

Protein kinase A (PKA) is a ubiquitous kinase that phosphorylates a broad variety of substrates. A-kinase anchoring proteins (AKAPs) confer specificity to PKA signaling by tethering the kinase to distinct cellular compartments, thereby limiting the access of PKA to a defined pool of its substrates.[1] Interactions between AKAPs and PKA play key roles in a plethora of physiologically relevant processes such as arginine-vasopressin (AVP) mediated water reabsorption in renal principal cells; AVP triggers PKA phosphorylation of the water channel aquaporin-2 (AQP2). AQP2 then redistributes from intracellular vesicles into the plasma membrane, thereby facilitating water reabsorption from primary urine. The redistribution only occurs if PKA interacts with AKAPs.[2] Dysregulation of cellular processes that depend on AKAP–PKA interactions causes or is associated with diseases.[1b, 3] For example, in heart failure, elevated AVP levels contribute to the excessive water retention by the above-described mechanism. Cardiac myocyte contractility is decreased in the failing heart; the control of contractility crucially depends on AKAP–PKA interactions.[4]
PKA holoenzyme is a tetramer consisting of a dimer of regulatory (RIα, RIβ, RIIα, or RIIβ) and two catalytic (C) subunits each bound to an R protomer. Upon binding of cAMP to the R subunits, the C subunits dissociate and phosphorylate their substrates. Interactions of PKA with AKAPs are mediated by the dimerization/docking (D/D) domains of R subunit dimers and the RII binding domain (RBD) of AKAPs. Dimerized D/D domains form a hydrophobic pocket that directly interacts with the 14–25 amino acid long α-helical RBD.[5] Synthetic peptides derived from RBDs of different AKAPs bind R subunits with nanomolar affinity, for example, AKAP18δ-L314E from AKAP18δ (KD=4 nm; see Figures S1 and S2 as well as Table S2 in the Supporting Information).[6] Such peptides effectively inhibit AKAP–PKA interactions.[1] For example, membrane-permeable versions of AKAP18δ-L314E[7] and Ht31[2a] (from AKAP-Lbc) abolish the AVP-induced redistribution of AQP2 in cultured principal cells. Thus disruption of AKAP–PKA interactions even appears beneficial for the treatment of diseases, such as heart failure, that are associated with AVP-dependent excessive water retention.[3]
As a consequence of their generally low membrane permeability and stability, peptides have limitations with regard to their use in cell and animal studies and for drug development. Small molecules and nonpeptide helix mimetics are considered as alternatives to peptides, and thus the small molecule FMP-API-1 was developed that inhibits AKAP–PKA interactions. However, FMP-API-1 also activates PKA, which suggests that it causes side effects in complex systems such as whole animals.[8]
Nonpeptide helix mimetics including terphenyl and terpyridine scaffolds[9] mimic α-helixes by presenting amino acid derived side chains from a rodlike axis and are expected to combine the desired properties of peptides, that is, specificity/target selectivity, and those of small molecules, that is, stability and membrane permeability. We developed polypyridines as helix mimetics of peptide AKAP18δ-L314E,[10] taking into account the favorable solubility, conformational orientation, and biological activity of helix mimetics of this class. The starting point was an interaction model in which hydrophobic residues (L301, L304, L308) of the peptide interact with the hydrophobic bottom of the D/D domain dimer of RIIα and where the hydrophilic side chains such as of E300 of the peptide interact with hydrophilic residues such as Q4 at the peripheral rim of the cavity formed by the D/D dimer.[6] Several terpyridines, 1 a–f (Schemes 1 and 4), were designed in silico as synthetic targets. The hydrophobic phase of the AKAP18δ-L314E helix in molecule 1 b corresponds to the meta-methyl group of the second pyridine ring, the condensed cyclopentyl ring of the third pyridine moiety, and the following two benzyl rings. Thus, they interact with the hydrophobic core (yellow in Figure 4 and Figure S1 in the Supporting Information) of the binding cavity. Other substituents address hydrophilic residues of the rim at the D/D domain such as the carboxy group of the external pyridine ring that mimics the side chain of E300 and is likely to interact with Q4 of the D/D domain.
Scheme 1.

Planned synthesis of terpyridines 1 a–f by a sequence of Suzuki coupling reactions.
Scheme 4.

Synthesis of the terpyridines 12 a, 12 b, and 12 c by microwave-assisted, “one-pot” Suzuki–Miyaura coupling followed by base- or acid-catalyzed hydrolysis to the terpyridylpropionic acids 1 a–f. Reagents and conditions: a) NaOH, EtOH/H2O, 85 °C, 4 h; b) 6 m HCl, EtOH, 95 °C, 1 h; c) NaOH, EtOH/H2O, 85 °C, 1 h. dba=trans,trans-dibenzylideneacetone, MW=microwaves.
Figure 4.
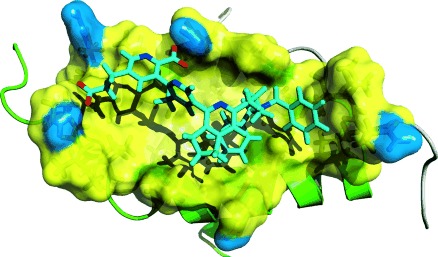
Model of the interaction between terpyridine 1 b (cyan) and the D/D domain of human regulatory RIIα subunits of PKA. The primary structure of the D/D domain is SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREAR (gene bank entry accession NP_004148); the residues of the pocket are shown with side chains and a translucent surface (yellow: hydrophobic, blue: hydrophilic). The binding pocket is rather hydrophobic (I5, P6, L9, T10, L13, T17, L21) and contains the possible hydrogen-bond donors Q4 and Q14 at the rim (blue); Q4 and Q14 are predicted to interact with the carboxy groups of the terpyridine moiety of 1b. For details see the Supporting Information.
The synthesis of the potential ligands 1 a–f involved two successive Suzuki cross-coupling reactions of 2,5-dibromopyridines 3 a,b (Scheme 2). The key to this strategy was the distinctive reactivity of the 2-bromo and 5-bromo substituents, which allowed for the regioselective Suzuki coupling of the dibromides 3 a,b.[11]
Scheme 2.

Synthesis of bipyridyl bromides 6 a,b, the building blocks for the final coupling reaction. TMP=2,2,6,6-tetramethylpiperidide.
The ortho-borylation of cyanopyridines 5 was adapted for the synthesis of the first pyridyl building block—the diisopropylboronic acid ester 2. An ortho-directed metalation reaction (DoM) involves the cyano function, which reacts with the first equivalent of LiTMP as base and most likely forms a lithiun amidinate that serves as the donor-metalation group (DMG) and directs deprotonation to the C3-site of the pyridine ring by the second equivalent of base.[12] A third equivalent of the base was required for effective metalation, presumably to transform the aliphatic ester into the enolate and thereby also protecting the neighboring benzylic position from deprotonation. The addition of four equivalents of the base increased the yield without any side reaction. Since the isolation of the diisopropylboronic ester was not possible and isolation of the boronic acid was associated with protodeboronation, we esterified the acid in situ by adding pinacol to the reaction mixture.[13] Synthesis of ester 2 required adding pinacol and heating the reaction mixture to 40 °C (Scheme 2).
The Pd-catalyzed coupling of 2,5-dibromo-3-methylpyridines 3 a,b and crude boronic acid ester 2 was carried out in the presence of potassium phosphate and yielded bipyridines 6 a,b in low amounts. When boronic acid ester 2 was added slowly through a dropping funnel over 90 min, the yield increased to 74 % (Scheme 2).
The cyclopentapyridine moiety of building blocks 4 a,b was synthesized by a RhII-catalyzed [2+2+2] cycloaddition of 1,6-heptadiyne (7) with succinonitrile (8; Scheme 3).[14] Reducing the nitrile moiety with borane-dimethyl sulfide led to amine 10. Monobenzylated amine 10-Bn was obtained by reductive amination followed by dibenzylation with Et3N as the base. The mixed lithium base aggregates approach was used for the regioselective halogenation at the C1-position of cyclopentapyridine 11 a.[15] No conversion of the starting material occurred when n-hexane was used as the solvent for the lithiation;[15] only the addition of diethyl ether as a co-solvent during the metalation process led to the formation of 4 a,b. CBr4 and C2Cl6 were used as electrophiles. Since the chlorination was more efficient, the final cross-coupling reaction was optimized using pyridyl chlorides 4 a,b.
Scheme 3.

Synthesis of cyclopentapyridyl chlorides 4 a,b.
The synthesis of cyclopentapyridine 11 b requires the separate introduction of the two different benzyl moieties. The secondary amine was obtained under harsh base conditions using NaOH, whereby the substituted benzyl group was introduced in a microwave-assisted aqueous N-alkylation.[16] Surprisingly, the employment of four equivalents of Li base was not sufficient for the lithiation of pyridine 4 a (59 % yield); increasing amounts of the base raised the yield of chloride 4 b fourfold to 78 %.
We developed a microwave-assisted “one-pot” procedure for the Suzuki–Miyaura coupling reaction for synthesis of terpyridines 12 a,b. For this, the borylation and C–C coupling reactions were optimized in terms of solvent, Pd0 catalyst, phosphine ligand, and reaction temperature. The best yields (12 a=46 %, 12 b=50 %) were obtained with [Pd2(dba)3] as the Pd0 source in combination with XPhos as a ligand and using dioxane as solvent. The use of aqueous potassium phosphate as a base was more effective than potassium carbonate. The borylation step resulted in complete consumption of the starting material when KOAc was used as the base, whereas the cross-coupling demanded the addition of a stronger base.[15] The last step of the synthesis was a hydrolysis of 2-cyanoterpyridines 12 a,b. Unexpectedly, the base-catalyzed hydrolysis (Scheme 4, conditions a) led to only partial hydrolysis of the nitrile function. The amidoterpyridylpropionic acids 1 a and 1 c were obtained from the ethyl esters 12 a and 12 b, respectively. The cyanoterpyridylpropionic acid 1 e was obtained when the hydrolysis time was reduced to one hour (Scheme 4, conditions c). Acid-catalyzed hydrolysis furnished the expected dicarboxylic acids 1 b and 1 d (Scheme 4, conditions b).
The binding of the new compounds to the D/D domain was evaluated in 15N-SOFAST-HMQC experiments.[17] Compounds 1 a–f caused similar shifts or disappearances of signals as the peptides derived from AKAP18δ, thus indicating that they bind to the same site on the D/D domain (see the Supporting Information).
Isothermal titration calorimetry revealed that 1 b and 1 f bind to the D/D domain with an estimated KD=148 μm and KD=31 μm, respectively (Figure 1). The binding is mainly driven by a large positive entropy change, presumably because of the release of water molecules from the binding interface. Compound 1 d bound to the D/D with a KD value similar to that of 1 f (10–60 μm). However, the calorimetric data were of lower quality because of the small enthalpy change associated with this binding event (see Figure S4 in the Supporting Information).
Figure 1.

Isothermal titration calorimetric measurements for determination of the KD values for interactions of 1 b and 1 f with the D/D domain of RIIα. Measurements consisted of successive 10 μL injections of the D/D domain into a less concentrated solution of compound in a MicroCal VP-ITC instrument, with 1.0 mm D/D domain and 0.05 mm 1 b, or 0.6 mm D/D domain and 0.03 mm 1 f. The data were fitted to a one-site binding model.
The influence of our compounds on AKAP–PKA interactions was determined quantitatively in homogeneous time-resolved fluorescence (HTRF) assays. HTRF measurements (at pH 7) with the D/D domain of RIIα and AKAP18α revealed that 1 b and 1 e decreased the FRET signal and thus inhibited the interaction (IC50=38 μm and 138 μm, respectively; Figure 2). As expected, the peptide AKAP18δ-L314E inhibited the interaction at a nanomolar concentration (12.7 nm),[6] while the inactive control peptide AKAP18δ-PP did not affect the interaction (Figure 2). The inhibitory potency of 1 a and 1 f was too low to determine IC50 values.
Figure 2.

Compounds 1 b and 1 e and peptide AKAP18δ-L314E inhibit the interaction of the D/D domain of RIIα with AKAP18α. AKAP18α lacks the 10 N-terminal amino acid residues which encode the membrane-targeting domain. Peptide AKAP18δ-PP represents the RII binding domain of AKAP18δ, but contains two proline residues which prevent interaction with the D/D domain. This peptide does not affect the interaction.[6] Homogeneous time-resolved fluorescence (HTRF) assays were carried out using the proteins in concentrations of 50 nm each.
Next, we examined the ability of 1 b to interfere with AKAP–PKA interactions in cells. HEK293 cells possess a negative feedback loop for terminating prostaglandin E1 (PGE1) induced cAMP synthesis, a pathway that depends on AKAP–PKA interactions. Stimulation of the cells with PGE1 leads to activation of adenylyl cyclase and the synthesis of cAMP; cAMP activates PKA, which phosphorylates cAMP phosphodiesterase PDE4D and thereby increases PDE activity and thus inhibits cAMP accumulation. PDE4D is phosphorylated by PKA bound to the AKAP gravin.[18] In addition, AKAP150 tethers PKA to adenylyl cyclase and facilitates PKA phosphorylation of the cyclase and thus its inactivation.[19] Disruption of AKAP–PKA interactions with peptides (Ht31), the small molecule FMP-API-1, or by PKA inhibition with H89 prevents the PGE1-dependent negative feedback and maintains high adenylyl cyclase activity in the presence of PGE1.[8,18,19] 1 b also inhibits the feedback inhibition, thus indicating that it interferes with AKAP–PKA interactions in cells (Figure 3). Although 1 a binds to the D/D domain (see Figure S3 in the Supporting Information), it did not interfere with the feedback inhibition, presumably because it does not reach sufficient concentrations in cells. The terpyridines do not inhibit the catalytic activity of PKA.
Figure 3.

HEK293 cells were transfected to express cAMP-dependent cyclic nucleotide-gated channels (CNGC) and loaded with the Ca2+ indicator Fura-2. The cells were left untreated, incubated with the PKA inhibitor H89, FMP-API-1, or compounds 1 a or 1 b (each 20 μm) for 30 min. The cells were stimulated with prostaglandin E1 (PGE1) to trigger cAMP synthesis at a time point of 60 s. cAMP opens the CNGCs, and then Ca2+ enters the cells and binds to Fura-2. This binding generates fluorescence signals, which were imaged. Numbers in brackets indicate the numbers of individual cells measured.
We have developed a set of terpyridines as α-helix mimetics of the PKA anchoring disruptor peptide AKAP18δ-L314E. Similar to the original peptide, the compounds bind to the D/D domain of RII subunits of PKA. One of them, 1 b, is the first nonpeptidic agent that binds to the D/D domain of RII subunits and inhibits AKAP–PKA interactions in vitro as well as in vivo. A model of the interaction is depicted in Figure 4. Evaluation of the structure–activity relationship (SAR) indicates that a COOH group as R2 in 1 b is critically involved in defining its inhibitory potency; NH2, as in 1 a, is not tolerated at this position. Both H and OMe seem to be tolerated as R1, as 1 b and 1 e inhibit AKAP–PKA interactions (Figure 2). However, the influence of CN as R2 in 1 e is not clear. The COOH group as R2 allows for the synthesis of more derivatives and thereby provides the basis for novel high affinity compounds to disrupt AKAP–PKA interactions. These SAR data together with our NMR data support a model of the interaction of 1 b with the D/D domain of RIIα subunits (Figure 4) in which 1 b assumes a conformation that mimics the α-helical AKAP18δ fragment that interacts with the D/D domain (see Figure S1 in the Supporting Information).
In summary, the new terpyridine scaffolds represent the first biologically active and nonpeptidic compounds that block the binding site of AKAPs on regulatory subunits of PKA and interfere with AKAP–PKA interactions.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Scott JD, Dessauer CW, Tasken K. Annu. Rev. Pharmacol. Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E. Int. Rev. Cell Mol. Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 2a.Klussmann E, Maric K, Wiesner B, Beyermann M, Rosenthal W. J. Biol. Chem. 1999;274:4934–4938. doi: 10.1074/jbc.274.8.4934. [DOI] [PubMed] [Google Scholar]
- 2b.Okutsu R, Rai T, Kikuchi A, Ohno M, Uchida K, Sasaki S, Uchida S. Kidney Int. 2008;74:1429–1433. doi: 10.1038/ki.2008.402. [DOI] [PubMed] [Google Scholar]
- 3.Tröger J, Moutty MC, Skroblin P, Klussmann E. Br. J. Pharmacol. 2012;166:420–433. doi: 10.1111/j.1476-5381.2011.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diviani D, Dodge-Kafka KL, Li J, Kapiloff MS. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1742–1753. doi: 10.1152/ajpheart.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5a.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b.Taylor SS, Ilouz R, Zhang P, Kornev AP. Nat. Rev. Mol. Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hundsrucker C, et al. Biochem. J. 2006;396:297–306. doi: 10.1042/BJ20051970. see the Supporting Information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szaszák M, Christian F, Rosenthal W, Klussmann E. Cell Signalling. 2008;20:590–601. doi: 10.1016/j.cellsig.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Christian F, et al. J. Biol. Chem. 2011;286:9079–9096. doi: 10.1074/jbc.M110.160614. see the Supporting Information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew. Chem. 2005;117:2764–2767. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2005;44:2704–2707. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- 9b.Davis JM, Tsou LK, Hamilton AD. Chem. Soc. Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 9c.Peters M, Trobe M, Breinbauer R. Chem. Eur. J. 2013;19:2450–2456. doi: 10.1002/chem.201203006. [DOI] [PubMed] [Google Scholar]
- 9d.Thompson S, Hamilton AD. Org. Biomol. Chem. 2012;10:5780–5782. doi: 10.1039/c2ob25273b. [DOI] [PubMed] [Google Scholar]
- 10.Davis JM, Truong A, Hamilton AD. Org. Lett. 2005;7:5405–5408. doi: 10.1021/ol0521228. [DOI] [PubMed] [Google Scholar]
- 11.Burzicki G, Voisin-Chiret AS, Santos JSD, Rault S. Tetrahedron. 2009;65:5413–5417. [Google Scholar]
- 12a.Cailly T, Fabis F, Bouillon A, Lemaitre S, Sopkova J, de Santos O, Rault S. Synlett. 2006:53–56. [Google Scholar]
- 12b.Cailly T, Fabis F, Lemaitre S, Bouillon A, Rault S. Tetrahedron Lett. 2005;46:135–137. [Google Scholar]
- 13.Alessi M, Larkin AL, Ogilvie KA, Green LA, Lai S, Lopez S, Snieckus V. J. Org. Chem. 2007;72:1588–1594. doi: 10.1021/jo0620359. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto Y, Kinpara K, Ogawa R, Nishiyama H, Itoh K. Chem. Eur. J. 2006;12:5618–5631. doi: 10.1002/chem.200600176. [DOI] [PubMed] [Google Scholar]
- 15.Kaminski T, Gros P, Fort Y. Eur. J. Org. Chem. 2003:3855–3860. [Google Scholar]
- 16.Ju YH, Varma RS. Green Chem. 2004;6:219–221. [Google Scholar]
- 17.Schanda P, Brutscher B. J. Am. Chem. Soc. 2005;127:8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 18.Willoughby D, Wong W, Schaack J, Scott JD, Cooper DM. EMBO J. 2006;25:2051–2061. doi: 10.1038/sj.emboj.7601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauman AL, et al. Mol. Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. see die Supporting Information. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information