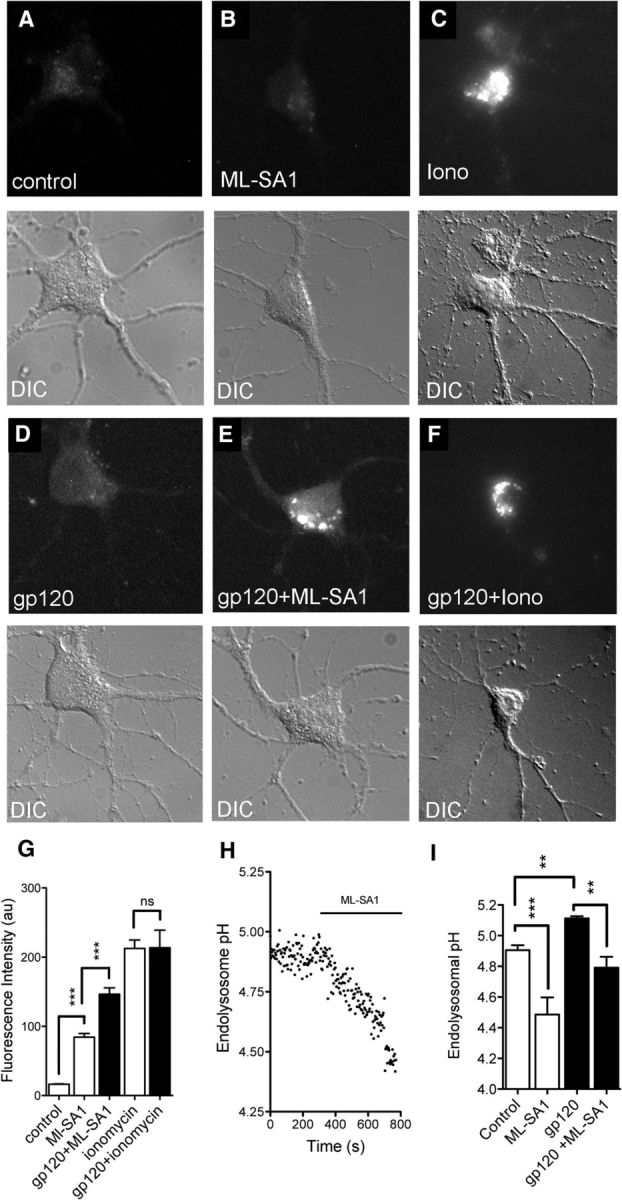
Calcium rapidly accumulates in the lysosomes of neurons treated with gp120. Representative images are of primary neurons transfected with the single wavelength genetically encoded calcium indicator GCaMP3-TRPML1. Representative images show peak lysosomal calcium release for the indicated conditions and corresponding differential interference contrast images (DIC). Time-lapse images were acquired at baseline, and following simulation with the TRPML1 agonist ML-SA1 (10 μm) in (A–C) control cells, or (D–F) neurons treated with gp120 (100 pm) for 6 h. G, Quantitation of maximal fluorescence intensity for the indicated conditions showing that ML-SA1-induced calcium release from lysosomes was increased following pretreatment with gp120. Applications of the calcium ionophore ionomycin produced similar maximal calcium increases in response to ML-SA1, suggesting that the probe was similarly expressed in all cells. Data are mean ± SEM of 21 cells/condition from three separate experiments. ANOVA with Tukey post hoc comparisons; ***p < 0.001 compared with control; ###p < 0.001 compared with gp120. H, Data from time-lapse experiments showing that ML-SA1 produced a rapid decline in endolysosomal pH. I, Summary data showing that gp120 increases endolysosomal pH, and that ML-SA1 reduces pH in control and gp120 pretreated cultures. Data are mean ± SEM of three cultures/condition. ANOVA with Tukey post hoc comparisons; **p < 0.01, ***p < 0.001.
Figure 12.
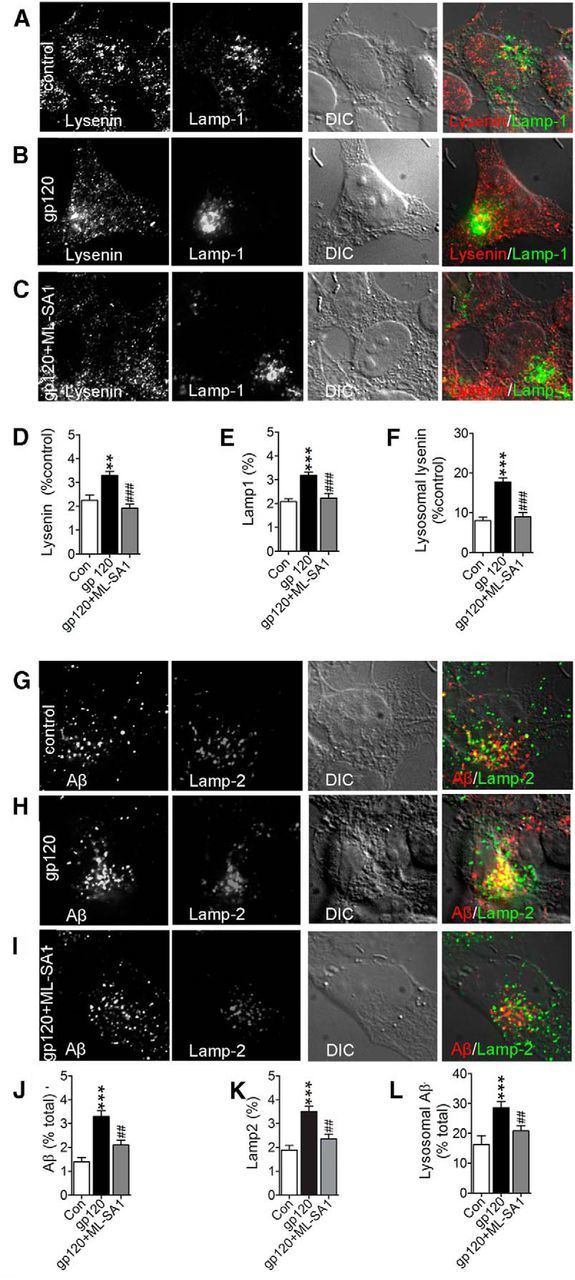
Activation of TRPML1 clears sphingomyelin and Aβ from lysosomes. Representative fluorescent images from SHSY5Y cells stably expressing human APP showing the sphingomyelin binding protein lysenin, the lysosomal associated membrane protein 1 (Lamp-1), DIC, and the merged images for (A) control, (B) cells treated for 6 h with gp120 (250 pm), and (C) cells treated for 3 h with gp120 then stimulated for 3 h with ML-SA1 in the continued presence of gp120. Colocalized lysenin and Lamp-1 appear yellow in merged images. Quantitative immunofluorescence for (D) lysenin, (E) Lamp-1, and (F) lysenin colocalized with Lamp-1 show that pretreatment with gp120 increased and ML-SA1 treatment reduced the amount of sphingomyelin localized to lysosomes. Representative fluorescent images showing Aβ (82E1), Lamp-2, DIC, and the merged images for (G) control, (H) cells treated for 24 h with gp120 (250 pm), and (I) cells exposed to gp120 for 24 h followed by 6 h with ML-SA1 in the continued presence of gp120. Colocalized Aβ and Lamp-2 in merged images appear yellow. Quantitative immunofluorescence for (J) Aβ, (K) Lamp-2, and (L) Aβ colocalized with Lamp-2 show that gp120 increased, and ML-SA1 decreased the amount of Aβ localized to lysosomes. Quantitative fluorescence data are mean ± SEM of 30–50 cells per experimental condition obtained from three independent experiments. ANOVA with Tukey post hoc comparisons; **p < 0.01, ***p < 0.001 compared with control; ##p < 0.01, ###p < 0.001 compared with gp120.
Discussion
Examination of autopsy brains from HIV-infected patients suggests that Aβ deposition is accelerated in this population (Esiri et al., 1998; Green et al., 2005; Achim et al., 2009). However, brain imaging studies with the Aβ-binding agent 11C-Pittsburg compound B (11C-PiB) have thus far been inconclusive (Ances et al., 2010, 2012b). Differences in the patterns of Aβ deposition in AD compared with HIV may help explain these discrepant findings. Although soluble and intraneuronal Aβ are commonly found in AD, senile plaques are the defining neuropathological observation (Haass and Selkoe, 2007). The pattern of Aβ deposition in HIV appears to be distinct from AD, with Aβ readily apparent as intraneuronal deposits located in the soma and axonal processes of neurons, with relatively small numbers of defined plaques (Esiri et al., 1998; Green et al., 2005; Achim et al., 2009). Because 11C-PiB was developed to identify Aβ plaques (Klunk et al., 2004; Wang et al., 2004), this compound may not efficiently bind intracellular forms of Aβ commonly found in HIV.
A number of other demographic and clinical factors are also likely to influence the deposition patterns of Aβ in HIV patients including genetic susceptibility, age, dyslipidemia, illicit drug use, and ART, each of which have been demonstrated to influence the production and/or clearance of Aβ (Cutler et al., 2004; Bandaru et al., 2009; Anthony et al., 2010; Giunta et al., 2011; Soontornniyomkij et al., 2012; Tanzi, 2012; Liu et al., 2013). For example, history of ART appears to increase the likelihood that Aβ deposits will be present (Green et al., 2005). Although the exact mechanisms for this potential drug interaction are not currently known, all classes of ART medications increase the formation and release of Aβ peptides (Giunta et al., 2011).
Aβ peptides are readily formed in the brains of healthy individuals, but are also efficiently cleared from the CNS (Edland and Galasko, 2011). The clearance of Aβ is impaired in AD, and Aβ accumulates in the extracellular space in oligomeric, and fibril forms (Mawuenyega et al., 2010). The formation of Aβ plaques requires an initial seeding event that involves formation of an ordered core, and a conformational shift from a α-helix-rich structure to β-sheet-rich structure that readily oligomerizes (Come et al., 1993). This process of fibril formation occurs most efficiently at the exterior surface of the plasma membrane, when Aβ is bound to the ganglioside GM1 (Kakio et al., 2003). Thus, the impairment of Aβ clearance in AD appears to be after secretion of Aβ peptides into the extracellular space. In contrast, impairments of Aβ clearance in HIV occur at the cytoplasmic level, and appears to involve deficits in endolysosomal trafficking.
Principal component analysis of the CSF proteome of HIV patients suggested that the severity of cognitive impairment in HIV-infected individuals is related to the integrity of complex signaling networks which govern neuroplasticity, cell survival, and innate immunity, with APP as a central contributing node (Angel et al., 2012). APP is a large type I transmembrane protein that can be sequentially processed by either α- or β-secretases followed by γ-secretase. The cleavage of APP by α-secretase precludes Aβ generation, as the cleavage site is within the Aβ domain. The α-cleavage of APP releases a large soluble ectodomain called sAPPα that has been implicated in neuronal survival, neurite outgrowth, synaptic maintenance, and plasticity (Roch et al., 1994; Morimoto et al., 1998; Bell et al., 2008; Bailey et al., 2011). Cleavage by β-secretase releases a soluble APP ectodomain (sAPPβ), and a membrane-bound 99-residue C-terminal fragment (Estus et al., 1992; Seubert et al., 1993). Subsequent cleavage of C99 fragment by the γ-secretase complex (comprised of presenilin, nicastrin, APH-1, and PEN-2) liberates Aβ-peptides. The γ-secretase cleaves at two primary sites of APP producing Aβ1–40 or Aβ1–42 (Walsh and Selkoe, 2007). Current evidence suggests that the α-cleavage of APP occurs outside of lipid rafts, where the α-secretase ADAM10 is exclusively located (Harris et al., 2009). Amyloidogenic processing of APP is thought to occur primarily in lipid rafts where all relevant proteins appear to be concentrated including: BACE1, presenilins, nicastrin, APH-1, PEN-2, and APP (Lee et al., 1998; Parkin et al., 1999; Wahrle et al., 2002; Ehehalt et al., 2003; Marlow et al., 2003; Kawarabayashi et al., 2004; Watanabe et al., 2004). Thus, an increased stabilization of membrane microdomains by gp120 involving the enhanced formation of ceramide (Jana and Pahan, 2004; Xu et al., 2011b) may be an initiating event that facilitates the formation of Aβ. Indeed, metabolic perturbations that result in accumulations of ceramide in brain and CSF occur early in the course of HIV-infection, and worsen with the onset or progression of cognitive impairment (Haughey et al., 2004; Bandaru et al., 2010; Mielke et al., 2010). These observations are consistent with our current findings that show gp120 increases Aβ production by stabilizing the interaction of APP with BACE1 in membrane microdomains. Inhibiting the formation of ceramide or disrupting the structure of lipid rafts prevented gp120 from increasing Aβ production, suggesting that therapeutics which target ceramide metabolism may block these early events that lead to aberrant processing of APP. These data also suggest that the increased processing of APP to Aβ in the setting of HIV-infection could decrease neurotrophic support by reducing α-secretase processing of APP to the trophic product sAPPα. Although the effects of HIV infection on the production of sAPPα remain to be determined, dystrophic neurites and dendritic pruning consistent with a reduced trophic environment are common neuropathological features associated with HAND (Pelle et al., 2008; Gelman et al., 2012; Desplats et al., 2013).
Aβ peptides are generated from the sequential cleavage of APP by β- and γ-secretases during endocytic trafficking. This Aβ can enter into recycling compartments and secreted, or can be degraded in lysosomes and autophagosomes (Small and Gandy, 2006) (Koo and Squazzo, 1994). Endolysosomal trafficking involves the formation, fusion and fission of vesicles. This biophysical process can be perturbed by alterations in the lipid content of membranes. In particular, increases in the content of ceramide or sphingomyelin can promote stiffness of membranes, and slow or prevent fusion and fission events, as evidenced by genetic disorders of lipid metabolism (Futerman and van Meer, 2004). In lysosomal storage disorders Aβ accumulates in endolysosomal compartments (Yamazaki et al., 2001; Keilani et al., 2012; Mattsson et al., 2012). Sphingolipid storage negatively affects the autophagic metabolism of APP and increases Aβ production (Tamboli et al., 2011a,b). Thus, HIV-associated accumulations of ceramide and sphingomyelin may promote the intraneuronal accumulation of Aβ by slowing or preventing fusion and fission events that are required for endolysosomal trafficking. This conclusion is consistent with enlargements in lysosomal systems and impairments in autophagy that have been reported in autopsy brains from HIV-infected subjects, in simian immunodeficiency virus-infected microglia (Gelman et al., 2005; Alirezaei et al., 2008; Zhou et al., 2011; Fields et al., 2013), and in brains of gp120 and APP/PS1/gp120 mice from the current study. These data suggest that therapeutics designed to restore lysosome function could protect the CNS in HIV-infected patients.
TRPML1 is located to late endosomal/lysosomal compartments. This cation channel is required for the formation and transport of vesicles to the trans Golgi network, and for the reformation of lysosomes from the late endosomal/lysosomal hybrid organelles and autolysosomes (Piper and Luzio, 2004; Treusch et al., 2004; Thompson et al., 2007). Because these membrane fusion and fission events are dependent on the release of luminal calcium (Piper and Luzio, 2004), neuropathological events that perturb TRPML1 function could have profound effects on the endolysosomal system. Indeed, neurodegeneration is a prominent feature of mucolipidosis type 4, a genetic disorder caused by a deficiency in TRPML1 (Sun et al., 2000). Likewise, neuronal damage and cell death are readily apparent in TRPML1 KO mice and in TRPML flies (Treusch et al., 2004; Shen et al., 2012). Although the endogenous regulators of TRPML1 are not known, it was recently demonstrated that sphingomyelin negatively regulates TRPML1 and that inducing calcium release with agonists of TRPML1 reduced lysosome storage. In addition to regulating fusion and fission events TRPML1 is important for regulating lysosome pH. Calcium can accumulate in the intraluminal space when cellular energetics are compromised, or when sphingomyelin is overproduced (Wang et al., 2006; Lloyd-Evans et al., 2008; Shen et al., 2012). This buildup of positive charges in the intraluminal space can prevent acidification by impairing H+ transport. By releasing the buildup of positive charges in the intraluminal space, TRPML1 agonists ease the ionic gradient thus allowing luminal acidification through restoration of H+ transport (Shen et al., 2012). Indeed, the TRPML1 agonist ML-SA1 cleared calcium, sphingomyelin and Aβ from lysosomal compartments in neurons with evidence of lysosome storage induced by HIV gp120.
The findings from this study suggest that therapeutics designed to restore lysosome function may protect the CNS in HIV-infected patients. Because deficits in lysosome function are common to a number of neurodegenerative conditions, further development and testing of TRPML1 agonists in vivo may reveal neuroprotective effects with application for a variety of neurodegenerative conditions.
Footnotes
This work was supported by NIH Grants MH077542, AG034849, AA0017408, and MH075673 to N.J.H., and by the intramural research program of the National Institute on Aging, GM103329 to M.G., K.T.-Y., and M.P.M., and AG043338, P30GM103329, and R01MH100972 to J.D.G. We thank Jacqueline Lovette for technical assistance.
The authors declare no competing financial interests.
References
- Achim CL, Adame A, Dumaop W, Everall IP, Masliah E. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J NeuroImmune Pharmacol. 2009;4:190–199. doi: 10.1007/s11481-009-9152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akay C, Lindl KA, Shyam N, Nabet B, Goenaga-Vazquez Y, Ruzbarsky J, Wang Y, Kolson DL, Jordan-Sciutto KL. Activation status of integrated stress response pathways in neurones and astrocytes of HIV-associated neurocognitive disorders (HAND) cortex. Neuropathol Appl Neurobiol. 2012;38:175–200. doi: 10.1111/j.1365-2990.2011.01215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PloS One. 2008;3:e2906. doi: 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances BM, Christensen JJ, Teshome M, Taylor J, Xiong C, Aldea P, Fagan AM, Holtzman DM, Morris JC, Mintun MA, Clifford DB. Cognitively unimpaired HIV-positive subjects do not have increased 11C-PiB: a case-control study. Neurology. 2010;75:111–115. doi: 10.1212/WNL.0b013e3181e7b66e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances BM, Ortega M, Vaida F, Heaps J, Paul R. Independent effects of HIV, aging, and HAART on brain volumetric measures. J Acquir Immune Defic Syndr. 2012a;59:469–477. doi: 10.1097/QAI.0b013e318249db17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances BM, Benzinger TL, Christensen JJ, Thomas J, Venkat R, Teshome M, Aldea P, Fagan AM, Holtzman DM, Morris JC, Clifford DB. 11C-PiB imaging of human immunodeficiency virus-associated neurocognitive disorder. Arch Neurol. 2012b;69:72–77. doi: 10.1001/archneurol.2011.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson L, Blennow K, Fuchs D, Svennerholm B, Gisslén M. Increased cerebrospinal fluid protein tau concentration in neuro-AIDS. J Neurol Sci. 1999;171:92–96. doi: 10.1016/S0022-510X(99)00253-1. [DOI] [PubMed] [Google Scholar]
- Angel TE, Jacobs JM, Spudich SS, Gritsenko MA, Fuchs D, Liegler T, Zetterberg H, Camp DG, 2nd, Price RW, Smith RD. The cerebrospinal fluid proteome in HIV infection: change associated with disease severity. Clin Proteomics. 2012;9:3. doi: 10.1186/1559-0275-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006;111:529–538. doi: 10.1007/s00401-006-0037-0. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Norrby KE, Dingwall T, Carnie FW, Millar T, Arango JC, Robertson R, Bell JE. Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain. 2010;133:3685–3698. doi: 10.1093/brain/awq263. [DOI] [PubMed] [Google Scholar]
- Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci. 2012;32:9477–9484. doi: 10.1523/JNEUROSCI.0865-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Ray B, Greig NH, Lahiri DK. Rivastigmine lowers Aβ and increases sAPPalpha levels, which parallel elevated synaptic markers and metabolic activity in degenerating primary rat neurons. PloS One. 2011;6:e21954. doi: 10.1371/journal.pone.0021954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ. Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology. 2007;68:1481–1487. doi: 10.1212/01.wnl.0000260610.79853.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, Troncoso J, Wheeler D, Pletnikova O, Wang J, Conant K, Haughey NJ. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer's but not normal brain. Neurobiol Aging. 2009;30:591–599. doi: 10.1016/j.neurobiolaging.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VVR, Mielke M, Chu M, Lentz M, McArthur JC, Sacktor N, Chang L, Ernst T, Wojna V, Pardo C, Pomper M, Haughey NJ the CHARTER group. CSF sphingolipids are biomarkers for neurocognitive status in HIV-infected patients. International Society for NeuroVirology. Milan. 2010 [Google Scholar]
- Bandaru VV, Patel N, Ewaleifoh O, Haughey NJ. A failure to normalize biochemical and metabolic insults during morphine withdrawal disrupts synaptic repair in mice transgenic for HIV-gp120. J NeuroImmune Pharmacol. 2011;6:640–649. doi: 10.1007/s11481-011-9289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KF, Zheng L, Fahrenholz F, Cuello AC. ADAM-10 over-expression increases cortical synaptogenesis. Neurobiol Aging. 2008;29:554–565. doi: 10.1016/j.neurobiolaging.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Bhatt KH, Sodhi A, Chakraborty R. Peptidoglycan induced expression of peroxisome proliferator-activated receptor gamma in mouse peritoneal macrophages: role of ERK and JNK MAP kinases. Cytokine. 2012;60:778–786. doi: 10.1016/j.cyto.2012.07.036. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Pemberton L, Blennow K, Wallin A, Hagberg L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology. 2005;65:1490–1492. doi: 10.1212/01.wnl.0000183293.95787.b7. [DOI] [PubMed] [Google Scholar]
- Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Yakupov R, Nakama H, Stokes B, Ernst T. Antiretroviral treatment is associated with increased attentional load-dependent brain activation in HIV patients. J NeuroImmune Pharmacol. 2008;3:95–104. doi: 10.1007/s11481-007-9092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Holt JL, Yakupov R, Jiang CS, Ernst T. Lower cognitive reserve in the aging human immunodeficiency virus-infected brain. Neurobiol Aging. 2013;34:1240–1253. doi: 10.1016/j.neurobiolaging.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi EY, Frey SL, Lee KY. Ganglioside G(M1)-mediated amyloid-beta fibrillogenesis and membrane disruption. Biochemistry. 2007;46:1913–1924. doi: 10.1021/bi062177x. [DOI] [PubMed] [Google Scholar]
- Come JH, Fraser PE, Lansbury PT., Jr A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci U S A. 1993;90:5959–5963. doi: 10.1073/pnas.90.13.5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi PJ, Abu-Jawdeh GM, Green RM, Green D. Amyloidosis in association with human immunodeficiency virus infection. Clin Infect Dis. 1992;14:189–191. doi: 10.1093/clinids/14.1.189. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Haughey NJ, Tammara A, McArthur JC, Nath A, Reid R, Vargas DL, Pardo CA, Mattson MP. Dysregulation of sphingolipid and sterol metabolism by ApoE4 in HIV dementia. Neurology. 2004;63:626–630. doi: 10.1212/01.WNL.0000134662.19883.06. [DOI] [PubMed] [Google Scholar]
- Desplats P, Dumaop W, Smith D, Adame A, Everall I, Letendre S, Ellis R, Cherner M, Grant I, Masliah E. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology. 2013;80:1415–1423. doi: 10.1212/WNL.0b013e31828c2e9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon NK, Peng F, Bokhari S, Callen S, Shin SH, Zhu X, Kim KJ, Buch SJ. Cocaine-mediated alteration in tight junction protein expression and modulation of CCL2/CCR2 axis across the blood-brain barrier: implications for HIV-dementia. J NeuroImmune Pharmacol. 2008;3:52–56. doi: 10.1007/s11481-007-9091-1. [DOI] [PubMed] [Google Scholar]
- Edland SD, Galasko DR. Fractional synthesis and clearance rates for amyloid β. Nat Med. 2011;17:1178–1179. doi: 10.1038/nm.2495. author reply 1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Biddolph SC, Morris CS. Prevalence of Alzheimer plaques in AIDS. J Neurol Neurosurg Psychiatry. 1998;65:29–33. doi: 10.1136/jnnp.65.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu XM, Tabira T, Greenberg BD. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992;255:726–728. doi: 10.1126/science.1738846. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood–brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31:9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, Masliah E. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol. 2013;19:89–101. doi: 10.1007/s13365-012-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008;14:318–326. doi: 10.1080/13550280802132857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, He J, Ohagen A, Vallat AV. Chemokine receptors in HIV-1 infection of the central nervous system. Semin Immunol. 1998;10:203–213. doi: 10.1006/smim.1998.0133. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Schuenke K. Brain aging in acquired immunodeficiency syndrome: increased ubiquitin-protein conjugate is correlated with decreased synaptic protein but not amyloid plaque accumulation. J Neurovirol. 2004;10:98–108. doi: 10.1080/13550280490279816. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Soukup VM, Holzer CE, 3rd, Fabian RH, Schuenke KW, Keherly MJ, Richey FJ, Lahart CJ. Potential role for white matter lysosome expansion in HIV-associated dementia. J Acquir Immune Defic Syndr. 2005;39:422–425. doi: 10.1097/01.qai.0000164250.41475.f2. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, Masliah E, Commins DL, Brandt D, Grant I, Singer EJ, Levine AJ, Miller J, Winkler JM, Fox HS, Luxon BA, Morgello S. The national NeuroAIDS tissue consortium brain gene array: two types of HIV-associated neurocognitive impairment. PloS One. 2012;7:e46178. doi: 10.1371/journal.pone.0046178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Ehrhart J, Obregon DF, Lam L, Le L, Jin J, Fernandez F, Tan J, Shytle RD. Antiretroviral medications disrupt microglial phagocytosis of beta-amyloid and increase its production by neurons: implications for HIV-associated neurocognitive disorders. Mol Brain. 2011;4:23. doi: 10.1186/1756-6606-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Ramos P, Asunción Morán M. Ultrastructural localization of intraneuronal Aβ-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007;11:53–59. doi: 10.3233/jad-2007-11109. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, Schols D, De Clercq E, Kunstman K, Wolinsky SM, Gabuzda D. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J Virol. 2001;75:10073–10089. doi: 10.1128/JVI.75.21.10073-10089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DA, Woulfe J. Lipofuscin and aging: a matter of toxic waste. Sci Aging Knowledge Environ. 2005;2005:re1. doi: 10.1126/sageke.2005.5.re1. [DOI] [PubMed] [Google Scholar]
- Gray F, Bazille C, Adle-Biassette H, Mikol J, Moulignier A, Scaravilli F. Central nervous system immune reconstitution disease in acquired immunodeficiency syndrome patients receiving highly active antiretroviral treatment. J Neurovirol. 2005;11:16–22. doi: 10.1080/13550280500511741. [DOI] [PubMed] [Google Scholar]
- Gray L, Roche M, Churchill MJ, Sterjovski J, Ellett A, Poumbourios P, Sheffief S, Wang B, Saksena N, Purcell DF, Wesselingh S, Cunningham AL, Brew BJ, Gabuzda D, Gorry PR. Tissue-specific sequence alterations in the human immunodeficiency virus type 1 envelope favoring CCR5 usage contribute to persistence of dual-tropic virus in the brain. J Virol. 2009;83:5430–5441. doi: 10.1128/JVI.02648-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AJ, Giovannoni G, Hall-Craggs MA, Thompson EJ, Miller RF. Cerebrospinal fluid tau concentrations in HIV infected patients with suspected neurological disease. Sex Transm Infect. 2000;76:443–446. doi: 10.1136/sti.76.6.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. Aids. 2005;19:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- Guglielmotto M, Monteleone D, Giliberto L, Fornaro M, Borghi R, Tamagno E, Tabaton M. Amyloid-β(4)(2) activates the expression of BACE1 through the JNK pathway. J Alzheimers Dis. 2011;27:871–883. doi: 10.3233/JAD-2011-110884. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Harris B, Pereira I, Parkin E. Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 2009;1296:203–215. doi: 10.1016/j.brainres.2009.07.105. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–267. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JL, Kraft-Terry SD, Chang L. Neuroimaging studies of the aging HIV-1-infected brain. J Neurovirol. 2012;18:291–302. doi: 10.1007/s13365-012-0114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izycka-Swieszewska E, Zóltowska A, Rzepko R, Gross M, Borowska-Lehman J. Vasculopathy and amyloid beta reactivity in brains of patients with acquired immune deficiency (AIDS) Folia Neuropathol. 2000;38:175–182. [PubMed] [Google Scholar]
- Jahanshad N, Valcour VG, Nir TM, Kohannim O, Busovaca E, Nicolas K, Thompson PM. Disrupted brain networks in the aging HIV+ population. Brain Connect. 2012;2:335–344. doi: 10.1089/brain.2012.0105-Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Xu G, Fromholt D, Gonzales V, Borchelt DR. Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:1220–1227. doi: 10.1093/jnen/62.12.1220. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto S, Kozutsumi Y, Matsuzaki K. Formation of a membrane-active form of amyloid beta-protein in raft-like model membranes. Biochem Biophys Res Commun. 2003;303:514–518. doi: 10.1016/S0006-291X(03)00386-3. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, Valenzano KJ, Checler F, Buxbaum JD, Yanagisawa K, Lockhart DJ, Wustman BA, Gandy S. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32:5223–5236. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Lee EK, Kim HH, Kuwano Y, Abdelmohsen K, Srikantan S, Subaran SS, Gleichmann M, Mughal MR, Martindale JL, Yang X, Worley PF, Mattson MP, Gorospe M. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol. 2010;17:732–739. doi: 10.1038/nsmb.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain. J Neurovirol. 2000;6:S70–S81. [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Mao Y, Shang Z, Imai Y, Hoshino T, Tero R, Tanaka M, Yamamoto N, Yanagisawa K, Urisu T. Surface-induced phase separation of a sphingomyelin/cholesterol/ganglioside GM1-planar bilayer on mica surfaces and microdomain molecular conformation that accelerates Aβ oligomerization. Biochimica et biophysica acta. 2010;1798:1090–1099. doi: 10.1016/j.bbamem.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Marlow L, Cain M, Pappolla MA, Sambamurti K. Beta-secretase processing of the Alzheimer's amyloid protein precursor (APP) J Mol Neurosci. 2003;20:233–239. doi: 10.1385/JMN:20:3:233. [DOI] [PubMed] [Google Scholar]
- Masci AM, Galgani M, Cassano S, De Simone S, Gallo A, De Rosa V, Zappacosta S, Racioppi L. HIV-1 gp120 induces anergy in naive T lymphocytes through CD4-independent protein kinase-A-mediated signaling. J Leukoc Biol. 2003;74:1117–1124. doi: 10.1189/jlb.0503239. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Olsson M, Gustavsson MK, Kosicek M, Malnar M, Månsson JE, Blomqvist M, Gobom J, Andreasson U, Brinkmalm G, Vite C, Hecimovic S, Hastings C, Blennow K, Zetterberg H, Portelius E. Amyloid-beta metabolism in Niemann-Pick C disease models and patients. Metab Brain Dis. 2012;27:573–585. doi: 10.1007/s11011-012-9332-8. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke MM, Bandaru VV, McArthur JC, Chu M, Haughey NJ. Disturbance in cerebral spinal fluid sphingolipid content is associated with memory impairment in subjects infected with the human immunodeficiency virus. J Neurovirol. 2010;16:445–456. doi: 10.1007/BF03210850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RF, Isaacson PG, Hall-Craggs M, Lucas S, Gray F, Scaravilli F, An SF. Cerebral CD8+ lymphocytosis in HIV-1 infected patients with immune restoration induced by HAART. Acta Neuropathol. 2004;108:17–23. doi: 10.1007/s00401-004-0852-0. [DOI] [PubMed] [Google Scholar]
- Morimoto T, Ohsawa I, Takamura C, Ishiguro M, Kohsaka S. Involvement of amyloid precursor protein in functional synapse formation in cultured hippocampal neurons. J Neurosci Res. 1998;51:185–195. doi: 10.1002/(SICI)1097-4547(19980115)51:2<185::AID-JNR7>3.0.CO%3B2-9. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Tsukuda M, Yamaguchi T, Ikeda K, Okada T, Yano Y, Hoshino M, Matsuzaki K. Ganglioside-mediated aggregation of amyloid beta-proteins (Aβ): comparison between Aβ-(1–42) and Aβ-(1–40) J Neurochem. 2011;116:851–857. doi: 10.1111/j.1471-4159.2010.06997.x. [DOI] [PubMed] [Google Scholar]
- Okada T, Ikeda K, Wakabayashi M, Ogawa M, Matsuzaki K. Formation of toxic Aβ(1–40) fibrils on GM1 ganglioside-containing membranes mimicking lipid rafts: polymorphisms in Aβ(1–40) fibrils. J Mol Biol. 2008;382:1066–1074. doi: 10.1016/j.jmb.2008.07.072. [DOI] [PubMed] [Google Scholar]
- Parkin ET, Hussain I, Karran EH, Turner AJ, Hooper NM. Characterization of detergent-insoluble complexes containing the familial Alzheimer's disease-associated presenilins. J Neurochem. 1999;72:1534–1543. doi: 10.1046/j.1471-4159.1999.721534.x. [DOI] [PubMed] [Google Scholar]
- Pelle MT, Bazille C, Gray F. Neuropathology and HIV dementia. Handb Clin Neurol. 2008;89:807–818. doi: 10.1016/S0072-9752(07)01270-5. [DOI] [PubMed] [Google Scholar]
- Piper RC, Luzio JP. CUPpling calcium to lysosomal biogenesis. Trends Cell Biol. 2004;14:471–473. doi: 10.1016/j.tcb.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Ragin AB, Du H, Ochs R, Wu Y, Sammet CL, Shoukry A, Epstein LG. Structural brain alterations can be detected early in HIV infection. Neurology. 2012;79:2328–2334. doi: 10.1212/WNL.0b013e318278b5b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan LE, Westmark CJ, Jarzembowski JA, Malter JS. hnRNP C increases amyloid precursor protein (APP) production by stabilizing APP mRNA. Nucleic Acids Res. 1998;26:3418–3423. doi: 10.1093/nar/26.14.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:1933–1945. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Fan S, Dykstra H, Reichenbach N, Del Valle L, Potula R, Phipps RP, Maggirwar SB, Persidsky Y. Dyad of CD40/CD40 ligand fosters neuroinflammation at the blood–brain barrier and is regulated via JNK signaling: implications for HIV-1 encephalitis. J Neurosci. 2010;30:9454–9464. doi: 10.1523/JNEUROSCI.5796-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riga D, Riga S, Halalau F, Schneider F. Brain lipopigment accumulation in normal and pathological aging. Ann N Y Acad Sci. 2006;1067:158–163. doi: 10.1196/annals.1354.019. [DOI] [PubMed] [Google Scholar]
- Roch JM, Masliah E, Roch-Levecq AC, Sundsmo MP, Otero DA, Veinbergs I, Saitoh T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid beta/A4 protein precursor. Proc Natl Acad Sci U S A. 1994;91:7450–7454. doi: 10.1073/pnas.91.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomquist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thal LJ. Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature. 1993;361:260–263. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soontornniyomkij V, Moore DJ, Gouaux B, Soontornniyomkij B, Tatro ET, Umlauf A, Masliah E, Levine AJ, Singer EJ, Vinters HV, Gelman BB, Morgello S, Cherner M, Grant I, Achim CL. Cerebral beta-amyloid deposition predicts HIV-associated neurocognitive disorders in APOE epsilon4 carriers. Aids. 2012;26:2327–2335. doi: 10.1097/QAD.0b013e32835a117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe-Drger B, Deppe M, Mohammadi S, Keller SS, Kugel H, Gregor N, Evers S, Young P, Ringelstein EB, Arendt G, Knecht S, Husstedt IW. Early microstructural white matter changes in patients with HIV: a diffusion tensor imaging study. BMC Neurol. 2012;12:23. doi: 10.1186/1471-2377-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS, Jr, Bove C, Kaneski CR, Nagle J, Bromley MC, Colman M, Schiffmann R, Slaugenhaupt SA. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- Tamboli IY, Prager K, Barth E, Heneka M, Sandhoff K, Walter J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the beta-amyloid precursor protein and amyloid beta-peptide. J Biol Chem. 2005;280:28110–28117. doi: 10.1074/jbc.M414525200. [DOI] [PubMed] [Google Scholar]
- Tamboli IY, Tien NT, Walter J. Sphingolipid storage impairs autophagic clearance of Alzheimer-associated proteins. Autophagy. 2011a;7:645–646. doi: 10.4161/auto.7.6.15122. [DOI] [PubMed] [Google Scholar]
- Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM, Saftig P, Sandhoff K, Walter J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Aβ generation. J Neurosci. 2011b;31:1837–1849. doi: 10.1523/JNEUROSCI.2954-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006296. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EG, Schaheen L, Dang H, Fares H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol. 2007;8:54. doi: 10.1186/1471-2121-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Treusch S, Knuth S, Slaugenhaupt SA, Goldin E, Grant BD, Fares H. Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc Natl Acad Sci U S A. 2004;101:4483–4488. doi: 10.1073/pnas.0400709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers- a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao HF, Zhang GJ. Mechanism of cytosol phospholipase C and sphingomyelinase-induced lysosome destabilization. Biochimie. 2006;88:913–922. doi: 10.1016/j.biochi.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klunk WE, Debnath ML, Huang GF, Holt DP, Shao L, Mathis CA. Development of a PET/SPECT agent for amyloid imaging in Alzheimer's disease. J Mol Neurosci. 2004;24:55–62. doi: 10.1385/JMN:24:1:055. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Araki W, Chui DH, Makifuchi T, Ihara Y, Tabira T. Glypican-1 as an Aβ binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer's disease. FASEB J. 2004;18:1013–1015. doi: 10.1096/fj.03-1040fje. [DOI] [PubMed] [Google Scholar]
- Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, Mattson MP, Geiger JD, Haughey NJ. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–1249. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Liu J, Chen L, Liang S, Fujii N, Tamamura H, Xiong H. HIV-1 gp120 enhances outward potassium current via CXCR4 and cAMP-dependent protein kinase A signaling in cultured rat microglia. Glia. 2011a;59:997–1007. doi: 10.1002/glia.21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Bae M, Tovar-y-Romo LB, Patel N, Bandaru VV, Pomerantz D, Steiner JP, Haughey NJ. The human immunodeficiency virus coat protein gp120 promotes forward trafficking and surface clustering of NMDA receptors in membrane microdomains. J Neurosci. 2011b;31:17074–17090. doi: 10.1523/JNEUROSCI.4072-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Chang TY, Haass C, Ihara Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-Pick type C cells. J Biol Chem. 2001;276:4454–4460. doi: 10.1074/jbc.M009598200. [DOI] [PubMed] [Google Scholar]
- Yao H, Duan M, Buch S. Cocaine-mediated induction of platelet-derived growth factor: implication for increased vascular permeability. Blood. 2011;117:2538–2547. doi: 10.1182/blood-2010-10-313593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, Toborek M. Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci. 2008;28:7788–7796. doi: 10.1523/JNEUROSCI.0061-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011;203:1647–1657. doi: 10.1093/infdis/jir163. [DOI] [PMC free article] [PubMed] [Google Scholar]