

Abstract
Type 1 diabetes (T1D) results from autoimmune destruction of islet β-cells, but the underlying mechanisms that contribute to this process are incompletely understood, especially the role of lipid signals generated by β-cells. Proinflammatory cytokines induce ER stress in β-cells and we previously found that the Ca2+-independent phospholipase A2β (iPLA2β) participates in ER stress-induced β-cell apoptosis. In view of reports of elevated iPLA2β in T1D, we examined if iPLA2β participates in cytokine-mediated islet β-cell apoptosis. We find that the proinflammatory cytokine combination IL-1β+IFNγ, induces: a) ER stress, mSREBP-1, and iPLA2β, b) lysophosphatidylcholine (LPC) generation, c) neutral sphingomyelinase-2 (NSMase2), d) ceramide accumulation, e) mitochondrial membrane decompensation, f) caspase-3 activation, and g) β-cell apoptosis. The presence of a sterol regulatory element in the iPLA2β gene raises the possibility that activation of SREBP-1 after proinflammatory cytokine exposure contributes to iPLA2β induction. The IL-1β+IFNγ-induced outcomes (b–g) are all inhibited by iPLA2β inactivation, suggesting that iPLA2β-derived lipid signals contribute to consequential islet β-cell death. Consistent with this possibility, ER stress and β-cell apoptosis induced by proinflammatory cytokines are exacerbated in islets from RIP-iPLA2β-Tg mice and blunted in islets from iPLA2β-KO mice. These observations suggest that iPLA2β-mediated events participate in amplifying β-cell apoptosis due to proinflammatory cytokines and also that iPLA2β activation may have a reciprocal impact on ER stress development. They raise the possibility that iPLA2β inhibition, leading to ameliorations in ER stress, apoptosis, and immune responses resulting from LPC-stimulated immune cell chemotaxis, may be beneficial in preserving β-cell mass and delaying/preventing T1D evolution.
Type 1 diabetes (T1D) results from a decrease in β-cell mass and function due to autoimmune destruction of β-cells (1), but the underlying mechanisms that cause β-cell apoptosis associated with T1D are not well understood. The destruction of β-cells can be mediated by many factors but cytokines produced by inflammatory cells are recognized to contribute to losses in β-cell function and viability by inducing β-cell necrosis and apoptosis during T1D evolution (2, 3). Hence, if the development of T1D is to be blunted or delayed, it is important to understand the mechanism(s) underlying proinflammatory cytokine-mediated β-cell loss.
In addition to extrinsic and intrinsic pathways, endoplasmic reticulum (ER) stress can cause apoptosis and is thought to give rise to several disease states, including diabetes (4). ER stress has now also been implicated in autoimmune destruction of β-cells in T1D (5). Because the secretory function of β-cells endows them with a highly developed ER (6), it is not unexpected that β-cells exhibit a heightened susceptibility to autoimmune-mediated ER stress (7). Whereas there is general agreement in the literature that cytokines induce ER stress in β-cells (8–13), it is still controversial if such induction contributes to β-cell apoptosis in T1D.
Our findings reveal that the group VIA Ca2+-independent phospholipase A2 (iPLA2β) participates in β-cell apoptosis due to ER stress (14–19). Furthermore, we found that this process involves triggering of the intrinsic apoptotic pathway by ceramides, generated via iPLA2β-mediated induction of neural sphingomyelinase-2 (NSMase2), and subsequent hydrolysis of sphingomyelins (15, 16). The iPLA2β (84–88 kDa) is part of a diverse family of PLA2 enzymes that catalyze hydrolysis of the sn-2 substituent from glycerophospholipid substrates to yield a free fatty acid and a 2-lysophospholipid in the absence of Ca2+, and is involved in a multitude of biological processes (20). The iPLA2β is expressed in many cells and in the islet, iPLA2β is predominantly localized in β-cells (19) and β-cell subcellular membranes are enriched in substrates for iPLA2β (21).
Recent reports reveal that diabetes is associated with iPLA2β induction. For instance, excessive superoxide production in human neutrophils, vascular smooth muscle hypercontractility in mice, and ischemia-induced conduction slowing in rat heart, that are all associated with diabetes, were attenuated by chemical inhibition or genetic ablation of iPLA2β (22–24). In view of these observations and reports of ER stress induction in β-cells by cytokines (8–13), we considered the possibility that inhibition of iPLA2β may be beneficial in protecting β-cells from death due to proinflammatory cytokines. We addressed this using islets and find that the combination of proinflammatory cytokines IL-1β+IFNγ, promotes ER stress, mSREBP-1 (mature form of sterol regulatory element binding protein-1), and induces iPLA2β, leading to iPLA2β-mediated lysophosphatidylcholine (LPC) generation, ceramide accumulation, mitochondrial decompensation, caspase-3 activation, and β-cell apoptosis. These findings suggest that lipids derived from activation of iPLA2β in β-cells play a role in autoimmune destruction of the β-cells during T1D evolution.
Materials and Methods
Materials
Human islets were obtained through the Islet Cell Resource Basic Science Islet Distribution Program, Juvenile Diabetes Research Foundation, and the University of Alabama at Birmingham Islet Resource Facility. The islets were isolated at various procurement centers from subjects with the following features: males, n = 6, age = 33.3 ± 4.5 years, body mass index = 25.3 ± 3.3, islet viability = 91 ± 1%, and islet purity = 82 ± 6%; females, n = 4, age = 39.7 ± 4.0 years, body mass index = 23.9 ± 1.5, islet viability = 92 ± 3%, and islet purity = 81 ± 7%. Causes of death were head trauma/intracerebral hemorrhage (50%), cardiovascular (20%), anoxia (10%), and stroke (20%). The islets were isolated from donors within 24 hours of death and delivered to us within the next 24 hours. Upon receipt, the islets were cultured 24 to 48 hours at 37°C under an atmosphere of 5%CO2/95% air prior to use. Breeders to generate wild-type (WT), iPLA2β-knockout(KO), and RIP-iPLA2β-Tg mice were generously provided by Dr John Turk (Washington University School of Medicine) and genotype of progeny was confirmed, as described (18).
Other materials were obtained from the following sources: insulin antibody (Abcam); rainbow molecular mass standards and enhanced chemiluminescence reagent (Amersham); SYBR Green PCR Kit (Applied Biosystems); lipid standards (Avanti Polar Lipids); bromoenol lactone suicide inhibitor of iPLA2β (S-BEL) (Cayman Chemicals); Coomassie reagent, SDS-PAGE supplies, and Triton X-100 (BioRad); cleaved casp-3 rabbit mAb, IRE1α (inositol-requiring enzyme 1), pPERK (phosphorylated form of ER-stress transducer pancreatic ER kinase) antibody (Cell Signaling); ΔΨ detection kit (Cell Tech Inc); paraformaldehyde (Electron Microscopy Sciences); DNase-free RNase A (Gentra Systems Inc); oligonucleotides (Integrated DNA Technologies); Accumax (Innovative Cell Technologies, Inc.); Alexa Fluor 594 goat antirabbit, SuperScriptII and SYBR Green PCR kits, and SYBR Gold nucleic acid gel stain (Invitrogen); fluorescein (FITC)-conjugated donkey anti-guinea pig IgG (Jackson ImmunoResearch Laboratories, Inc); Immobilin-P PVDF membrane (Millipore); DiOC6(3), Slow Fade light antifade kit (Molecular Probes); Effectene and RNeasy kit (Qiagen); TUNEL kit (Roche Diagnostics Corporation); 1° antibody for actin, activating transcription factor 6 (ATF6α), 78 kDa glucose-regulated protein (GRP78), iNOS, iPLA2β, mSREBP-1, and tubulin, and goat anti-guinea pig IgG-TR and rabbit antigoat IgG (Santa Cruz Biotechnology Inc); caspase-3 colorimetric assay kit, protease inhibitor cocktail, common reagents, and salts (Sigma Chemical Co); and Vectashield with DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories).
Culture and treatment of human islets
Each islet shipment was from a single donor (< 40-years-old) and contained 2000 to 3000 islets, which did not permit us to perform all presented analyses (requiring 500 islets/condition) with every shipment. Each donor islet preparation was therefore used for certain analyses that were repeated in subsequent shipments to achieve a minimum of 3 replicates. Islet viability was verified after arrival and periodically glucose-stimulated insulin secretion was measured. Because of limited islet abundance and time constraints, glucose-stimulated insulin secretion was not performed with each preparation. In consideration of the variability between donor preparations, the effects of cytokine treatment in each experiment were compared against vehicle dimethyl sulfoxide (DMSO)-treated islets from the same donor. Because islets from different donors were not available at one time and the analyses required fresh islets, pooling of islet shipments was not feasible. Islet preparations of at least 80% purity and 90% viability were processed for experimentation.
Upon receipt, the islets were immediately cleaned of non-islet material under a microscope and cultured in CMRL 1066 (containing 10% fetal calf serum, 200 mmo/L glutamine, 1% of 100× penicillin/streptomycin, 25 mmo/L HEPES) at 37°C under an atmosphere of 5%CO2/95% air for up to 48 hours prior to treatment. Islets (500) were treated with either vehicle (DMSO, 1 μL/mL) alone or with IL-1β (100 U/mL) + IFNγ (300 U/mL) and cultured for up to 48 hours. Because of limited availability of human islets, our studies were confined to testing a combination of proinflammatory cytokines (IL-1β+IFNγ, designated CTK) that has previously been shown to be sufficient in impacting human islet β-cell function/survival and promoting insulitis in T1D (25–30). In some experiments, the islets were pretreated with inhibitors of iPLA2β (S-BEL, 10 μmol/L) for 30 minutes or NSMase2 (GW4869, 20 μmol/L) for 1 hour, prior to treatment with DMSO or CTK. Islets treated with DMSO only for the longest duration in a given protocol served as the controls.
Immunoblotting analyses
Islets were harvested at various times (0–48 h) after exposure to IL-1β+IFNγ and sonicated to obtain an islet homogenate. An aliquot (containing 30 μg protein) of homogenate was processed for immunoblotting analyses, as described (14, 17, 19). The targeted proteins and the (1°/2°) antibody concentrations were ATF6α, GRP78, IRE1α, iNOS, iPLA2β, pPERK, and SREBP-1 (1:1000/1:10,000). Tubulin in human islet and actin in mouse islet analyses were used as loading controls (1:5000/1:10,000). Immunoreactive bands were visualized by enhanced chemiluminescence.
iPLA2β enzymatic activity assay in islets
Cytosol was prepared from islets, protein concentration determined, and iPLA2β catalytic activity (in a 30 μg protein aliquot) was assayed and quantified, as described (17).
Immunostaining of islet sections
Islets were fixed in 10% formalin containing 5 μL of tissue marking dye prior to addition of low-melting Agarose (150 μL). The mixture was spun down quickly to settle the islets at one surface of the Agarose, which was then allowed to solidify. The islet-containing blocks were then processed and paraffin sections (10 μm) prepared for insulin and activated caspase-3 immunostaining, as described (19). DAPI (25 μL) was then added to stain nuclei, and the sections were sealed with coverslip using nail polish. Fluorescence was recorded using a Nikon Eclipse TE300 microscope and images were captured at 20× magnification.
Ceramide, sphingomyelin, and lysophosphatidylcholine (LPC) analyses by electrospray ionization mass spectrometry (ESI/MS/MS)
Islets were harvested, lipids extracted, as described (19), and internal standard (IS) not endogenous to β-cells [C8-ceramide (m/z 432) for ceramides, 14:0/14:0-glycerophosphocholine (m/z 684) for sphingomyelins, or 19:0-LPC (m/z = 544) for LPCs], added. Relative abundances of individual ceramide, sphingomyelin, and LPC molecular species, relative to the respective IS, were assessed by ESI/MS/MS and normalized to lipid phosphorous, as described (14–16, 19).
Quantitative RT-PCR (RT-qPCR)
Total RNA was isolated from human islets using RNeasy kit and cDNA was synthesized for RT-qPCR analyses of iPLA2β, NSMase2, and SPT1 (serine palmitoyl transferase), as described (19). The primers were designed based on known human sequences for iPLA2β (gi:52486250), NSMase2 (gi:92859617), SPT1 (gi:30474867), and internal control 18S (gi:337376). The sense/antisense primer sets were as follows: iPLA2β, gcaatgctcggtgcaacat/acaccccttctgagagaacttca; NSMase2, ggctgctgcctgctgaa/gcccttgaagtcccgagttt; SPT1, gcgcgctacttggagaaaga/tgttccaccgtgaccacaac; and 18S, gccgctagaggtgaaattcttg/cattcttggcaaatgctttcg.
Assessment of mitochondrial membrane potential (ΔΨ)
Islets were dispersed into cells by incubating them in equal volumes of PBS and Accumax reagent (2 h, 37°C) and ΔΨ was assessed, as described (19). Fluorescence images were captured by confocal microscopy and analyzed using ImageJ software, as described (http://sciencetechblog.com/2011/05/24/measuring-cell-fluorescence-using-imagej/) (31).
Indices of apoptosis
In situ detection of DNA cleavage by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
Islets were harvested and processed for TUNEL analyses and counterstained with DAPI (1 μg/mL) to identify nuclei, as described (19). Incidence of apoptosis was assessed under a fluorescence microscope using a FITC filter and cells with TUNEL-positive nuclei were considered apoptotic. DAPI staining was used to determine the total number of islet cells in a field. At least six fields per slide were used to calculate the percent of apoptotic islet cells.
Cleaved caspase-3 activity.
To obtain a quantitative measure of apoptosis in isolated islets, cleaved (activated) caspase-3 (aC3) activity was determined in islet cell lysates by a colorimetric assay based on aC3-catalyzed generation of p-nitroaniline (pNA) from Ac-DEVD-pNA substrate, as described (19). Absorbance of released pNA was read at 405 nm and normalized to sample protein.
Mouse islet isolation and treatment
Islets were isolated from WT, RIP-iPLA2β-TG (overexpressing iPLA2β in only the β-cells), and iPLA2β-deficient (KO) mice, as described (18). After an overnight equilibration, islets (500) were treated with either vehicle (DMSO, 1 μL/mL) alone or with IL-1β (100 U/mL)+IFNγ (300 U/mL) and cultured for 48 hours. The islets were then harvested and prepared for various analyses, as described (18, 19). All mouse studies were performed according to protocols approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Statistical analyses
Data were converted to mean ± SEM and the Student's t test was used to determine significant differences between groups (P < .05).
Results
IL-1β+IFNγ (CTK) induces ER stress factors and iNOS in human islets
Immunoblotting (Figure 1A, representative blots) and quantitative analyses (Figure 1B) of ER stress markers revealed increases in GRP78 and pPERK in islets exposed to IL-1β+IFNγ between 24 to 48 hours, relative to vehicle (DMSO)-treated islets. Consistent with responses to the cytokines (25), iNOS was induced between 24 to 48 hours by IL-1β+IFNγ (Figure 1, A and B). These findings suggest that proinflammatory cytokines induce ER stress in human islets. Because initial studies revealed that the incidence of β-cells apoptosis occurred between 24 to 48 hours, subsequent analyses were done up to 24 hours after IL-1β+IFNγ treatment. In addition to promoting ER stress, IL-1β+IFNγ induced iPLA2β in islets and this was associated with SREBP-1 activation (Figure 1, C and D), raising the possibility that CTK effects in β-cells are, in part, mediated by iPLA2β activation.
Figure 1.

IL-1β+IFNγ (CTK) induces ER stress factors, iPLA2β, and mSREBP-1 in human pancreatic islets. Islets (500/condition) were treated with either vehicle (DMSO) or IL-1β+IFNγ (CTK), harvested at 24 and 48 hours, and processed for immunoblotting analyses for GRP78, pPERK, iNOS, iPLA2β, and mSREBP-1. Tubulin was used as loading control. A and C, Analyses were repeated 3 times and representative blots are presented. B and D, Densitometry quantitation. (*Significantly different vs corresponding DMSO groups, P < .05; §vs other groups, P < .05; # and † vs corresponding DMSO groups, P < .01 and P < .05, respectively, n = 3.)
Pretreatment of islets with S-BEL inhibits IL-1β+IFNγ (CTK)-induced iPLA2β activity but not iPLA2β protein in islets
Because mSREBP-1 induces iPLA2β gene transcription in β-cells (14), we further examined the effects of IL-1β+IFNγ on islet iPLA2β induction. CTK exposure promoted a temporal increase in iPLA2β message that was 2-fold greater at 16 hours than control message level (Figure 2A) and reflected by higher iPLA2β activity (Figure 2B). Pretreatment of islets with S-BEL that has a greater specificity for β-isoform of iPLA2 (32), inhibited iPLA2β activity but had no significant effect on iPLA2β protein expression (Figures 2, C and D). Furthermore, S-BEL did not alter SREBP-1 activation suggesting that this event is upstream of iPLA2β.
Figure 2.

IL-1β+IFNγ (CTK) induces iPLA2β and mSREBP-1 in islets. Islets (500/condition, n = 3–5) were treated as in Figure 1 for 24 hours. A, Human islet iPLA2β mRNA was determined by RT-qPCR. (*Significantly different from DMSO, P < .05). B, iPLA2β specific activity (SA) was determined in cytosol of islets after treatment with vehicle or CTK ± S-BEL and presented as mean pmol/min/mg protein ± SEM: DMSO, 11 ± 0.5; S-BEL, 3 ± 3; CTK and CTK, 25 ± 6.5; CTK + S-BEL, 1.5 ± 1.5. (*†Significantly different from other groups, P < .05). C, iPLA2β and mSREBP-1 protein. Cytosol of human and mouse islets (n = 3 each) after treatment with vehicle or CTK ± S-BEL was processed for immunoblotting analyses and representative blots from mouse islets are presented (actin was used as loading control). D, Densitometry quantitation. (* and †Significantly different from corresponding DMSO and S-BEL groups, P < .005 and P < .05, respectively, n = 6.)
IL-1β+IFNγ (CTK)-promotes ceramide generation in human islets by an iPLA2β/neutral sphingomyelinase-2 (NSMase2)-dependent mechanism
Because we found that IL-1β+IFNγ induces ER stress, we examined the possibility that the proinflammatory cytokines also promote ceramide generation. As shown in Figure 3A, ESI/MS/MS analyses identified 16:0 (m/z 544), 18:0 (m/z 572), 20:0 (m/z 600), 22:0 (m/z 628), 24:1 (m/z 654), and 24:0 (m/z 656) as the fatty amide substituents of the major ceramide species endogenous to human islets. After exposure to CTK, ion intensity of each ceramide molecular species, relative to C8-ceramide IS (m/z 544), increased in comparison with vehicle (DMSO) treatment (Figure 3B). This is reflected by the higher ratio (indicated in parenthesis) of each ion, relative to IS, in the CTK spectrum. However, after treatment of islets with S-BEL, the relative intensity of all ceramide molecular species decreased (lower ratio of ion, relative to IS) back to control levels (Figure 3C). In addition, treatment of islets with the NSMase2 inhibitor, GW4869, prevented the increases in ceramide molecular species (Figure 3D). These findings suggest that proinflammatory cytokines induce ceramide generation via a mechanism involving iPLA2β and NSMase2-catalyzed sphingomyelin hydrolysis.
Figure 3.

IL-1β+IFNγ (CTK) promotes ceramide generation in human islets by an iPLA2β-mediated induction of NSMase2. Islets (500/condition) were treated as in Figure 1 for 24 hours and processed for ceramide analyses by ESI/MS/MS. Representative spectra from (3–5 replicates) are presented, where individual ceramide molecular species ions (m/z) and their (ratios), relative to internal standard (IS, 14:0/14:0-CM), are identified in each spectrum. A, DMSO vehicle. B, CTK. C, CTK + S-BEL (iPLA2β inhibitor). D, CTK + GW4869 (NSMase2 inhibitor). Quantitation of these analyses is presented in Figure 4.
IL-1β+IFNγ (CTK) induces NSMase2 (but not SPT1) and ceramide and LPC generation in human islets in an iPLA2β-dependent manner
To verify the involvement of NSMase2 in ceramide generation in islets after exposure to the proinflammatory cytokines, NSMase2 mRNA was determined. As illustrated in Figure 4A, NSMase2 message increased after CTK exposure. However, pretreatment of islets with S-BEL decreased both the basal and CTK-induced increases in NSMase2. In contrast, exposure to CTK did not increase mRNA of SPT1 that catalyzes the rate-limiting step in de novo synthesis of ceramides, and S-BEL was without effect (Supplemental Figure 1). These findings suggest that proinflammatory cytokines induce NSMase2 by an iPLA2β-dependent mechanism. Consistent with these findings, pretreatment of islets with S-BEL or GW4869 prevented CTK-induced increases in the ceramide pool (Figure 4B). Surprisingly, however, the pool of sphingomyelins was unaffected by either S-BEL or GW4869 (Figure 4C).
Figure 4.

IL-1β+IFNγ (CTK) induces neutral sphingomyelinase-2 (NSMase2), and ceramide and LPC generation by an iPLA2β-dependent manner in human islets. Islets (500/condition) were processed after 24 hours exposure to CTK for NSMase2 mRNA by RT-qPCR (A), and for ceramide (B), sphingomyelin (C), and LPC (D) molecular species ± S-BEL by ESI/MS/MS analyses. To determine the content of each species, standard curves were generated from a series of samples containing a fixed amount of internal standard (IS) and varied amounts of individual molecular species standards. The relative abundance of individual species, relative to IS, was measured by ESI-MS/MS scanning. Standard curves were then plotted as a ratio of individual species relative to the IS and a linear regression equation generated. From a given experimental condition, the ratio of a molecular species to the added IS was calculated and applied to the equation. The same amount of IS was added to each of the samples and the highest intensity ion (IS or a sample molecular species) was taken as 100%. The ratio of individual molecular species to IS was contained within the linear range of the standard curves. This value was then divided by the molecular species mass and normalized to lipid phosphorus content in the given sample to obtain amount of molecular species/nmol PO4 for each molecular species identified, as described (14–16, 19). These were then summed to obtain a pooled value for each lipid class. Basal ceramide, sphingomyelin, and LPC pools ranged from 4 to 24 nmol/nmol PO4, 123 to 409 pmol/nmol PO4, and 0.03 to 0.25 pmol/nmol PO4 respectively. Because of subject-to-subject variation in basal ceramide, sphingomyelin, and LPC molecular species in human islets, the values for each subject were normalized to corresponding DMSO control and the data are presented as % change, relative to control. (*Significantly different from corresponding minus S-BEL groups, P < .05; †significantly different from DMSO minus S-BEL groups, P < .05; and §significantly different from other groups, P < .05, n = 3–5).
Activation of PLA2s promotes release of a free fatty acid and the generation of lysophospholipids. To verify the increased iPLA2β induction after CTK exposure, islet phospholipids were extracted and the abundances of LPCs were determined. ESI/MS/MS revealed 3 prominent LPC molecular species in the islets at m/z 502, 528, and 530, representing 16:1-LPC, 18:1-LPC, and 18:0-LPC, respectively (Supplemental Figure 2A). Exposure to CTK promoted accumulations in all three species (Supplemental Figure 1B) in the islets, as reflected by the higher signal intensity ratio of each ion species relative to the IS. Whereas pretreatment of islets with S-BEL alone had no significant effect (Supplemental Figure 1C), it attenuated CTK-induced increases in the LPCs (Supplemental Figure 1D). As shown in Figure 4D, the total pool of LPCs was increased 2.5-fold in CTK-treated islets, relative to vehicle-treated islets, and this was prevented by S-BEL. These findings are consistent with induction of islet iPLA2β activity by proinflammatory cytokines, leading to the generation of NSMase2-catalyzed ceramides and LPCs.
IL-1β+IFNγ (CTK) induces ΔΨ loss in human islet cells in an iPLA2β-dependent manner
Loss of ΔΨ is characteristic of intrinsic (mitochondrial) apoptotic pathway activation (33, 34). We therefore monitored ΔΨ in dispersed islet cells loaded with a mitochondrial fluorescence dye that is retained in healthy but not in compromised cells. In flow cytometry analyses, this is reflected by the appearance of second peak on the left, where M1 represents the percentage of cells that have lost ΔΨ. Representative spectra and quantitation (Figure 5A) reveal similar percentage of compromised cells in the vehicle and S-BEL only treated islets. However, after CTK exposure, the incidence of islet cells with ΔΨ loss increased nearly 3-fold. In contrast, pretreatment of islets with S-BEL prevented the CTK-induced ΔΨ loss. Assessment of mitochondrial dye DiOC6(3) fluorescence in individual cells (14, 18, 19) confirmed the loss in mitochondria fluorescence with CTK and its preservation by pretreatment of islets with S-BEL (Figure 5B). Both analyses revealed minimal effects of S-BEL alone. Because β-cells are the islet cells that express iPLA2β, these findings suggest that proinflammatory cytokines cause decompensation of β-cell mitochondria and that this occurs via an iPLA2β-dependent mechanism.
Figure 5.

IL-1β+IFNγ (CTK) induces mitochondrial membrane potential (ΔΨ) loss in human islet cells in an iPLA2β-dependent manner. Islets (500/condition) were treated as in Figure 1 for 24 hours, dispersed into individual cells, and ΔΨ monitored by MitoFlow (stable analog of TMRE, tetramethylrhodamine, ethyl ester) fluorescence or DiOC6(3) staining. A, Flow cytometry analyses. Representative spectra (top panels) were obtained from analyses of 10,000 cells and the percentage of cells with compromised ΔΨ is indicated by M1. Quantitation of cells with MMP loss ± S-BEL (bottom panel). The percentage of cells with ΔΨ loss, relative to total number of cells, is presented as mean ± SEM. (*Significantly different from other groups, P < .05, n = 3–5). B, DiOC6(3) staining. Cells were loaded with green mitochondrial [DiOC6(3)] stains and examined by confocal microscopy. Representative images are shown (top panels). DiOC6(3) fluorescence intensity was quantitated using ImageJ Software and is presented as mean ± SEM (bottom panel). (*Significantly different from other groups, P < .05, n = 6).
IL-1β+IFNγ (CTK) induces activated caspase-3 (aC3) in and apoptosis of human islet β-cells in an iPLA2β-dependent manner
Cleavage of caspase-3 to an activated form (aC3) is essential for the execution of apoptosis. We therefore assessed induction of aC3 by proinflammatory cytokines in islets using immunofluorescence analyses. As illustrated in Figure 6A (top panels), aC3 red fluorescence is evident in islet cells after CTK exposure in the absence of S-BEL and it colocalizes with insulin green fluorescence, as reflected by the yellow fluorescence in the merged image (insulin + aC3). In contrast, pretreatment of islets with S-BEL nearly completely inhibited aC3 generation (Figure 6A, bottom panels). To obtain a quantitative measure of apoptosis, TUNEL analyses were performed in dispersed cells prepared from islets treated with IL-1β+IFNγ ± S-BEL for 48 hours (Figure 6B). The incidence of islet-cell TUNEL-positivity is increased nearly 3-fold after exposure to CTK and this is significantly attenuated by pretreatment of islets with S-BEL. These findings suggest that proinflammatory cytokines induce caspase-3 activation leading to β-cell apoptosis and that this occurs via an iPLA2β-dependent mechanism.
Figure 6.
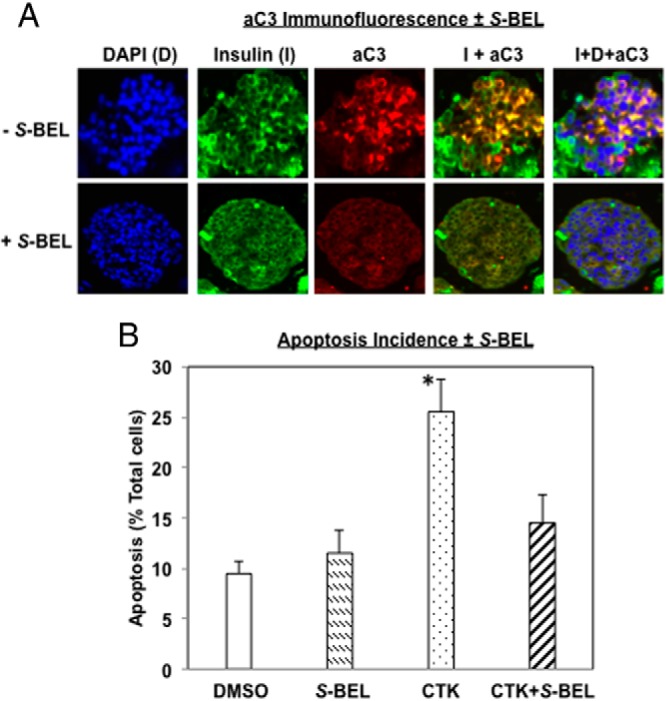
IL-1β+IFNγ (CTK) induces activated caspase-3 (aC3) and TUNEL-positivity in human islet β-cells in an iPLA2β-dependent manner. Islets (500/condition) were treated with CTK ± S-BEL for 48 hours and then processed for aC3 and apoptosis analyses. A, Activated casp-3 (aC3) was assessed by immunofluorescence (DAPI, blue; insulin, green; and aC3, red). Representative images of individual and merged fluorescence islet images are presented. B, Apoptosis incidence was assessed by TUNEL staining and the percentage of TUNEL-positive cells, relative to total number of cells (DAPI-stained), was determined in a minimum of 6 fields on each slide from separate experiments and presented as mean ± SEM. (* Significantly different from groups, P < .05, n = 4–5).
IL-1β+IFNγ (CTK)-induced ER stress and β-cell apoptosis is modulated by iPLA2β expression
To preclude nonspecific chemical inhibition of S-BEL, the effects of proinflammatory cytokines were examined in islets isolated from mice in which iPLA2β is increased only in β-cells (RIP-iPLA2β-Tg) or is globally-deficient (iPLA2β-KO) (18, 35, 36). Examination of ER stress markers revealed that all three signaling arms (pPERK, IRE1α, and ATFα) of the unfolded protein response are activated in WT islets by IL-1β+IFNγ (Figure 7A and B). Induction of all three is amplified in islets from RIP-iPLA2β-Tg mice and mitigated in islets from iPLA2β-KO mice, in comparison with WT islets. Assessment of apoptosis indices, aC3 activity (Figure 7C) and TUNEL positivity (Figure 7D), revealed that apoptosis was exacerbated in islets from RIP-iPLA2β-Tg and similar in islets from iPLA2β-KO mice, as compared with WT islets. These findings are consistent with a role for iPLA2β in amplifying proinflammatory cytokine-mediated β-cell apoptosis and also suggest a reciprocal impact of iPLA2β activation on ER stress development.
Figure 7.

iPLA2β expression modulates ER stress and islet cell apoptosis induced by IL-1β+IFNγ (CTK). Islets (500/condition) isolated from RIP-iPLA2β-Tg and iPLA2β-KO mice were treated with vehicle (DMSO) alone or CTK for up to 48 hours and then processed for ER stress markers (pPERK, IRE1α, and ATF6α) and apoptosis indices [activated caspase-3 (aC3) and TUNEL positivity] analyses. A, Representative immunoblots of pPERK, IRE1α, ATF6α, and loading control actin. B, Densitometry quantitation, abundances of the ER stress markers, relative to corresponding actin bands ± CTK in each group, are plotted as mean ± SEM (n = 3). (pPERK, *, #, ¶, and † significantly different from corresponding DMSO group, P < .001; P < .0001; P < .01; and P < .05, respectively, a and b significantly different from corresponding WT group, P < .001 and P < .05, respectively; IRE1α, †, *, and # significantly different from corresponding DMSO group, P < .05; P < .001; and P < .0001, respectively; c significantly different from corresponding WT group, P < .0005; and ATF6α, † and § significantly different from corresponding DMSO group, P < .05 and P < .005, respectively; d and b significantly different from corresponding WT group, P < .05 and P < .01, respectively.) C, aC3 activity. Islet lysates were prepared and activity in 30 μg aliquot of protein was assayed using a colorimetric-based protocol. (§ and † significantly different from corresponding DMSO groups, P < .01 and P < .005, respectively; *significantly different from corresponding WT and KO groups, P < .05; and # significantly different from Tg-DMSO and Tg-48h groups, P < .05, n = 3–6.) D, TUNEL analyses. The percentage of TUNEL-positive cells relative to total number of cells (DAPI-stained) was determined in a minimum of 6 fields on each slide from 3 separate experiment and are presented as mean ± SEM. (#* Significantly different from corresponding DMSO group, P < .01 and P < .05, respectively; † and § significantly different from corresponding WT and KO groups, P < .005 and P < .01, respectively.)
Discussion
T1D is a consequence of autoimmune destruction of β-cells and autoreactive T-cells (CD4+ and CD8+) play a key role in this process. These cells, in addition to secreting perforins and granzymes that promote apoptosis and necrosis, produce proinflammatory cytokines that activate macrophages leading to β-cell death (37). Furthermore, reactive oxygen species (ROS) that are essential for proinflammatory cytokine production are becoming recognized as being crucial for T-cell maturation (38, 39). Examination of the underlying mechanisms by which proinflammatory cytokines induce β-cell death in T1D suggests involvement of the intrinsic apoptotic pathway, ER stress, and nitric oxide-dependent necrosis (40). Missing from this literature, however, is the role that lipid signals may play in the autoimmune destruction of β-cells.
In view of an observed link between iPLA2β and ER stress-induced β-cell apoptosis (14–19) and findings of elevated iPLA2β in T1D (22–24), we explored the possibility that iPLA2β-derived lipid signals participate in proinflammatory cytokine-induced islet β-cell apoptosis. Three factors were considered during the design of the present studies: a) model for testing - whereas we previously used insulinoma cells to establish a link between iPLA2β and β-cell apoptosis that was subsequently verified in rodent and then human islet β-cells, here, we chose human islets for study. In addition to being more physiologically relevant, the yield of islets is greater from donor preparations thus providing more starting material for a given analysis; b) age of human islet donors - our previous assessments in human islets revealed that basal activation of caspase-3 increases with age (19), we therefore focused our studies in islets isolated from 20 to 40-year-old donors; and c) choice of cytokines - complicated by the recognized impact of different cytokines on β-cell function and survival (41) and taking into account the availability, within the needed age-range, and expense of human islets we tested the proinflammatory cytokine combination of IL-1β+IFNγ, previously demonstrated to be appropriate for examining human islet β-cell responses (25–30).
Our findings reveal that exposure of human islets to IL-1β+IFNγ leads to ER stress, as evidenced by induction of ER stress factors GRP78 and pPERK (42, 43). Unexpectedly, the proinflammatory cytokines induced iPLA2β message and protein. This was reflected by higher iPLA2β enzymatic activity and increased generation of LPCs that were attenuated by S-BEL, a specific inhibitor of the β-isoform of iPLA2 (32). Inhibition of the accompanying increases in NSMase2 mRNA, ceramide accumulation, mitochondrial decompensation, activation of caspase-3, and β-cell apoptosis by S-BEL, collectively, provide strong evidence for iPLA2β mediation of proinflammatory cytokine-induced β-cell death. Our earlier demonstrations that in islets, iPLA2β is predominantly expressed in the β-cells (19, 44), taken together with the current detection of activated caspase-3, the executioner of apoptosis, predominantly in insulin-containing cells of the islet, reinforces this possibility.
Whereas there has been a long-standing debate on whether ER stress contributes to β-cell apoptosis caused by proinflammatory cytokines (8–13), it was recently reported that β-cell dysfunction in the diabetes-prone autoimmune non-obese diabetic (NOD) model of T1D was accompanied by age-dependent increases in ER stress factors (45). This study also recapitulated the induction of ER stress in MIN-6 insulinoma cells after exposure to cytokines that was similar in profile to one after exposure to the ER stressor, thapsigargin. It is therefore noteworthy that the outcomes of proinflammatory cytokine exposure we describe here, including iPLA2β induction and β-cell apoptosis, are analogous to ER stress-induced effects in β-cells (14–19). Taken together, these findings demonstrate that ER stress contributes to β-cell dysfunction and subsequent β-cell apoptosis due to proinflammatory cytokines and that this mechanism is integral to the evolution of autoimmune destruction of β-cells leading to T1D. Consistent with a role for iPLA2β in this process, preliminary findings in our laboratory reveal age-dependent increases in iPLA2β expression in diabetes-prone NOD mouse islets (in revision).
Because S-BEL has been reported to inhibit non-iPLA2β proteins as well [reviewed in (46)], the impact of proinflammatory cytokine exposure on islets from WT, RIP-iPLA2β-Tg, and iPLA2β-KO mice was compared to preclude potential nonspecific effects of S-BEL. The RIP-iPLA2β-Tg model further allowed assessment of altered iPLA2β expression specifically in β-cells. Comparison of outcomes between the three genotypes revealed activation of all 3 signaling pathways (PERK, IRE1, and ATF6) of the unfolded protein response and of caspase-3, and induction of β-cell apoptosis in the presence of IL-1β+IFNγ, in the WT islets. These were all amplified in RIP-iPLA2β-Tg islets but mitigated in iPLA2β-KO islets, suggesting iPLA2β-derived lipid signals modulate these events. Interestingly in the KO islets, whereas pPERK and IRE1α were induced, there were no changes in ATF6α from basal levels and activated caspase-3 was modestly (but not significantly) elevated at 48 hours. However, the incidence of apoptosis in KO islets after IL-1β+IFNγ exposure was similar to that in WT islets. These findings for the first time suggest a) iPLA2β activation has a reciprocal impact on the full complement of unfolded protein response, where the ATF6α pathway appears to be spared from induction in its absence, and b) proinflammatory cytokines cause β-cell apoptosis via iPLA2β-dependent and iPLA2β-independent mechanisms. Thus, the iPLA2β-derived lipids seem to amplify processes that are activated during proinflammatory cytokine-mediated β-cell apoptosis; the plausibility of iPLA2β contributing to autoimmune destruction of β-cells in this manner is supported by reports of increased iPLA2β in T1D (22–24) and in the islets of diabetes-prone NOD mice (Bone et al, in revision).
Intriguingly, we find that proinflammatory cytokines also promote ceramide accumulation in islets. Ceramides are complex lipids that can suppress cell growth and induce apoptosis and are recognized activators of the mitochondrial apoptotic pathway (47). To date, the link between cytokines and ceramides is circumstantial, as demonstrated by the mimicking of cytokine-mediated effects on β-cell function and survival by exogenous ceramides or, inconsistent, as in studies with insulinoma cells (reviewed in Ref. 48). To the best of our knowledge, the present observations are the first demonstration of proinflammatory cytokine-induced ceramide generation in human islets and that such an increase is inhibited by S-BEL, suggesting that it occurs via an iPLA2β-mediated mechanism. Additionally, ceramide accumulation with IL-1β+IFNγ exposure is prevented by inhibition of NSMase2, consistent with hydrolysis of sphingomyelins as the ceramide-generating pathway activated by proinflammatory cytokines. It is recognized that sphingomyelin-derived ceramides play important roles in decreasing cell proliferation and increasing apoptosis (49). Our findings are also consistent with proinflammatory cytokine-induced NSMase2 expression, but not SPT1, the rate-limiting enzyme in de novo synthesis of ceramides increased during lipoapoptosis of β-cells (50). As previously observed under ER stress (15), the increased expression of NSMase2 in islets after exposure to proinflammatory cytokines is mediated by iPLA2β, as reflected by its decrease with S-BEL.
Although NSMase2 expression is increased and its inhibition prevents ceramide accumulations, the pool of sphingomyelins is unchanged in islets after the proinflammatory cytokine exposure. One potential explanation for this is that only sphingomyelins in specific subcellular β-cell organelles are subject to hydrolysis by NSMase2 induced by proinflammatory cytokines and that changes in those limited pools are masked by the more abundant total islet pool of sphingomyelins. In support of this possibility are earlier observations that sphingomyelin pools in the ER and mitochondria are decreased in INS-1 cells treated with the ER stressor, thapsigargin (16). Alternatively, it has been reported that proinflammatory cytokines in addition to increasing ceramides also increase sphingomyelins (51). It is therefore likely that after proinflammatory cytokine exposure, hydrolysis of sphingomyelins by NSMase2 leads to generation of ceramides whereas other undefined processes targeted by the cytokines lead to replenishment of the sphingomyelin pools.
Based on our collection of findings, the steps after induction of ER stress leading to β-cell apoptosis, appear to be mediated via increases in iPLA2β expression and activity. In all models we have examined to date, a decrease in iPLA2β reduces and an increase in iPLA2β amplifies ceramide generation via NSMase2-catalyzed hydrolysis of sphingomyelins, mitochondrial perturbations, activation of caspase-3, and β-cell apoptosis (14–19). However, the exact mechanism(s) by which proinflammatory cytokines promote iPLA2β expression remains to be elucidated. In this regard, it is recognized that stresses (ie, ER, glucolipotoxicity, and oxidative) promote the generation of mSREBP-1 (14, 52, 53) that can translocate into the nucleus to induce gene transcription, including that of iPLA2β (14, 54). The present finding of mSREBP-1 increase in human islets after proinflammatory cytokine exposure raises the possibility that this could be a mechanism by which proinflammatory cytokines induce iPLA2β. Elucidation of transcription factor recognition sites on the iPLA2β gene is currently at the infant stage and continued exploration in this area may lead to identification of additional sites that are targets of downstream products arising from the actions of proinflammatory cytokines in β-cells.
Alternatively, cytokines are linked to ROS generation and oxidative stress in the β-cells (40) that exhibit decreased levels of antioxidants that normally scavenge free radicals (55). Oxidative stress leads to β-cell apoptosis and antioxidant enzymes protect β-cells from cytokine-induced apoptosis (56, 57). The islets, however, express γ-glutamylcysteine synthetase, the rate-limiting enzyme for the intracellular oxidant glutathione (GSH) synthesis, and cytokines decrease γ-glutamylcysteine synthetase expression and GSH levels (58), raising ROS abundance. In non-β-cells, ROS are reported to induce iPLA2β (59) and decreases in GSH have been associated with diabetes and NSMase induction (60). Furthermore, increases in arachidonic acid and LPC, as might occur with increased iPLA2β activation, decrease GSH (61). Consistent with their generation via induction of iPLA2β by proinflammatory cytokines, accumulations in LPC molecular species in islets exposed to IL-1β+IFNγ are decreased in S-BEL-pretreated islets. The significance of this finding is increased by the well-recognized role of LPCs to act as chemoattractants during elicitation of immune responses (62–65). This raises the possibility that LPCs derived from iPLA2β activation promotes islet infiltration by immune cells to initiate the autoimmune β-cell destruction process.
Given that autoimmune T1D is associated with β-cell apoptosis, that cytokines play a pivotal role, and the recent demonstration of ER stress in β-cells of autoimmune diabetes-prone NOD mice (45), the motivation for the present study was to determine if ER stress is induced by cytokines and if this becomes linked downstream to iPLA2β-mediated processes. Our evidence supports this possibility, as we report for the first time that proinflammatory cytokines induce ER stress, mSREBP-1, and iPLA2β in human pancreatic islet β-cells, leading to ceramide generation, mitochondrial perturbations, caspase-3 activation, and β-cell apoptosis. That these outcomes are, in part, mediated by iPLA2β is consistent with their attenuation by chemical inactivation or KO of iPLA2β and amplification in RIP-iPLA2β-Tg islets. These findings suggest that ER stress and iPLA2β activation contribute to autoimmune destruction of β-cells during T1D evolution, as illustrated in Figure 8. This model, modified from an earlier version (59), includes sequence of proposed events after proinflammatory cytokine stimulation and raises the possibility that inhibition of iPLA2β may be beneficial in preserving β-cell mass and delaying or preventing T1D. The infrequent availability of human islets from donors of appropriate age precluded more detailed studies to elucidate the mechanisms of proinflammatory cytokines-mediated induction of iPLA2β leading to β-cell apoptosis. However, as we have identified a link between the two and an iPLA2β inhibitor that is amenable for use in vivo is now available (46), studies are underway to examine the underlying involved processes, in the context of T1D. The findings described here are the first demonstration of a link between proinflammatory cytokines-mediated β-cell death and iPLA2β activation in β-cells. Furthermore, they offer new directions in the study of iPLA2β biology, especially in the context of disease states that are associated with autoimmunity and ER stress.
Figure 8.

Proposed model of iPLA2β involvement in the autoimmune destruction of β-cells in T1D. Based on our present findings, we propose that proinflammatory cytokines cause ER stress leading to SREBP-1 activation and induction of iPLA2β. Alternatively, iPLA2β induction may also be promoted by downstream products of proinflammatory cytokines activation. It is suggested that the iPLA2β-derived lipid signals promote β-cell death by a) triggering the intrinsic apoptotic pathway via ceramide generation and by serving as chemoattractants to enhance islet infiltration of immune cells and b) exacerbating ER stress and consequential downstream events. It is speculated that the iPLA2β-derived lipid signals amplify proinflammatory cytokine-induced β-cell apoptosis and that inhibition of iPLA2β may be beneficial in reducing the impact of proinflammatory cytokines on β-cells and mitigating autoimmune destruction of β-cells, thus preventing or delaying the onset and/or progression of T1D. (Dashed arrows reflect undefined pathways.)
Acknowledgments
We would like to thank the National Institute of Diabetes and Digestive and Kidney Diseases-sponsored Integrated Islet Distribution Program, Washington University/Juvenile Diabetes Research Foundation (Award 31–2008-382 to Dr Thalachallour Mohanakumar), and University of Alabama at Birmingham (UAB) Islet Resource Facility, supported by UAB Diabetes Research and Training Center Grant (NIH P60-DK079626) awarded to the UAB Comprehensive Diabetes Center for providing (to approved user S.R.) the human islets. The authors would like to thank the expert technical assistance of Ms. Karen Goodwin and Ms. Ying Gai (UAB), and Washington University Diabetes Research and Training Center supported Morphology Core, UAB Comprehensive Diabetes Center Islet Biology Core, and the Diabetes Research Center at UAB.
This work was supported by the National Institutes of Health (R01-DK69455, DK34388, P01-HL57278, P41-RR00954, P60-DK20579, and P30-DK56341) and the American Diabetes Association (1–09-RA-147 and 7–12-CD-11).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATF6
- activating transcription factor 6
- S-BEL
- bromoenol lactone suicide inhibitor of iPLA2β
- CTK
- IL-1β+IFNγ
- DAPI
- 4′,6′-diamidino-2-phenylindole
- DMSO
- dimethyl sulfoxide
- ER
- endoplasmic reticulum
- ESI
- electrospray ionization
- GRP78
- 78 kDa glucose-regulated protein
- GSH
- glutathione
- iNOS
- inducible nitric oxide synthase
- iPLA2β
- β-isoform of group VIA calcium-independent phospholipase A2
- iPLA2β-KO
- globally iPLA2β-deficient
- IRE1
- inositol-requiring enzyme 1
- KO
- knockout
- LPC
- lysophosphatidylcholine
- ΔΨ
- mitochondrial membrane potential
- MS
- mass spectrometry
- mSREBP-1
- mature form of sterol regulatory element binding protein-1
- NOD
- non-obese diabetic
- NSMase2
- neutral sphingomyelinase-2
- pPERK
- phosphorylated form of ER-stress transducer pancreatic ER kinase
- PLA2
- phospholipase A2
- pNA
- p-nitroaniline
- RIP-iPLA2β-Tg
- iPLA2β overexpressed in only β-cells
- ROS
- reactive oxygen species
- RT-qPCR
- quantitative RT-PCR
- SPT1
- serine palmitoyl transferase
- T1D
- type 1 diabetes
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
- wild-type.
References
- 1. Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297 [DOI] [PubMed] [Google Scholar]
- 2. Rabinovitch A, Suarez-Pinzon WL. Cytokines and their roles in pancreatic islet beta-cell destruction and insulin-dependent diabetes mellitus. Biochem Pharmacol. 1998;55:1139–1149 [DOI] [PubMed] [Google Scholar]
- 3. Mandrup-Poulsen T. Beta-cell apoptosis: stimuli and signaling. Diabetes. 2001;50:S58–S63 [DOI] [PubMed] [Google Scholar]
- 4. Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in PERK−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163 [DOI] [PubMed] [Google Scholar]
- 5. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345 [DOI] [PubMed] [Google Scholar]
- 6. Kroncke K-D, Brenner H-H, Rodriguez M-L, Etzkorn K, Noack EA, Kolb H, et al. Pancreatic islet cells are highly susceptible towards the cytotoxic effects of chemically generated nitric oxide. Biochimic Biophys Acta. 1993;1182:221–229 [DOI] [PubMed] [Google Scholar]
- 7. Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39:1005–1029 [DOI] [PubMed] [Google Scholar]
- 8. Akerfeldt MC, Howes J, Chan JY, et al. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes. 2008;57:3034–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61 [DOI] [PubMed] [Google Scholar]
- 10. Kacheva S, Lenzen S, Gurgul-Convey E. Differential effects of proinflammatory cytokines on cell death and ER stress in insulin-secreting INS1E cells and the involvement of nitric oxide. Cytokine. 2011;55:195–201 [DOI] [PubMed] [Google Scholar]
- 11. Wang Q, Zhang H, Zhao B, Fei H. IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol Cell Biochem. 2009;324:183–190 [DOI] [PubMed] [Google Scholar]
- 12. Weber SM, Chambers KT, Bensch KG, Scarim AL, Corbett JA. PPARgamma ligands induce ER stress in pancreatic beta-cells: ER stress activation results in attenuation of cytokine signaling. Am J Physiol Endocrinol Metab. 2004;287:E1171–E1177 [DOI] [PubMed] [Google Scholar]
- 13. Zhong J, Rao X, Xu JF, Yang P, Wang CY. The role of endoplasmic reticulum stress in autoimmune-mediated beta-cell destruction in type 1 diabetes. Exp Diabetes Res. 2012;2012:238980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lei X, Zhang S, Barbour SE, et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem. 2010;285:6693–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. The Group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry. 2007;46:10170–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lei X, Zhang S, Bohrer A, Ramanadham S. Calcium-independent phospholipase A2 (iPLA2beta)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem. 2008;283:34819–34832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramanadham S, Hsu FF, Zhang S, et al. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2 beta) and suppressed by inhibition of iPLA2 beta. Biochemistry. 2004;43:918–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lei X, Bone RN, Ali T, et al. Genetic modulation of islet β-cell iPLA2β expression provides evidence for its impact on β-cell apoptosis and autophagy. Islets. 2013;5:29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lei X, Zhang S, Bohrer A, Barbour SE, Ramanadham S. Role of calcium-independent phospholipase A2β in human pancreatic islet β-cell apoptosis. Am J Physiol Endocrinol Metab. 2012;303:E1386–E1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilkins WP, 3rd, Barbour SE. Group VI phospholipases A2: homeostatic phospholipases with significant potential as targets for novel therapeutics. Curr Drug Targets. 2008;9:683–697 [DOI] [PubMed] [Google Scholar]
- 21. Ramanadham S, Bohrer A, Gross RW, Turk J. Mass spectrometric characterization of arachidonate-containing plasmalogens in human pancreatic islets and in rat islet beta-cells and subcellular membranes. Biochemistry. 1993;32:13499–13509 [DOI] [PubMed] [Google Scholar]
- 22. Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, et al. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J Immunol. 2010;184:1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rahnema P, Shimoni Y, Nygren A. Reduced conduction reserve in the diabetic rat heart: role of iPLA2 activation in the response to ischemia. Am J Physiol. 2011;300:H326–H334 [DOI] [PubMed] [Google Scholar]
- 24. Xie Z, Gong MC, Su W, Xie D, Turk J, Guo Z. Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem. 2010;285:8628–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heitmeier MR, Kelly CB, Ensor NJ, et al. Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. J Biol Chem. 2004;279:53145–53151 [DOI] [PubMed] [Google Scholar]
- 26. Hindlycke H, Lu T, Welsh N. Cytokine-induced human islet cell death in vitro correlates with a persistently high phosphorylation of STAT-1, but not with NF-κB activation. Biochem Biophys Res Commun. 2012;418:845–850 [DOI] [PubMed] [Google Scholar]
- 27. Cardozo AK, Proost P, Gysemans C, Chen MC, Mathieu C, Eizirik DL. IL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia. 2003;46:255–266 [DOI] [PubMed] [Google Scholar]
- 28. Heitmeier MR, Scarim AL, Corbett JA. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem. 1997;272:13697–13704 [DOI] [PubMed] [Google Scholar]
- 29. Corbett JA, Kwon G, Marino MH, et al. Tyrosine kinase inhibitors prevent cytokine-induced expression of iNOS and COX-2 by human islets. Am J Physiol. 1996;270:C1581–C1587 [DOI] [PubMed] [Google Scholar]
- 30. Corbett JA, Sweetland MA, Wang JL, Lancaster JR, Jr, McDaniel ML. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci USA. 1993;90:1731–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci USA. 2010;107:12564–12569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem. 2002;277:32807–32814 [DOI] [PubMed] [Google Scholar]
- 33. Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6:118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Desagher S, Osen-Sand A, Nichols A, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bao S, Miller DJ, Ma Z, et al. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao S, Jacobson DA, Wohltmann M, et al. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2β in pancreatic beta-cells and in iPLA2beta-null mice. Am J Physiol Endocrinol Metab. 2008;294:E217–E229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118 [DOI] [PubMed] [Google Scholar]
- 38. Tse HM, Milton MJ, Schreiner S, Profozich JL, Trucco M, Piganelli JD. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 2007;178:908–917 [DOI] [PubMed] [Google Scholar]
- 39. Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med. 2004;36:233–247 [DOI] [PubMed] [Google Scholar]
- 40. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci. 2013;1281:16–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mandrup-Poulsen T. Beta cell death and protection. Ann N Y Acad Sci. 2003;1005:32–42 [DOI] [PubMed] [Google Scholar]
- 42. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274 [DOI] [PubMed] [Google Scholar]
- 43. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924 [DOI] [PubMed] [Google Scholar]
- 44. Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry. 1993;32:327–336 [DOI] [PubMed] [Google Scholar]
- 45. Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ali T, Kokotos G, Magrioti V, et al. Characterization of FKGK18 as inhibitor of group VIA Ca2+-Independent Phospholipase A2 (iPLA2β): candidate drug for preventing beta-cell apoptosis and diabetes. PloS one. 2013;8:e71748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529 Pt 1:57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boslem E, Meikle PJ, Biden TJ. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets. 2012;4:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859 [DOI] [PubMed] [Google Scholar]
- 50. Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490 [DOI] [PubMed] [Google Scholar]
- 51. Memon RA, Holleran WM, Moser AH, et al. Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler Thromb Vasc Biol. 1998;18:1257–1265 [DOI] [PubMed] [Google Scholar]
- 52. Kaplan M, Aviram M, Hayek T. Oxidative stress and macrophage foam cell formation during diabetes mellitus-induced atherogenesis: role of insulin therapy. Pharmacol Ther. 2012;136:175–185 [DOI] [PubMed] [Google Scholar]
- 53. Wang H, Kouri G, Wollheim CB. ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J Cell Sci. 2005;118:3905–3915 [DOI] [PubMed] [Google Scholar]
- 54. Seashols SJ, del Castillo Olivares A, Gil G, Barbour SE. Regulation of group VIA phospholipase A2 expression by sterol availability. Biochim Biophys Acta. 2004;1684:29–37 [DOI] [PubMed] [Google Scholar]
- 55. Grankvist K, Marklund SL, Täljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199:393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang M, Crager M, Pugazhenthi S. Modulation of apoptosis pathways by oxidative stress and autophagy in β-cells. Exp Diabetes Res. 2012;2012:647914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao F, Wang Q. The protective effect of peroxiredoxin II on oxidative stress induced apoptosis in pancreatic β-cells. Cell Biosci. 2012;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rutkute K, Asmis RH, Nikolova-Karakashian MN. Regulation of neutral sphingomyelinase-2 by GSH: a new insight to the role of oxidative stress in aging-associated inflammation. J Lipid Res. 2007;48:2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lei X, Barbour SE, Ramanadham S. Group VIA Ca2+-independent phospholipase A2 (iPLA2beta) and its role in beta-cell programmed cell death. Biochimie. 2010;92:627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murakami K, Kondo T, Ohtsuka Y, Fujiwara Y, Shimada M, Kawakami Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism. 1989;38:753–758 [DOI] [PubMed] [Google Scholar]
- 61. Hayter HL, Pettus BJ, Ito F, Obeid LM, Hannun YA. TNFalpha-induced glutathione depletion lies downstream of cPLA2 in L929 cells. FEBS Lett. 2001;507:151–156 [DOI] [PubMed] [Google Scholar]
- 62. Lauber K, Bohn E, Kröber SM, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730 [DOI] [PubMed] [Google Scholar]
- 63. Peter C, Waibel M, Keppeler H, et al. Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity. 2012;45:568–573 [DOI] [PubMed] [Google Scholar]
- 64. Peter C, Waibel M, Radu CG, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. 2008;283:5296–5305 [DOI] [PubMed] [Google Scholar]
- 65. Weaver JR, Holman TR, Imai Y, Jadhav A, Kenyon V, Maloney DJ, et al. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol. 2012;358:88–95 [DOI] [PubMed] [Google Scholar]