

Abstract
Glucose transporter isoform 4 (GLUT4) is the insulin-responsive glucose transporter mediating glucose uptake in adipose and skeletal muscle. Reduced GLUT4 translocation from intracellular storage compartments to the plasma membrane is a cause of peripheral insulin resistance. Using a chronic hyperinsulinemia (CHI)-induced cell model of insulin resistance and Rab5 mutant overexpression, we determined these manipulations altered endosomal sorting of GLUT4, thus contributing to the development of insulin resistance. We found that CHI induced insulin resistance in 3T3-L1 adipocytes by retaining GLUT4 in a Rab5-activity-dependent compartment that is unable to equilibrate with the cell surface in response to insulin. Furthermore, CHI-mediated retention of GLUT4 in this non-insulin-responsive compartment impaired filling of the transferrin receptor (TfR)-positive and TfR-negative insulin-responsive storage compartments. Our data suggest that hyperinsulinemia may inhibit GLUT4 by chronically maintaining GLUT4 in the Rab5 activity-dependent endosomal pathway and impairing formation of the TfR-negative and TfR-positive insulin-responsive GLUT4 pools. This model suggests that an early event in the development of insulin-resistant glucose transport in adipose tissue is to alter the intracellular localization of GLUT4 to a compartment that does not efficiently equilibrate with the cell surface when insulin levels are elevated for prolonged periods of time.
One of the primary actions of insulin on adipose and skeletal muscle is to increase glucose uptake from the blood plasma. This process is a result of the translocation of the insulin-responsive glucose transporter 4 (GLUT4) from specialized intracellular retention compartments to the plasma membrane (1–6). GLUT4 translocation, and the resulting glucose flux, is the rate-limiting step in reestablishing euglycemia after a meal (7–9). The development of peripheral insulin resistance is accompanied by decreased GLUT4 at the cell surface in response to insulin (9–16). In obesity and type 2 diabetes mellitus, changes in the transcriptional regulation ultimately result in the loss of GLUT4 expression and insulin resistance in adipose tissue (6, 16–18). However, previous reports suggest that the loss of surface GLUT4 precedes the loss of GLUT4 expression and significantly contributes to the early onset of peripheral insulin resistance (19–25).
Several lines of evidence suggest that chronic insulin exposure, as a result of over nutrition, is sufficient to induce insulin resistance (14, 16, 26, 27). Chronic insulin exposure leads to the desensitization of the insulin-signaling pathway through the down-regulation of the upstream insulin signaling adaptor, insulin receptor substrate-1/2 (28–31). Interestingly, chronic insulin exposure also alters the intracellular traffic and half-life of GLUT4 in 3T3-L1 adipocytes (24, 25, 32). It is uncertain whether the down-regulation of insulin signaling or whether alteration in GLUT4 traffic is the initiating molecular event in the development of insulin resistance. In the current study, we used a 3T3-L1 adipocyte model of hyperinsulinemia-induced insulin resistance to investigate the temporal relationship between impaired insulin-mediated GLUT4 translocation, intracellular GLUT4 traffic, and insulin signaling as insulin resistance develops. Our findings suggest that chronic hyperinsulinemia in early-onset insulin resistance significantly altered intracellular GLUT4 traffic, which contributed to impaired surface GLUT4 levels.
GLUT4 traffics through several intracellular compartments including the early (sorting) endosome, endosome recycling center (ERC), trans-Golgi network (TGN), and specialized insulin-responsive storage vesicles (IRSVs) (reviewed in reference 33). The relative distribution of GLUT4 within each of these compartments plays an important role in maintaining basal retention and establishing insulin-dependent, steady-state redistribution of GLUT4 at the cell surface. Kinetic analysis of GLUT4 translocation determined that two insulin-responsive GLUT4 compartments are necessary to establish and maintain the elevated insulin-mediated GLUT4 redistribution associated with insulin stimulation: the IRSVs, which do not contain transferrin receptor (TfR) and a TfR-positive subdomain of the ERC (7, 34–44). Although the IRSVs are thought to provide the initial release of GLUT4 to the plasma membrane in response to insulin (translocation), the TfR-positive compartment establishes and maintains the elevated steady-state redistribution of GLUT4 at the plasma membrane (7, 34, 39, 44).
Several studies of insulin-resistant GLUT4 translocation have focused on the signaling and trafficking events involved in the exocytic arm of GLUT4 traffic. Insulin signaling through the serine kinase, Akt, is thought to be the major pathway leading to GLUT4 translocation (reviewed in references 45 and 46). In adipocytes, Akt phosphorylates a putative Rab GAP (TBC1D4/AS160), allowing GLUT4 to be released from the IRSVs (47, 48). In adipocytes, most evidence suggests that the physiological substrate for TBC1D4/AS160 is Rab10, and this Rab is responsible for exocytosis from the IRSVs (47–54). Recently Rab14 has also been shown to be part of the Rab10 pathway and appears to mediate endosomal sorting into the Rab10-responsive GLUT4 compartment (54). In view of the complex intracellular GLUT4 itinerary, it is likely that many Rab proteins are involved with discrete steps in GLUT4 traffic (54–56). Identification of the specific Rabs has been hindered by our lack of knowledge and sensitive assays to identify and measure all the discrete trafficking steps GLUT4 is able to use. Because GLUT4 is known to traffic through the early endosome and intracellular compartmentalization of GLUT4 is altered in models of insulin resistance, we sought to determine how slowing early endosome sorting and intracellular compartmentalization would alter GLUT4 availability during insulin resistance (34, 35, 57–61). Herein we demonstrate that chronic exposure to insulin alters GLUT4 trafficking through the early endosome resulting in sorting to an endosomal compartment that is less responsive to insulin.
Materials and Methods
Plasmids, ligands, and antibodies
Stratagene QuikChange site-directed mutagenesis kit was used to generate a single-nucleotide mutation at F5, E499, and E501 of wild-type (WT) hemagglutinin (HA)-GLUT4-green fluorescent protein (GFP) (35, 62). Mutations were verified by sequencing. The pTetOff and pTetOn gene expression systems were obtained from CLONTECH. The pBI Rab5S34N-myc, pBI Rab5WT-FLAG, and pBI Rab5Q79L-FLAG constructs were provided by Dr Guangpu Li (63). Predesigned scrambled and Rab5a (sc-36345) small interfering RNA (siRNA) constructs were obtained from Santa Cruz Biotechnologies.
Monoclonal mouse α-HA.11 (16B12) antibody was purchased from Covance. Alexa-647 (A647)- and Alexa-680 (A680)-conjugated secondary anti-IgG antibodies were purchased from Invitrogen. Unlabeled goat α-mouse IgG antibody was purchased from Pierce. Rabbit α-Akt, rabbit α-phospho-Akt (Ser473), rabbit α-AS160, rabbit α-phospho-AS160 (Thr642), and rabbit α-Rab5 antibodies were purchased from Cell Signaling. Monoclonal mouse α-c-Myc antibodies were purchased from Santa Cruz Biotechnologies. Peroxidase-conjugated ChromPure human transferrin was purchased from Jackson ImmunoResearch Laboratories.
Cell culture and transfection
3T3-L1 fibroblasts purchased from the American Type Tissue Culture repository were cultured and maintained as previously described (64). Differentiation of confluent cultures was induced by incubating in DMEM plus 25 mM glucose, 10% fetal bovine serum (FBS), 175 nM insulin, 1 μM dexamethasone, 0.5 mM isobutyl-1-methylxanthine (pH 7.4) medium. After 3.5 days, the medium was switched to DMEM plus 25 mM glucose, 10% FBS (pH 7.4) (10% FBS/DMEM). Confluent day 5 postdifferentiated 3T3-L1 adipocytes were transfected via electroporation as previously described (18, 64).
For siRNA knockdown, day 2 postdifferentiating 3T3-L1 adipocytes were transfected via electroporation as described above with 50 nM siRNA and plated on 60-mm tissue culture plates with the medium of 175 nM insulin, 1 μM dexamethasone, 0.5 mM isobutyl-1-methylxanthine. For immunoblots, cells were recovered for 72 hours and then lysed and separated by SDS-PAGE as described below. For translocation assay, cells were recovered for 48 hours and then subjected to a second transfection via electroporation as described above with 50 μg of HA-GLUT4-GFP.
HA-GLUT4-GFP translocation assay
Day 5 postdifferentiated 3T3-L1 adipocytes were electroporated as described above with HA-GLUT4-GFP [WT, loss-of-function mutation, F5A (FA), or E499A, E501A (EEAA)] without or with pTetOff -or pTetOn-inducible expression system and pBI Rab5S34N-myc. Cells were plated with 10% FBS/DMEM on four-chamber BD Falcon culture slides overnight. For chronic hyperinsulin (CHI) experiments, the medium was supplemented with 100 nM insulin. For delayed Rab5S34N expression, cells expressing pTetOn/pBI Rab5S34N-myc were stimulated immediately or 6 hours after electroporation with 1 mg/mL doxycycline. Twenty-hours after electroporation, cells were serum starved for 2 hours in F-12 Ham's plus 0.5% BSA and then treated for 30 minutes without or with 10 nM insulin and then washed and fixed as described (7, 57). Cells were blocked for 15 minutes with 10% calf serum, labeled for 1 hour with 1 μg/mL mouse α-HA.11 antibody at room temperature, and then visualized by indirect immunofluorescence with 5 μg/mL A647 goat α-mouse IgG antibody (Invitrogen) for 30 minutes at 37°C. Images were collected using a Leica SP2 MP confocal microscope with a ×63, 1.4 numerical aperture oil immersion objective (Leica Microsystems). Maximum-intensity projection images were compiled from 13 sections, 15–25 μm thick, and represent the entire z-axis of the cell (65). Leica LCS Lite software was used for quantification of surface Alexa-647 and total GFP intensities. Thirty to 40 cells from four different experiments were imaged per condition. Surface to total (A647 to GFP) intensity ratios were calculated and normalized to the insulin-stimulated control. Outliers were determined as greater or less than the mean plus or minus 3 times the SD. Final data were reported as the normalized mean A647 to GFP ratio plus SE.
HA-GLUT4-GFP recycling pool assay
Day 5 postdifferentiated 3T3-L1 adipocytes were electroporated and cultured in chambered culture slides as described above. After recovery, cells were serum starved for 2 hours in F-12 Ham's plus 0.5% BSA and then incubated for 2 hours with 2 μg/mL mouse α-HA.11 antibody and 10 nM insulin in 10% FBS/DMEM. Cells were transferred to ice, washed, fixed for 20 minutes with 3.7% paraformaldehyde, and blocked for 15 minutes with 10% calf serum. Surface-bound mouse α-HA.11 was blocked with 20 μg/mL unlabeled goat α-mouse IgG antibody for 30 minutes at 37°C. Cells were refixed, permeabilized for 20 minutes with 250 μg/mL saponin, quenched on ice with sodium borohydride, and then reblocked with 10% calf serum. Internalized mouse α-HA.11 was labeled with 5 μg/mL A647 goat α-mouse IgG antibody for 30 minutes at 37°C. Cell images were collected and analyzed as described above.
Whole-cell lysates and immunoblots
For CHI experiments, day 5 postdifferentiated 3T3-L1 adipocytes cultured in 100-mm tissue culture plates were treated as described above. For delayed Rab5S34N expression experiments, cells transfected without or with pTetOn/pBI Rab5S34N-myc were plated on 100-mm tissue culture plates and treated with 1 mg/mL doxycycline as described above. Twenty hours after treatment, whole-cell extracts were prepared with ice-cold Nonidet p-40 (NP-40) lysis buffer (1% NP-40; 2 mM EDTA; 100 mM sodium fluoride; 10 mM sodium pyrophosphate; 2 mM sodium vanadate; 12 mM molybdate; 0.5 mM phenylmethylsulfonyl fluoride; 1× protease inhibitor cocktail (Roche Applied Science); and 20 mM HEPES, pH 7.4). Total protein concentrations were determined with Coomassie Plus protein assay (Pierce) per the manufacturer's protocol. Proteins were transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore) and immunolabeled with specified primary antibodies. Membranes were visualized using the appropriate A680-conjugated secondary α-IgG antibodies (Invitrogen) and analyzed using the Odyssey infrared imaging system (LiCor). Data were reported as mean relative densitometry plus SE.
Fluorescence ablation assay
Day 5 postdifferentiated 3T3-L1 adipocytes were transiently transfected, plated on chamber slides, and treated as described above. Twenty hours after electroporation, cells were stimulated for 30 minutes with 170 nM insulin in ablation medium (DMEM plus 25 mM glucose; 20 mM bicarbonate; 20 mM HEPES, pH 7.4; and 10% FBS) at 37°C, 5% CO2. Insulin was removed by washing with a mild acid buffer (pH 5.0) (50 mM MES and 200 mM NaCl). Cells were reequilibrated with an ablation wash buffer (20 mM HEPES, pH 7.4; 150 mM NaCl; 1 mM CaCl2; 5 mM KCl; 1 mM MgCl2) and then returned to basal in ablation medium plus 2 μg/mL mouse α-HA.11 without or with 20 μg/mL transferrin (Tf)-horseradish peroxidase (HRP) for 4 hours at 37°C, 5% CO2. Cells were washed with ablation wash buffer to remove excess antibody and ligand. For acute insulin restimulation, cells were treated with 100 nM insulin for 30 minutes. Fluorescent quenching by diaminobenzidine polymerization was done by incubating cells in 300 μg/mL diaminobenzidine plus 0.02% H2O2 diluted in ablation wash buffer for 30 minutes in the dark, on ice. Cells were washed with ablation wash buffer, fixed, and blocked, and then surface-bound mouse α-HA.11 was blocked as described above. Cells were refixed, permeabilized, quenched, reblocked, and internalized, and unablated mouse α-HA.11 was detected with 5 μg/mL A647 goat α-mouse IgG antibody as described above. Cell images were collected and analyzed as described above.
Colocalization of HA-GLUT4-mCherry and Rab-GFP
Day 5 postdifferentiated 3T3-L1 adipocytes were transfected with HA-GLUT4-red fluorescent protein (mCherry) (provided by Dr Joshua Zimmerman, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD) and either a Rab5 wild-type GFP or a Rab5-S34N-GFP construct. Eighteen hours after transfection, cells were fixed and imaged by confocal microscopy as described for the HA-GLUT4-GFP translocation assay as described above.
Rab5-GTP activation assay
Day 5 postdifferentiated 3T3-L1 adipocytes were stimulated acutely with 10 nM insulin for 30 minutes at 37°C or treated without or with CHI as described above with acute restimulation with 10 nM insulin. Whole-cell lysates were prepared in lysis buffer (25 mM Tris, pH 7.4; 30 mM MgCl2; 150 mM NaCl; 1% NP-40; 2 mM sodium vanadate complexes; 1 mM phenylmethylsulfonyl fluoride; and 1× protease Inhibitors). Equal total protein was immunoprecipitated with α-active Rab5 mouse monoclonal antibody (Neweast Biosciences). Complexes were separated by SDS-PAGE and prepared for immunoblot as described above.
Results
Chronic exposure to insulin inhibits GLUT4 translocation
To quantify GLUT4 redistribution, we used a dual-labeled GLUT4 reporter (HA-GLUT4-GFP) carrying an HA epitope in the first exofacial loop and a GFP fused in frame at the C terminus (Figure 1A) (62). In nonpermeabilized cells, the HA-GLUT4-GFP reporter allowed for detection and quantification of surface-exposed GLUT4 (HA) normalized to total GLUT4 (GFP) (32, 38). To induce insulin resistance, 3T3-L1 cells transfected with the HA-GLUT4-GFP reporter were treated with 100 nM insulin for 20 hours (referred hereafter as chronic hyperinsulin), a model that has been widely used in cultured adipocytes (23–25, 32). The effect of CHI on GLUT4 redistribution was measured after a 2-hour serum starvation and subsequent acute restimulation with 10 nM insulin for 30 minutes. Cells were fixed without permeabilization and stained for the surface-exposed HA epitope. Consistent with previous reports, CHI significantly impaired the subsequent, acute insulin-stimulated HA-GLUT4-GFP redistribution (Figure 1, B and C) (25, 32). The effect of CHI on GLUT4 translocation was dose dependent (Figure 1D), supporting the use of CHI treatment of 3T3-L1 adipocytes to investigate the effects of insulin resistance on GLUT4 traffic. For the remaining studies, we used the maximal dose of insulin to optimize the effects.
Figure 1.

Chronic exposure to insulin inhibits GLUT4 translocation, but not downstream AS160 phosphorylation. A, Schematic diagram of WT HA-GLUT4-GFP reporter with HA epitope located on the first exofacial loop and GFP conjugated in frame at the C terminus. B, 3T3-L1 cells were transfected with HA-GLUT4-GFP and treated without or with 100 nM insulin for 20 hours (CHI). After 20 hours, cells were serum starved for 2 hours and then stimulated for 30 minutes without or with 10 nM insulin. Cells were fixed without permeabilizing and stained for surface-exposed HA as described in Materials and Methods. HA epitope staining (A647) is a measure of surface GLUT4 and GFP is a measure of total GLUT4. C, Quantification of surface (A647)-to-total (GFP) fluorescence intensity in panel B was reported as mean and SE for 30–40 cells normalized to insulin control. D, 3T3-L1 cells transfected with HA-GLUT4-GFP were treated with CHI dose response for 20 hours. Cells were treated and acutely stimulated with insulin as described in panel B. Quantification of A647-to-GFP fluorescence intensity is reported as mean and SE for 10–12 cells. E and F, Whole-cell lysates prepared from 3T3-L1 cells treated with CHI and then serum starved and restimulated with insulin as described in panel B were immunoblotted using antibodies against total Akt (E), phospho-Ser473 Akt (E), total AS160 (F), and phospho-Thr642 AS160 (F) as described in Materials and Methods. E and F, Quantification of phospho-Ser473 Akt-to-total Akt (E) and phospho-Thr642 AS160-to-total AS160 (F) was determined by densitometry. Data were reported as mean and SE from n = 3. All data were analyzed using a Student's t test for two-sample experiments or one-way ANOVA with pair-wise comparison. *, P < .05 for comparisons designated by bracket; #, P < .05 for comparison of basal vs insulin.
To determine whether CHI modified acute insulin signaling events, whole-cell lysates from similarly treated 3T3-L1 cells were immunoblotted for total and phosphorylated Akt and TCB1D4/AS160. The 2-hour serum starvation was sufficient to reduce phosphorylated Akt (Ser473) and AS160 (pAS160; Thr642) to basal levels (Figure 1, E and F). As previously reported, CHI inhibited Akt phosphorylation by approximately 40% in response to insulin restimulation (Figure 1E) (32). Although Akt has several downstream signaling events involved in insulin signaling, TBC1D4/AS160 is the only known downstream target of Akt directly involved in the acute insulin-mediated release and translocation of GLUT4 to the plasma membrane (35, 47, 49, 66, 67). Despite the loss of phosphorylated Akt, CHI did not significantly reduce the phosphorylation of TBC1D4/AS160 (Figure 1F), suggesting that the CHI-mediated loss of GLUT4 redistribution is not solely due to impaired downstream insulin signaling. Furthermore, because TBC1D4/AS160 is involved in the insulin-mediated exocytosis of GLUT4, we hypothesized that the CHI-mediated reduction in GLUT4 redistribution may be due, in part, to alterations in GLUT4 endocytosis.
Chronic exposure to insulin alters GLUT4 translocation through altered GLUT4 endocytosis and sorting
During and after acute insulin stimulation, internalized GLUT4 is sorted for either recycling (during acute stimulation) or retention (after acute stimulation) by way of the sorting early endosome (33–35, 57, 58, 68). Rab5 is an important regulator of early endosome sorting and has been implicated in the regulation of GLUT4 traffic (56, 58–61, 69, 70). To determine whether slowing internalization and traffic through the early endosome alternatively affects GLUT4 redistribution in CHI-treated cells, we overexpressed a dominant inhibitory Rab5 (Rab5 S34N) mutant in combination with our HA-GLUT4-GFP reporter (63, 71–75). The expression level of the Rab5 S34N mutant is shown in Figure 2A. We found that CHI did not significantly alter acute insulin-mediated HA-GLUT4-GFP redistribution after a 2-hour starvation in the presence of Rab5 S34N expression, compared with that of Rab5 S34N alone (Figure 2, B and C). The lack of an additive affect between Rab5 S34N overexpression and CHI indicates that CHI alters GLUT4 sorting at a step that is distal to Rab5 activity-dependent sorting steps. This is consistent with our hypothesis that CHI alters GLUT4 endosomal sorting.
Figure 2.

CHI alters intracellular GLUT4 traffic via a Rab5 activity-dependent endosomal pathway. A–C, 3T3-L1 cells were transfected with HA-GLUT4-GFP reporter and either pcDNA3/pTetOff or pBI Rab5S34N-myc/pTetOff. A, Whole-cell lysates were prepared 20 hours after transfection and immunoblotted with a Rab5-specific antibody as described in Materials and Methods. B and C, Transfected 3T3-L1 cells were treated without or with CHI for 20 hours and then serum starved for 2 hours and restimulated for 30 minutes without or with 10 nM insulin. Cells were fixed and stained for surface-exposed HA as described in Materials and Methods. B, Images are representative maximum intensity projections of the z-axis (see Materials and Methods). C and D, Quantification of A647-to-GFP fluorescence intensity was reported as mean and SE for 25–35 cells normalized to the insulin control (C) or basal control (D). Data were analyzed using a Student's t test. *, P < .05. E, Whole-cell lysates were prepared 72 hours after transfection with Rab5a targeting siRNA and then immunoblotted with Rab5 or α/β-tubulin-specific antibodies as described in Materials and Methods. F, 3T3-L1 cells transfected with HA-GLUT4-GFP and Rab5a-targeting siRNA as described in panel E were serum starved for 2 hours and then fixed and stained for surface-exposed HA as described in Materials and Methods. Quantification of A647 to GFP fluorescence intensity ratio was reported as mean and SE for 20–25 cells normalized to the basal control. G, 3T3-L1 cells were transfected with pBI Rab5S34N-myc/pTetOn and then treated without or with 1 μg/mL Dox 0 or 6 hours after transfection. Whole-cell lysates were prepared 24 hours after transfection and immunoblotted using antibodies against c-Myc as described in Materials and Methods. Data are a representative image from n = 3. H, 3T3-L1 cells transfected with HA-GLUT4-GFP reporter and pBI Rab5S34N-myc/pTetOn were treated as described in panel G. After 24 hours, cells were serum starved and then fixed and stained for surface-exposed HA as described in Materials and Methods. A647-to-GFP fluorescence intensity was measured as described in Materials and Methods and reported as normalized basal mean and SE for 30–40 cells. All data were analyzed using a Student's t test. *, P < .05 for comparisons designated by bracket; #, P < .05 for comparison of basal vs insulin. IB, immunoblotting.
We observed that Rab5 S34N overexpression alone significantly impaired the basal retention of HA-GLUT4-GFP, compared with control cells (Figure 2D). To control for the off-target effects of Rab5 S34N overexpression, we used siRNA knockdown to deplete Rab5 in 3T3-L1 adipocytes. Rab5a depletion was confirmed by immunoblot (Figure 2E). Consistent with the Rab5 S34N overexpression, Rab5a depletion significantly increased basal GLUT4 on the cell surface (Figure 2F), thus indicating the specificity of the Rab5 S34N inhibition of Rab5 in the 3T3-L1 adipocytes. These findings suggest that Rab5 activity-dependent sorting during early endocytosis is required to maintain the low basal levels of GLUT4 on the cell surface.
In addition, we used a doxycycline (Dox)-inducible expression system (pTetOn) to delay Rab5 S34N mutant expression to ensure that the Rab5 S34N-mediated loss in basal GLUT4 retention was not due to altered GLUT4 sorting from the biosynthetic pathway. Dox was added immediately or 6 hours after transfection, allowing sufficient time for the HA-GLUT4-GFP reporter to exit the TGN and form the insulin responsive storage compartments (76). Delaying Dox treatment did not hinder the accumulation of Rab5 S34N 20 hours after transfection (Figure 2G). Furthermore, delaying the onset of Rab5 S34N expression similarly increased the basal appearance of HA-GLUT4-GFP on the cell surface (Figure 2H), confirming that altering Rab5 function disturbs GLUT4 traffic independent of the biosynthetic pathway.
Chronic exposure to insulin alters GLUT4 traffic independent of GLUT4 internalization or sorting to IRSVs
Although Rab5 S34N overexpression specifically altered basal HA-GLUT4-GFP retention and reduced insulin-mediated redistribution, it completely prevented a CHI-mediated loss in insulin-mediated GLUT4 redistribution. For this reason we hypothesized that CHI alters GLUT4 traffic downstream of a Rab5-dependent sorting event. To narrow down the trafficking step(s) altered by CHI, we combined the Rab5 S34N overexpression or CHI treatment with previously characterized HA-GLUT4-GFP sorting mutants that carry loss-of-function mutations in the N-terminal F5QQI and/or C-terminal TE499LE501Y motifs (Figure 3A).
Figure 3.

Rab5 regulates the formation of cellular compartments required for normal GLUT4 maintaining basal levels of GLUT4 on the cell surface. A, Schematic diagram of HA-GLUT4-GFP with loss-of-function mutations shown in red: A5QQI (FA) or TA499LA501Y (EEAA). B, 3T3-L1 cells were transfected with FA, EEAA, or FA/EEAA double mutant HA-GLUT4-GFP reporter in combination with either empty vector/pTetOff or pBI Rab5S34N-myc/pTetOff. After 20 hours of recovery, cells were serum starved for 2 hours and stimulated for 30 minutes with 10 nM insulin. Cells were fixed and stained for surface-exposed HA as described in Materials and Methods. Quantification of A647-to-GFP fluorescence was reported as mean and SE for 30–40 cells normalized to insulin alone. Two-sample data were analyzed using a Student's t test. *, P < .05 for comparisons indicated; #, P < .05 for basal vs insulin for each condition. Comparisons between multiple conditions were analyzed by a one-way ANOVA where a indicates P < .05 for comparison of the mutant GLUT4 to WT in basal conditions, and b indicates P < .05 for mutant GLUT4 compared with WT under insulin conditions. C–E, 3T3-L1 cells transfected with FA (C) or EEAA (D) or FA/EEAA double mutant (E) HA-GLUT4-GFP were treated without or with CHI and then serum starved for 2 hours and restimulated without or with 10 nM insulin for 30 minutes. Cells were fixed and stained as described in panel B. A647-to-GFP fluorescence was measured as described in panel B and reported as mean and SE for 30–40 cells normalized to acute insulin. All data were analyzed using a Student's t test. *, P < .05 for comparisons indicated by brackets; #, P < .05 for comparison between basal and insulin for each construct.
F5QQI is required for GLUT4 incorporation into clathrin-coated vesicles through its association with μ-adaptor proteins (36, 68). The loss-of-function mutation, FA, impairs μ-adaptor binding resulting in both the loss of GLUT4 retention within the TGN and impaired internalization from the plasma membrane (35, 77). Consistent with previous reports, FA HA-GLUT4-GFP increased both basal and insulin-mediated HA-GLUT4-GFP redistribution (35). Rab5 S34N coexpression further increased the basal levels of FA HA-GLUT4-GFP in the plasma membrane (Figure 3B). The additive effect on basal appearance of GLUT4 on the cell surface suggests Rab5 S34N impaired GLUT4 traffic at an endocytic step independent or downstream of the impaired clathrin-mediated internalization caused by the FA mutation. The compartment(s) in which the FA HA-GLUT4-GFP mutant is retained without and with Rab S34N are insulin responsive because insulin treatment increased the surface-exposed HA epitope in both cases (Figure 3B).
In contrast, when cells expressing FA HA-GLUT4-GFP were treated with CHI, insulin-mediated FA HA-GLUT4 GFP redistribution was significantly impaired with no effect on basal levels of GLUT4 on the cell surface (Figure 3C). This finding suggested that CHI also impaired GLUT4 traffic independent of GLUT4 internalization. Importantly, because Rab5 S34N overexpression, but not FA HA-GLUT4-GFP mutant, protected against CHI-mediated loss of insulin-dependent GLUT4 redistribution, we hypothesized that CHI treatment altered GLUT4 traffic downstream of both internalization and early endosome sorting.
The TE499LE501Y motif of GLUT4 has been demonstrated to regulate the sorting of GLUT4 into the specialized, TfR-deficient IRSVs (78, 79). A loss-of-function mutation within this motif, TA499LA501Y (EEAA) retained GLUT4 within an insulin-responsive, TfR-positive ERC and impaired formation of the IRSVs (35). When we expressed the EEAA mutant with Rab5 S34N, basal redistribution to the cell surface increased, however, there was no change in the insulin response (Figure 3B), consistent with Rab5 S34N slowing endosomal traffic upstream of the impaired sorting of EEAA HA-GLUT4-GFP into IRSVs, but allowing entry into an insulin-responsive ERC. Next, we investigated whether the altered EEAA trafficking altered CHI-dependent sorting. When the EEAA HA-GLUT4-GFP was expressed in combination with CHI, the insulin-mediated redistribution was significantly impaired without further altering the basal levels of GLUT4 on the cell surface (Figure 3D). This finding supports that CHI altered GLUT4 traffic at a step that is independent (or downstream) of sorting into the IRSVs.
Expression of the double FA/EEAA mutant significantly increased basal fusion with the cell surface and significantly reduced the insulin response (Figure 3B), and coexpression of the double mutant with Rab5 S34N increased both basal and insulin-dependent appearance of the reporter on the cell surface (Figure 3B). More importantly, even though the insulin response was blunted, CHI treatment of cells expressing the double FA/EEAA mutant had a further reduction in insulin response (Figure 3E). Taken together these data suggest that CHI-dependent sorting is independent (or downstream) of both the IRSVs and the insulin-responsive ERC.
Rab5 S34N overexpression and chronic exposure to insulin alter the GLUT4 recycling pool size
To visualize alterations in the distribution of GLUT4 within endosomal compartments, we used compartmental ablation of the TfR-positive ERC in combination with acute insulin stimulation, CHI, or Rab5 S34N overexpression (23, 35). In this assay, live cells expressed WT HA-GLUT4-GFP and were acutely treated with 170 nM insulin for 30 minutes to recruit HA-GLUT4-GFP to the plasma membrane. After the acute insulin stimulation, the cells were bathed in media containing anti-HA antibody in the presence or absence of HRP-labeled transferrin (HRP-Tf) as the cells returned to basal. This allowed us to label the pool of GLUT4 molecules that were able to fuse, or equilibrate, with the cell surface in response to insulin (total pool). Then the cells were restimulated without or with 100 nM insulin for 30 minutes. Next, the cells were subjected to chemical ablation, rendering HA-GLUT4-GFP undetectable due to cross-linking of HRP-TfR with HA-GLUT4-GFP within a shared lumen. The residual labeled HA-GLUT4-GFP was a measure of the internalized GLUT4 sorted into non-TfR-containing, insulin-responsive compartments, presumably the IRSVs (TfR-neg pool).
To make sure that our strategy of blocking the cell surface worked, we performed a control experiment in which we blocked the surface-exposed HA antibody-labeled HA-GLUT4-GFP in fixed, nonpermeabilized cells with unlabeled secondary antibody followed by refixing and staining with Alexa647-conjugated secondary antibody (Figure 4Ac). This demonstrated that we could effectively block the cell surface. In a parallel experiment, the surface-blocked cells were then refixed and permeabilized to label the internal HA antibody from the total pool or the TfR-negative pool of HA-GLUT4-GFP-containing vesicles (Figure 4Bh). In basal cells, the total pool appears as a punctate, perinuclear compartment (Figure 4Be). In contrast, acute insulin restimulation resulted in the accumulation of GLUT4-containing, TfR-negative vesicles near the edges of the cell (Figure 4Ah, indicated by arrows). Consistently, quantification of the internal A647-to-GFP fluorescence ratio revealed a 2-fold significant expansion of the non-TfR-containing compartments (Figure 4C). Similarly, a 2.2-fold increase in the total recycling pool was also observed. Because no additional antibody was added to the cells upon restimulation, the expansion of the pool was not due to the additional uptake of the antibody. These findings indicate that acute insulin restimulation facilitates a change in how GLUT4 vesicles were distributed in the cell that altered the fluorescence measurements. For this reason, subsequent experiments tested alterations in size and appearance of the recycling compartment in basal cells only.
Figure 4.

GLUT4 recyclable pool is reduced by CHI. 3T3-L1 cells transfected with WT HA-GLUT4-GFP (A–E) were treated without (A–C) or with CHI (D and E). After 20 hours, cells were stimulated with 170 nM insulin for 30 minutes and then returned to basal for 4 hours in the presence of α-HA.11 antibody without or with HRP-Tf. Cells were restimulated without (panels Ba, Bb, Be, Bf, Bi, Bj, and D) or with (panels A, Bc, Bd, Bg, Bh, Bk, and Bl) 100 nM insulin for 30 minutes. HRP-Tf-containing compartments were ablated and then fixed as described in Materials and Methods. Surface-bound α-HA.11 antibody was blocked with a saturating concentration of unlabeled secondary α-mouse IgG antibody, and then cells were refixed, permeabilized, and stained with Alexa-647-conjugated goat α-mouse antibody to label internalized α-HA.11 antibody (see Materials and Methods). The efficacy of blocking cell surface HA with unlabeled secondary antibody is demonstrated in panel A using insulin-restimulated cells. B and D, Images are representative maximum-intensity projections of the z-axis. White arrows indicate nonablated HA-GLUT4-GFP-containing peripheral endocytic vesicles in insulin-restimulated cells (panel Bh). C and E, HA-epitope staining (A647) to total (GFP) ratio is a measure of the total HA-GLUT4-GFP recycling pool in the absence of HRP-Tf (panels Bi, Bk, Di, and Dk) or a measure of the recycling HA-GLUT4-GFP pool in TfR-negative compartments in the presence of HRP-Tf (panels Bj, Bl, Dj, and Dl). Mean internalized α-HA.11 (A647)-to-total HA-GLUT4-GFP expression (GFP) for 30–40 cells was normalized to nonablated basal control and reported as mean and SE. All data were analyzed using a Student's t test. *, P < .05 for comparisons indicated by brackets; #, P < .05 for total vs TfR negative.
The observed changes in insulin-dependent GLUT4 redistribution brought about by CHI could be due to alterations in trafficking to one or both of the insulin-responsive compartments. To begin testing this prediction, we monitored the colocalization of the recyclable pool of HA-GLUT4-GFP in basal cells that had been treated previously without or with CHI. Consistent with our hypothesis that CHI diverted GLUT4 into noninsulin-dependent recycling compartments, the total recycling pool size prior to compartmental ablation was significantly reduced after CHI treatment (Figure 4, D and E). Importantly, the GLUT4 pool residing in TfR-negative compartments (after ablation) remained unchanged, suggesting that the loss in total recycling GLUT4 pool was due to altered traffic away from TfR-positive compartments. These findings support a hypothesis in which CHI impaired insulin-mediated GLUT4 redistribution by diverting GLUT4 into a nonrecycling retention compartment.
In contrast to CHI treatment, Rab5 S34N overexpression significantly altered the size of the TfR-negative GLUT4 recycling pool with minimal effect on the total recycling pool size (Figure 5, A and B). Interestingly, we observed that the nonablated, TfR-negative GLUT4 pool resides at the cell periphery (Figure 5Ah, indicated by arrows), a morphology similar to that of acute insulin. These findings suggest that Rab5 S34N may slow the internal sorting of GLUT4-containing endocytic vesicles. Furthermore, the fact that Rab5 S34N increased basal levels of GLUT4 on the cell surface suggests that the accumulation of peripheral vesicles, both with Rab5 S34N and during acute insulin stimulation, may promote rapid recycling to the plasma membrane to help maintain elevated steady-state GLUT4 redistribution.
Figure 5.
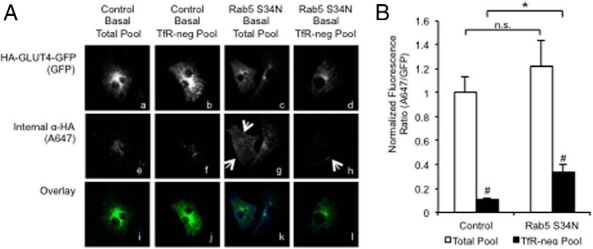
Rab5S34N reduces GLUT4 colocalization with TfR. 3T3-L1 cells transfected with WT HA-GLUT4-GFP with or without pBI Rab5S34N-myc/pTetOff. After 20 hours, cells were stimulated with 170 nM insulin for 30 minutes and then returned to basal for 4 hours in the presence of α-HA.11 antibody without or with Tf-HRP. Tf-HRP-containing compartments were ablated then fixed as described in Materials and Methods. Surface-bound α-HA.11 antibody was blocked with a saturating concentration of unlabeled secondary α-mouse IgG antibody, and then cells were refixed and permeabilized (see Materials and Methods). The efficacy of blocking cell surface HA with unlabeled secondary antibody is demonstrated in panel A. This control was carried out insulin-stimulated 3T3-L1 cells transfected with HA-GLUT4-GFP. Thirty minutes after insulin stimulation, cells were fixed and stained with HA antibody and then followed by an unlabeled goat-antimouse secondary antibody. The cells were washed, refixed, permeabilized, and then stained with Alexa-647-conjugated goat-antimouse secondary antibody to confirm complete blockage of surface HA. This control experiment was performed with each compartmental ablation experiment to verify blocking of surface HA. Internalized, nonablated α-HA.11 antibody was detected using Alexa647 (A647) secondary α-mouse IgG antibody. Images are representative maximum intensity projections of the z-axis. White arrows indicate nonablated HA-GLUT4-GFP-containing peripheral endocytic vesicles (panels Ag and Ah). B, HA epitope staining (A647) to total (GFP) is a measure of the HA-GLUT4-GFP recycling pool in the absence of Tf-HRP or a measure of the recycling HA-GLUT4-GFP in TfR− compartments in the presence of Tf-HRP. Mean internalized α-HA.11 (A647)-to-total HA-GLUT4-GFP expression (GFP) for 30–40 cells was normalized to nonablated basal control and reported as mean and SE. All data were analyzed using a Student's t test. *, P < .05 for comparisons indicated by brackets; #, P < .05 for total pool vs TfR-negative.
Acute insulin stimulation down-regulates Rab5-GTP loading
Given the similarities in the morphological distribution of intracellular GLUT4 during both acute insulin stimulation and basal Rab5 S34N overexpression, we hypothesized that acute insulin may reduce Rab5 activity to slow the intracellular traffic of internalized GLUT4 to promote rapid recycling to the plasma membrane. To begin testing this hypothesis, we coexpressed a wild-type Rab5-GFP fusion protein with an HA-GLUT4-mCherry reporter to assess distribution of these reporters. Both fusion proteins were each localized in the perinuclear region of the cells (Figure 6Ad). When the HA-GLUT4 mCherry was coexpressed with a Rab5 S34N-GFP fusion protein, the morphology of the labeled vesicles was altered. The stained vesicles appeared smaller, and there was greater staining at the edges of the cell as indicated by the white arrows (Figure 6Ah). These data are consistent with the formation of a basal GLUT4-containing recycling compartment in those cells containing a block in Rab5 function.
Figure 6.

Acute insulin stimulation promotes Rab5 activity-dependent formation of rapid recycling peripheral GLUT4 pool. A, Colocalization of HA-GLUT4-mCherry and GFP-Rab5 WT (panels Aa–Ad) and GFP-Rab5 S34N mutant (panels Ae–Ah) were analyzed by confocal microscopy. The arrows indicate the accumulation of small HA-GLUT4-mCherry vesicles at the margins of the cell (panel Ah). B–E, 3T3-L1 adipocytes expressing Rab5-GTP WT were acutely stimulated with insulin (panels B and C) or treated with CHI followed by restimulation with 10 nM insulin for 30 minutes (panels D and E), and then Rab5-GTP was immunoprecipitated from whole-cell lysates as described in Materials and Methods using a Rab5-GTP-specific antibody. Immunoprecipitated Rab5-GTP was analyzed by immunoblot using antibody against Rab5 as described in Materials and Methods. Representative images from n = 3 are shown. Quantification of Rab5-GTP was reported as mean and SE. All data were analyzed using a Student's t test. *, P < .05. Ins, insulin; IP, immunoprecipitation; WCL, whole-cell lysate.
The peripheral distribution of GLUT4 vesicles in basal cells that expressed Rab5 S34N resembled the peripheral distribution of GLUT4 vesicles in acute insulin-treated cells. We hypothesized that acute insulin treatment may lead to peripheral distribution by reducing Rab5-GTP loading. To test this, we used a Rab5-GTP-specific immunoprecipitation assay. Consistent with our hypothesis, we found that acute insulin stimulation significantly reduced Rab5-GTP loading (Figure 6, B and C).
Given this observation and that Rab5 S34N expression protected against CHI-mediated loss of acute insulin-stimulated GLUT4 redistribution, we further hypothesized that the insulin-dependent reduction in Rab5-GTP did not occur in CHI-treated cells. This was supported by the finding that acute restimulation with insulin did not significantly reduce Rab5-GTP loading in CHI-treated cells (Figure 6, D and E). In conclusion, our findings suggest that CHI impairs GLUT4 redistribution downstream of Rab5 activity-dependent early endosome sorting.
Discussion
Chronic hyperinsulinemia is an early event in the development of insulin resistance that often precedes the development of fasting hyperglycemia (14, 16, 26). How this leads to insulin resistance is not clearly understood. It has been hypothesized that chronic insulin exposure impairs GLUT4 exocytosis by reducing insulin signaling, altering intracellular GLUT4 traffic, or both. Identifying the specific target(s) that contribute to insulin-resistant GLUT4 redistribution has been difficult (24, 28, 29, 31, 32). It has been established that insulin-dependent GLUT4 redistribution requires insulin signaling to and downstream of Akt. Although the loss of Akt phosphorylation is a well-established effect of chronic insulin stimulation, Hoehn et al (29) in 2008 demonstrated that upstream signaling between insulin receptor substrate-1 and Akt activation was not responsible for insulin resistant glucose transport. Furthermore, the pathway from Akt to glucose transport is a complex and highly branched one, affecting both transcriptional and posttranscriptional processes, and chronic insulin treatment reportedly affects these pathways differently (80). For example, CHI treatment of 3T3-L1 adipocytes was shown to impair insulin-mediated GLUT4 translocation but not Forkhead box O1 nuclear exclusion (32). Using TIRF microscopy, Xiong et al (81) demonstrated that this selective insulin-resistant signaling impaired a discrete GLUT4 vesicle trafficking step near the plasma membrane resulting in impaired GLUT4 vesicle accumulation in the evanescent field. In contrast, Lizunov et al (82) recently demonstrated that live human adipocytes from insulin-resistant donors have deficient vesicle tethering and fusion. Although both studies led to decreased fusion, the mechanisms seem different. This could be accounted for by differences in the source of the insulin-resistant adipocytes or due to experimental differences between the fixed cells (Xiong et al) and the live cells (Lizunov et al). Consistent between these studies of insulin-resistant adipocytes is that steady-state levels of GLUT4 fused with the plasma membrane were diminished after acute insulin stimulation. It is possible in both cases that impaired GLUT4 fusion is a result of altered GLUT4 sorting into internal compartments with a reduced insulin sensitivity, a model consistent with our current data.
Zeigerer et al (38) described two insulin-responsive sorting steps, one that sorts from the endosome into the insulin-responsive compartment and one that sorts from the insulin-responsive compartment to the cell surface. The latter step appears to be regulated by AS160 and Rab10 under the control of insulin (54). Importantly, TBC1D4/AS160 is currently the only Rab GAP identified to regulate GLUT4 traffic downstream of Akt signaling, and, although Akt phosphorylation is reduced, the signal amplification from Akt to TBC1D4/AS160 phosphorylation is sufficient to achieve similar pAS160 levels after CHI exposure in our system (Figure 1). Therefore, we concluded that TBC1D4/AS160 phosphorylation after CHI should be sufficient to mediate GLUT4 exocytosis. In further support, GLUT4 exhibited normal plasma membrane insertion after CHI treatment of Rab5 S34N-expressing cells (Figure 1). These findings support a model in which the impaired steady-state GLUT4 redistribution is due, at least in part, to alterations in intracellular GLUT4 traffic, specifically the endocytic pathway of GLUT4.
Herein we present evidence supporting altered endosomal sorting as a mechanism for the instigation of insulin resistance. Importantly, we prevented insulin-resistant GLUT4 translocation after the chronic exposure to insulin by blocking Rab5 function with overexpression of a dominant-negative Rab5 S34N mutant cDNA (Figure 2C). In addition, by using two, well-characterized GLUT4 sorting mutants, we demonstrated that the CHI-dependent alteration did not simply block sorting into a specific insulin responsive storage compartment. The fact that both of these mutations, and a double mutant, were equally affected by CHI indicates that GLUT4 traffic was altered by sorting GLUT4 into a non-insulin-responsive compartment at the expense of each of the two insulin-responsive compartments (Figure 3). This finding was further supported by our observation that the size of the pool of GLUT4 capable of equilibrating with the cell surface was significantly reduced by chronic insulin exposure (Figure 4).
We used compartmental ablation of the TfR-positive compartments to visualize how the insulin-responsive GLUT4 retention compartments were compromised as a result of CHI treatment. This allowed us to morphologically identify an acute insulin-dependent accumulation of internalized GLUT4 in TfR-negative peripheral endosomes, which may contribute to the observed expansion of the GLUT4 recycling pool size (Figure 4) (38, 39). Importantly, these endosomes were not present after CHI exposure and were dependent on Rab5 inactivation. These data suggest that GLUT4 is internalized and resides separately from TfR until entering the early endosome, a finding supported by other studies showing similar separation of internalized membrane proteins (70). Furthermore, the accumulation of peripheral vesicles after both acute insulin stimulation and Rab5S34N overexpression is consistent with previously reported effects of inhibiting Rab5 function on early endosome formation (74). These findings, in combination with our Rab5-GTP pull-down assay, suggest that acute insulin stimulation temporally inhibits Rab5 activity to promote the accumulation and possibly the rapid recycling of GLUT4 at the periphery to help facilitate and maintain elevated steady-state GLUT4 redistribution.
In summary, this study used a combination approach of altered Rab5 function and GLUT4 trafficking mutants to begin elucidating the effects on GLUT4 sorting and insulin-mediated redistribution. This approach demonstrated that chronic insulin exposure altered GLUT4 endosomal sorting in addition to its previously established effects on insulin signaling and GLUT4 exocytosis. Our findings are consistent with the existence of a non-insulin-responsive endosomal compartment that can be occupied by GLUT4. Although the characterization of this compartment has yet to be determined, the compartmental ablation data suggest that the insulin-insensitive compartment is a subset of TfR-positive endosomes. Lastly, our study supports previous findings that acute insulin temporally inhibits Rab5 function to promote GLUT4 redistribution and further provides a potential mechanism in which inhibiting fusion at the early endosome promotes rapid recycling, possibly facilitated by activated membrane fusion machinery, to establish and maintain steady state GLUT4 redistribution during acute insulin stimulation.
Acknowledgments
This work was supported by Grant DK081545 from the National Institutes of Health and Grant HR09-075 from the Oklahoma Center for the Advancement of Science and Technology (to A.L.O.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CHI
- chronic hyperinsulin
- Dox
- doxycycline
- ERC
- endosome recycling center
- FA
- loss-of-function mutation, F5A
- FBS
- fetal bovine serum
- GFP
- green fluorescent protein
- GLUT4
- glucose transporter 4
- HA
- hemagglutinin
- HRP
- horseradish peroxidase
- IRSV
- insulin-responsive storage vesicle
- mCherry
- red fluorescent protein
- NP-40
- Nonidet P-40
- siRNA
- small interfering RNA
- Tf
- transferrin
- TfR
- Tf receptor
- TGN
- trans-Golgi network
- WT
- wild type.
References
- 1. Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255:4758–4762 [PubMed] [Google Scholar]
- 2. Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci USA. 1980;77:2542–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. James DE, Brown R, Navarro J, Pilch PF. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature. 1988;333:183–185 [DOI] [PubMed] [Google Scholar]
- 4. Birnbaum MJ. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell. 1989;57:305–315 [DOI] [PubMed] [Google Scholar]
- 5. Charron MJ, Brosius FC, 3rd, Alper SL, Lodish HF. A glucose transport protein expressed predominately in insulin-responsive tissues. Proc Natl Acad Sci USA. 1989;86:2535–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olson AL, Liu ML, Moye-Rowley WS, Buse JB, Bell GI, Pessin JE. Hormonal/metabolic regulation of the human GLUT4/muscle-fat facilitative glucose transporter gene in transgenic mice. J Biol Chem. 1993;268:9839–9846 [PubMed] [Google Scholar]
- 7. Govers R, Coster AC, James DE. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol Cell Biol. 2004;24:6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karlsson HK, Chibalin AV, Koistinen HA, et al. Kinetics of GLUT4 trafficking in rat and human skeletal muscle. Diabetes. 2009;58:847–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rossetti L, Stenbit AE, Chen W, et al. Peripheral but not hepatic insulin resistance in mice with one disrupted allele of the glucose transporter type 4 (GLUT4) gene. J Clin Invest. 1997;100:1831–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733 [DOI] [PubMed] [Google Scholar]
- 11. Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rothman DL, Magnusson I, Cline G, et al. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci USA. 1995;92:983–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith U. Impaired (‘diabetic’) insulin signaling and action occur in fat cells long before glucose intolerance–is insulin resistance initiated in the adipose tissue? Int J Obes Relat Metab Disord. 2002;26:897–904 [DOI] [PubMed] [Google Scholar]
- 14. Brozinick JT, Jr, McCoid SC, Reynolds TH, et al. GLUT4 overexpression in db/db mice dose-dependently ameliorates diabetes but is not a lifelong cure. Diabetes. 2001;50:593–600 [DOI] [PubMed] [Google Scholar]
- 15. Koranyi L, James D, Mueckler M, Permutt MA. Glucose transporter levels in spontaneously obese (db/db) insulin-resistant mice. J Clin Invest. 1990;85:962–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Atkinson BJ, Griesel BA, King CD, Josey MA, Olson AL. Moderate GLUT4 overexpression improves insulin sensitivity and fasting triglyceridemia in high fat fed transgenic mice. Diabetes. 2013;62(7):2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sparling DP, Griesel BA, Weems J, Olson AL. GLUT4 enhancer factor (GEF) interacts with MEF2A and HDAC5 to regulate the GLUT4 promoter in adipocytes. J Biol Chem. 2008;283:7429–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weems J, Olson AL. Class II histone deacetylases limit GLUT4 gene expression during adipocyte differentiation. J Biol Chem. 2011;286:460–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garvey WT, Maianu L, Hancock JA, Golichowski AM, Baron A. Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 1992;41:465–475 [DOI] [PubMed] [Google Scholar]
- 20. Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garvey WT, Maianu L, Zhu JH, Hancock JA, Golichowski AM. Multiple defects in the adipocyte glucose transport system cause cellular insulin resistance in gestational diabetes. Heterogeneity in the number and a novel abnormality in subcellular localization of GLUT4 glucose transporters. Diabetes. 1993;42:1773–1785 [DOI] [PubMed] [Google Scholar]
- 22. Maianu L, Keller SR, Garvey WT. Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J Clin Endocrinol Metab. 2001;86:5450–5456 [DOI] [PubMed] [Google Scholar]
- 23. Livingstone C, James DE, Rice JE, Hanpeter D, Gould GW. Compartment ablation analysis of the insulin-responsive glucose transporter (GLUT4) in 3T3-L1 adipocytes. Biochem J. 1996;315(Pt 2):487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sargeant RJ, Paquet MR. Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem J. 1993;290(Pt 3):913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maier VH, Gould GW. Long-term insulin treatment of 3T3-L1 adipocytes results in mis-targeting of GLUT4: implications for insulin-stimulated glucose transport. Diabetologia. 2000;43:1273–1281 [DOI] [PubMed] [Google Scholar]
- 26. Hatori M, Vollmers C, Zarrinpar A, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dubuc PU. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism. 1976;25:1567–1574 [DOI] [PubMed] [Google Scholar]
- 28. Haruta T, Uno T, Kawahara J, et al. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794 [DOI] [PubMed] [Google Scholar]
- 29. Hoehn KL, Hohnen-Behrens C, Cederberg A, et al. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science. 1996;271:665–668 [DOI] [PubMed] [Google Scholar]
- 31. Clark SF, Molero JC, James DE. Release of insulin receptor substrate proteins from an intracellular complex coincides with the development of insulin resistance. J Biol Chem. 2000;275:3819–3826 [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez E, Flier E, Molle D, Accili D, McGraw TE. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc Natl Acad Sci USA. 2011;108:10162–10167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277 [DOI] [PubMed] [Google Scholar]
- 34. Coster AC, Govers R, James DE. Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic. 2004;5:763–771 [DOI] [PubMed] [Google Scholar]
- 35. Blot V, McGraw TE. Molecular mechanisms controlling GLUT4 intracellular retention. Mol Biol Cell. 2008;19:3477–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palacios S, Lalioti V, Martinez-Arca S, Chattopadhyay S, Sandoval IV. Recycling of the insulin-sensitive glucose transporter GLUT4. Access of surface internalized GLUT4 molecules to the perinuclear storage compartment is mediated by the Phe5-Gln6-Gln7-Ile8 motif. J Biol Chem. 2001;276:3371–3383 [DOI] [PubMed] [Google Scholar]
- 37. Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol Biol Cell. 2001;12:3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeigerer A, Lampson MA, Karylowski O, et al. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol Biol Cell. 2002;13:2421–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin OJ, Lee A, McGraw TE. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J Biol Chem. 2006;281:484–490 [DOI] [PubMed] [Google Scholar]
- 40. El-Jack AK, Kandror KV, Pilch PF. The formation of an insulin-responsive vesicular cargo compartment is an early event in 3T3-L1 adipocyte differentiation. Mol Biol Cell. 1999;10:1581–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Millar CA, Meerloo T, Martin S, et al. Adipsin and the glucose transporter GLUT4 traffic to the cell surface via independent pathways in adipocytes. Traffic. 2000;1:141–151 [DOI] [PubMed] [Google Scholar]
- 42. Habtemichael EN, Brewer PD, Romenskaia I, Mastick CC. Kinetic evidence that Glut4 follows different endocytic pathways than the receptors for transferrin and α-2-macroglobulin. J Biol Chem. 2011;286(12):10115–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jhun BH, Rampal AL, Liu H, Lachaal M, Jung CY. Effects of insulin on steady state kinetics of GLUT4 subcellular distribution in rat adipocytes. Evidence of constitutive GLUT4 recycling. J Biol Chem. 1992;267:17710–17715 [PubMed] [Google Scholar]
- 44. Xu Y, Rubin BR, Orme CM, et al. Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J Cell Biol. 2011;193:643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Welsh GI, Hers I, Berwick DC, et al. Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans. 2005;33:346–349 [DOI] [PubMed] [Google Scholar]
- 47. Miinea CP, Sano H, Kane S, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kane S, Sano H, Liu SC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118 [DOI] [PubMed] [Google Scholar]
- 49. Larance M, Ramm G, Stockli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813 [DOI] [PubMed] [Google Scholar]
- 50. Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J. 2007;403:353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sano H, Eguez L, Teruel MN, et al. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5:293–303 [DOI] [PubMed] [Google Scholar]
- 52. Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411:89–95 [DOI] [PubMed] [Google Scholar]
- 53. Chen Y, Wang Y, Zhang J, et al. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol. 2012;198:545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol Biol Cell. 2013;24:2544–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zaid H, Antonescu CN, Randhawa VK, Klip A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J. 2008;413:201–215 [DOI] [PubMed] [Google Scholar]
- 56. Bao S, Zhu J, Garvey WT. Cloning of Rab GTPases expressed in human skeletal muscle: studies in insulin-resistant subjects. Horm Metab Res. 1998;30:656–662 [DOI] [PubMed] [Google Scholar]
- 57. Govers R, James DE, Coster AC. High-throughput analysis of the dynamics of recycling cell surface proteins. Methods Mol Biol. 2008;440:129–146 [DOI] [PubMed] [Google Scholar]
- 58. Huang J, Imamura T, Olefsky JM. Insulin can regulate GLUT4 internalization by signaling to Rab5 and the motor protein dynein. Proc Natl Acad Sci USA. 2001;98:13084–13089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cormont M, Van Obberghen E, Zerial M, Le Marchand-Brustel Y. Insulin induces a change in Rab5 subcellular localization in adipocytes independently of phosphatidylinositol 3-kinase activation. Endocrinology. 1996;137:3408–3415 [DOI] [PubMed] [Google Scholar]
- 60. Lodhi IJ, Bridges D, Chiang SH, et al. Insulin stimulates phosphatidylinositol 3-phosphate production via the activation of Rab5. Mol Biol Cell. 2008;19:2718–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Su X, Lodhi IJ, Saltiel AR, Stahl PD. Insulin-stimulated Interaction between insulin receptor substrate 1 and p85α and activation of protein kinase B/Akt require Rab5. J Biol Chem. 2006;281:27982–27990 [DOI] [PubMed] [Google Scholar]
- 62. Dawson K, Aviles-Hernandez A, Cushman SW, Malide D. Insulin-regulated trafficking of dual-labeled glucose transporter 4 in primary rat adipose cells. Biochem Biophys Res Commun. 2001;287:445–454 [DOI] [PubMed] [Google Scholar]
- 63. Liu J, Lamb D, Chou MM, Liu YJ, Li G. Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol Biol Cell. 2007;18:1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eyster CA, Duggins QS, Olson AL. Expression of constitutively active Akt/protein kinase B signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J Biol Chem. 2005;280:17978–17985 [DOI] [PubMed] [Google Scholar]
- 65. Bowen JR, Hwang D, Bai X, Roy D, Spiliotis ET. Septin GTPases spatially guide microtubule organization and plus end dynamics in polarizing epithelia. J Cell Biol. 2011;194:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell. 2006;17:4484–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602 [DOI] [PubMed] [Google Scholar]
- 68. Al-Hasani H, Kunamneni RK, Dawson K, Hinck CS, Muller-Wieland D, Cushman SW. Roles of the N- and C-termini of GLUT4 in endocytosis. J Cell Sci. 2002;115:131–140 [DOI] [PubMed] [Google Scholar]
- 69. Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25:99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leonard D, Hayakawa A, Lawe D, et al. Sorting of EGF and transferrin at the plasma membrane and by cargo-specific signaling to EEA1-enriched endosomes. J Cell Sci. 2008;121:3445–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749 [DOI] [PubMed] [Google Scholar]
- 72. Li G, Stahl PD. Post-translational processing and membrane association of the two early endosome-associated rab GTP-binding proteins (rab4 and rab5). Arch Biochem Biophys. 1993;304:471–478 [DOI] [PubMed] [Google Scholar]
- 73. Flinn RJ, Backer JM. mTORC1 signals from late endosomes: taking a TOR of the endocytic system. Cell Cycle. 2010;9:1869–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wegner CS, Malerod L, Pedersen NM, et al. Ultrastructural characterization of giant endosomes induced by GTPase-deficient Rab5. Histochem Cell Biol. 2010;133:41–55 [DOI] [PubMed] [Google Scholar]
- 75. Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Williams D, Pessin JE. Mapping of R-SNARE function at distinct intracellular GLUT4 trafficking steps in adipocytes. J Cell Biol. 2008;180:375–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bernhardt U, Carlotti F, Hoeben RC, Joost HG, Al-Hasani H. A dual role of the N-terminal FQQI motif in GLUT4 trafficking. Biol Chem. 2009;390:883–892 [DOI] [PubMed] [Google Scholar]
- 78. Shewan AM, Marsh BJ, Melvin DR, Martin S, Gould GW, James DE. The cytosolic C-terminus of the glucose transporter GLUT4 contains an acidic cluster endosomal targeting motif distal to the dileucine signal. Biochem J. 2000;350(Pt 1):99–107 [PMC free article] [PubMed] [Google Scholar]
- 79. Shewan AM, van Dam EM, Martin S, et al. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tan SX, Ng Y, Meoli CC, et al. Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J Biol Chem. 2012;287:6128–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xiong W, Jordens I, Gonzalez E, McGraw TE. GLUT4 is sorted to vesicles whose accumulation beneath and insertion into the plasma membrane are differentially regulated by insulin and selectively affected by insulin resistance. Mol Biol Cell. 2010;21:1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lizunov VA, Lee JP, Skarulis MC, Zimmerberg J, Cushman SW, Stenkula KG. Impaired tethering and fusion of GLUT4 vesicles in insulin-resistant human adipose cells. Diabetes. 2013;62(9):3114–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]