

Abstract
The interplay between inflammation and cancer progression is a growing area of research. A combination of clinical, epidemiological, and basic science investigations indicate that there is a relationship between inflammatory changes in the pancreas and neoplastic progression. Diets high in ω-6 polyunsaturated fatty acids provide increased substrate for arachidonic acid metabolism by cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) to form eicosanoids. These eicosanoids directly contribute to pancreatic cancer cell proliferation. Both COX-2 and 5-LOX are upregulated in multiple cancer types, including pancreatic cancer. In vitro studies using pancreatic cancer cell lines have demonstrated upregulation of COX-2 and 5-LOX at both the mRNA and protein levels. When COX-2 and 5-LOX are blocked via a variety of mechanisms, cancer cell proliferation is abrogated both in vitro and in vivo. The mechanism of COX-2 has been shown to include effects on apoptosis as well as angiogenesis. 5-LOX has been implicated in apoptosis. The use of COX-2 and 5-LOX inhibitors in clinical studies in patients with pancreatic cancer has been limited. Patient enrollment has been restricted to those with advanced disease which makes evaluation of these drugs as chemopreventive agents difficult. COX-2 and 5-LOX expression have been shown to be present during the early neoplastic changes of pancreatic cancer, well before progression to invasive disease. This indicates that the ideal role for these interventions is early in the disease process as preventive agents, perhaps in patients with chronic pancreatitis or hereditary pancreatitis.
Keywords: Arachidonic acid, Eicosanoid, Cyclooxygenase-2, 5-lipoxygenase, Pancreatic cancer, Inflammation
Core tip: This review article highlights the relationship between inflammation and pancreatic cancer, specifically focusing on the enzymes cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX). The role of inflammation and tumor progression is a burgeoning area of research. This review delves into the research that has been conducted investigating COX-2 and 5-LOX and their relationship to pancreatic cancer both in vivo and in vitro. We discuss a variety of investigations including basic science, epidemiological, and clinical as they relate to pancreatic inflammation and eicosanoids.
INTRODUCTION
The relationship between inflammation and cancer is well established. Rudolf Virchow noticed leukocytes in cancerous tissue as early as 1863 and conjectured that there was a link between chronic inflammation and neoplasia[1]. This theory has been validated by clinical examples such as Marjolin’s ulcers which are squamous cell carcinomas that form in sites of chronic inflammation such as burn scars or chronic ulcers[2]. Other examples of inflammatory conditions with correlative cancers are inflammatory bowel disease and colorectal cancer, gastritis caused by Helicobacter pylori and gastric cancer, hepatitis and hepatocellular carcinoma, and chronic pancreatitis and pancreatic cancer. These examples highlight the impact of inflammation on the neoplastic process though the mechanism is unclear.
The inflammatory response is marked by cytokine release from epithelial cells which attract and activate inflammatory cells. When macrophages, neutrophils, fibroblasts, and mast cells are attracted to this inflammatory microenvironment, they produce reactive oxygen species (ROS) and stimulate epithelial cell proliferation[3]. The infiltration of these cells into the tumor microenvironment has been implicated in pancreatic tumor progression (Figure 1)[4-7]. ROS can directly cause DNA damage by increasing the probability that genetic mutation will occur. Combined with their effects on cellular proliferation, ROS increase the likelihood of neoplastic transformation[3,8]. A key step in the inflammatory process is the activation of the arachidonic acid pathway that produces eicosanoids. The purpose of this paper will be to review inflammatory mechanisms as they relate to pancreatic cancer, specifically the roles of cyclooxygenase (COX) and lipoxygenase (LOX), and how their metabolites contribute to carcinogenesis.
Figure 1.
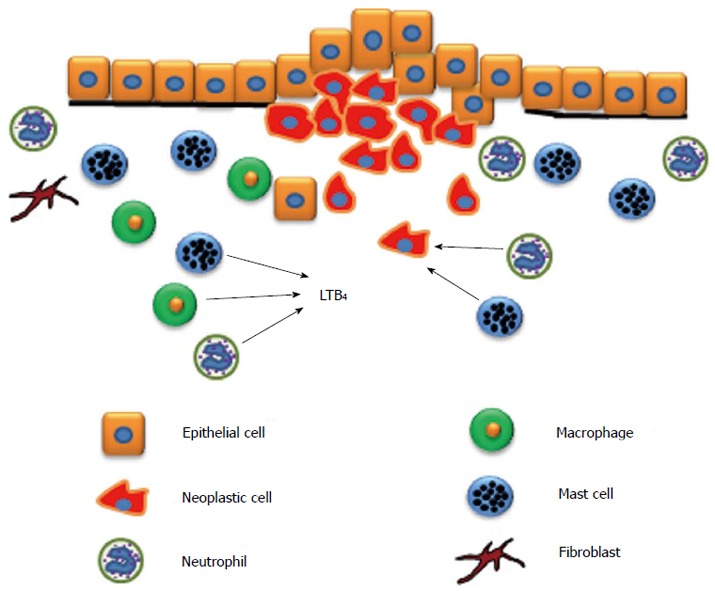
Inflammatory cell infiltration into the tumor microenvironment. As pancreatic adenocarcinoma progresses, inflammatory cells such as mast cells, neutrophils, and macrophages are attracted to the tumor microenvironment and enhance tumor growth. Leukotriene B4 (LTB4) is a chemotactic factor for macrophages, neutrophils, and mast cells. Fibroblasts are also activated and enhance collagen production.
INFLAMMATION AND PANCREATIC CANCER
Pancreatic cancer is the fourth leading cause of cancer-related death in the United States, and the vast majority of those afflicted succumb to this disease. The 5-year survival rate is about 5%-6%[9]. Since the majority of pancreatic cancer is discovered late in the disease process, well after potentially curative surgery is an option, understanding the early oncogenic changes is necessary to aid in prevention. Since inflammation has been shown to be a key factor in the neoplastic process as it contributes to genetic changes and DNA damage, its role in pancreatic cancer is of particular interest.
Studying the mechanisms of pancreatitis in patients can be helpful for understanding inflammation as it relates to pancreatic cancer development. Patients with hereditary pancreatitis, a rare disease responsible for less than 1% of pancreatitis cases, have frequent episodes of acute inflammation[10]. Repeated episodes of pancreatitis result in fibrosis, chronic inflammation, and the eventual destruction of the gland[11]. This chronic inflammatory environment is thought to contribute to malignant transformation of pancreatic ductal cells. In patients with hereditary pancreatitis, the risk of developing pancreatic cancer is 53 times higher than unaffected individuals, and by 70 years of age, approximately 40% of these patients will develop pancreatic cancer[10]. Patients afflicted with non-hereditary chronic pancreatitis also have an increased risk of pancreatic cancer. Population studies suggest that patients with chronic pancreatitis are 17 times more likely to develop pancreatic cancer compared to age matched controls, and the risk is correlated with the duration of inflammation[12]. Therefore it will be important to understand the mechanisms that link pancreatitis to the development of pancreatic cancer.
The inflammatory process begins with the inappropriate release of proteolytic pancreatic enzymes that cause acinar cell injury[13]. This generates an immune response in which inflammatory cells are attracted to cytokines released from the cells at the site of injury. Our lab, as well as others, previously investigated the relationship between one of the major inflammatory cell types, mast cells, and pancreatic cancer[6,14]. We have shown that mast cell infiltration in pancreatic cancer specimens correlates with worse prognosis[6]. Ma demonstrated that pancreatic ductal adenocarcinoma (PDAC) cells promote mast cell migration and activation in vitro. The study also showed that blocking mast cell migration in an orthotopic PDAC mouse model decreased PDAC growth in vivo[15]. Similarly, Soucek demonstrated in an islet-cell tumor mouse model that mast cells mediate expansion of these tumors and are essential for tumor maintenance[5].
The generation of ROS and activation of the arachidonic acid pathway are also key steps in potentiating the inflammatory response[13]. The body mounts a natural response to chronic insults to the pancreas by releasing growth factors such as platelet-derived growth factor and transforming growth factor beta. This stimulates cell proliferation, which can potentially worsen DNA damage and increase genetic mutations[16].
EPIDEMIOLOGICAL STUDIES
Epidemiological studies have shown that high-fat diets, specifically with a high proportion of polyunsaturated omega-6 fatty acids, are associated with increased cancer rates, particularly in breast, pancreas, and prostate cancers[17-22]. Studies have shown that cancer incidence in an ethnic group often changes after migration and drastic dietary changes. An example is the migration of the Japanese to Western countries that have relatively higher fat diets compared to Japanese diets. Studies have reported increased colon, pancreas, breast, and prostate cancer incidence in individuals migrating to Western countries from Japan[21]. The relationship between a high-fat diet and pancreatic cancer was evaluated by a prospective study investigating obesity in various age groups including early adulthood, midlife, and older age. There were significant positive associations between pancreatic cancer and obesity in all age groups studied[23]. Patients with the longest duration of obesity and diabetes were at the greatest risk for pancreatic cancer[23]. One of the mechanisms proposed for this association is the high content of arachidonic acid in animal fats. Arachidonic acid is metabolized to biologically active lipids by COX, LOX, and epoxygenase pathways to generate eicosanoids[24]. Eicosanoids have been implicated in various carcinogenic mechanisms including tumor progression and metastasis[25]. Studies conducted in EL-Kras transgenic mice fed a high ω-6 fatty acid diet demonstrated increased frequency and size of pancreatic neoplastic lesions as well as increased pancreatic mast cell densities[26]. In a related study, a high ω-3 fatty acid diet in EL-Kras transgenic mice was found to have a protective effect against the formation of pancreatic lesions. These mice had reduced incidence, frequency, and proliferative index of pancreatic precancer compared to those fed standard chow[27]. In unpublished findings by our lab, we demonstrate that EL-Kras transgenic mice fed high ω-6 fatty acid diets had increased PGE2 and LTB4 compared to their counterparts fed a high ω-3 fatty acid diet. Therefore, ω-3 and ω-6 fatty acids are involved in carcinogenic mechanisms and have opposing effects on pancreatic neoplasia, which is hypothesized to be mediated through the regulation of eicosanoid production.
Further evidence to support the role of eicosanoids in the carcinogenic process are epidemiological studies indicating that the use of non-steroidal anti-inflammatory drugs (NSAIDs) reduces the incidence of various solid tumors[24,28]. One study used a meta-analysis to examine the effect of regular NSAID use on colon, lung, breast, and prostate cancers. The results indicated that there is a risk reduction of 43% for colon cancer, 28% for lung cancer, 25% for breast cancer, and 27% for prostate cancer[28]. The role of NSAIDs and pancreatic cancer is not clear. Anderson conducted a prospective study with 28000 post-menopausal women and demonstrated a decreasing trend in pancreatic cancer incidence in women with more frequent aspirin use[29]. Alternatively, a study among United States adults followed for 18 years found no association between aspirin use and pancreatic cancer mortality[30]. A different prospective study in a large cohort of women with an 18 year follow-up showed an association with long-term aspirin use and pancreatic cancer although there was a higher prevalence of obesity and diabetes mellitus among patients who reported regular aspirin use[31]. A study conducted in the United Kingdom demonstrated that NSAID use for more than 773 d in the 5 years prior to diagnosis was associated with a 20% risk reduction of pancreatic cancer, although increasing doses did not have an impact on risk[32]. A meta-analysis involving 11 studies analyzing the association between pancreatic cancer and aspirin and other NSAIDS did not find a conclusive association[33]. The summary relative risk did not find an association between aspirin or other NSAIDS and pancreatic cancer, nor an association between regular use vs irregular use, nor frequency of aspirin or NSAID use[33].
BIOCHEMISTRY OF COX AND LOX
The ability of NSAIDs to exert their anti-inflammatory and anti-tumor effects by inhibiting the COX enzyme, which results in decreased prostanoid production, demonstrates the intimate relationship between inflammation and cancer[24]. There is evidence to suggest that 5-LOX, a close relative of COX-2, is essential for eicosanoid production and tumor pathogenesis. The precursors of eicosanoids are arachidonic acids. Both prostaglandins (PG) and leukotrienes (LT) are members of the eicosanoid family, which are lipid mediators made of a 20 carbon fatty acid derivative[34]. Eicosanoids are vital due to their distinct biological activity in the body and effectiveness in the nanomolar concentration range[34]. The two eicosanoid members that will be discussed in detail here are prostaglandins and leukotrienes.
Prostaglandins are made by most cells in the body, and they act as both paracrine and autocrine mediators[34]. Arachidonic acid is released from the membrane by the phospholipase cPLA2 and acted on by prostaglandin G/H synthase (known as COX) to become an intermediate known as PGH2[24] (Figure 2). There are two main forms of COX: COX-1 and COX-2. COX-1 is generally thought of as the constitutively expressed enzyme that is responsible for basal production of prostanoids for tissue homeostasis, and COX-2 is induced by cytokines and growth factors, particularly at sites of inflammation and neoplasia[13]. Therefore, COX-2 has a key role in the setting of inflammation and the tumor microenvironment[24].
Figure 2.
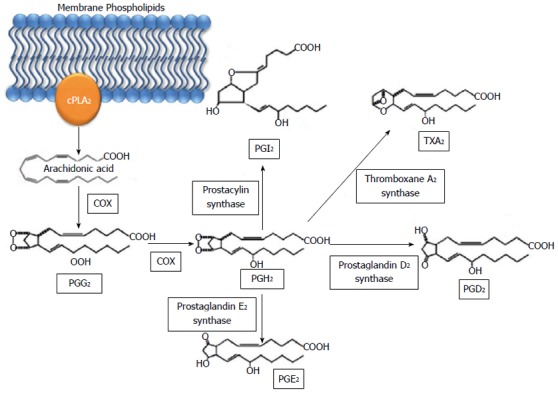
Metabolic pathway of prostaglandins via cyclooxygenase. Arachidonic acid is released from membrane phospholipids by phospholipase A2 and converted to PGG2 and subsequently PGH2 by COX. PGH2 is then converted to PGI2, TXA2, PGD2, and PGE2. cPLA2: Cytosolic phospholipase A2; COX: Cyclooxygenase; PG: Prostaglandin; TX: Thromboxane.
Leukotrienes, while derived from the same precursor as prostaglandins, are functionally distinct. Leukotrienes are predominately produced by inflammatory cells, and once cellular activation occurs, cPLA2 and 5-lipoxygenase (5-LOX) are translocated to the nuclear envelope[34]. LOX enzymes are a family of nonheme iron-containing dioxygenases with labeling based on the location of oxygen insertion at the carbon position of arachidonic acid[25]. The most common LOX enzymes are 5-, 8-, 12-, and 15-LOX[25]. These then form the corresponding hydroperoxyeicosatetraenoic acids (HPETE)[25]. Specifically, 5-LOX transforms arachidonic acid via a dehydration reaction to the unstable epoxide LTA4[25]. LTA4 is further oxidized to form either 5-HETE or the leukotrienes[25]. LTA4 can be hydrolyzed by leukotriene A4 hydrolase in the cytoplasm or nucleus resulting in LTB4 (Figure 3). LTB4 is known as a potent chemoattractant, and its receptors are upregulated in pancreatic cancer[35]. LTA4 can also be conjugated with glutathione to form LTC4 by LTC4 synthase. LTC4 can then undergo extracellular metabolism resulting in LTD4 and LTE4[34]. The activation of 5-LOX is dependent upon the 5-LOX-activating protein (FLAP).
Figure 3.
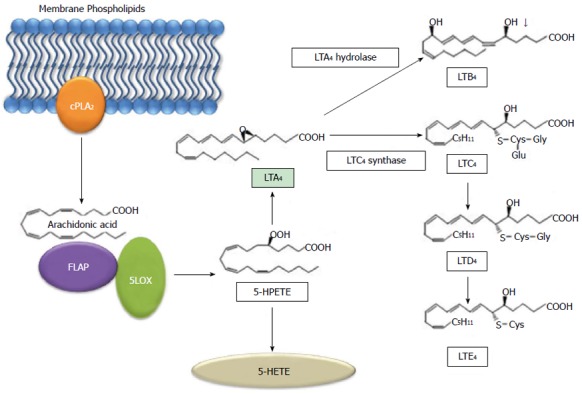
Metabolic pathway of arachidonic acid via 5- lipoxygenase. Arachidonic acid is released from membrane phospholipids by phospholipase A2 and converted to 5-HPETE by 5-LOX and 5-LOX activating protein (FLAP). 5-HPETE can then form either 5-HETE or LTA4. LTA4 then becomes LTB4 or LTC4. LTC4 can then form LTD4 and subsequently LTE4. cPLA2: Cytosolic phospholipase A2; LOX: Lipoxygenase; FLAP: 5-LOX activating protein; HPETE: Hydroperoxyeicosatetraenoic acid; HETE: Hydroxyl 6 trans 8, 11, 14 cis eicosatetraenoic acid; LT: Leukotriene.
One of the ways in which LTB4 directs chemotaxis and regulates neutrophil adhesion is by activating integrin receptors[34,36-39]. It has been demonstrated that local cell death causes “swarm-like” interstitial neutrophil clustering and LTB4 plays an important role in intercellular communication between neutrophils and facilitates neutrophil movement through tissue[39]. In the tumor microenvironment, LTB4 has been shown in vivo to enhance leukocyte recruitment into the tumor stroma[40].
ROLE OF COX IN PANCREATIC NEOPLASIA AND CANCER
COX-2 expression is upregulated in a variety of malignancies including colon, esophagus, breast, and pancreatic cancer[41-43]. Multiple studies have indicated that COX-2 is also important in carcinogenesis. One example in a murine model of familial adenomatous polyposis showed a marked reduction in the number and size of intestinal polyps in COX-2 null mice with an APC mutation[44].
The relationship between COX-2 and pancreatic cancer has been evaluated in multiple studies with the majority of the evidence demonstrating upregulated COX-2 expression in pancreatic cancer at both the mRNA and protein levels. One study showed that levels of COX-2 mRNA were increased 60-fold in pancreatic cancer compared to normal tissue. In addition, COX-2 protein was expressed in 9 out of 10 pancreatic cancer samples, while nontumor samples had no COX-2 expression[45]. Immunohistochemistry (IHC) confirmed COX-2 expression in malignant epithelial cells[45]. A different study demonstrated an increase in COX-2 expression using IHC when pancreatic carcinoma was compared to normal pancreas[43]. Five pancreatic cancer cell lines were studied, and COX-2 protein expression was detected in BxPC-3, Capan-1, and MDAPanc-3 cells, and increased levels of COX-2 mRNA were detected in 4 of the 5 cell lines[43]. When an NSAID was used, a dose-dependent inhibition of cellular proliferation was observed in all cell lines studied[43]. Kokawa et al[46] used different pancreatic cancer cell lines (KP-2, PNS-1, MiaPaca-2, and Panc-1) to show that COX-2 expression was upregulated in all 4 of them, and NSAID inhibition of cellular proliferation was correlated with the expression of COX-2. Maitra used automated cellular imaging to evaluate COX-2 expression not only in pancreatic adenocarcinoma but also its precursor, pancreatic intraepithelial neoplasia (PanIN). This showed an increase in the overall average number of positive cells from 19.2% in normal ducts to 36.3% in PanINs to 47.3% in adenocarcinomas[47]. This study suggests tumorigenic activity of COX-2 in preinvasive pancreatic lesions and a potential role for chemopreventive agents such as COX-2 inhibitors in pancreatic cancer.
While multiple studies have shown the association between pancreatic cancer and COX-2 expression, few have investigated the underlying mechanism of COX-2 and how it promotes neoplastic changes. Overexpression of COX-2 leads to increased tumor prostanoid levels, and PGE2 is known to have several tumorigenic effects. PGE2 has been implicated in the inhibition of apoptosis and the induction of proliferation and angiogenesis[48]. One group investigated the relationship between high-mobility group A1 (HMGA1) and COX-2 in pancreatic cancer. The authors proposed that the HMGA1-COX-2 axis is a key molecular pathway in pancreatic cancer because the upregulation of COX-2 expression is HMGA1 dependent in various pancreatic cancer cell lines. It was first demonstrated that a positive correlation between HMGA1 and COX-2 expression in six pancreatic cancer cell lines (BxPC-3, HPAF-II, MiaPaCa-2, Panc1, PL45, and XPA-3) existed[49]. COX-2 expression after knock-down of HMGA1 in two pancreatic cancer cell lines was evaluated and showed that HMGA1 binds to the COX-2 promoter to induce its expression[49]. A significant reduction in COX-2 expression after using an HMGA1 siRNA was observed, and COX-2 inhibitors blocked tumorigenesis in human pancreatic cancer xenografts that overexpressed HMGA1[49].
Another potential mechanism proposed for the involvement of COX-2 in tumorigenesis is its effect on angiogenesis. Chu compared the angiogenic effects of a COX-2 expressing pancreatic cancer cell line BxPC-3 with the COX-2 negative AsPC-1 cell line. The group found a significant increase in endothelial cell migration induced by BxPC-3 migration compared with AsPC-1. These findings were supported by data demonstrating that BxPC-3 treatment with a COX-2 inhibitor decreased the angiogenic responses of the endothelial cells[50]. Eibl et al[51] showed in a subset of pancreatic cancer cell lines that COX-2 increased PGE2 which subsequently increased VEGF secretion. In a subsequent in vivo study, an orthotopic pancreatic cancer model in nude mice was used to demonstrate the effects of nimesulide, a selective COX-2 inhibitor, on angiogenesis. In mice with COX-2 positive tumors, nimesulide resulted in an increase in VEGF production by malignant cells but a compensatory decrease in production by nonmalignant cells, ultimately leading to reduced tumor angiogenesis and growth[52].
Ito’s study on the effect of COX-2 on tumor invasion found that PGE2 mediated pancreatic cancer cell invasion through induction of matrix metalloproteinase-2 expression. This induction was found to be dependent on an extracellular signal-regulated kinase (ERK)/Ets-1-dependent mechanism[53].
Another study investigated the expression of COX-2 on clinical outcomes and found no correlation between global COX-2 expression and clinical outcome. The clinical outcomes studied were survival, stage, tumor size, or vascular invasion[54]. The expression of COX-2 was related to an increase in perineural invasion[54].
Several preclinical mouse models evaluating pancreatic lesions have been reported. One particular transgenic model, LSL-KRASG12D; PDX-1-Cre, is a mouse with a KRAS mutation expressed in pancreatic progenitor cells. This model results in PanIN lesions which eventually develop through advanced PanIN lesions into adenocarcinoma[55]. The efficacy of a selective COX-2 inhibitor, nimesulide, was evaluated in this mouse model. Animals treated with nimesulide demonstrated significantly fewer PanIN lesions and decreased intrapancreatic prostaglandin E2 levels compared to mice on a control diet[55]. In two unpublished works from our group, another mouse model with mutant Kras expression targeted to acinar cells (EL-Kras)[56] have been crossed with COX-2 knock-out mice to generate cohorts of EL-Kras/COX-2-/- mice. These mice have a significantly reduced frequency of cystic papillary neoplasms compared with EL-Kras mice with wild-type COX-2. Also, mice that overexpress COX-2 in acinar cells develop hyperplastic, mildly dysplastic ducts with accompanying focal fibrosis and lymphocytic infiltration[57]. A different transgenic mouse model, BK5.COX2, results in COX-2 overexpression in the exocrine pancreas[58]. The resulting histology demonstrated pancreatitis-like changes with acinar-to-ductal metaplasia by 3 mo, and at 6-8 mo strongly dysplastic features. The described phenotype was completely prevented by maintaining the mice on a COX-2 inhibitor. Cell lines derived from lesions in these mice were tumorigenic when injected into nude mice. Both of these mouse models highlight the relationship between COX-2 and pancreatic cancer and will be important in future studies.
ROLE OF LOX IN PANCREATIC NEOPLASIA AND CANCER
Similar to COX-2, LOX has been implicated in several human cancers including lung, prostate, colon, breast, and pancreatic; however, relatively little research has been conducted to elucidate its role in cancer progression[59-61]. 5-LOX expression is upregulated in both pancreatic adenocarcinoma as well as in neoplastic lesions of the pancreas[25]. In a study by Hennig, three pancreatic cancer cell lines, AsPC-1, PANC-1, and MiaPaCa2, were found to have 5-LOX mRNA expression while normal human pancreatic cells did not express 5-LOX[35]. They also confirmed that 5-LOX protein was expressed in these cell lines and in two additional cell lines, Capan-1 and HPAF[35]. Moreover, the expression levels of both 5-LOX and its downstream metabolite LTB4 were found to be significantly upregulated in pancreatic tumors compared with normal pancreatic tissue[35]. Interestingly, staining was evident in both the cancer cells as well as the ductal cells and adjacent islets. A follow-up study by Hennig et al[62] investigated 5-LOX expression in PanIN lesions. Greater than 90% of the ductal cells had strong positive 5-LOX staining in all grades of PanINs with no significant difference between grades of PanINs. This was compared to normal pancreatic specimens that had 0 to 7.5% of the ductal cells showing 5-LOX staining[62]. This study also reported that 5-LOX expression was present in pancreatic PanIN-like lesions in N-nitroso-bis (2-oxopropyl)-amine (BOP) treated hamsters as well as EL-Kras transgenic mice[62]. Ding reported similar results showing increased 5-LOX expression in MiaPaCa2, PANC-1, AsPC-1, and Capan2 pancreatic cancer cell lines at the mRNA level[63]. The general LOX inhibitor (NDGA), a 5-LOX inhibitor (Rev5901), and a FLAP inhibitor (MK-886), all inhibited thymidine incorporation in MiaPaCa2 cells indicating that these compounds induced growth inhibition in pancreatic cancer cells. Finally, it was demonstrated that arachidonic acid and linoleic acid induced pancreatic cancer cell proliferation[63].
While there have been no studies published to date examining mouse models deficient in 5-LOX, our lab is currently investigating this mouse model. We have developed a EL-Kras/5-LOX null mouse and preliminary results have indicated a decrease in pancreatic lesions in the 5-LOX null mice compared with their wildtype counterparts.
While it is well established that 5-LOX plays an important role in pancreatic tumor progression, fewer studies have investigated its underlying mechanism in this disease. Ding showed that the 5-LOX metabolite, 5(S)-hydroxyeicosatetraenoic acid [5(S)-HETE], stimulates pancreatic cancer cell proliferation in a time- and concentration-dependent manner[63]. In a subsequent study, Ding demonstrated that 5-(S)-HETE has mitogenic effects due to its role in the MEK/ERK and PI3 kinase/AKT pathways[64]. In an additional study, this group demonstrated that both the general LOX inhibitor (NDGA) and the 5-LOX inhibitor (Rev5901) induced apoptosis in four different pancreatic cancer cell lines[65]. Apoptosis was confirmed using three different methods including DNA propidium iodide staining, DNA fragmentation, and terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay in PANC-1, MiaPaCa2, Capan2, and HPAF cell lines[65]. A follow-up study performed by Tong further delineated the mechanism behind the LOX inhibitor-induced apoptosis showing that it is a mitochondria-mediated pathway[66]. Specifically, LOX inhibitors (NDGA and Rev5901) decreased Bcl-2 and Mcl-1 and increased Bax expression in human pancreatic cancer cells[66]. LOX inhibitors also induced cytochrome-c release and caspase-9 activation. The effect of the LOX inhibitors was also demonstrated in vivo where it blocked pancreatic cancer cell growth and induced apoptosis in athymic mice[66]. These studies suggest the relationship between 5-LOX and its role in apoptosis in the tumor microenvironment.
LTB4 AND PANCREATIC CANCER
LTB4 is a metabolite of 5-LOX and an important inflammatory mediator. LTB4 is involved in recruiting inflammatory cells and is a potent chemokine for monocytes, neutrophils, and eosinophils. It also enhances adhesion and migration of neutrophils across the vascular endothelium[67]. BLT1 and BLT2 are two G-protein-coupled receptors that have a high and low affinity, respectively, for LTB4[68]. LTB4 is secreted from human pancreatic cancer cells and its receptors are upregulated in pancreatic cancer tissue as well as in multiple cell lines[35,69]. Similar to COX-2 and 5-LOX, BLT1 and BLT2 have also been found to be upregulated in PanIN lesions which suggests a potential role of LTB4 and its receptors in chemoprevention[70].
Multiple LTB4 receptor antagonists have been developed but earlier compounds had poor oral bioavailability[68]. A more stable and orally bioavailable compound was later developed, LY293111, which blocks LTB4-mediated kinase phosphorylation[67]. LY293111 inhibits pancreatic cancer growth in vivo and in vitro through inhibition of proliferation and induction of apoptosis in a variety of pancreatic cancer cell lines (MiaPaCa-2, HPAC, Capan-1, Capan-2, PANC-1, and AsPC-1) in a time- and concentration-dependent manner[69,71]. When LTB4 was added to the cancer cell lines, it stimulated proliferation and induced ERK1/2 phosphorylation in all six cell lines[69]. In a different study, LY293111 was found to cause cell cycle arrest in S phase and suppress cyclin A, cyclin E, and cdk2 expression[71]. When LY293111 was administered to athymic mice with human pancreatic cancer xenografts, the LTB4 receptor antagonist suppressed growth of the subcutaneous xenografts[69].
CLINICAL CORRELATION
COX inhibitors
Multiple studies have been conducted evaluating the use of COX-2 inhibitors combined with different chemotherapy regimens. A phase II trial of Uracil/Tegafur plus Leucovorin and Celecoxib combined with radiotherapy in patients with locally advanced pancreatic cancer did not show a significant response and resulted in substantial gastrointestinal toxicity[72]. A study of Celecoxib and 5-fluorouracil in patients with advanced pancreatic cancer who had progressed after gemcitabine-based chemotherapy showed promising results in that the Celecoxib was well tolerated and capable of inducing durable responses[73]. In a phase II trial of gemcitabine, Irinotecan, and Celecoxib in patients with inoperable pancreatic cancer, the addition of Celecoxib was found to increase the percentage of patients achieving a one-year overall survival from about 3 mo to 9 mo and increased overall survival from about 6 mo to 18 mo[74]. Other studies in patients with advanced pancreatic cancer evaluated the combination of Celecoxib and gemcitabine or the combination of Gemcitabine, Celecoxib, and Cisplatin, but Celecoxib did not increase the efficacy of either chemotherapy regimen (Table 1)[75,76]. While the idea of using a COX-2 inhibitor is promising in patients with pancreatic cancer, it will likely be most effective as a preventive agent very early in the disease process as opposed to improving survival in those patients with advanced disease.
Table 1.
Clinical trials
| Drug | Type | Trial | Type | Cancer | Outcome | Toxicity |
| Celecoxib | COX-2 inhibitor | Uracil/Tegafur, Leucovorin, Celecoxib + RT[72] | II | Pancreatic; locally advanced unresectable | No significant partial or complete response | Significant GI toxicity |
| Celecoxib, 5-FU[73] | Pilot study | Pancreatic; advanced after Gemcitabine treatment | Durable response | Well tolerated | ||
| Gemcitabine, irinotecan, celecoxib[74] | II | Pancreatic; unresectable | Increased OS from 6 m to 18 m | Well tolerated | ||
| Gemcitabine, celecoxib[76] | II | Pancreatic; locally advanced or metastatic | No significant response | Well tolerated | ||
| Gemcitabine, cisplatin, celecoxib[75] | II | Pancreatic; metastatic | No significant response | Well tolerated | ||
| LY293111 | LTB4 receptor antagonist | Irinotecan, LY293111[80] | I | Solid tumors (including pancreatic); locally advanced or metastatic | No significant response | Significant GI toxicity |
| Gemcitabine, LY293111[81] | II | Pancreatic; locally advanced or metastatic | No significant response | Significant GI toxicity |
Cox: Cyclooxygenase; LTB4: Leukotriene B4; RT: Radiation therapy; FU: Fluorouracil; OS: Overall survival; GI: Gastrointestinal.
LOX inhibitors
Zileuton is a 5-LOX inhibitor of the N-hydroxyurea series, approved by the Food and Drug Administration in 1996 for the treatment of asthma[25]. It was shown in clinical trials to produce moderate airway improvement in asthmatics. While Zileuton has had promising effects for airway disease, this drug has not yet been tested in patients with cancer.
Several studies have investigated Zileuton in animal studies and shown promising results for multiple cancers including carcinoma of the colon, lung, and pancreas. Zileuton was shown to reduce cell proliferation in murine colon adenocarcinoma cell lines[77]. In a xenograft model using human colon cancer cells, Zileuton inhibited tumor growth and reduced tumor mass[78]. In pancreatic cancer studies using the Syrian hamster model with BOP-induced pancreatic cancer, Zyflo (an extended release formulation of Zileuton) was found to reduce the incidence and size of the pancreatic cancer both alone and in combination with a COX-2 inhibitor[79].
LTB4 RECEPTOR ANTAGONIST
A few clinical trials have been conducted using LY293111 in patients with pancreatic cancer. A phase I study demonstrated that LY29311 was well tolerated in combination with Irinotecan although no responses were seen[80]. A different study randomized patients with pancreatic cancer to gemcitabine and LY293111 vs gemcitabine and placebo. There was no significant difference in six-month survival or progression-free survival[81]. Finally, a study conducted in patients with non-small cell lung cancer receiving LY293111 and Cisplatin/Gemcitabine also did not show a survival benefit[82]. Similar to COX-2 inhibitors, an LTB4 receptor antagonist would probably be most efficacious early in the disease process.
FLAP inhibitors
MK-886 is a FLAP inhibitor and inhibits leukotriene biosynthesis. It was first developed for use in asthma although clinical development was halted due to only a 50% inhibition of leukotriene production when used[83]. A second-generation FLAP inhibitor, MK-0591, had more potent inhibitory effects on leukotriene production, although it did not clinically perform as expected and was also discontinued[84].
Similar to Zileuton, MK-886 has shown promising results in vitro and in vivo. As mentioned above, MK-866 was shown to promote growth inhibition in a pancreatic cancer cell line. It was also shown in vivo to reduce pancreatic cancer development in a hamster model[85].
CONCLUSION
The inflammatory pathway is an important process in cancer progression. A combination of clinical studies, epidemiological studies, and basic science investigations indicate that there is a relationship between inflammatory changes in the pancreas and neoplastic progression. Intake of ω-6 polyunsaturated fatty acids provides increased substrate for COX and LOX mediated metabolism of arachidonic acid into eicosanoids. These eicosanoids directly contribute to pancreatic cancer cell proliferation. When COX-2 and 5-LOX are blocked via a variety of mechanisms, cancer cell proliferation is abrogated both in vitro and in vivo. The use of COX-2 and 5-LOX inhibitors in clinical studies in patients with pancreatic cancer has been limited. Patient enrollment has been restricted to patients with advanced disease which makes evaluation of these drugs as chemopreventive agents difficult. COX and LOX expression have been shown to be present during the early neoplastic changes of pancreatic cancer, well before progression to invasive disease. This indicates that the ideal role for these interventions is early in the disease process as preventive agents, perhaps in patients with chronic pancreatitis or hereditary pancreatitis. Further investigation is needed to broaden our understanding of the complex relationship between inflammation and pancreatic cancer and how these inflammatory pathways can be targeted to treat this deadly disease.
Footnotes
P- Reviewer: Golovko MY, Salh B, Yang GY S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 2.Phillips TJ, Salman SM, Bhawan J, Rogers GS. Burn scar carcinoma. Diagnosis and management. Dermatol Surg. 1998;24:561–565. [PubMed] [Google Scholar]
- 3.Cohen SM, Ellwein LB. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 1991;51:6493–6505. [PubMed] [Google Scholar]
- 4.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 6.Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, Dangi-Garimella S, Wang E, Munshi HG, Khazaie K, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res. 2010;16:2257–2265. doi: 10.1158/1078-0432.CCR-09-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56:1279–1282. [PubMed] [Google Scholar]
- 9.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 10.Whitcomb DC, Applebaum S, Martin SP. Hereditary pancreatitis and pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:201–209. doi: 10.1111/j.1749-6632.1999.tb09524.x. [DOI] [PubMed] [Google Scholar]
- 11.Martin SP, Ulrich CD. Pancreatic cancer surveillance in a high-risk cohort. Is it worth the cost? Med Clin North Am. 2000;84:739–747, xii-xiii. doi: 10.1016/s0025-7125(05)70255-8. [DOI] [PubMed] [Google Scholar]
- 12.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 13.Farrow B, Albo D, Berger DH. The role of the tumor microenvironment in the progression of pancreatic cancer. J Surg Res. 2008;149:319–328. doi: 10.1016/j.jss.2007.12.757. [DOI] [PubMed] [Google Scholar]
- 14.Chang DZ, Ma Y, Ji B, Wang H, Deng D, Liu Y, Abbruzzese JL, Liu YJ, Logsdon CD, Hwu P. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:7015–7023. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013;73:3927–3937. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friess H, Guo XZ, Nan BC, Kleeff J, Büchler MW. Growth factors and cytokines in pancreatic carcinogenesis. Ann N Y Acad Sci. 1999;880:110–121. doi: 10.1111/j.1749-6632.1999.tb09515.x. [DOI] [PubMed] [Google Scholar]
- 17.Van't Veer P, Kok FJ, Brants HA, Ockhuizen T, Sturmans F, Hermus RJ. Dietary fat and the risk of breast cancer. Int J Epidemiol. 1990;19:12–18. doi: 10.1093/ije/19.1.12. [DOI] [PubMed] [Google Scholar]
- 18.Thind IS. Diet and cancer--an international study. Int J Epidemiol. 1986;15:160–163. doi: 10.1093/ije/15.2.160. [DOI] [PubMed] [Google Scholar]
- 19.Howe GR, Jain M, Miller AB. Dietary factors and risk of pancreatic cancer: results of a Canadian population-based case-control study. Int J Cancer. 1990;45:604–608. doi: 10.1002/ijc.2910450405. [DOI] [PubMed] [Google Scholar]
- 20.Wynder EL. An epidemiological evaluation of the causes of cancer of the pancreas. Cancer Res. 1975;35:2228–2233. [PubMed] [Google Scholar]
- 21.Woutersen RA, Appel MJ, van Garderen-Hoetmer A, Wijnands MV. Dietary fat and carcinogenesis. Mutat Res. 1999;443:111–127. doi: 10.1016/s1383-5742(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 22.Stamler J. Assessing diets to improve world health: nutritional research on disease causation in populations. Am J Clin Nutr. 1994;59:146S–156S. doi: 10.1093/ajcn/59.1.146S. [DOI] [PubMed] [Google Scholar]
- 23.Stolzenberg-Solomon RZ, Schairer C, Moore S, Hollenbeck A, Silverman DT. Lifetime adiposity and risk of pancreatic cancer in the NIH-AARP Diet and Health Study cohort. Am J Clin Nutr. 2013;98:1057–1065. doi: 10.3945/ajcn.113.058123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy TJ, Chan CY, Ding XZ, Adrian TE. Lipoxygenase inhibitors for the treatment of pancreatic cancer. Expert Rev Anticancer Ther. 2003;3:525–536. doi: 10.1586/14737140.3.4.525. [DOI] [PubMed] [Google Scholar]
- 26.Cheon EC, Strouch MJ, Barron MR, Ding Y, Melstrom LG, Krantz SB, Mullapudi B, Adrian K, Rao S, Adrian TE, et al. Alteration of strain background and a high omega-6 fat diet induces earlier onset of pancreatic neoplasia in EL-Kras transgenic mice. Int J Cancer. 2011;128:2783–2792. doi: 10.1002/ijc.25622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strouch MJ, Ding Y, Salabat MR, Melstrom LG, Adrian K, Quinn C, Pelham C, Rao S, Adrian TE, Bentrem DJ, et al. A high omega-3 fatty acid diet mitigates murine pancreatic precancer development. J Surg Res. 2011;165:75–81. doi: 10.1016/j.jss.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17:55–67. doi: 10.1007/s10787-009-8049-8. [DOI] [PubMed] [Google Scholar]
- 29.Anderson KE, Johnson TW, Lazovich D, Folsom AR. Association between nonsteroidal anti-inflammatory drug use and the incidence of pancreatic cancer. J Natl Cancer Inst. 2002;94:1168–1171. doi: 10.1093/jnci/94.15.1168. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs EJ, Connell CJ, Rodriguez C, Patel AV, Calle EE, Thun MJ. Aspirin use and pancreatic cancer mortality in a large United States cohort. J Natl Cancer Inst. 2004;96:524–528. doi: 10.1093/jnci/djh084. [DOI] [PubMed] [Google Scholar]
- 31.Schernhammer ES, Kang JH, Chan AT, Michaud DS, Skinner HG, Giovannucci E, Colditz GA, Fuchs CS. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst. 2004;96:22–28. doi: 10.1093/jnci/djh001. [DOI] [PubMed] [Google Scholar]
- 32.Bradley MC, Hughes CM, Cantwell MM, Napolitano G, Murray LJ. Non-steroidal anti-inflammatory drugs and pancreatic cancer risk: a nested case-control study. Br J Cancer. 2010;102:1415–1421. doi: 10.1038/sj.bjc.6605636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larsson SC, Giovannucci E, Bergkvist L, Wolk A. Aspirin and nonsteroidal anti-inflammatory drug use and risk of pancreatic cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:2561–2564. doi: 10.1158/1055-9965.EPI-06-0574. [DOI] [PubMed] [Google Scholar]
- 34.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 35.Hennig R, Ding XZ, Tong WG, Schneider MB, Standop J, Friess H, Büchler MW, Pour PM, Adrian TE. 5-Lipoxygenase and leukotriene B(4) receptor are expressed in human pancreatic cancers but not in pancreatic ducts in normal tissue. Am J Pathol. 2002;161:421–428. doi: 10.1016/S0002-9440(10)64198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 37.Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568–575. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 38.Peters-Golden M, Brock TG. Intracellular compartmentalization of leukotriene synthesis: unexpected nuclear secrets. FEBS Lett. 2001;487:323–326. doi: 10.1016/s0014-5793(00)02374-7. [DOI] [PubMed] [Google Scholar]
- 39.Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, Germain RN. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borgström P, Hughes GK, Hansell P, Wolitsky BA, Sriramarao P. Leukocyte adhesion in angiogenic blood vessels. Role of E-selectin, P-selectin, and beta2 integrin in lymphotoxin-mediated leukocyte recruitment in tumor microvessels. J Clin Invest. 1997;99:2246–2253. doi: 10.1172/JCI119399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin DT, Subbaramaiah K, Shah JP, Dannenberg AJ, Boyle JO. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck. 2002;24:792–799. doi: 10.1002/hed.10108. [DOI] [PubMed] [Google Scholar]
- 42.Howe LR, Dannenberg AJ. A role for cyclooxygenase-2 inhibitors in the prevention and treatment of cancer. Semin Oncol. 2002;29:111–119. doi: 10.1053/sonc.2002.34063. [DOI] [PubMed] [Google Scholar]
- 43.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999;59:4356–4362. [PubMed] [Google Scholar]
- 44.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 45.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey TJ. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59:987–990. [PubMed] [Google Scholar]
- 46.Kokawa A, Kondo H, Gotoda T, Ono H, Saito D, Nakadaira S, Kosuge T, Yoshida S. Increased expression of cyclooxygenase-2 in human pancreatic neoplasms and potential for chemoprevention by cyclooxygenase inhibitors. Cancer. 2001;91:333–338. doi: 10.1002/1097-0142(20010115)91:2<333::aid-cncr1006>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 47.Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA, Cameron JL, Hruban RH, Wilentz RE. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118:194–201. doi: 10.1309/TPG4-CK1C-9V8V-8AWC. [DOI] [PubMed] [Google Scholar]
- 48.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–436. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 49.Hillion J, Smail SS, Di Cello F, Belton A, Shah SN, Huso T, Schuldenfrei A, Nelson DM, Cope L, Campbell N, et al. The HMGA1-COX-2 axis: a key molecular pathway and potential target in pancreatic adenocarcinoma. Pancreatology. 2012;12:372–379. doi: 10.1016/j.pan.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu J, Lloyd FL, Trifan OC, Knapp B, Rizzo MT. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Mol Cancer Ther. 2003;2:1–7. [PubMed] [Google Scholar]
- 51.Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, Hines OJ. PGE(2) is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun. 2003;306:887–897. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- 52.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, Hines OJ. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–990. [PubMed] [Google Scholar]
- 53.Ito H, Duxbury M, Benoit E, Clancy TE, Zinner MJ, Ashley SW, Whang EE. Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets-1-dependent induction of matrix metalloproteinase-2. Cancer Res. 2004;64:7439–7446. doi: 10.1158/0008-5472.CAN-04-1177. [DOI] [PubMed] [Google Scholar]
- 54.Merati K, said Siadaty M, Andea A, Sarkar F, Ben-Josef E, Mohammad R, Philip P, Shields AF, Vaitkevicius V, Grignon DJ, et al. Expression of inflammatory modulator COX-2 in pancreatic ductal adenocarcinoma and its relationship to pathologic and clinical parameters. Am J Clin Oncol. 2001;24:447–452. doi: 10.1097/00000421-200110000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Funahashi H, Satake M, Dawson D, Huynh NA, Reber HA, Hines OJ, Eibl G. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–7071. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 56.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- 57.Dangi-Garimella S, Munshi HG. A Novel Microrna Signaling Cascade Regulates Pancreatic Cancer Cell Invasion in Collagen. Pancreas. 2008;37:466–467. doi: 10.1097/01.Mpa.0000335439.14797.A8. [DOI] [Google Scholar]
- 58.Colby JK, Klein RD, McArthur MJ, Conti CJ, Kiguchi K, Kawamoto T, Riggs PK, Pavone AI, Sawicki J, Fischer SM. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia. 2008;10:782–796. doi: 10.1593/neo.08330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Avis I, Hong SH, Martinez A, Moody T, Choi YH, Trepel J, Das R, Jett M, Mulshine JL. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001;15:2007–2009. doi: 10.1096/fj.00-0866fje. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci USA. 1998;95:13182–13187. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shureiqi I, Lippman SM. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001;61:6307–6312. [PubMed] [Google Scholar]
- 62.Hennig R, Grippo P, Ding XZ, Rao SM, Buchler MW, Friess H, Talamonti MS, Bell RH, Adrian TE. 5-Lipoxygenase, a marker for early pancreatic intraepithelial neoplastic lesions. Cancer Res. 2005;65:6011–6016. doi: 10.1158/0008-5472.CAN-04-4090. [DOI] [PubMed] [Google Scholar]
- 63.Ding XZ, Iversen P, Cluck MW, Knezetic JA, Adrian TE. Lipoxygenase inhibitors abolish proliferation of human pancreatic cancer cells. Biochem Biophys Res Commun. 1999;261:218–223. doi: 10.1006/bbrc.1999.1012. [DOI] [PubMed] [Google Scholar]
- 64.Ding XZ, Tong WG, Adrian TE. Multiple signal pathways are involved in the mitogenic effect of 5(S)-HETE in human pancreatic cancer. Oncology. 2003;65:285–294. doi: 10.1159/000074640. [DOI] [PubMed] [Google Scholar]
- 65.Ding XZ, Kuszynski CA, El-Metwally TH, Adrian TE. Lipoxygenase inhibition induced apoptosis, morphological changes, and carbonic anhydrase expression in human pancreatic cancer cells. Biochem Biophys Res Commun. 1999;266:392–399. doi: 10.1006/bbrc.1999.1824. [DOI] [PubMed] [Google Scholar]
- 66.Tong WG, Ding XZ, Witt RC, Adrian TE. Lipoxygenase inhibitors attenuate growth of human pancreatic cancer xenografts and induce apoptosis through the mitochondrial pathway. Mol Cancer Ther. 2002;1:929–935. [PubMed] [Google Scholar]
- 67.Ding XZ, Talamonti MS, Bell RH, Adrian TE. A novel anti-pancreatic cancer agent, LY293111. Anticancer Drugs. 2005;16:467–473. doi: 10.1097/00001813-200506000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Adrian TE, Hennig R, Friess H, Ding X. The Role of PPARgamma Receptors and Leukotriene B(4) Receptors in Mediating the Effects of LY293111 in Pancreatic Cancer. PPAR Res. 2008;2008:827096. doi: 10.1155/2008/827096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tong WG, Ding XZ, Hennig R, Witt RC, Standop J, Pour PM, Adrian TE. Leukotriene B4 receptor antagonist LY293111 inhibits proliferation and induces apoptosis in human pancreatic cancer cells. Clin Cancer Res. 2002;8:3232–3242. [PubMed] [Google Scholar]
- 70.Hennig R, Osman T, Esposito I, Giese N, Rao SM, Ding XZ, Tong WG, Büchler MW, Yokomizo T, Friess H, et al. BLT2 is expressed in PanINs, IPMNs, pancreatic cancer and stimulates tumour cell proliferation. Br J Cancer. 2008;99:1064–1073. doi: 10.1038/sj.bjc.6604655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tong WG, Ding XZ, Talamonti MS, Bell RH, Adrian TE. Leukotriene B4 receptor antagonist LY293111 induces S-phase cell cycle arrest and apoptosis in human pancreatic cancer cells. Anticancer Drugs. 2007;18:535–541. doi: 10.1097/01.cad.0000231477.22901.8a. [DOI] [PubMed] [Google Scholar]
- 72.Morak MJ, Richel DJ, van Eijck CH, Nuyttens JJ, van der Gaast A, Vervenne WL, Padmos EE, Schaake EE, Busch OR, van Tienhoven G. Phase II trial of Uracil/Tegafur plus leucovorin and celecoxib combined with radiotherapy in locally advanced pancreatic cancer. Radiother Oncol. 2011;98:261–264. doi: 10.1016/j.radonc.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 73.Milella M, Gelibter A, Di Cosimo S, Bria E, Ruggeri EM, Carlini P, Malaguti P, Pellicciotta M, Terzoli E, Cognetti F. Pilot study of celecoxib and infusional 5-fluorouracil as second-line treatment for advanced pancreatic carcinoma. Cancer. 2004;101:133–138. doi: 10.1002/cncr.20338. [DOI] [PubMed] [Google Scholar]
- 74.Lipton A, Campbell-Baird C, Witters L, Harvey H, Ali S. Phase II trial of gemcitabine, irinotecan, and celecoxib in patients with advanced pancreatic cancer. J Clin Gastroenterol. 2010;44:286–288. doi: 10.1097/MCG.0b013e3181cda097. [DOI] [PubMed] [Google Scholar]
- 75.El-Rayes BF, Zalupski MM, Shields AF, Ferris AM, Vaishampayan U, Heilbrun LK, Venkatramanamoorthy R, Adsay V, Philip PA. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs. 2005;23:583–590. doi: 10.1007/s10637-005-1028-z. [DOI] [PubMed] [Google Scholar]
- 76.Dragovich T, Burris H, Loehrer P, Von Hoff DD, Chow S, Stratton S, Green S, Obregon Y, Alvarez I, Gordon M. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: results of a phase II trial. Am J Clin Oncol. 2008;31:157–162. doi: 10.1097/COC.0b013e31815878c9. [DOI] [PubMed] [Google Scholar]
- 77.Hussey HJ, Tisdale MJ. Inhibition of tumour growth by lipoxygenase inhibitors. Br J Cancer. 1996;74:683–687. doi: 10.1038/bjc.1996.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melstrom LG, Bentrem DJ, Salabat MR, Kennedy TJ, Ding XZ, Strouch M, Rao SM, Witt RC, Ternent CA, Talamonti MS, et al. Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clin Cancer Res. 2008;14:6525–6530. doi: 10.1158/1078-0432.CCR-07-4631. [DOI] [PubMed] [Google Scholar]
- 79.Wenger FA, Kilian M, Achucarro P, Heinicken D, Schimke I, Guski H, Jacobi CA, Müller JM. Effects of Celebrex and Zyflo on BOP-induced pancreatic cancer in Syrian hamsters. Pancreatology. 2002;2:54–60. doi: 10.1159/000049449. [DOI] [PubMed] [Google Scholar]
- 80.Baetz T, Eisenhauer E, Siu L, MacLean M, Doppler K, Walsh W, Fisher B, Khan AZ, de Alwis DP, Weitzman A, et al. A phase I study of oral LY293111 given daily in combination with irinotecan in patients with solid tumours. Invest New Drugs. 2007;25:217–225. doi: 10.1007/s10637-006-9021-8. [DOI] [PubMed] [Google Scholar]
- 81.Saif MW, Oettle H, Vervenne WL, Thomas JP, Spitzer G, Visseren-Grul C, Enas N, Richards DA. Randomized double-blind phase II trial comparing gemcitabine plus LY293111 versus gemcitabine plus placebo in advanced adenocarcinoma of the pancreas. Cancer J. 2009;15:339–343. doi: 10.1097/PPO.0b013e3181b36264. [DOI] [PubMed] [Google Scholar]
- 82.Janne P, Gottried AR, Randomized phase II trial of cisplatin/gemcitabine with or without LY293111, a multiple eicosonaid pathway modulator, in patients with chemotherapy naïve advanced non-small cell lung carcinoma. J Clin Oncol. 2006;24:18S. [Google Scholar]
- 83.Friedman BS, Bel EH, Buntinx A, Tanaka W, Han YH, Shingo S, Spector R, Sterk P. Oral leukotriene inhibitor (MK-886) blocks allergen-induced airway responses. Am Rev Respir Dis. 1993;147:839–844. doi: 10.1164/ajrccm/147.4.839. [DOI] [PubMed] [Google Scholar]
- 84.Brooks CD, Summers JB. Modulators of leukotriene biosynthesis and receptor activation. J Med Chem. 1996;39:2629–2654. doi: 10.1021/jm960088k. [DOI] [PubMed] [Google Scholar]
- 85.Schuller HM, Zhang L, Weddle DL, Castonguay A, Walker K, Miller MS. The cyclooxygenase inhibitor ibuprofen and the FLAP inhibitor MK886 inhibit pancreatic carcinogenesis induced in hamsters by transplacental exposure to ethanol and the tobacco carcinogen NNK. J Cancer Res Clin Oncol. 2002;128:525–532. doi: 10.1007/s00432-002-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]