

Abstract
Botulinum neurotoxin serotype A (BoNT/A) is the most lethal toxin among the Tier 1 Select Agents. Development of potent and selective small molecule inhibitors against BoNT/A zinc metalloprotease remains a challenging problem due to its exceptionally large substrate binding surface and conformational plasticity. The exosites of the catalytic domain of BoNT/A are intriguing alternative sites for small molecule intervention, but their suitability for inhibitor design remains largely unexplored. In this study, we employed two recently identified exosite inhibitors, D-chicoric acid and lomofungin, to probe the structural features of the exosites and molecular mechanisms of synergistic inhibition. The results showed that D-chicoric acid favors binding at the α-exosite, whereas lomofungin preferentially binds at the β-exosite by mimicking the substrate β-sheet binding interaction. Molecular dynamics simulations and binding interaction analysis of the exosite inhibitors with BoNT/A revealed key elements and hot-spots that likely contribute to the inhibitor binding and synergistic inhibition. Finally, we performed database virtual screening for novel inhibitors of BoNT/A targeting the exosites. Hits C1 and C2 showed non-competitive inhibition and likely target the α- and β-exosites, respectively. The identified exosite inhibitors may provide novel candidates for structure-based development of therapeutics against BoNT/A intoxication.
Keywords: Botulinum neurotoxin, exosite, synergistic inhibition, binding free energy calculations, structure-based drug design, virtual screening
Introduction
Clostridium botulinum neurotoxins (BoNTs) are classified as Tier 1 Select Agent toxins by the Centers for Disease Control and Prevention [1,2]. Serotype A (BoNT/A) is one of seven known serotypes of botulinum neurotoxins (A–G), and has an estimated human LD50 of only 1 ng/kg [3]. The toxin consists of a single 150 kDa polypeptide chain that is post-translationally proteolysed into a ~100 kDa heavy chain (HC) and a ~50 kDa light chain (LC) [4]. The toxin’s mechanism of action is known to involve cleavage of one of the three soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE), thereby blocking neurotransmitter release and inducing flaccid paralysis [5]. The holotoxin consists of three structurally distinct domains. The binding and translocation domains on the HC are responsible for receptor binding and translocation of the protein into the cytosol, respectively, while the cleavage of the substrate is catalyzed by the LC, a zinc metalloprotease [6]. A number of structures of BoNT/A and other BoNTs have been determined experimentally including structures of substrate and inhibitor complexes, and have revealed the structural basis for the catalytic mechanism and substrate recognition [7,8]. The structure of the catalytic domain of BoNT/A in complex with SNAP-25 (synaptosomal-associated protein, 25 kDa) shows ~60 amino acids of the substrate bound in an extended form wrapping around the enzyme from the active site to the rear surface with extensive binding interactions in the distant exosites [9].
Currently, there is no effective post-intoxication small molecule therapeutic to counteract BoNT/A LC-mediated paralysis. Vaccination and passive immunization antibody therapy are used, but have significant limitations in practice, as they cannot exert an effect on toxin that has already entered the neuron [10,11]. Intracellularly, the toxin is stable for months. Intoxication occurs within hours and antibodies must administered within this period [12]. A number of small molecule inhibitors of BoNT/A have been developed in the past, and have primarily targeted the zinc in the LC active site [13–16]. Most of these active-site inhibitors (e.g. hydroxamate) utilize metal-chelating motifs that are not selective against other metalloproteinases [17]. Hydroxamates have been shown to have efficacy in mice, but have relatively short in vivo half-lives [18]. Developing tight binding non-chelating inhibitors of BoNT/A has proven to be a difficult task in part due to the high conformational plasticity of the binding pocket and induced conformational changes in adjacent loops upon substrate or inhibitor binding [19]. The exceptionally large substrate binding surface of BoNT/A poses an extremely challenging problem to design effective small molecule inhibitors that are capable of disrupting the extensive protein-protein interactions within the substrate binding interface.
The α- and β-exosites of BoNT/A, which were first addressed by Breidenbach and Brunger, provide intriguing alternatives for small molecule inhibition of enzyme-substrate interactions [9]. The α-exosite is located on the rear surface of the protein (relative to the active site) and consists of four helices, while the β-exosite lies in a dynamic loop region adjacent to the active site and forms the hallmark three-stranded antiparallel β-sheet interaction involving the substrate SNAP-25 [9]. While studies have indicated that these exosites play an important role in substrate recognition and catalysis, the potential for small molecule binding and structure-based inhibitor design at these sites has been largely unexplored. Compared to the deep pocket of the active site, these regions appear to be relatively shallow and undefined. Therefore, questions still remain as to whether the exosites are amenable to small molecule binding. A single domain antibody was recently shown to inhibit SNAP-25 cleavage in vitro and bind to a small crevice in the α-exosite with a low-nM Kd, suggesting that low nM inhibition may be possible [20]. Recently, studies from Janda’s group showed that the natural products of phenolic caffeoyl derivatives such as D/L-chicoric acid exhibited non-competitive partial inhibition of BoNT/A [21]. The combination of D-chicoric acid with an active-site inhibitor, 2,4-dichlorocinnamic hydroxamate, displayed nonmutually exclusive inhibition. More interestingly, another non-competitive inhibitor, lomofungin, was identified which also exhibited synergistic inhibition against BoNT/A when used in combination with 2,4-dichlorocinnamic hydroxamate and chicoric acid [22]. While no structural evidence has been generated, it has been speculated based upon kinetic data that the binding regions of the two small molecules might map to the α- and β-exosites [22].
The discovery of exosite inhibitors of BoNT/A inspired us to further investigate the small molecule binding interactions and molecular mechanisms of inhibition at the exosites. The synergy of exosite inhibition provides a valuable approach for designing novel inhibitors against BoNT intoxication. Herein, we applied computational approaches to explore the structural features of the exosites of BoNT/A using chicoric acid and lomofungin as model probes. The potential binding interactions of these small molecules at the exosites were investigated using an unbiased ensemble docking search and stepwise binding mode analysis. To gain insight into the structural basis of synergistic inhibition, we modeled a tripartite inhibitor binding complex of BoNT/A with a hydroxamate inhibitor bound at the active site, D-chicoric acid bound at the α-exosite, and lomofungin bound at the β-exosite. The tripartite inhibitor binding complex was analyzed in comparison with the substrate SNAP-25 binding complex, and revealed details of the binding site preferences and key residue determinants contributing to synergistic inhibition at the exosites. Finally, based upon the predicted binding models of the two exosite inhibitors, we performed high throughput in silico screening to identify novel inhibitors targeting the exosites of BoNT/A.
Materials and methods
Structure and inhibitors of BoNT/A
The crystallographic coordinates of BoNT/A in complex with the substrate, SNAP-25, (PDB code 1XTG) was used as the initial model [9]. The SNAP-25 was removed and the two mutated residues, E224Q and Y366F, were changed back to the wild type. The resulting structure was energy-minimized prior to MD simulations. Three inhibitors of BoNT/A were used as probes in this study (Figure 1). 2,4-dichlorocinnamic hydroxamate (DCH) is the smallest, most potent BoNT/A inhibitor yet (Ki = 0.30 μM) and the crystal structure in complex with BoNT/A has been determined (PDB code 2IMA) [23]. The D-chicoric acid and lomofungin are exosite inhibitors of BoNT/A that have been reported from studies of Janda [21,22]. The structures of compounds were built using Discovery Studio (Accelrys, San Diego, CA) and energy minimized prior to docking.
Figure 1.
Inhibitors of BoNT/A used as probes in this study. 2,4-dichlorocinnamic hydroxamate (DCH) is a potent BoNT/A active site inhibitor which chelates the zinc [23]. D-chicoric acid (DCA) and Lomofungin are exosite inhibitors of BoNT/A which have been reported from studies by Janda et al [21,22].
REMD simulations
REMD simulations were conducted for the apo BoNT/A in implicit solvent to investigate the dynamics of the protein. Ten replicas were used with temperatures ranging from 300 to 395 K (300, 310, 320, 330, 340, 351, 361, 372, 384, and 395 K). The simulated system for each replica was first subjected to a 500-ps equilibration by heating them to their respective temperatures using the Langevin thermostat, followed by a production run of 1-ns length in total. The SANDER module of the AMBER 12 package was used for the simulations with the ff99SB force field [24]. The resulting trajectories were analyzed using the PTRAJ module and the weighted histogram analysis method (WHAM) [25]. The root-mean-square deviations (RMSDs) of the protein backbone were calculated from the trajectories at 1-ps interval using the initial structure as the reference. The root-mean-square positional fluctuations (RMSFs) were calculated from the trajectory at room temperature (300 K) based upon the superposition of all Cα atoms. The averaged structure of trajectories was used as the reference and compared with the experimental B-factors from the crystal structure.
Essential dynamics analysis
Essential dynamics (ED) analysis is a technique which reduces the complexity of the data, and extracts the concerted motion in simulations that are essentially correlated and presumably meaningful for biological function. In the ED analysis, a variance/covariance matrix was constructed from the trajectories after removal of the rotational and translational movements. A set of eigenvectors and eigenvalues were identified by diagonalizing the matrix. The eigenvalues represented the amplitude of the eigenvectors along the multidimensional space, and the displacements of atoms along each eigenvector showed the concerted motions of protein along each direction. An assumption of ED analysis is that the correlated motions for the function of the protein are described by eigenvectors with large eigenvalues. The movements of protein in the essential subspace were identified by projecting the Cartesian trajectory coordinates along the most important eigenvectors from the analysis. In this case the ED analysis was performed for the REMD trajectories at room temperature and a total of 10 essential modes were generated and analyzed using the PTRAJ module in the Amber 12 package [24].
Ensemble docking and stepwise binding mode analysis
We applied an approach of stepwise binding mode analysis combining ensemble docking, clustering, MD simulations, and binding free energy calculations to probe the small molecule binding interactions with BoNT/A. The ensemble docking was employed to account for protein flexibility, especially for the dynamic loop regions surrounding the active site. As outlined in Figure 2A, ensembles of the BoNT/A structure were generated from five REMD trajectories (300–340 K). The means method in the PTRAJ module was used for clustering analysis, and a total of 10 cluster representatives of BoNT/A conformations were obtained from the MD trajectory. The AutoDock-based DOVIS program [26] was used for the ensemble docking of selected BoNT/A inhibitors. The active site of the protein was defined by a grid of 120 × 120 × 120 points with a grid spacing of 0.5 Å centered at the catalytic zinc ion, which encompassed the entire protein for an unbiased docking search. The default parameters of zinc in AutoDock 4.2 (R-eqm = 1.48 Å and weighted epsilon = 0.091) were used and a charge of + 2 was assigned [27]. The Lamarckian Genetic Algorithm (LGA) [28] was applied with 100 runs and the maximum number of energy evaluations was set to 2 × 106. All poses from each ensemble docking were retained for clustering and binding mode analysis.
Figure 2.
(A) Stepwise binding mode analysis combining an unbiased docking search, clustering analysis, MD simulation and binding free energy calculations to probe the binding interaction of BoNT/A inhibitors. (B) Flowchart of the virtual screening protocol.
Analysis of the binding modes was performed in a stepwise process using the AutoDockTool package [27]. First, docked poses with each protein conformation were merged and clustered with a fine-tuned criteria for the RMSD (10 Å). The major clusters were identified as potential sites of inhibitor binding. Second, sub-clustering of docked poses at each binding site was applied to identify the most plausible binding mode (RMSD = 2.0 Å). Representatives of the top clusters at each binding pocket were inspected and the most favorable binding modes were selected. Third, the representative binding conformations from each sub-cluster were refined with MD simulations, and the binding free energies were calculated using the MM/PBSA method as described below for the optimal binding mode selection. Finally, binding free energies were decomposed to residues at the binding site to examine their contributions to the inhibitor binding affinity.
MD simulations and binding free energy calculations
MD simulations were performed for the predicted binding complexes of D-chicoric acid and lomofungin with BoNT/A using the AMBER 12 package and the ff99SB force field [24]. The solvated protein systems were subjected to a thorough energy minimization prior to MD simulations. Bond lengths involving hydrogen were constrained with SHAKE and the time step for all MD simulations was set to 2 fs. A non-bonded cutoff of 10 Å was used, and the non-bonded pair list was updated every 25 time steps. Periodic boundary conditions were applied to simulate a continuous system. The periodic box defined in the simulated system was 94.6 × 85.8 × 94.7 Å and the charges were neutralized by adding Cl− counter ions. The particle mesh Ewald (PME) method was employed to calculate the long-range electrostatic interactions [29]. The simulated system was first subjected to a gradual temperature increase from 0 K to 300 K over 100 ps, and then equilibrated for 500 ps at 300 K, followed by production runs of 2-ns length in total. The Langevin theromostat was used to maintain the temperature in the MD simulations.
The binding free energies were calculated using the MM-PBSA method [30]. A set of 200 snapshots was extracted at 10 ps intervals from trajectories of each binding complex simulation. The polar contribution (GPB) was calculated using the Poisson Boltzmann equation. The nonpolar contributions (GSA) were estimated using the MSMS algorithm according to the equation: GSA = γ × SASA + b kcal/mol, with and b set to 0.00542 kcal/mol·Å−2 and 0.92 kcal/mol [31], and the probe radius used to calculate the solvent accessible surface area (SASA) was set to 1.4 Å. The entropy contribution was neglected in the free energy calculations. Decomposition of the calculated binding free energies was performed using the same MM-PBSA module in AMBER 12 package [24].
Virtual screening
Virtual screening was performed to search the MLSMR library consisting of over 350,000 druglike compounds using a stepwise VS protocol combining a ligand-based 3D similarity search and structure-based docking (Figure 2B). The predicted binding modes of D-chicoric acid and lomofungin were used as queries for searching the entire database using the shape-based program ROCS, followed by docking the top 5% of the hits to the structure target of BoNT/A (PDB code: 1XTG) using the AutoDock-based virtual screen program DOVIS (26). The active site of BoNT/A was occupied with the hydroxamate inhibitor DCH in the docking process, thus allowing an extensive search of potential exosite binders. The docked poses at the α- and β-exosites were analyzed in terms of binding interactions with the hotspot residues identified from D-chicoric acid and lomofungin binding models. The final hits were cherry-picked based upon an overall scoring schema from the post-docking process, including consensus scoring, clustering and chemotype analysis.
FRET assay
Two substrates, SNAPtide and a yellow and cyan fluorescent protein (YFP-(141-206)-CFP) substrate were used in the continuous and discontinuous FRET assays. Assays mixtures contained 2.7 M YFP-(141-206)-CFP in 20 mM HEPES pH 8.0, ±1.25 mM DTT, 0.01% Tween-20, and 10–30 nM BoNT/A(1-448). All inhibitors were dissolved in DMSO, and the DMSO concentration was held at 2%. Reactions (50 L) were monitored using excitation and emission wavelengths of 485/520 or 435/525 nm for SNAPtide and the YFP-(141-206)-CFP substrates, respectively. The continuous assays were used as the primary assays for testing compounds selected in silico, and for IC50 and Ki measurements. For the Ki measurement the data was collected in triplicate. The discontinuous gel-based assay was used as a secondary assay to rule out artifactual hits due to fluorescence quenching or autofluorescence.
Gel-based assay
SDS-PAGE gel electrophoresis (Pierce 8–16% Tris-HEPES gradient gels from Thermo Scientific Inc.) was used to verify inhibition (20–24 M YFP-(141-206)-CFP substrate, 0.23–0.24 M BoNT/A 1-448 or 0.33 M BoNT/A 1-425, 200 M inhibitor). The BoNT/A 1-448 reactions were run for 10–12 minutes at room temperature, and BoNTA 1-425 reactions were run for 22 hours until cleavage was nearly complete in the no-inhibitor control. Reactions were quenched with Laemlli loading buffer (1:1), and heated for at least 3 minutes at 70+ °C. The products were separated by gel electrophoresis, and band densities were quantitated using a BioRad imager.
The percentage of cleaved or uncleaved substrate could be calculated using the intensities of the bands for the control reactions (no enzyme control or no inhibitor control, upper or lower bands). The intensity of the uncut or fully cut bands were then used to measure the percent uncleaved and cleaved. IN is the intensity of substrate band in the reaction containing inhibitor, enzyme and substrate. Io is the intensity of the substrate band in the presence of enzyme and absence of inhibitor, and IU is the intensity of the substrate band in the absence of enzyme.
Results and Discussion
Dynamics of substrate binding site of BoNT/A
We applied replica-exchange MD simulations (REMD), which provided a more efficient approach to sample the conformational changes of the large substrate binding interface of BoNT/A, to investigate the dynamics of BoNT/A associated with substrate and inhibitor binding. As shown in Figure 3A, the active site of BoNT/A contains a large hydrophobic cavity with a zinc ion at the center bordered by flexible loop residues 60–70 (loop 60) and a small loop 170. The 200 and 250 loops at the β-exosite are known to undergo large conformational changes to accommodate substrate and inhibitor binding [32]. Figure 3B depicts the atomic positional fluctuations of the residues (Cα) calculated from the REMD simulations at room temperature. In agreement with experimental observations (Suppl. Figure S1), the active-site (loop 60 and 170) and the β-exosite loop 200 and 250 exhibited high fluctuations in the apo state, whereas these dynamic loops were significantly stabilized upon substrate binding. The β-exosite loop 200 had the largest conformational changes as expected. Interestingly, loop 170 also exhibited high degree of dynamics in the MD simulations. This is not evident from experimental structures. The loop is located on top of the active site near a deep groove, which serves as an anchor site for the substrate wrapping around from the rear α-exosite to the catalytic active site (the groove is referred to as the anchor exosite) (Figure 3A).
Figure 3.

(A) Structure of the BoNT/A LC catalytic domain in ribbon representation. The catalytic binding site with Zn located at the center (red), the α-exosite (yellow), the β-exosite sheet (magenta), the anchor site and other loops surrounding these binding sites are shown. (B) Calculated B-factors from MD simulations in comparison with the experimental B-factors. (C) Conformations of BoNT/A observed from essential dynamic analysis. The first essential mode is associated with loops 200, 250, and 370 at the β-exosite, while the second dynamical mode is found with loops 60 and 170 surrounding the active site.
In contrast to the active site and β-exosite, the α-exosite was more stable during the simulations. Two methionine residues, Met-106 and Met-344 from helices H1 and H3, participated in extensive hydrophobic interactions with residues Phe-341 and Ile-348 at the center of the α-exosite, and thereby, significantly stabilized the movement of these two helices. The α-exosite binding pocket is surrounded by a number of polar and charged residues including Arg-113, Asp-102, Lys-337, Lys-340, and Lys-343. A closer examination of these residues showed that they were more flexible with conformational changes surrounding the α-exosite. The conformational flexibility is likely associated with their functional role in facilitating substrate binding.
To characterize the dynamical properties of the binding site, we performed essential dynamic (ED) analysis to extract the concerted motions and their correlations from the trajectories. The first essential mode showed that the major motions occurred at the β-exosite, which was associated with two distinct conformations of loops 200 and 250 (Figure 3C). Loop 200 adopted an extended, open conformation at the β-exosite and moved towards loop 250 in a closed form with the largest distance of up to 12 Å. Along with this remarkable movement, the second essential mode was found to be related to loops 60/70 and 170 surrounding the active site. The ED analysis indicated that these dynamical modes were highly correlated with loop 200 and 250 motions at the β-exosite. Loop 200 moved towards loop 250 in the MD simulations and switched between an open and closed form. Loop 60/70 as well as loop 170 underwent conformational changes, and the active site adopted a more open conformation, which may be more favorable for substrate binding and product release. Such an active conformation of the catalytic domain of BoNT/A was observed in the structure of the substrate complex[33]. We hypothesize that loops 200 and 250 in the closed conformation are associated with the active form of BoNT/A, whereas the dynamic and disordered conformation of the β-exosite loops are correlated with the inactive state of the enzyme.
Binding mode of chicoric acid at the α-exosite
We first investigated the binding interaction of D-chicoric acid with BoNT/A using the stepwise binding mode analysis. The ensemble docking of D-chicoric acid to BoNT/A was performed in the presence of DCH bound in the catalytic binding pocket. Clustering analysis of the docked poses showed that major clusters were located at three distinct binding regions, which accounted for 70% of the docked poses in total (Figures 4A and 4B). Remarkably, the largest cluster was found at the α-exosite, with a population of 35% of all binding poses and the lowest docking scores. The second cluster was found at the β-exosite, where the inhibitor was inserted into the deep pocket of the β-exosite in a closed form. The third binding cluster was observed at the anchor exosite. The small molecule was positioned in a deep narrow groove along the substrate binding interface with various binding conformations.
Figure 4.

(A) Cluster analysis of binding modes of D-chicoric acid docked to BoNT/A using AutoDock 4.2. Members of docked poses in the cluster are shown on the Y-axis. The X-axis shows the lowest docked score found in the cluster. (B) Three major clusters of D-chicoric acid bound to the α-exosite (red), the β-exosite (green), and the anchor exosite (magenta) of BoNT/A. The DCH (cyan) interacts with Zn (red) at the active site. (C) Binding interaction of D-chicoric acid at the α-exosite. The protein is shown as ribbons and key residues are shown as sticks. D-chicoric acid (dark yellow) is shown as ball and stick. (D) Hot-spot residues at the α-exosite identified from the binding free energy decomposition.
The clustering results indicated that D-chicoric acid bound to the α-exosite more preferably than at the other exosites. To further probe this, the top-ranked binding conformations from each cluster were optimized and the binding free energies were calculated (Table 1). The lowest binding free energies of D-chicoric acid at the α-exosite were approximately -32.6kcal/mol, as compared to −20.3 and −15.5 kcal/mol of the best binding complex at the β-exosite and the anchor exosite, respectively. Figure 4C shows the optimal binding mode of D-chicoric acid at the α-exosite. The small molecule was accommodated in the α-exosite in such a manner that the di-carboxylate group was oriented at the center forming interactions with residues Lys-337 and Lys-340. One of the di-hydroxyphenyl groups was inserted into a cavity adjacent to residue Arg-113, while the other di-carboxylate group pointed outward to a hydrophobic region and formed extensive interactions with residues mainly from helix H2 including Asp-102, Leu-103, and Tyr-223. Such a U-shaped binding conformation apparently fits well at the α-exosite along the groove of helices H2 and H3. Moreover, docking analysis of D-chicoric acid to other serotypes of BoNTs showed that the small molecule bound to BoNT/B and BoNT/E in a similar manner to BoNT/A (Suppl. Figure S2), suggesting that these serotypes are more structurally similar at the α-exosite in terms of small molecule inhibition.
Table 1.
Calculated binding free energies of D-chicoric acid and lomofungin at the exosites of BoNT/A.
| Probe | Binding Cluster | Binding site | Binding mode | ΔG (kcal/mol) |
|---|---|---|---|---|
| D-chicoric acid | Cluster 1 | α-exosite | 1 | −32.6 |
| 2 | −22.0 | |||
| 3 | −23.7 | |||
| Cluster 2 | β-exosite (A) | 1 | −17.3 | |
| 2 | −27.1 | |||
| 3 | −8.1 | |||
| Cluster 3 | anchor exosite | 1 | −26.6 | |
| 2 | −15.9 | |||
| 3 | −11.0 | |||
| lomofungin | Cluster 1 | β-exosite (A) | 1 | −22.5 |
| 2 | −22.0 | |||
| 3 | −13.7 | |||
| Cluster 2 | β-exosite (B) | 1 | −21.1 | |
| 2 | −20.9 | |||
| 3 | −10.8 | |||
| Cluster 3 | anchor exosite | 1 | −12.8 | |
| 2 | −15.5 | |||
| 3 | −16.1 |
MD simulations and binding free energy decomposition revealed key residues involved in inhibitor binding at the α-exosite (Figure 4D). Residues Asp-102, Lys-340, Arg-113 and Lys-337 had major contributions to the calculated free energies, indicating that the inhibitor binding was mainly driven by the polar and charged interactions. These residues form a highly polar wall surrounding the α-exosite, and exhibited a high degree of flexibility to accommodate inhibitor binding. Asp-102 made the remarkably largest binding energy contribution. This residue formed hydrogen bonds with the hydroxyphenyl group of D-chicoric acid at the bottom of the α-exosite, which apparently governed the U-shaped binding conformation. The second largest contribution of binding energy was found with a number of hydrophobic residues located at the center of the pocket including two methionines, Met-106 and Met-344. Analysis of BoNT/A-substrate binding complex revealed that the SNAP-25 bound to BoNT/A at the α-exosite mainly through α-helix interactions at this hydrophobic region. Residues Ile-156 and Met-167 of SNAP-25 are buried in the pocket and make extensive interactions with Met-106, Arg-113, and Met-344 of BoNT/A. Mutations of residues Ile-156 and Met-167 that disrupted the interactions with Met-106 and Arg-113 substantially decreased the substrate binding affinities [9].
Binding mode of lomofungin at the β-exosite
Figure 5A shows the major binding clusters of lomofungin docked to BoNT/A from the stepwise binding model analysis. The ensemble docking generated a large number of binding conformations of the small molecule around the protein surface. Clustering analysis showed that the top two major clusters were predominantly located at the β-exosite, which accounted for 38% of binding poses in total. The first binding cluster was found at the β-exosite region as observed in the crystal structure of the substrate binding complex (PDB 1XTG), where a three-stranded antiparallel β-sheet interaction was formed between loop 250 and SNAP-25. The second cluster was located in the β-exosite pocket, similar to the D-chicoric acid binding. However, the tricyclic small molecule was positioned into the pocket more deeply by forming hydrogen bonds with Lys-371 and extensive aromatic and non-polar interactions with residue Leu-200, Leu-207, Tyr-250 and Phe-369. The lowest binding free energies of the two binding modes were −22.5 and −21.1 kcal/mol2 (Table 1), indicating that binding affinities of the inhibitor at the two β-exosites are comparable. Besides the β-exosite, several small clusters were also found at the anchor site along the substrate binding groove and the N-terminal region adjacent to the α-exosite. It is interesting to note that, unlike D-chicoric acid, no binding cluster was observed with lomofungin at the α-exosite, suggesting that the rigid small molecule did not fit into the α-exosite binding region.
Figure 5.
(A) Binding Clusters of lomofungin from SBMA clustering analysis. (B) Two binding modes of lomofungin bound at the β-exosite. Structure of BoNT/A is shown as ribbons. Lomofungin and key residues involved in binding (shown as sticks). Pairs of atoms involved in H-bonding are connected with dashed lines.
Our docking and clustering analysis support the experimental finding that lomofungin displays non-competitive binding with both the active site inhibitor DCH and D-chioric acid [21]. Moreover, the small molecule appeared to bind more preferably to the β-exosite. As shown in Figure 5B, lomofungin was orientated in line with the two strands of the β-sheet involving loop 250 and mimicked the antiparallel β-sheet substrate binding interaction. Similar to the SNAP-25 complex, the lomofungin binding complex was stabilized by forming two hydrogen bonds with the backbone of Leu-256 and Gly-255, and extensive vdW and hydrophobic interactions with residues Tyr-250, Phe-369, Leu-256, and Met-253. These residues at the β-exosite form a well-defined hydrophobic pocket at the periphery of the active site, which is also referred to as the S5′ binding pocket for residues 201–204 of SNAP-25 [34]. Mutagenesis studies indicated that the binding site and residues within loop 370 such as Tyr-366 and Asp-370 have significant effects on the catalytic activity [35,36]. The binding preference of lomofungin at the loop pocket was also seen in the MD simulations and binding free energy calculations (Table 1). The binding complex at the β-exosite was more stable and had the lowest binding free energies when compared to binding interactions at other exosites. Because the dynamics of the β-exosite loops are highly associated with the active site, synergistic inhibition by lomofungin binding at the β-exosite likely prevents conformational changes within the active site, locking the protein in an inactive state.
The synergistic binding model
To gain insight into the synergy of exosite inhibition, we modeled a tripartite binding complex of BoNT/A bound with three inhibitors: a hydroxamate inhibitor, DCH, bound at the catalytic site, D-chicoric acid bound at the α-exosite, and lomofungin bound at the β-exosite (Figure 6A). MD simulations showed that the dynamical changes of BoNT/A in the tripartite inhibitor binding were similar to those observed in the BoNT/A-SNAP-25 complex (Figure 6B), suggesting that the three inhibitors interact with BoNT/A cooperatively in a manner that is comparable to the substrate binding. The calculated binding energies of the tripartite binding interactions were approximately −133.9 kcal/mol, whereas the calculated binding energies of SNAP-25 with BoNT/A were approximately −331.2 kcal/mol. Given the extremely large substrate binding interface of BoNT/A, the higher binding affinity of the substrate compared with that of small molecule binding was not unexpected. In comparison with the predicted binding energies of the three inhibitors from their separate binding complexes, which were −54.5, −32.6, and −22.5 kcal/mol for DCH, D-chicoric acid, and lomofungin respectively (Table 1), the total binding energies of the three inhibitors in the tripartite complex were much lower (−133.9 vs −110.0 kcal/mol). The gain of binding affinity appeared to result from a synergy of inhibitor binding.
Figure 6.

(A) The tripartite binding model of BoNT/A in complex with hydroxamate inhibitor DCH (cyan) bound at the active site, D-chicoric acid (red) at the α-exosite, and lomofungin (green) at the β-exosite. The substrate SNAP-25 (yellow) is also shown in ribbons. (B) Calculated RMSD of the tripartite binding complex (red) in comparison with the substrate-binding complex with SNAP-25 (blue) from the MD simulations. (C) and (D) Contributions of hot-spot residues at the active site and the exosites of BoNT/A identified from the binding free energy decomposition for the substrate SNAP-25 binding complex and the tripartite inhibitor binding complex.
The tripartite binding model provided a plausible explanation for the structural basis of the binding preference associated with the active-site and exosite inhibitors. Analysis of binding energy contributions showed that more than 40 residues of BoNT/A contribute to substrate binding interactions (Figure 6C). These hot-spot residues were predominantly clustered at four binding sites, which, according to their total binding energy contributions, are the active site (30%), the α-exosite (18%), the anchor exosite (15%), and the β-exosite (12%). Such binding site preferences were indeed in agreement with that predicted from docking analysis using the small molecule probes. The identified hot-spots at the α- and β-exosite from the tripartite binding complex were also consistent with those involved in substrate binding interactions (Figure 6D), suggesting that the inhibitor binding at the exosites largely mimicked the substrate interaction. It is postulated that the D-chicoric acid binds at the α-exosite that is capable of blocking the protein-substrate interaction, while lomofungin binds at the β-exosite to stabilize the protein in an inactive form. The two exosite inhibitors bind in a synergistic manner with the active-site inhibitor DCH, thereby achieving high binding stability and maximum inhibitory activity.
Novel exosite inhibitor identified from virtual screening
Based on the predicted binding models of D-chicoric acid and lomofungin, we performed high throughput in silico screening to identify novel inhibitors targeting the exosites of BoNT/A. The MLSMR library consisting of over 350,000 drug-like compounds was screened using a protocol combining ligand-based similarity searches and structure-based docking (Figure 3B). The hits bound at the α- and β-exosites were analyzed in terms of binding interactions with the hotspot residues identified from the binding mode analysis. A total of 167 compounds were selected for experimental evaluation using the continuous assay and FRET substrates. In order to differentiate potential inhibitors at the exosites rather than the active site, two different substrates, the SNAPtide (12-mer) and the long substrate of YFP-(141-206)-CFP (66-mer), were utilized in the primary assay since exosite inhibitors may compete with the longer substrates, but not the short substrates.
The results showed a number of compounds with inhibitory activities in the primary SNAPtide and YFP-(141-206)-CFP assay (Suppl. Figure S3). The most promising hits were examined in the discontinuous assay to eliminate artifacts due to fluorescence quenching or autofluorescence (Suppl. Figure S4). We also tested the compounds using both the full length BoNT/A and the truncated enzyme as inhibitors of BoNT/A were found to display different activities with the two enzyme forms [37]. The identified inhibitors were also confirmed in the gel-based assay with and without DTT (Suppl. Figure S4). Some interesting hits are shown in Table 2. These compounds represent a diversity of chemotypes for the BoNT/A inhibitors that have not been previously reported. Inhibitor C1 was found to be active in the YFP-(141-206)-CFP assay, but not in the SNAPtide assay. It displayed complete inhibition in the gel assay. Inhibition persisted even when >90% of the substrate was cut in the no-inhibitor control lane. Additionally, inhibitor C2 and C4 exhibited high potency in the FRET assay as compared to other hits, but only displayed partial inhibition in the gel assay (Suppl. Figure S4).
Table 2.
Identified inhibitors of BoNT/A from in silico screening. IC50 values were measured using BoNT/A 1-448 and the listed FRET substrate (continuous assay).
| Inhibitor | CID | Structure | SNAPtide IC50 ( M) | YFP-(141-206)-CFP IC50 ( M) |
|---|---|---|---|---|
| C1 | 24761252 |
|
>200 | 56 ± 21 (Ki = 31 ± 4 M)1 130 ± 30 |
| C2 | 16192427 |
|
0.5 ± 0.2 | 0.10 ± 0.06 (Ki = 0.09 ± 0.01 M) |
| C3 | 2917753 |
|
130 ± 801 | 160 ± 201 |
| C4 | 16745487 |
|
ND* | 0.4 ± 0.2 |
| C5 | 24790597 |
|
26 ± 11 | >100 |
| C6 | 1742967 |
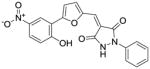
|
8 ± 3 | 43 ± 7 |
| C7 | 5515977 |
|
50 ± 20 | 70 ± 30 |
| C8 | 9659590 |
|
6 ± 6 | >200 |
partial inhibition
Buffer used for measurement contained reducing agent (20mM HEPES pH 8.0, 1.25mM DTT, 0.01% Tween-20).
C1 and C2 are putative exosite inhibitors
We followed up on the top two inhibitors - C1 and C2. C1 contains several appealing functionalities: isoxazole, benzodioxol, and 3-carboxyquinoline. The quinoline-based compounds are known inhibitors of BoNT/A [38]. Lai et al reported that the 8-hydroxyquinol and derivatives showed non-competitive inhibition on BoNT/A [14]. C2 is a piperidine-based chemotype and also contains an interesting benzodioxol group. To further characterize the binding mechanism of the two inhibitors, we performed in vitro kinetic analysis using the full-length BoNT/A. The data for both compounds was best fit to a model for non-competitive inhibition (Figure 7A and 7B). The non-competitive inhibition by these two inhibitors is consistent with exosite inhibition. However, for C1 (the weaker binding inhibitor of the two) other modes of inhibition could not be ruled out. Both inhibitors were susceptible to reducing agents and reduced potency was observed after just a few freeze-thaw cycles of stock solutions (in DMSO).
Figure 7.

(A) Kinetic analysis of inhibitor C1 using CFP-(141-206)-YFP and BoNT/A 1-448. DTT was included in the assay buffer. Global fits (using all points) to models for competitive and non-competitive inhibition were compared. The data was best fit to the model describing non-competitive inhibition. (B) Kinetic analysis of inhibitor C2 using CFP-(141-206)-YFP and BoNT/A 1-448 in the absence of reducing agents. (C) Predicted binding model of inhibitor C1 bound at the α-exosite. (D) Predicted binding model of inhibitor C2 bound at the β-exosite.
We attempted to determine the binding mode of the inhibitors by co-crystallization, but have not yet obtained adequate crystals (likely due to inhibitor binding near crystal-packing interfaces). We therefore performed a refined docking analysis to examine the putative binding interactions of C1 and C2 with BoNT/A. Interestingly, C1 preferably bound at the α-exosite of BoNT/A, whereas C2 favored the β-exosite. As shown in Figure 7C, the isoxazole of C1 was positioned at the α-exosite by forming H-bonding interactions with residues Lys-106 and Lys-346, whereas the carboxyquinoline group pointed down towards the hydrophobic region and formed H-bonding interactions with Asp-112. The carboxyquinoline group appeared to play an important role in facilitating inhibitor binding. Several analogs of C1 were tested in the screen; however, none showed inhibition at 200 μM. The predicted binding model of C2 showed that, while the benzodioxol group inserted into the S1 pocket, the bipiperidine ring together with the phenyl formed the β-sheet binding interaction at the β-exosite by mimicking the substrate binding (Figure 7D).
We should point out that, owing to the large substrate binding interface of BoNT/A and the intrinsic nature of protein mobility, inhibition at the exosites and the synergistic effects on the activity of the catalytic domain of BoNT/A remain elusive. Some of the inhibitors identified from virtual screening (Table 2) are Michael acceptors and could inhibit BoNT/A by forming covalent bond with cysteine residue at the active site or exosite sites. It is possible that the two exosite inhibitors C1 and C2 bind to a region that is distant from the substrate binding interface to generate long-range effects on the active site, and consequently the catalytic process. Subtle changes in the metal coordination geometry or metal binding affinity could significantly impact activity. Recent studies from Janda’s group showed that the C-terminus of full length BoNT/A led to additional flexibility at the active site and profoundly affected inhibitor binding when compared with the truncated form of BoNT/A. We have also observed differences in binding inhibition with the inhibitors using the full length 1-448 and truncated 1-424 enzymes. Unlike the D-chicoric acid, which showed partial inhibition of the full length BoNT/A, inhibitor C1 exhibited non-competitive and complete inhibition. Compared with C1, inhibitor C2 was remarkably potent in the FRET assay, but appeared to be affected by DTT. The inhibitory mechanism of the compounds at the exosites and their potential synergistic activities with other known inhibitors are currently under investigation.
Conclusion
The overarching goal of the study was to identify novel and potent small molecule inhibitors of BoNT/A targeting the exosites. To this end, we first performed extensive computational analysis to probe the structural features of the exosites in the context of binding interactions with lomofungin and D-chicoric acid. Characterization of the binding modes of these two putative inhibitors provided a useful guide for structure-based in silico screening of novel exosite inhibitors of BoNT/A. The identified inhibitor C1 in this study represents a novel chemotype of BoNT/A inhibitor that likely targets the α-exosite. Further investigation of the binding interaction and inhibition mechanism with the exosite inhibitors could offer promising candidates for the development of therapeutics against BoNT/A intoxication.
Supplementary Material
Acknowledgments
This work was supported by Defense Threat Reduction Agency award CBS.MEDBIO.01.10.WR.001. The opinions or assertions contained herein belong to the authors and are not necessarily the official views of the U.S. Navy or the U.S. Department of Defense.
References
- 1.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. JAMA : the journal of the American Medical Association. 2001;285(8):1059. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 2.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. Nat Rev Drug Discov. 2005;4(4):281. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson LL. Annu Rev Pharmacol Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 4.Montecucco C, Schiavo G. Quarterly reviews of biophysics. 1995;28(4):423. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 5.Schiavo G, Matteoli M, Montecucco C. Physiological reviews. 2000;80(2):717. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 6.Brunger AT, Rummel A. Toxicon : official journal of the International Society on Toxinology. 2009;54(5):550. doi: 10.1016/j.toxicon.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat Struct Biol. 1998;5(10):898. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 8.Swaminathan S. The FEBS journal. 2011;278(23):4467. doi: 10.1111/j.1742-4658.2011.08183.x. [DOI] [PubMed] [Google Scholar]
- 9.Breidenbach MA, Brunger AT. Nature. 2004;432(7019):925. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- 10.Byrne MP, Smith LA. Biochimie. 2000;82(9–10):955. doi: 10.1016/s0300-9084(00)01173-1. [DOI] [PubMed] [Google Scholar]
- 11.Smith LA, Rusnak JM. Crit Rev Immunol. 2007;27(4):303. doi: 10.1615/critrevimmunol.v27.i4.20. [DOI] [PubMed] [Google Scholar]
- 12.Sepulveda J, Mukherjee J, Tzipori S, Simpson LL, Shoemaker CB. Infect Immun. 78(2):756. doi: 10.1128/IAI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merino I, Thompson JD, Millard CB, Schmidt JJ, Pang YP. Bioorg Med Chem. 2006;14(10):3583. doi: 10.1016/j.bmc.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Lai H, Feng M, Roxas-Duncan V, Dakshanamurthy S, Smith LA, Yang DC. Arch Biochem Biophys. 2009;491(1–2):75. doi: 10.1016/j.abb.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Capkova K, Yoneda Y, Dickerson TJ, Janda KD. Bioorg Med Chem Lett. 2007;17(23):6463. doi: 10.1016/j.bmcl.2007.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstrom JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Biochem Biophys Res Commun. 2003;310(1):84. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- 17.Kumaran D, Rawat R, Ludivico ML, Ahmed SA, Swaminathan S. J Biol Chem. 2008;283(27):18883. doi: 10.1074/jbc.M801240200. [DOI] [PubMed] [Google Scholar]
- 18.Pang YP, Davis J, Wang S, Park JG, Nambiar MP, Schmidt JJ, Millard CB. PloS one. 2010;5(4):e10129. doi: 10.1371/journal.pone.0010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson AA, Jiao GS, Kim S, Thai A, Cregar-Hernandez L, Margosiak SA, Johnson AT, Han GW, O’Malley S, Stevens RC. Biochemistry. 2011;50(19):4019. doi: 10.1021/bi2001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong J, Thompson AA, Fan Y, Lou J, Conrad F, Ho M, Pires-Alves M, Wilson BA, Stevens RC, Marks JD. J Mol Biol. 397(4):1106. doi: 10.1016/j.jmb.2010.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Šilhár P, Čapková K, Salzameda NT, Barbieri JT, Hixon MS, Janda KD. Journal of the American Chemical Society. 2010;132(9):2868. doi: 10.1021/ja910761y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eubanks LM, Šilhár P, Salzameda NT, Zakhari JS, Xiaochuan F, Barbieri JT, Shoemaker CB, Hixon MS, Janda KD. ACS medicinal chemistry letters. 2010;1(6):268. doi: 10.1021/ml100074s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem Biol. 2007;14(5):533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Case DA, Darden T, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 11. 2010. University of California; San Francisco: 2010. [Google Scholar]
- 25.Case DA, Darden T, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 26.Jiang X, Kumar K, Hu X, Wallqvist A, Reifman J. Chem Cent J. 2008;2:18. doi: 10.1186/1752-153X-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30(16):2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Journal of Computational Chemistry. 1998;19(14):1639. [Google Scholar]
- 29.Darden T, York D, Pedersen L. Journal of Chemical Physics. 1993;98(12):10089. [Google Scholar]
- 30.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., 3rd Accounts of chemical research. 2000;33(12):889. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 31.Sanner MF, Olson AJ, Spehner JC. Biopolymers. 1996;38(3):305. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Burnett JC, Schmidt JJ, McGrath CF, Nguyen TL, Hermone AR, Panchal RG, Vennerstrom JL, Kodukula K, Zaharevitz DW, Gussio R, Bavari S. Bioorg Med Chem. 2005;13(2):333. doi: 10.1016/j.bmc.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 33.Kumaran D, Rawat R, Ahmed SA, Swaminathan S. PLoS Pathog. 2008;4(9):e1000165. doi: 10.1371/journal.ppat.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Kim JJ, Barbieri JT. J Biol Chem. 2007;282(13):9621. doi: 10.1074/jbc.M611211200. [DOI] [PubMed] [Google Scholar]
- 35.Binz T, Bade S, Rummel A, Kollewe A, Alves J. Biochemistry. 2002;41(6):1717. doi: 10.1021/bi0157969. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed SA, Olson MA, Ludivico ML, Gilsdorf J, Smith LA. Protein J. 2008;27(3):151. doi: 10.1007/s10930-007-9118-8. [DOI] [PubMed] [Google Scholar]
- 37.Šilhár P, Lardy MA, Hixon MS, Shoemaker CB, Barbieri JT, Struss AK, Lively JM, Javor S, Janda KD. ACS medicinal chemistry letters. 2013;4(2):283. doi: 10.1021/ml300428s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roxas-Duncan V, Enyedy I, Montgomery VA, Eccard VS, Carrington MA, Lai H, Gul N, Yang DC, Smith LA. Antimicrob Agents Chemother. 2009;53(8):3478. doi: 10.1128/AAC.00141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.