

Abstract
A re-examination of the mechanism controlling eating, locomotion, and metabolism prompts formulation of a new explanatory model containing five features: a coordinating joint role of the (1) autonomic nervous system (ANS); (2) the suprachiasmatic (SCN) master clock in counterbalancing parasympathetic digestive and absorptive functions and feeding with sympathetic locomotor and thermogenic energy expenditure within a circadian framework; (3) interaction of the ANS/SCN command with brain substrates of reward encompassing dopaminergic projections to ventral striatum and limbic and cortical forebrain. These drive the nonhomeostatic feeding and locomotor motivated behaviors in interaction with circulating ghrelin and lateral hypothalamic neurons signaling through melanin concentrating hormone and orexin-hypocretin peptides; (4) counterregulation of insulin by leptin of both gastric and adipose tissue origin through: potentiation by leptin of cholecystokinin-mediated satiation, inhibition of insulin secretion, suppression of insulin lipogenesis by leptin lipolysis, and modulation of peripheral tissue and brain sensitivity to insulin action. Thus weight-loss induced hypoleptimia raises insulin sensitivity and promotes its parasympathetic anabolic actions while obesity-induced hyperleptinemia supresses insulin lipogenic action; and (5) inhibition by leptin of bone mineral accrual suggesting that leptin may contribute to the maintenance of stability of skeletal, lean-body, as well as adipose tissue masses.
Keywords: Insulin, Leptin, Weight regulation, Autonomic, Circadian
Core tip: The novel proposal for the mechanism of body weight regulation deals with all three components of body mass: bone, lean tissue, and fat depots. It attributes the central control of counterbalancing energy expenditure and intake to an autonomic nervous system-circadian clock command center that encompases brain reward substrates, lateral hypothalamic peptidergic circuits and areas of the cortex. The nonhomeostatic character of feeding and locomotion is driven and controlled by the reward circuits and modulated by shifts in insulin sensitivity induced by counterregulation by leptin of insulin as weight deviates between underweight and overweight and alters basal leptin concentrations.
INTRODUCTION
Finding and ingesting food and drink are intermittent behaviors essential for individual and species survival against continuous energy cost of staying alive. Our complex physiological design insures that opportunities to ingest food are not missed and that drive to seek food increases and compensatory processes are deployed to counteract substantial losses of body mass. That this behavior supports both growth of body mass as well as its maintenance when statural growth has ceased only adds to its complexity and challenges our ability to understand its mechanism. Therefore, the transformation from a system in which food abundance drives the acquisition of body mass during statural growth to a system where energy intake is matched to each finite adult physique requires an explanation that integrates both phenomena. In addition, feeding behavior is coupled to spontaneous variations in movement and locomotion in ways that are imperfectly understood, and the two behaviors and control of metabolic heat production also contribute to regulation of body mass. A satisfactory model for the regulation of stable adult body mass must integrate central neural, autonomic, and endocrine controls of feeding, locomotion, and metabolic heat production. But it also needs to account for the prospect that some humans[1,2] and animals[3] can deviate from body mass stability and predictably become obese[1,2] under conditions providing abundant foods of high energy density and palatability along with limited opportunities and incentives for physical activity.
The quest for understanding what guides intermittent meal-to-meal eating and body mass maintainance as well as increased hunger and food intake responding to substantial losses of body mass, has a long history but no satisfactory closure or consensus. Because of its complexity, and relevance to professionals in disconnected fields of psychology, nutrition, gastrointestinal physiology, endocrinology, exercise science, neuroscience, and physiology among others, the wealth of information about the neural, autonomic, and hormonal mechanism of feeding, physical activity, and thermogenesis in body mass regulation has not been satisfactorily integrated. A prevailing preference for a unitary deductive model of body mass regulation has placed emphasis on the presumed metering and matching of energy consumed to energy expended and to the energy content of body fat mass under both ad libitum and underweight conditions[4-9]. The core feature of this model is operation of a negative feedback exerted by adipokine leptin (and in some variations of the hypothesis, also by insulin) over feeding behavior and energy expenditure in response to changes in body fat mass. This widely accepted hypothesis is not supported by the empirical data under conditions of intact neuroendocrine system, environmental abundance of food, reduced opportunities for physical exertion, and rising levels of body fat. Obesity coexists with high basal concentrations of leptin and insulin. Further, administration of leptin to obese humans is ineffective in suppressing feeding and reducing the body or fat mass[10]. On the other hand, two robust findings regarding leptin actions on feeding and body energy status need to be reconciled with its inability to reduce body fat mass in a negative feedback fashion in neurologically normal obese individuals under the ad-libitum feeding conditions. The first finding is a consistent proportional relationship between distributed body fat mass and basal leptin (and insulin) concentrations in humans and animals first clearly demonstrated in humans by Considine[11] and postulated to exert sustained inhibition over feeding and facilitation of energy expenditure[4-9]. The second finding is capacity of leptin to inhibit pronounced and consistently high hunger and suppress high fat mass in freely feeding humans and animals that lack leptin signaling capacity. This was first reported in humans by Farooqi[12,13] and in animals by Pelleymounter[14].
A unitary mechanism of weight regulation that can account for eating and weight changes leading to obesity and in non-deprivation as well as weight-loss conditions needs to account for (1) central neural coordination of this process; (2) interactions of this mechanism with the biological clock in structuring ultradian and nycthemeral rhythms of intermittent hunger and feeding; (3) opportunistic as opposed to homeostatic control of food intake and locomotion; (4) counterregulation by leptin of insulin secretion and actions to fluctuations of short-term energy availability and deviations in body fat mass; and (5) inclusion of skeletal and lean body masses along with the fat mass in the energy regulatory process. The proposed mechanism accounts for these processes in a novel way that differs from the currently prevailing view[4-9]. Its main propositions are that : (1) the autonomic brain centers activate hunger drive in; (2) a circadian pattern suppressed by intermittent inhibition from gastrointestinal (GI) filling and food processing that coordinate anabolic and catabolic processes to produce weight stability; (3) meal-to-meal eating and spontaneous physical activity represent non-homeostatic behaviors motivated through activation of a common brain substrates of reward that are connected to, and controlled by, the autonomic centers and circadian clock and responsive to short-term variations in the filling of the GI tract with food and fluctuations in body fat reserves and body mass; (4) autonomic nervous system (ANS) controls counterregulation by leptin of insulin secretion and tissue sensitivity to insulin actions to yoke leptin’s thermogenesis and catabolic metabolism to insulin’s anabolic actions; gastric leptin participates in GI processing of ingested nutrients and thus contributes to defining meal size through both anabolic digestive and restrictive satiating effects. It does so in conjunction with leptin of adipose tissue origin to regulate peripheral tissue and ANS/circadian command center sensitivity in response to body fat and body mass deviations from the adult setpoint; and (5) brain defends skeletal and lean body masses along with body fat mass against losses demonstrating that these body components should be integrated along with the adipose tissue in the regulation of adult mammalian body weight. The proposed postulates of this novel formulation of weight regulatory mechanism reconcile the conundrum of central and peripheral resistance to actions of insulin and leptin in obesity that is inherent in the homeostatic negative feedback view and the dichotomy of absence-of-protection model of energy regulation in non-deprivation eating with the central-resistance model of homeostatic leptin negative feedback[15].
CENTRAL COORDINATING ROLE OF THE ANS IN THE CONTROL OF FEEDING
Coordination of parasympathetic functions of nutrient intake, digestion, absorption, storage, and behavioral quiescence with sympathetic control of behavioral and metabolic energy expenditure has been recognized for over half a century. In 1947, Adolph[16] reported that body weight in rats stabilizes and is defended at a given plateau at the end of the growth period when mature rats with unrestricted access to food eat daily an amount of standard lab chow sufficient to maintain a stable weight plateau. Application of various methods of localized brain damage[17] and transsections of neural pathways[18,19] has revealed that ventromedial (VMH) and arcuate (ARC) hypothalamic lesions result in transient hyperphagia and hyperinsulinemia, permanent hypoactivity, and defective postprandial and cold-exposure thermogenesis. This has been interpreted by some to reflect an imbalanced parasympathetic overactivation because insulin oversecretion[20], hyperphagia, deficient thermogenesis[21,22], and spontaneous hypoactivity[23] were preventable by subdiaphragmatic vagotomy. In support of this interpretation, electrical stimulation of ventromedial hypothalamus[24-27] or administration of sympathomimetics[28,29] to neurologically intact animals elicited fuel mobilization and energy expenditure. After the weight in lesioned animals stabilizes, the same amount of food per unit weight is consumed as in intact rats, and the new weight plateau is defended against weight loss[30] indicating that regulation of stable weight is a consequence of balance between parasympathetic and sympathetic actions that is only reset by lesions to a new plateau by damage to the sympathetic controls or pathways. Although the relatively crude methods of brain lesions and neural tract transection initially singled out the VMH in the medial basal hypothalamus as the source of sympathetic actions[31], other lines of evidence identified the paraventricular hypothalamic nucleus (PVN) as the control center of sympathetic outflow and, by inference, dorsal motor nucleus of the vagus as the site of parasympathetic control of visceral actions other than cardiac function. Interest in the role of parasympathetic nervous system in the control of feeding has taken a back seat compared to the focus on leptin actions in the ARC and VMH nuclei. Nevertheless, pharmacological and denervation approaches have shown that suppression of sympathetic tone reduces thermogenesis[32] and increases parasympathetic functions of white adipose tissue (WAT) cell proliferation and body fat accumulation[33].
THE CENTRAL CLOCK COORDINATES ANS CONTROL OF FEEDING
One of the missing pieces in our understanding of energy regulation is the causative stimulus of hunger and meal initiation. The proposition that ghrelin is the key initiator of hunger and feeding[34-37] is challenged by normal food intake and weight maintenance in animals with deficient ghrelin signaling[38] and by a correlational and transient changes in ghrelin concentration and hunger sensations in the course of a meal[34,39]. On the other hand, the proposition that an autonomic controller coordinated by the circadian master clock regulates meal taking, locomotion, and thermogenesis is supported by a wealth of both behavioral, lesioning, and anatomical evidence.
Meal eating is intermittent in contrast to continuous behavioral and metabolic energy expenditure. Its ultradian and circadian patterning is a universal feature of mammalian feeding behavior. Rodents take meals at ultradian intervals of 3 to 4 h with a circadian segregation of eating to only the waking portion of the day[40]. Humans also eat during their nycthemeral wakeful period at 3-h intervals if snacks are included and at about 6-h intervals if more substantive main meals are considered. Circadian control of feeding in mammals is supported by extensive neuroanatomical evidence. Suprachiasmatic nucleus (SCN), the master circadian clock, has multiple ANS interconnections with structures that are implicated in weight regulation. Neural pathways through which the photo-entrainable SCN controls behavioral, endocrine, and metabolic rhythms related to energy balance include direct projections to subparaventricular zone (SPZ), an anterior hypothalamic region that receives innervation from both the PVN and SCN and is therefore thought to integrate circadian and metabolic information[41]. Additional areas receiving SCN innervation include medial preoptic area and dorsomedial hypothalamic nucleus (DMN)[42,43]. DMN, which is innervated both by the SCN and the SPZ, also controls circadian pattern of feeding, sleep-wakefulness, and locomotor activity. SCN also influences the circadian control of food intake, locomotion, and metabolic energy expenditure through its fibers projecting to the ARC, the VMH, and the ventral part of the lateral hypothalamus (LH), all areas implicated in the control of feeding and energy regulation. Interneurons from the SCN inhibit the PVN through γ-aminobutyric acid neurotransmission to facilitate parasympathetic functions. Consequently, most viscera receive SCN-dependent circadian time cues via their parasympathetic and/or sympathetic innervations that reflect metabolic and digestive events at peripheral sites[43]. Besides the obligatory periodicity of meal eating, nycthemeral patterning of feeding is necessary for the maintenance of stable body and fat masses. When the nocturnal part of the circadian sleep-wake cycle in humans is truncated, inappropriate overeating during extended wakeful periods ensues contributing to obesity and associated health risk factors[44,45]. Similarly, a seasonal change in the length of circadian exposure to light produces changes in feeding and body fat accumulation in some mammals[46].
Additional evidence for a functional interaction between the circadian clock and the ANS energy regulatory circuits involves loss of feeding, locomotor, and thermogenic periodicities when either the ANS or SCN circuits are disrupted. Destruction of SCN results in the loss of all bioenergetic circadian responses including circadian pattern of drinking and locomotor activity[47,48]. Destruction of VMH and ARC nuclei within the medial basal hypothalamus disrupts circadian alternation between active and inactive periods of food seeking and eating and results in protracted 24-h extension of meal taking and obesity[49,50]. Postprandial[51] and general thermogenesis also display circadian[52,53] and ultradian[54] rhythms of activation that have an acrophase during the active portion of the day and a nadir during the inactive phase. Metabolic and thermogenic gene expression in brown adipose tissue (BAT) and WAT also follows circadian periodicity[55]. The activation is attributable to stimulation of BAT by sympathetic nerves that originate in PVN, SCN, and DMN[31]. And thermogenesis can be elicited by electrical stimulation of sympathetic nerves to BAT[56], application of sympathomimetics[28,29,57,58] and activation by leptin of sympathetic nerves to BAT[28,29,31,59] when the hormone is applied to DMN, one of key sites involved in circadian components of energy regulation[59]. Leptin itself exhibits a prominent circadian pattern of secretion in humans with an acrophase around midnight and a nadir during mid-day[60-62]. This diurnal pattern is entrained to meal taking and phase shifts by the same number of hours with temporal displacement of meals[62]. In addition to its circadian pattern, leptin secretion is pulsatile with 32 pulses over 24 h, and a mean pulse duration of 33 min[63]. Circadian control of several aspects of energy regulation is seen in circadian changes in postprandial BAT thermogenesis in response to olfactory and gustatory stimulation by hedonic properties of palatable diets. The importance of stimulation of olfactory and gustatory receptors in eliciting postprandial BAT thermogenesis is demonstrated by diet-induced thermogenesis (DIT) attenuation when oral route of food administration is bypassed by tube feeding[64], or by administration of endocannabinoid blocker rimonabant[65,66]. Similarly, overeating palatable diets elicits a greater DIT than does eating diets of lesser olfactory and gustatory appeal[67]. Olfactory responsiveness[68] and hedonic responses to food and associated increases in DIT[67] show a diurnal rhythm with an acrophase during the active portion of the circadian period. The rhythm and the magnitude of thermogenic response are abolished by SCN lesion, sympathetic denervation of BAT[66], or deletion of β1 receptors in BAT[60]. Endocannabinoid blockade of DIT thermogenesis is more effective during the active that during the inactive phase of the circadian cycle[66].
Circadian influence in human meal eating is evident by comparing the effect of energy expenditure during long nocturnal inter-meal interval (IMI) on morning hunger[69]. We determined that the nocturnal IMI generated expenditure of between 710 and 750 Kcal in healthy postmenopausal women as compared to 340 to 450 Kcal expended during diurnal sedentary 6-h IMIs. Yet hunger rating at the end of 11 to 12-h long nocturnal IMI was only half as large as the hunger rating recorded at the end of individual diurnal IMIs and approximately as low as the evening hunger rating. Even more remarkably, the quantity of food consumed at the end of two mid-diurnal IMIs bore no relationship to the magnitude of preceding energy expenditure (Figure 1). These data support the operation of a circadian control of hunger with an acrophase at mid-day, a presumed nadir in the middle of sleep period, and transitional effects at dawn and dusk. They also indicate that the quantity of food eaten at a meal bears no homeostatic relationship to preceding energy balance but is influenced by time of day.
Figure 1.

Correlation between energy expenditure and peak hunger in two studies in which exercise energy expenditure of between 2300 and 2500 KJ was inserted between the morning and midday meal (A) or between both morning and midday, and midday and afternoon, meals (B). Correlation coefficient between energy expenditure during overnight fast before the morning meal, and during morning intermeal interval that included exercise on one hand and peak hunger at the next meal was 0.06 in A. In B, the correlation coefficient between intermeal intervals that included exercise and peak hunger at the subsequent meal was 0.0002. Data from Ref. [69].
The universal circadian and ultradian patterning of mammalian feeding behavior suggests the operation of a central circadian meal- and hunger-timing mechanism where the signals related to meal digestion may be entrained to an ultradian gastric-contraction oscillator. The circadian clock restricts the predisposition to seek and take food to the active portion of the day when it is interrupted only by the GI signals of fullness and suppresses it during the inactive phase. The uniformity and regularity in the postprandial rise in hunger and attainment of peak hunger regardless of the pre-meal energy balance is consistent with suppression by the GI stimuli of the influence of a central food-seeking command. Energy content of orally taken food appears responsible for partial suppression of hunger when the stomach is incompletely filled (Figure 2A and B). Here, GI nutrient sensing and the rate of stomach emptying according to the energy content of the meal may affect the predisposition for supplementary food intake. Circadian control of hunger and initiation of eating is inferred from low morning and evening hunger and a hunger acrophase between 10 and 19 h[69] that are independent of variations in pre-meal energy availability[39] (Figure 1). An empty stomach and completed GI transit of food generate peak pre-meal hunger during wakeful portion of diurnal cycle (Figure 2A and B) and could do so through removal of gastrointestinal inhibition over the central circadian command guiding the predisposition to eat.
Figure 2.
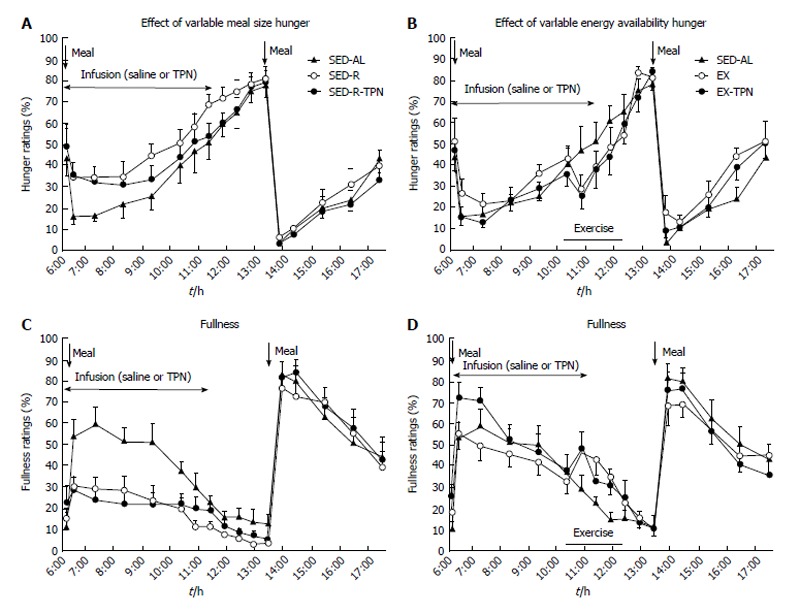
The effects of variable meal size (A) and energy availability (B) on the psychophysical ratings of hunger (A and B) and fullness (C and D) in 10 postmenopausal women subjected to a sedentary trial with a large morning meal (SED-AL), or a small morning meal SED-R, 2 h of moderate intensity exercise after a large morning meal (EX), and iv nutrient infusion (TPN) as a replacement of energy withheld from a morning meal (SED-R-TPN) or expended through exercise (EX-TPN). Meal size had a negative effect on hunger (Fdf4,36 = 39.3, P < 0.0001) and a positive effect on fullness (Fdf4,36 = 115.3, P < 0.0001). Exercise energy expenditure had a negative effect on hunger (Fdf4,36 = 25.5, P < 0.0001), and a positive effect on fullness (Fdf4,36 = 42.8; P < 0.0001). TPN had no effect on psychophysical ratings. Data from Ref. [39].
OPPORTUNISTIC AND HEDONISTIC CONTROL OF MEAL-TO-MEAL FEEDING: THE ROLES OF TASTE, OLFACTION, GI NUTRIENT SENSING, AND SOCIAL FACILITATION
In contrast to much of our physiology that operates automatically, we have an innate capacity to consciously detect and prefer foods with sweet and savory taste[70] that leads to predisposition for acceptance and intake of palatable food. Sweet and savory nutrients elicit swallowing even at a fetal stage of development[71], positive facial expressions and sucking in newborn infants[72], and acceptance of palatable foods by children[73]. Sampled nutrients bind to five different populations of taste receptors in the mouth. Their gustatory properties are signaled in the afferents of facial (VIIth), glossopharyngeal (IXth), and vagus (Xth) nerves and are relayed to the rostral two thirds of the nucleus of the solitary tract (NTS) in medulla oblongata[74]. Gustatory information also reaches parabrachial nucleus in the pons[75], ventral tegmental area[76], and several regions of the cortex to elicit hedonic appreciation of the properties of the food. The amygdala and medial and mid-anterior edge of orbitofrontal cortex, and anterior cingulate and insular cortex contribute the emotional component of hedonic responses. The nucleus accumbens (NA) in ventral pallidum contributes to hedonic reinforcement of intake of palatable food through the release of endocannabinoids[76-78]. These innate properties justify the hypothesis that non-homeostatic olfactory and gustatory stimuli provide incentives for non-homeostatic intake of food.
Olfactory and gustatory stimuli complement sensing by the GI tract of food properties and eliciting digestive and absorptive endocrine reflexes[79]. Chemosensory receptors for sugars, amino acids, and fatty acids are located in the neuroendocrine epithelium of the stomach, duodenum, and small intestine. By sensing ingested nutrients, chemosensory neuroendocrine cells in the stomach secrete gastrin from G cells. In the intestine, ghrelin is released from P or X/A cells, somatostatin from D cells, cholecystokinin (CCK) from I cells, serotonin from enterochromaffin cells, glucose-dependent insulinotropic peptide (GIP) from K cells in the proximal small intestine, while glucagon-like peptides (GLPs) and peptide tyrosine tyrosine (PYY) are released from L cells in the distal small intestine. These GI hormones bind to receptors on the afferent vagal fibers that are located in the lamina propria[80]. Stoichiometric GI endocrine responses to energy content of ingested nutrients affect the rate and duration of nutrient digestion and absorption. Some digestive hormones also elicit conscious sensation. Ghrelin increases olfactory salience of food stimuli, decreases olfactory detection threshold, and elicits sniffing[81] as its secretion rises in parallel with pre-meal appetite and declines with meal completion. This action is its most probable contribution to facilitation of the pre-meal appetite[34,35]. Besides their digestive roles in promoting enzyme release and slowing the rate of stomach emptying, CCK[82-85], GLP-1[86-88], and PYY[89], also contribute to the conscious detection of stomach fullness and therefore participate in short-term meal-associated control of post-meal satiation.
Opportunistic characteristic of feeding also is revealed in its responsiveness to the abundance of food and communal food setting. More fluid is consumed if presented in tall, rather than short, glasses[90]. Savory food is consumed in greater amounts from larger platters than from small ones[91]. More food is eaten in company of others[92-96], a social facilitation phenomenon widely shared by mammals[97-99] and even birds[100]. Further, increasing the number of palatable food choices in all-you-can-eat settings leads to overeating in animals[3] and humans[101-103]. In effect, that represents the basis for producing experimental obesity by providing animals fat-enriched, in addition to standard laboratory, diet[3].
A direct test of the homeostatic metering of energy during feeding requires either changing the caloric density of food or the magnitude of pre-meal energy expenditure (EE). Studies manipulating the energy content of food and the meal size indicate that sensations of fullness after the meal and the amount eaten in the subsequent meal are guided by the volume of food eaten rather than its energy content[104-106]. That such non-homeostatic eating bears no direct relationship to the energy content of ingested food also during a longer time frame was suggested by an 11-wk study in which 13 females were provided with either low-fat (20%-25% of energy as fat), or a higher fat, diet (35%-40% fat)[107]. The volume or weight of food eaten daily was comparable on the two diets resulting in a daily energy intake error of 1.22 KJ. Only 35% of this caloric error on a low-fat diet was compensated by the end of 11 wk resulting in a weight loss of 2.5 kg, twice the amount of weight lost on a higher-fat diet.
A more rigorous test of human ability to homeostatically sense energy availability in non-deprived state requires that hunger and food consumption show evidence of caloric compensation when oral, olfactory, and GI sensing is bypassed. Three circumstances that meet that criterion include already mentioned prolonged nocturnal period without food, exercise energy expenditure (EEE), and intravenous supplementation of withheld or expended calories in the form of total parenteral nutrition (TPN). Examination of the effects of between 2300 to 2500 KJ of EEE inserted during morning and afternoon IMIs reveals that this increase in energy expenditure does not influence peak hunger ratings at the onset of the next meal[69] (Figure 1). A similar lack of a relationship between pre-meal energy expenditure and the size of spontaneous meal was previously described in rats[40]. In another study, the search for compensatory changes in food intake was extended to manipulations of EEE, intravenous TPN supplementation for energy withheld in a small meal or for EEE, and the size of meals taken by oral and intragastric route. In this crossover study[39], ten overweight postmenopausal women were provided with a large breakfast containing 2100 KJ in three trials and a small one containing 420 KJ in two trials. The energy supply in the large breakfast in one trial was cancelled by 2270 KJ EEE in another, and EEE was largely replaced by intravenous infusion of 1530 KJ of TPN in the third trial. The low energy content in the small 420 KJ breakfast in the fourth trial, was supplemented with the intravenous infusion of 1530 KJ of TPN in the fifth. The results showed unequivocally that changes in the sensations of hunger (Figure 2A and B) and fullness (Figure 2C and D) were only elicited by the size of the meals taken by oral, and processed by GI, route but not by energy lost exercising or supplemented intravenously. Moreover, the quantity of food eaten, and peak hunger rating at the onset of the next ad-libitum meal is indistinguishable among the five conditions, two of which generated substantial negative energy balance (Figure 3). Furthermore, hormones insulin and leptin tracked accurately changes in energy balance that resulted from unequal meal size, energy lost exercising, and energy supplemented intravenously (Figure 4), but the changes in their plasma concentrations bore no apparent relationship to conscious sensations of hunger and fullness (Figure 2).
Figure 3.

Effect of the morning energy availability (A) on the energy consumed during the midday meal (B) and the residual postmeal energy balance (C) in 10 women subjected to small (SED-R) or large (SED) morning meals, exercise (EX), and TPN (SED-R-T, EX-TPN). Midday meal did not compensate for the significantly lower energy balance in SED-R and EX trials (Fdf4,45 = 77.2; P < 0.0001), which remained uncorrected after the meal (Fdf4,45 = 10.2; P < 0.0001). Data from Ref. [39].
Figure 4.

The effects of variable meal size (A) and energy variability (B) on the plasma concentrations of insulin (A and B), GIP (C and D) and leptin (E and F) under the conditions described in Figure 2. Insulin showed significant postprandial increases to meal size (A) and TPN (B) (Fdf4,45 = 25.7; P < 0.0001), whereas GIP responded only to meal size (Fdf4,45 = 42.3; P < 0.0001). Neither insulin nor GIP responded to exercise energy expenditure. Plasma leptin concentration slowly and progressively decreased in response to reduced energy availability caused by small meal (Fdf4,36 = 48.1; P < 0.0001) and exercise (Fdf4,36 = 39.1; P < 0.0001), and this response was abolished by large meal and TPN. Data from Ref. [39].
Collectively, the above studies support the hypothesis that intermittent meal-to-meal eating under unrestricted access to food is guided by cues provided by oral and GI processing of food. Hunger and fullness ratings, the conscious guides for food intake and meal termination, are affected by the size of the orally ingested nutrients (Figure 2) but not by fluctuation in short-term energy availability caused by intravenous nutrient infusion or by EEE, or by changes in the plasma concentrations of insulin and leptin[39]. Moreover, the peak hunger rating at the onset of the next meal, and the amount eaten during that meal are not responsive to preceding energy imbalance[39,69,104-107]. Stomach filling as a guide to meal size held true in the 11-wk study in which the subjects were largely unresponsive to the energy content of the food[107].
Additional supportive evidence for the role of GI signaling rather than for homeostatic metering of pre-meal caloric deficit in the control of ad-libitum meal-to-meal eating is available in the singular success of various forms of bariatric surgery in curbing hunger and reducing food intake. A common feature of several variants of bariatric surgery is reduction in stomach capacity to hold food and associated suppression of appetite and hunger[108] or increased nausea and vomiting[109]. The efficacy of stomach fullness as a deterrent for hunger and food intake is also evident in successful application of inflatable balloons to induce weight loss[110,111]. A century ago, Cannon and Washburn[112] demonstrated a striking concordance between episodic bursts of gastric contractions and intermittent sensations of hunger using intragastric balloons as pressure gauges. In addition to Cannon’s classic demonstration of the correlation between the ultradian periodicity of empty stomach contractions and hunger, connections of mechanosensitive elements in the smooth muscle of the stomach with the afferent vagus also have been documented more recently[113-115]. Further, these GI smooth muscle mechanoreceptors inhibit eating in response to volume of food introduced into the stomach without regard to its nutritional properties[116,117]. On the other hand, nutrient quality and energy content are sensed by vagal receptors in the intestine and lead to secretion of digestive hormones such as CCK/gastric leptin, GLP-1 and PYY[117]. More recently, pooled data from 8 studies on 67 healthy humans confirmed Cannon and Washburn observation by identifying pyloric pressure waves and peak CCK concentrations as predictors of food intake while finding intravenous nutrient infusions ineffective[118].
In its basic outline, the blueprint of human non-deprivation meal-to-meal eating bears a striking resemblance to the feeding mechanism of a blowfly[119,120]. The insect whose adult body mass is confined within a rigid exoskeleton, accepts sapid solutions whenever its crop is empty. Similar to ad-libitum feeding humans in whom termination of growth imposes a finite body mass, blowfly’s food acceptance operates on an opportunistic and hedonic principle, and feeding termination on a GI negative feedback. The fly will ingest to capacity higher concentrations of sweet solutions rather than larger quantities of more dilute solutions. It stops feeding when its full crop inhibits a brain mechanism responsible for predisposition to seek and ingest nutrients whenever the crop is empty. If the recurrent nerve that provides the negative feedback from the crop to the brain is severed, the animal overeats, and with sufficiently high sugar concentrations, will rupture its crop. Presented evidence supports the conclusion that a similar system of nonhomeostatic meal-to-meal eating operates in humans. However, these considerations still leave unanswered the question regarding the signal initiating hunger and food intake. The present reinterpretation of energy regulation proposes that a central ANS command mechanism, given temporal structure by the SCN master circadian clock is responsible for sustained food seeking and meal intake interrupted intermittently by the inhibition from the signaling of gastric distension as sensations of satiation and fullness associated with GI processing of food. This proposition is consistent with close anatomical connections between SCN and the ANS energy regulatory circuits, circadian and ultradian pattern of meal eating and sympathetic activation of BAT thermogenesis, and disruption of both feeding pattern and thermogenesis and DIT in particular with lesions of either the master clock or the ANS energy regulatory substrates.
COUNTERREGULATION BY LEPTIN OF INSULIN SECRETION, ACTION, AND SENSITIVITY
The key feature of the proposed novel view of body weight regulation is the counterregulation of insulin by leptin under the control of the ANS-circadian command mechanism. Leptin counterregulates insulin in four ways, by (1) acting as a gut peptide signaling satiating fullness and contributing to termination of meals; (2) suppressing insulin secretion; (3) counteracting insulin anabolic actions; and (4) regulating ANS and peripheral tissue sensitivity to insulin in response to downward or upward deflections in the components of body mass. Through these counterregulatory interactions with insulin, leptin matches its sympathetic energy expending actions to the parasympathetic energy conserving actions of insulin.
The sustained stoichiometric relationship between the body fat mass and basal leptin secretion[11] has strongly influenced formulation of a homeostatic lipostatic hypothesis of body fat regulation featuring leptin negative- feedback from WAT to the brain. Integration of short-term secretory responsiveness of leptin to fasting[121-124], meal intake[123,125,126], glucose[127-129], pyruvate[128], insulin secretion[121,130], and insulin-stimulated carbohydrate metabolism[39,127,131-133] with the long-term parallel shifts in plasma leptin concentration and body fat mass has largely escaped scrutiny. To update the understanding about leptin physiology, it should be pointed out that, besides the WAT[134], the hormone also is secreted from the stomach[135-143], placenta[144], and lactating mammary glands[145]. Since leptin of gastric origin is likely to react more rapidly to short-term fluctuations in prandial state than leptin of WAT origin, and both may contribute to short-term changes in circulating leptin concentration, it is useful to briefly review how gastric leptin secretion and appearance in circulation differs from that arising in WAT.
Gastric leptin is rapidly mobilized by cholinergic neurotransmission, nutrient entry into the stomach[139], and release of CCK[135]. Its release is distinctly regulated by these stimuli in contrast to the leptin release from the WAT which is predominantly released in a constitutive fashion[128,134,140,146,147]. Leptin is released into the stomach lumen in exocrine fashion from the chief cells in gastric mucosa. Complexing of gastric leptin with its soluble receptor (LepR) prior to being released from the Golgi apparatus protects it from denaturation by gastric acid[141]. It then is transported to the duodenum where it binds with LepR on the luminal membrane and is transcytozed into the Golgi apparatus of the duodenal enterocyte. There it again binds with LepR and leaves the intestinal mucosa for systemic circulation[139-141].
The first counterregulation of insulin by leptin is clearly of gastric origin and consists of its counteracting the absorptive actions of insulin during a meal. Gastric leptin, mobilized by ingested nutrients and CCK, potentiates the satiating effects of CCK[148,149] and GLP-1[150,151], actions that the two hormones exert in part by slowing the rate of gastric emptying[82-85,88,151], trigerring a sensation of fullness and thus contributing to the termination of a meal. The potentiation by leptin of satiating properties of CCK is mediated by vagal primary afferent neurons selectively responsive to both hormones and to gastric distension and transmitting gastric stretch information to NTS[152] via vagal sensory nodose ganglion[153]. Activation of gastric smooth muscle mechanoreceptors is sensitive only to volume of food introduced into the stomach without regard to its nutritional properties[116] while vagal intestinal receptors sense directly the nutrient quality and energy content of ingested food[117]. The potentiation by leptin of CCK satiating effect is activated by nutrient intake while fasting and obesity attenuate vagal afferent stretch signaling[154]. Repeated gastric overstretching, common in overeating and some eating disorders, delays onset of feeding, suppresses leptin concentration and reduces neuropeptide Y levels in ARC and NTS after meal intake as compared to no stomach overstretching[155]. The indirect involvement of leptin in the control of postprandial insulin response and the meal size explains the lack of a relationship between its postprandial concentration (Figure 4C and D) and sensation of fullness (Figure 2C and D). The role of gastric leptin in curtailing postprandial insulin actions may contribute to increased food consumption in free feeding individuals[12,13] and animals[14] who have an inability to produce leptin or leptin receptors. In line with the parasympathetic source of gastric leptin elicitation, the sympathetic actions of leptin suppress cardiac rate by acting on the rostral ventrolateral medullary heart pacer[156,157].
Since leptin of both gastric and WAT origin reaches systemic circulation, it is difficult to distinguish their relative role in the remaining three counterregulations of insulin by leptin. Similar to the responsiveness of gastric leptin to meal ingestion, secretion of leptin from WAT adipocytes also is responsive to short-term fluctuations in prandial state and a number of hormones[160,161]. Feeding increases leptin secretion from WAT cells[134,140,147] and fasting decreases both[147]. Endocrine secretagogues are insulin[128,158,159] and cortisol[158-160], and inhibitors are β-adrenergic stimulation, adrenocorticotropic hormone (ACTH), alpha melanocyte stimulating hormone (αMSH)[161] and testosterone[161,162]. Furthermore, carbohydrate metabolism has to be present for insulin to increase leptin secretion[129,133], linking the WAT cell responses to short-term metabolic changes. The uncertainty as to the origin of circulating leptin particularly arises when the hormone is being stimulated by systemic administration of insulin in hyperinsulinemic euglycemia. This stimulus applied for longer than 3 to 4 h increases leptin concentration in the plasma[130,163,164] but not if the duration of the clamp[165-170] or of the postprandial period[170,171] is shorter or if hyperinsulinemia is accompanied by hypoglycemia[124].
The second way that leptin counterregulates insulin is by suppressing its secretion in pancreatic β cells[172-176] as shown by insulin oversecretion after deletion of leptin receptors in these cells[176] (Figure 5, circle). Thus, after leptin gene deletion or pharmacological antagonism of leptin action, insulin secretion is supranormal, and leptin administration in ob/ob mice that are unable to produce leptin suppresses it[172-177]. Insulin oversecretion results from leptin counterregulation of insulin secretion and not from obesity because it occurs before any significant tissue fat accumulation takes place[176].
Figure 5.

The conceptual model of the autonomic regulation of body weight. Autonomic nervous system regulates the energy flux through the energy conserving actions of insulin that are counterbalanced by the energy expending actions of leptin to match energy intake (EI) and expenditure (EE) and maintain stable body weight (center circle). The counterbalancing is achieved by the upregulation of leptin by the glycolytic energy flux in the WAT stimulated by insulin. Leptin, in turn, inhibits insulin secretion and actions in several organs. If dieting or food scarcity cause weight loss (left arrow), energy conservation is achieved, in part, by reduced Symp activity and EE (EEm). Predominance of Parasymp actions are manifested in reduced fasting insulin and leptin concentrations, increased tissue sensitivities to insulin (Si) and leptin (Sl) along with increased sensitivity in enzymatic nutrient sensing (Se) of energy depletion. In addition, energy is conserved through reduced S activation of metabolism. When overeating and reduced physical activity result in obesity (right arrow), there is a reverse change in fasting insulin and leptin concentrations, tissues become resistant to both hormones as well as to S elicitation of metabolic EE. Due to insulin and leptin resistance, the ineffective compensatory increase in S activity to counteract further body fat, lean body mass, and bone accretion, mainly causes vasoconstriction and hypertension.
The third way that leptin counterregulates insulin is by suppressing its lipogenic and other anabolic actions. While the catecholamines and growth hormone facilitate lipolysis and lipid utilization to systemic signals of energy deficit[178,179] and actually decrease leptin gene expression in WAT[180-182] and its circulating concentration[183-187], leptin binds to adipocytes to selectively counteract insulin-stimulated lipogenesis and activate lipolysis and lipid utilization in WAT[188], especially in its visceral conpartment[189]. It similarly counterregulates insulin lipogenesis in other tissues and thus reduces triglyceride (TG) content in pancreas[190], liver[189-193], and the muscle[190,194-198]. In the liver[192,199], the skeletal muscle[197,200], the BAT[201] and WAT[202], leptin shifts the metabolism from insulin-mediated carbohydrate utilization and TG synthesis toward free fatty acid (FFA) uptake and increased lipid utilization. In the skeletal muscle, leptin activates the enzyme 5’-AMP-activated protein kinase (AMPK) that is capable of sensing metabolic energy depletion[190,194,195]. AMPK in turn inhibits fat synthesis and facilitates FFA entry into the mitochondria for fat oxidation[195-198,203,204]. While some of these metabolic leptin actions result from the hormone binding directly to its receptors in peripheral target organs such as the pancreas[190] the WAT[188,189], the liver[205,206], and the muscle[198,207], the same actions also can be achieved by leptin binding to its receptors in the brain. Suppression by leptin of lipogenic actions of insulin in the WAT[205,208-211] and liver[205,206] is controlled both by the brain, particularly the VMH[208-211] and also is effected at the tissue level[205], particularly in the liver[206].
Leptin counteracts insulin’s postprandial anabolic effects by stimulating DIT. It does so by upregulating the thermogenic uncoupling protein UCP1 in BAT by increasing sympathetic nerve activity[123,124,212,213] and norepinephrine turnover in BAT[214]. It also upregulates UCP2 in WAT[215,216], and UCP3 in skeletal muscle[217]. Leptin increases muscle thermogenesis by stimulating substrate cycling[218,219], both lipid and carbohydrate oxidation[200], and expression of genes for anaerobic glycolysis, a metabolic pathway that is bioenergetically less efficient than lipid oxidation[203,204]. While insulin increases postprandial metabolism and thermogenesis through its stimulation of carbohydrate oxidation and sympathetic activation of fat oxidation in BAT[220-222], thermogenic actions of leptin are yoked to postprandial insulin release.
The fourth way that leptin counterregulates insulin action is by controlling the sensitivity of peripheral tissues and the brain to insulin actions as body fat and body masses deviate from the adult plateau. Considering first the peripheral tissues, it is well established that insulin sensitivity increases with body fat and body mass losses, and insulin resistance increases with body fat and body mass gains. Tissues such as the liver, muscle and the WAT display direct autoregulatory increases in numbers of spare receptors, hormone-receptor binding[223], and enzyme sensitivity to nutrients as they are depleted of storage molecules and structural proteins. After glycogen-depleting exercise, activity of glycogen synthase increases in proportion to the magnitude of glycogen depletion which leads to a faster rate of glycogen resynthesis during recovery from exercise[224,225]. As they are depleted of storage nutrients, liver, muscle, and WAT develop direct and autoregulatory increases in sensitivities to the anabolic actions of insulin[191,223,225-227] and catabolic actions of catecholamines[228] some of which are induced by counterregulatory actions of leptin[190-193]. Changes in hormone sensitivities and responses are greater to more rapid rather than to gradual or prolonged reductions in energy availability. Insulin sensitivity (IS) increases more during the initial weight loss than during maintenance of reduced body weight[229]. Declines in leptin concentration are greater during faster weight loss over a two-day food restriction[230] than to a slower but cumulatively larger energy deficit extended over a 4-[231] or 7-d period[122]. During weight loss, sympathetic activation of metabolic EE is suppressed and only the release of adrenal epinephrine[232] regulates the metabolic shift to predominant lipid utilization[222].
The insulin sensitizing effect of leptin in peripheral tissues becomes manifest as body mass index (BMI) declines below 25 kg/m2 and fasting plasma leptin concentration drops below 15 ng/dL[233,234]. At its low plasma concentrations, leptin contributes to insulin’s parasympathetic actions by increasing muscle glucose uptake[201,235,236] achieved in part by inhibiting the expression of negative regulators of glucose transporter type 4 (GLUT4) translocation to the membrane[237]. By restraining visceral fat accumulation and insulin oversecretion[191-226], leptin preserves insulin sensitivity in the liver[191,226,238] implicating hyperinsulinemia in resistance to insulin action. When the visceral fat is surgically removed[226], reduced glycogenolysis and hepatic glucose production, increased glucose uptake, and reduced insulin requirements to maintain euglycemia are all markers of increased IS. In addition, metabolic gene expression in favor of reduced WAT fat synthesis also results from visceral fat removal[226]. In the oxidative skeletal muscle, leptin counteracts insulin facilitation of intramyocellular triglyceride synthesis and storage by activating AMPK[200]. Through this action, leptin preserves the sensitivity of muscle to insulin leading to increased glucose uptake and glycogen synthesis[175,225]. In addition to being able to exert some of these actions directly in respective tissues studied in vitro[175,209], most of leptin actions are contingent on its systemic counteraction of insulin secretion and actions.
The physiological significance of insulin sensitizing actions of low leptin concentrations in weight-reduced state is that it contributes to increased metabolic efficiency that facilitates weight regain and a shift in the ANS balance in favor of the parasympathetic activation[239] (Figure 5, left arrow). A rebound increase in carbohydrate utilization and insulin oversecretion in insulin-sensitive state during post-deprivation overeating in the rats[240,241] is comparable to the postlesion insulin oversecretion after VMH-ARC damage that is prevented by subdiaphragmatic vagotomy[20].
With weight gain at body mass indices above 25 to 27 kg/m2[233,234] caused by the oportunistic and hedonic design of human meal-to-meal eating where energy intake and expenditure are loosely coupled[242-245], rising basal plasma concentrations of insulin and leptin lead to peripheral tissue resistance to the two hormones[233,234,246,247]. Although adult human adipose tissue retains some capacity to expand both through hyperplasia and hypertrophy[248-250] and is refractory to reductions in adipocyte numbers[251], the parallel rises in obesity and tissue resistance to high plasma leptin and insulin concentrations limit additional body fat and mass accumulation. Resistance to both hormones[246,252] has several causes. An enzymatic resistance to anabolic actions of insulin and counterregulatory actions of leptin[198,207,253] develops in part due to downregulation of respective receptors exposed to prolonged high insulin[173] and leptin[179,181] concentrations. Insulin resistance (IR) also develops due to impaired hormone signaling that results from the action of intermediates of fat biosynthesis driven by high circulating lipid concentrations[253] and accumulation of TG in peripheral organs[200,209,252,253]. Although IR and leptin resistance (LR) increase in parallel with the rise in adiposity, they differ in the timing of their development and their relationship to WAT mass[163,254,255]. Hyperinsulinemia causes hyperleptinemia[163] and both lead to IR and LR. A decline in insulin signaling and IS is a consequence of hyperinsulinemia rather than of IR, since its correction with insulin-lowering diazoxide restores IS and prevents development of obesity while treatment of IR with metformin does not[173]. IR has received a lot of medical attention as a gateway to type 2 diabetes. However, development of IR and LR can also be viewed as an important compensatory processes in autonomic regulation of energy flux in the form of both enzymatic[15,256,257] and sympathetic resistance against additional accretion of body fat. The autonomic resistance to accretion of additional energy storage involves an increase in sympathetic activation of thermogenesis[258] (Figure 5, right arrow), the action of which is rendered ineffective by resistance of enlarged adipocytes to actions of catecholamines[193,228]. The deleterious health consequence of sympathetic overactivation and tissue resistance to hormones in obesity are increased vasoconstriction and hypertension[259-262]. Finally, peripheral LR is possibly dissociable from the resistance of the brain and ANS to leptin actions because of its origin from two different sources, stomach and the WAT, and different routes of accessing the brain, vagal transmission of gastric leptin signals to the NTS, and endocrine signaling of both gastric and WAT leptin to the hypothalamus. This dissociation is suggested by continued effectiveness of leptin when administered intracerebroventricularly at the time dietary obesity has rendered leptin applied intraperitoneally ineffective[263].
Remarkably and importantly leptin controls insulin sensitivity of the ANS energy regulatory command center as body fat and body masses deviate from the norm. The brain substrate that is responsive to changes in body fat and body mass is midbrain ventral tegmental (VTA) dopaminergic and opioidergic projection to the NA in the ventral striatum[76,77] that has rich interconnections with hypothalamic and cortical circuits responsible for activation and inhibition of feeding, voluntary activity, and thermogenesis. The key neurotransmitter mediating behavioral reinforcements is dopamine (DA)[264,265], originating in medial VTA and projecting to ventromedial striatum including medial olfactory tubercle and medial shell of the NA[265]. Activation of these midbrain DA neural projections to ventral striatum supports nonhomeostatic motivating, rewarding, and incentive properties of food and drives locomotor and eating behaviors[76,77,266-268]. Functional connections between the hypothalamus and this motivational circuitry is illustrated by the LH being the key effective site for behavioral self stimulation with mild electric current[269,270]. LH area also is responsible for arousal and incentive activation of locomotion probably linked to search for food through its component ghrelin[271], melanin concentrating hormone (MCH), and orexin/hypocretin[272-274] neural circuits. LH ghrelin is involved in anticipatory meal-associated increase in locomotion[271] and increases in olfactory stimulus salience during intermeal intervals[81]. MCH neurons regulate olfactory locomotor food-seeking behaviors[272]. In addition to motivating feeding[273], MCH neurons affect energy metabolism[274-277], and their secretion is regulated by gut peptide GLP-1[276], leptin[277], and β3 adrenergic stimulation[278]. Distinct presympathetic-premotor neurons in LH express both orexin and MCH[279]. Orexin-hypocretin neurotransmission elicits circadian periodicity of locomotion[280], locomotor food seeking, and sequencing of postprandial behavioral satiety and grooming[281,282]. Activation of LH orexin-hypocretin neurons is functionally connected to DA reward circuit[282]. Further, the hyperactivity in anorexia nervosa is hypothesized to be driven in part by increased ghrelin signaling to DA neurons in ventral tegmental area during underweight and hypoleptinemia[283].
At this point, the attention should be brought to the fact that spontaneous locomotion and physical activity levels are, like meal-to-meal eating, under nonhomeostatic control although their interaction brings about the stability of adult body weight[284]. Cross-sectional human data show that total non-basal energy expenditure normalized for body mass is inversely related to body fat[285,286], and that morbidly obese individuals are almost completely inactive[287]. On the other extreme, underweight subjects with anorexia nervosa are known for compulsive running, “drive for activity”, and “restlessness”[288,289]. This paradox where overweight and obese subjects reduce locomotor energy expenditure while the underweight ones are hyperactive, defies the homeostatic expectations. Several lines of experimental animal research confirm the inverse relationship between spontaneous physical activity and body fatness. Obesity induced by either VMH lesions in rats[23], rostromedial septal lesions[290] and hippocampal[291] or septo-hypothalamic transections[292] in hamsters, or cafeteria and high-fat diets in neurologically intact animals[3,293,294] reduce spontaneous running activity. On the other hand, severe dietary restriction consisting of only 2-h access to food, leads to weight loss in rats and up to 300% to 500% increase in spontaneous running activity to the point of emaciation[295]. Spontaneous running by rodents in wheels is a motivated behavior amplified by the device challenges[296] and mediated in part by μ opioids[297]. The inverse relationship between body fat and activity levels is associated with neurochemical changes in brain areas where damage produces obesity and hypoactivity[293]. Obesity-inducing brain lesions in hamsters abolish the inverse relationship to body fat[292]. This then indicates a neurochemical link between the nonhomeostatic physical activity and body fat and body mass.
The motivational basis of spontaneous activity can be demonstrated by placing obese and hypoactive lesioned animals on a motor-driven treadmill. Mild electrical shock at the base of treadmill provides external motivation for animals to keep running on the moving track. Compared to neurologically intact animals, obese hypoactive animals can be compelled by such external negative incentive to run on a treadmill as long and as fast as the intact controls[298]. In a similar vein, rats displaying hyperphagia during ad libitum access to food following a weight loss, display reduced willingness to run and need external incentives to increase their activity[241].
So how then do non-homeostatic feeding and non-homeostatic spontaneous physical activity add up to maintenance of stable adult body mass and composition? The evidence presented so far permits a conclusion that the intermittent feeding and locomotor and other physical activities are loosely coupled with continuous body energy drain[244-246,248]. The way they result in stable body mass setpoint is by sharing the same neural substrate which provides variable reward for these behaviors based on the changes in the brain substrate’s sensitivity to circulating concentrations of insulin and leptin. The brain substrate that supports motivations to locomote and search and ingest food is richly populated by insulin[299] and leptin[300] receptors and consists of dopaminergic projections from VTA to NA in the ventral striatum, to limbic forebrain structures and to orbitofrontal cortex[265-267]. Endogenous opiates and cannabinoids[301,302] also are components of this DA reward circuitry, with most of NA, but also some of its parts in particular, showing increased hedonic responding to sweets after stimulation of μ opioid receptors[303]. Mu opioids are also implicated in the motivation for spontaneous running[297]. LH circuits responsive to circulating ghrelin and signaling through MCH and orexin-hypocretin neurons[304] also are associated with DA reward system[273,282,283] in supporting behavioral activation and search for food. The basis of changes in incentive value for locomotor search and ingestion of food[76,77,264-268,302] is vested in changes in the brain substrate’s sensitivity to changes in the concentrations of the two hormones as body mass undergoes deviations from the adult norm. Withdrawal of leptin during weight loss reduces its counterregulation of insulin actions, increases the sensitivity of the brain reward substrates to locomotor, olfactory, and gustatory rewards and increases the efficiency of insulin actions leading to lipogenesis and recovery of depleted body energy reservoirs. Leptin administration to underweight humans and animals suppresses the motivation to eat[305], insulin metabolic efficiency[203,204,305], and motivation for spontaneous locomotion[306-309]. With body mass loss and declines in leptin and insulin concentration, increased parasympathetic activation and sensitivities of tissues to insulin and leptin action facilitate efficient energy storage. Insulin actions are enhanced by reduced leptin counterregulation of its secretion and actions (Figure 5). The parasympathetic dominance in underweight state is reflected in hyperphagia, insulin oversecretion to food intake, and increased efficiency of energy storage that prevail as long as peripheral and central insulin and leptin concentrations remain low and tissue and ANS sensitivity to their actions high.
As increased hunger and metabolic efficiency drive restoration of body fat and body stores to pre-deprivation plateau, the sensitivity of the brain reward circuit declines. The transport of both hormones into the brain also declines[310,311], a process that most likely signals that predeprivation body weight setpoint has been attained. Accrual of excess body fat and body mass along with increases in basal insulin and leptin concentrations leads to reduced motivation to locomote, while feeding is supported in part by palatability of food rather than responsiveness to hunger[76,77]. When excess fat is gained, increased basal concentrations of both insulin and leptin lead to reduced peripheral tissue sensitivity to their actions, and increased activation of sympathetic tone develops as a countermeasure against further body fat and body mass accretion (Figure 5). Thus the brain reward circuit is a component of the autonomic-circadian command center responsible for balancing of sympathetic and parasympathetic processes in part by controlling the secretion of insulin and leptin.
Alternating cycles of famine and feast very likely produced the evolutionary pressure toward coupling of nonhomeostatic search for food opportunities with variable incentive rewards associated with these behaviors[312]. Meal taking and meal processing represent shorter cycles of intermittent refueling of the body that expends energy continuously (Figure 6). Pre-meal behavioral arousal and increased nonhomeostatic locomotion may reflect the activation by the central ANS/circadian command center of lateral hypothalamic neurons responsive to ghrelin, and signaling through MCH and orexin/hypocretin neurons as well as ultradian contractile activity of the empty stomach. The activation of these processes increases locomotor behavior and responsiveness to olfactory gustatory and other signals of food availability. Meal eating inhibits the ANS/circadian command center by GI signals of fullness and satiation. Post-meal grooming in animals and somnolence is induced in part by postprandial insulin secretion[313] and activation of orexin-hypocretin circuits in the LH[280]. The inhibition of the ANS/circadian command center by volumetric and hormonal signals of GI repletion declines progressively as the GI processing of food is completed allowing the sensation of hunger to progressively rise (Figure 2).
Figure 6.

The conceptual model of the linkage between nonhomeostatic meal eating and nonhomeostatic facilitation of physical activity. Completion of gastrointestinal (GI) transit of food removes the inhibitory influence of volumetric and nutritional afferent information mediated by the vagus nerve from reaching nucleus tractus solitaries and DA and opioidergic brain centers of reward.This allows activation of sympathetic actions over fuel mobilization, full operation of behavioral arousal, nonhomeostatic increase in locomotion in quest of food associated with activation of orexin/hypocretin and ghrelin. Completion of nonhomeostatically controled meal results in filling of the stomach, activation of GI nutrient sensing, and increases in postprandial plasma concentrations of insulin and leptin. Vagal projections of this information to the brain reinstate the inhibition over autonomic sympathetic actions and activate parasympathetic control of food digestion and absorption and behavioral quiescence. It is probable that weight loss increases postprandial events linked by the left arrow, and that obesity increases the postprandial events linked by right arrow. Additional consequences of weight loss and weight gain are mediated by changes in tissue sensitivities to leptin and insulin actions altering the prevailing sympathovagal balance and illustrated in Figure 5. DA: Dopaminergic.
REGULATION OF SKELETAL, LEAN AND FAT BODY MASS
Body weight losses or gains along with accumulation of excess fat by either damage to the sympathetic and circadian components of the ANS or cafeteria or high-fat food are viewed by some as a pathological dysfunction of brain substrates where leptin and insulin fail to exert a negative feedback over feeding due to neural inflammation[9]. What this formulation fails to take into account is that weight regulatory mechanism is in full operation at either starvation or obesity extreme of energy balance. Animals rendered obese by medial basal (or in the case of hamsters, septal) lesions or by cafeteria and fat diets defend their new elevated body weight plateau after it has been attained against downward deflections[3,314,315]. The lesions and hedonic nonhomeostatic overeating therefore only raise the plateau at which WAT mass is defended and do not interfere with the body mass regulatory mechanism per se. The clearest demonstration of the integrity of the body mass regulatory defenses after VMH lesions is absence of hyperphagia and hyperinsulinemia (or even presence of hypophagia) if animals are rendered obese by prolonged insulin injections prior to VMH lesion. The change in their feeding behavior lasts until they attain the usual obese body mass plateau characteristic of lesioned animals and thus demonstrate its regulatory defense[316].
The origin of the signal for body mass recovery can therefore not reside exclusively in the size of WAT or adipocyte fat content but requires consideration of the role of the other two body components, the bone and lean tissues. The bone is the probable source of such signals as its mass changes in parallel with changes in body fat. With each kilogram of body fat gained or lost, 16.5 g of bone mineral is gained or lost[317], and changes in body fat level are accompanied by changes in lean mass. In the hamster, rostral septal lesions[290] and hippocampal[291] and septo-hypothalamic[292] transections increase obesity but also elicit bone and lean body mass growth and an upward displacement of regulated body mass setpoint. Remarkably, increases in body mass setpoint without obesity also can be triggered by voluntary running in this species[318] proving Gordon Kennedy right about the interconnectedness of voluntary activity, weight regulation, and body growth[30,319-321]. The facility of producing upward displacement of body mass setpoint by voluntary running in neurologically intact mature animals provides an exceptional opportunity to examine the location and nature of neural changes responsible for termination of statural growth and initiation of regulatory defenses of the adult body mass setpoint[322,323]. That this is a maturational event is seen by growth acceleration not being possible during animals’ natural early rapid growth[323]. The requirement for growth cessation before the defenses of stable adult weight against downward deflections are initiated is shown by the necessity of pituitary gland presence for acceleration of growth by exercise[324], increased pulsatile oversecretion of growth hormone during that growth[325,326], and increased pulsatile growth hormone secretion after the disruption of the brain substrates involved in the maintenance of weight stability in non-growing animals[290-292]. Finally, the permanence of neural changes involved in the defense of the growth-induced upward displacement of body mass setpoint is seen in the phenomenon of catch-up growth[325]. If the hamsters are not given enough food during exercise, their bones and other body components cannot grow as they do when the food is available in unlimited amounts[327]. When the unlimited food is restored, previously exercised hamsters now execute catch-up growth to the approximate body mass plateau they achieve in the absence of food restriction[325]. These data thus demonstrate that exercise has raised mature hamster weight setpoint, and the catch-up growth represents a compensatory process of attaining it.
The supporting inference that leptin is involved in the regulation of lean tissues of the adult body, in particular the bone, should be credited to Gerard Karsenty. He and his research team demonstrated that leptin and sympathetic nerves regulate bone mass in adult mammals by affecting the bioactivity of the bone hormone osteocalcin (OCN)[328]. Although his studies do not include measurements of physical activity, they suggest that signals from the bone osteoblasts influence ANS circuits involved in the regulation of adult body mass. The key finding of Karsenty research was that leptin-induced increase in sympathetic stimulation of the bone suppresses its mineralization and growth. It does so by blocking bioactivity of OCN by activation of an Esp gene in osteoblasts and gamma carboxylation of the hormone. Upregulation of this gene reduces osteoblast numbers and blocks increases in bone mineralization and size. The effect requires β adrenergic receptors on the osteoblasts in the absence of which a high-bone, obese, and hypoactive phenotype is observed similar to that of VMH lesioned animals or mice with deficient leptin signaling (ob/ob and db/db mice). These findings help explain why with each kg of body fat lost, 16.5 g of bone mineral is lost, and then gained back with body fat regain[317]. Acknowledgment that all three compartments of body mass are regulated extends our understanding of the scope of the roles of leptin and ANS both in short-term nonhomeostatic behaviors and in maintenance of adult weight stability.
The proposed re-interpretation of body weight regulation presents it as a counterpoint between the sympathetic and parasympathetic actions of the ANS/circadian command center in which counterregulation by leptin of insulin secretion and actions and change in tissue sensitivities to the two hormones influence nonhomeostatric locomotor and ingestive behaviors as body fat and body mass are displaced from the stable adult norm. This novel integration offers an opportunity to revise the prevailing homeostatic view of energy regulation and to refocus weight regulation research. The inclusion of body components other than fat stores in body weight regulation expands the scope of study of this mechanism. The proposal that the role of leptin is to counterbalance energy storage associated with insulin secretion as well as help guide lost body mass to pre-deprivation setpoint prompts new hypotheses and research about its possible role in termination of growth and initiation of the maintenance of a stable adult body mass.
Footnotes
P- Reviewer: Lehtonen SH, Liu EQ, Pirola L, Zhao D S- Editor: Wen LL L- Editor: A E- Editor: Liu SQ
References
- 1.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity among adults: United States, 2011-2012. NCHS Data Brief. 2013;(131):1–8. [PubMed] [Google Scholar]
- 2.Malik VS, Willett WC, Hu FB. Global obesity: trends, risk factors and policy implications. Nat Rev Endocrinol. 2013;9:13–27. doi: 10.1038/nrendo.2012.199. [DOI] [PubMed] [Google Scholar]
- 3.Sclafani A, Springer D. Dietary obesity in adult rats: Similarity to hypothalamic and human obesity syndromes. Physiol Behav. 1976;17:461–471. doi: 10.1016/0031-9384(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 5.Woods SC, Schwartz MW, Baskin DG, Seeley RJ. Food intake and the regulation of body weight. Annu Rev Psychol. 2000;51:255–277. doi: 10.1146/annurev.psych.51.1.255. [DOI] [PubMed] [Google Scholar]
- 6.Benoit SC, Clegg DJ, Seeley RJ, Woods SC. Insulin and leptin as adiposity signals. Recent Prog Horm Res. 2004;59:267–285. doi: 10.1210/rp.59.1.267. [DOI] [PubMed] [Google Scholar]
- 7.Woods SC, D’Alessio DA. Central control of body weight and appetite. J Clin Endocrinol Metab. 2008;93:S37–S50. doi: 10.1210/jc.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan KK, Woods SC, Seeley RJ. Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metab. 2012;15:137–149. doi: 10.1016/j.cmet.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guyenet SJ, Schwartz MW. Clinical review: Regulation of food intake, energy balance, and body fat mass: implications for the pathogenesis and treatment of obesity. J Clin Endocrinol Metab. 2012;97:745–755. doi: 10.1210/jc.2011-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 11.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 12.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 13.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 14.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz MW, Niswender KD. Adiposity signaling and biological defense against weight gain: absence of protection or central hormone resistance? J Clin Endocrinol Metab. 2004;89:5889–5897. doi: 10.1210/jc.2004-0906. [DOI] [PubMed] [Google Scholar]
- 16.Adolph EF. Urges to eat and drink in rats. Am J Physiol. 1947;151:110–125. doi: 10.1152/ajplegacy.1947.151.1.110. [DOI] [PubMed] [Google Scholar]
- 17.Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59:719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- 18.Bray GA, Sclafani A, Novin D. Obesity-inducing hypothalamic knife cuts: effects on lipolysis and blood insulin levels. Am J Physiol. 1982;243:R445–R449. doi: 10.1152/ajpregu.1982.243.3.R445. [DOI] [PubMed] [Google Scholar]
- 19.Gold RM. Hypothalamic hyperphagia produced by parasagittal knife cuts. Physiol Behav. 1970;5:23–25. doi: 10.1016/0031-9384(70)90007-7. [DOI] [PubMed] [Google Scholar]
- 20.Berthoud HR, Jeanrenaud B. Acute hyperinsulinemia and its reversal by vagotomy after lesions of the ventromedial hypothalamus in anesthetized rats. Endocrinology. 1979;105:146–151. doi: 10.1210/endo-105-1-146. [DOI] [PubMed] [Google Scholar]
- 21.Nishizawa Y, Bray GA. Ventromedial hypothalamic lesions and the mobilization of fatty acids. J Clin Invest. 1978;61:714–721. doi: 10.1172/JCI108984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue S, Bray GA. The effects of subdiaphragmatic vagotomy in rats with ventromedial hypothalamic obesity. Endocrinology. 1977;100:108–114. doi: 10.1210/endo-100-1-108. [DOI] [PubMed] [Google Scholar]
- 23.Eng R, Gold RM, Sawchenko PE. Hypothalamic hypoactivity prevented but not reversed by subdiaphragmatic vagotomy. Physiol Behav. 1978;20:637–641. doi: 10.1016/0031-9384(78)90257-3. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi A, Shimazu T. Hypothalamic regulation of lipid metabolism in the rat: effect of hypothalamic stimulation on lipolysis. J Auton Nerv Syst. 1981;4:195–205. doi: 10.1016/0165-1838(81)90044-8. [DOI] [PubMed] [Google Scholar]
- 25.Saito M, Minokoshi Y, Shimazu T. Ventromedial hypothalamic stimulation accelerates norepinephrine turnover in brown adipose tissue of rats. Life Sci. 1987;41:193–197. doi: 10.1016/0024-3205(87)90493-0. [DOI] [PubMed] [Google Scholar]
- 26.Iwai M, Hell NS, Shimazu T. Effect of ventromedial hypothalamic stimulation on blood flow of brown adipose tissue in rats. Pflugers Arch. 1987;410:44–47. doi: 10.1007/BF00581894. [DOI] [PubMed] [Google Scholar]
- 27.Sudo M, Minokoshi Y, Shimazu T. Ventromedial hypothalamic stimulation enhances peripheral glucose uptake in anesthetized rats. Am J Physiol. 1991;261:E298–E303. doi: 10.1152/ajpendo.1991.261.3.E298. [DOI] [PubMed] [Google Scholar]
- 28.Dulloo AG, Miller DS. The thermogenic properties of ephedrine/methylxanthine mixtures: animal studies. Am J Clin Nutr. 1986;43:388–394. doi: 10.1093/ajcn/43.3.388. [DOI] [PubMed] [Google Scholar]
- 29.Dulloo AG, Miller DS. The thermogenic properties of ephedrine/methylxanthine mixtures: human studies. Int J Obes. 1986;10:467–481. [PubMed] [Google Scholar]
- 30.Kennedy GC. The hypothalamic control of food intake in rats. Proc R Soc Lond B Biol Sci. 1950;137:535–549. doi: 10.1098/rspb.1950.0065. [DOI] [PubMed] [Google Scholar]
- 31.Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276:R1569–R1578. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- 32.Dulloo AG, Miller DS. The effect of parasympathetic drugs on energy expenditure: relevance to the autonomic hypothesis. Can J Physiol Pharmacol. 1986;64:586–591. doi: 10.1139/y86-097. [DOI] [PubMed] [Google Scholar]
- 33.Bowers RR, Festuccia WT, Song CK, Shi H, Migliorini RH, Bartness TJ. Sympathetic innervation of white adipose tissue and its regulation of fat cell number. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1167–R1175. doi: 10.1152/ajpregu.00558.2003. [DOI] [PubMed] [Google Scholar]
- 34.Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 35.Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992. doi: 10.1210/jcem.86.12.8111. [DOI] [PubMed] [Google Scholar]
- 36.Patterson M, Bloom SR, Gardiner JV. Ghrelin and appetite control in humans--potential application in the treatment of obesity. Peptides. 2011;32:2290–2294. doi: 10.1016/j.peptides.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 37.Parker JA, Bloom SR. Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology. 2012;63:18–30. doi: 10.1016/j.neuropharm.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, et al. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci USA. 2004;101:8227–8232. doi: 10.1073/pnas.0402763101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borer KT, Wuorinen E, Ku K, Burant C. Appetite responds to changes in meal content, whereas ghrelin, leptin, and insulin track changes in energy availability. J Clin Endocrinol Metab. 2009;94:2290–2298. doi: 10.1210/jc.2008-2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Magnen J. Peripheral and systemic actions of food in the caloric regulation of intake. Ann N Y Acad Sci. 1969;157:1126–1157. doi: 10.1111/j.1749-6632.1969.tb12940.x. [DOI] [PubMed] [Google Scholar]
- 41.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maywood ES, O’Neill JS, Chesham JE, Hastings MH. Minireview: The circadian clockwork of the suprachiasmatic nuclei--analysis of a cellular oscillator that drives endocrine rhythms. Endocrinology. 2007;148:5624–5634. doi: 10.1210/en.2007-0660. [DOI] [PubMed] [Google Scholar]
- 43.Froy O. Metabolism and circadian rhythms--implications for obesity. Endocr Rev. 2010;31:1–24. doi: 10.1210/er.2009-0014. [DOI] [PubMed] [Google Scholar]
- 44.Nedeltcheva AV, Kilkus JM, Imperial J, Schoeller DA, Penev PD. Insufficient sleep undermines dietary efforts to reduce adiposity. Ann Intern Med. 2010;153:435–441. doi: 10.1059/0003-4819-153-7-201010050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Cauter E, Spiegel K, Tasali E, Leproult R. Metabolic consequences of sleep and sleep loss. Sleep Med. 2008;9 Suppl 1:S23–S28. doi: 10.1016/S1389-9457(08)70013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dark J, Zucker I, Wade GN. Photoperiodic regulation of body mass, food intake, and reproduction in meadow voles. Am J Physiol. 1983;245:R334–R338. doi: 10.1152/ajpregu.1983.245.3.R334. [DOI] [PubMed] [Google Scholar]
- 47.Kriegsfeld LJ, LeSauter J, Silver R. Targeted microlesions reveal novel organization of the hamster suprachiasmatic nucleus. J Neurosci. 2004;24:2449–2457. doi: 10.1523/JNEUROSCI.5323-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA. 1972;69:1583–1586. doi: 10.1073/pnas.69.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egawa M, Inoue S, Satoh S, Takamura Y, Nagai K, Nakagawa H. Acute and chronic effects of VMH lesions on circadian rhythms in food intake and metabolites. Brain Res Bull. 1993;31:293–299. doi: 10.1016/0361-9230(93)90220-6. [DOI] [PubMed] [Google Scholar]
- 50.Kakolewski JW, Deaux E, Christensen J, Case B. Diurnal patterns in water and food intake and body weight changes in rats with hypothalamic lesions. Am J Physiol. 1971;221:711–718. doi: 10.1152/ajplegacy.1971.221.3.711. [DOI] [PubMed] [Google Scholar]
- 51.Sakaguchi T, Takahashi M, Bray GA. Diurnal changes in sympathetic activity. Relation to food intake and to insulin injected into the ventromedial or suprachiasmatic nucleus. J Clin Invest. 1988;82:282–286. doi: 10.1172/JCI113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Veen DR, Shao J, Chapman S, Leevy WM, Duffield GE. A diurnal rhythm in glucose uptake in brown adipose tissue revealed by in vivo PET-FDG imaging. Obesity (Silver Spring) 2012;20:1527–1529. doi: 10.1038/oby.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Redlin U, Nuesslein B, Schmidt I. Circadian changes of brown adipose tissue thermogenesis in juvenile rats. Am J Physiol. 1992;262:R504–R508. doi: 10.1152/ajpregu.1992.262.3.R504. [DOI] [PubMed] [Google Scholar]
- 54.Ootsuka Y, de Menezes RC, Zaretsky DV, Alimoradian A, Hunt J, Stefanidis A, Oldfield BJ, Blessing WW. Brown adipose tissue thermogenesis heats brain and body as part of the brain-coordinated ultradian basic rest-activity cycle. Neuroscience. 2009;164:849–861. doi: 10.1016/j.neuroscience.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zvonic S, Ptitsyn AA, Conrad SA, Scott LK, Floyd ZE, Kilroy G, Wu X, Goh BC, Mynatt RL, Gimble JM. Characterization of peripheral circadian clocks in adipose tissues. Diabetes. 2006;55:962–970. doi: 10.2337/diabetes.55.04.06.db05-0873. [DOI] [PubMed] [Google Scholar]
- 56.Freeman PH, Wellman PJ. Brown adipose tissue thermogenesis induced by low level electrical stimulation of hypothalamus in rats. Brain Res Bull. 1987;18:7–11. doi: 10.1016/0361-9230(87)90026-8. [DOI] [PubMed] [Google Scholar]
- 57.Ueta CB, Fernandes GW, Capelo LP, Fonseca TL, Maculan FD, Gouveia CH, Brum PC, Christoffolete MA, Aoki MS, Lancellotti CL, et al. β(1) Adrenergic receptor is key to cold- and diet-induced thermogenesis in mice. J Endocrinol. 2012;214:359–365. doi: 10.1530/JOE-12-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci. 2011;31:12189–12197. doi: 10.1523/JNEUROSCI.2336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sinha MK, Ohannesian JP, Heiman ML, Kriauciunas A, Stephens TW, Magosin S, Marco C, Caro JF. Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1996;97:1344–1347. doi: 10.1172/JCI118551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saad MF, Riad-Gabriel MG, Khan A, Sharma A, Michael R, Jinagouda SD, Boyadjian R, Steil GM. Diurnal and ultradian rhythmicity of plasma leptin: effects of gender and adiposity. J Clin Endocrinol Metab. 1998;83:453–459. doi: 10.1210/jcem.83.2.4532. [DOI] [PubMed] [Google Scholar]
- 62.Schoeller DA, Cella LK, Sinha MK, Caro JF. Entrainment of the diurnal rhythm of plasma leptin to meal timing. J Clin Invest. 1997;100:1882–1887. doi: 10.1172/JCI119717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Licinio J, Mantzoros C, Negrão AB, Cizza G, Wong ML, Bongiorno PB, Chrousos GP, Karp B, Allen C, Flier JS, et al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med. 1997;3:575–579. doi: 10.1038/nm0597-575. [DOI] [PubMed] [Google Scholar]
- 64.Saito M, Minokoshi Y, Shimazu T. Metabolic and sympathetic nerve activities of brown adipose tissue in tube-fed rats. Am J Physiol. 1989;257:E374–E378. doi: 10.1152/ajpendo.1989.257.3.E374. [DOI] [PubMed] [Google Scholar]
- 65.Bajzer M, Olivieri M, Haas MK, Pfluger PT, Magrisso IJ, Foster MT, Tschöp MH, Krawczewski-Carhuatanta KA, Cota D, Obici S. Cannabinoid receptor 1 (CB1) antagonism enhances glucose utilisation and activates brown adipose tissue in diet-induced obese mice. Diabetologia. 2011;54:3121–3131. doi: 10.1007/s00125-011-2302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verty AN, Allen AM, Oldfield BJ. The effects of rimonabant on brown adipose tissue in rat: implications for energy expenditure. Obesity (Silver Spring) 2009;17:254–261. doi: 10.1038/oby.2008.509. [DOI] [PubMed] [Google Scholar]
- 67.Rothwell NJ, Stock MJ, Warwick BP, Winter PD. Diurnal variations in circulating hormone levels and brown adipose tissue activity in “cafeteria”-fed rats. Comp Biochem Physiol A Comp Physiol. 1983;75:461–465. doi: 10.1016/0300-9629(83)90110-x. [DOI] [PubMed] [Google Scholar]
- 68.Tanida M, Shen J, Nakamura T, Niijima A, Nagai K. Day-night difference in thermoregulatory responses to olfactory stimulation. Neurosci Lett. 2008;439:192–197. doi: 10.1016/j.neulet.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 69.Wuorinen EC, Borer KT. Circadian and ultradian components of hunger in human non-homeostatic meal-to-meal eating. Physiol Behav. 2013;122:8–16. doi: 10.1016/j.physbeh.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 70.Steiner JE, Glaser D, Hawilo ME, Berridge KC. Comparative expression of hedonic impact: affective reactions to taste by human infants and other primates. Neurosci Biobehav Rev. 2001;25:53–74. doi: 10.1016/s0149-7634(00)00051-8. [DOI] [PubMed] [Google Scholar]
- 71.DeSnoo K. Das trinkende Kind im Uterus. Monats Geburtsh Gynaekol. 1937;105:88–97. [Google Scholar]
- 72.Desor JA, Maller O, Andrews K. Ingestive responses of human newborns to salty, sour, and bitter stimuli. J Comp Physiol Psychol. 1975;89:966–970. doi: 10.1037/h0077171. [DOI] [PubMed] [Google Scholar]
- 73.Lipchock SV, Reed DR, Mennella JA. The gustatory and olfactory systems during infancy: implications for development of feeding behaviors in the high-risk neonate. Clin Perinatol. 2011;38:627–641. doi: 10.1016/j.clp.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Norgren R. The gustatory system in mammals. Am J Otolaryngol. 1983;4:234–237. doi: 10.1016/s0196-0709(83)80064-7. [DOI] [PubMed] [Google Scholar]
- 75.Dutta TM, Josiah AF, Cronin CA, Wittenberg GF, Cole JW. Altered taste and stroke: a case report and literature review. Top Stroke Rehabil. 2013;20:78–86. doi: 10.1310/tsr2001-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berridge KC. ‘Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiol Behav. 2009;97:537–550. doi: 10.1016/j.physbeh.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berridge KC. Food reward: brain substrates of wanting and liking. Neurosci Biobehav Rev. 1996;20:1–25. doi: 10.1016/0149-7634(95)00033-b. [DOI] [PubMed] [Google Scholar]
- 78.Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. doi: 10.1016/s0149-7634(02)00007-6. [DOI] [PubMed] [Google Scholar]
- 79.Janssen S, Depoortere I. Nutrient sensing in the gut: new roads to therapeutics? Trends Endocrinol Metab. 2013;24:92–100. doi: 10.1016/j.tem.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 80.Berthoud HR, Kressel M, Raybould HE, Neuhuber WL. Vagal sensors in the rat duodenal mucosa: distribution and structure as revealed by in vivo DiI-tracing. Anat Embryol (Berl) 1995;191:203–212. doi: 10.1007/BF00187819. [DOI] [PubMed] [Google Scholar]
- 81.Tong J, Mannea E, Aimé P, Pfluger PT, Yi CX, Castaneda TR, Davis HW, Ren X, Pixley S, Benoit S, et al. Ghrelin enhances olfactory sensitivity and exploratory sniffing in rodents and humans. J Neurosci. 2011;31:5841–5846. doi: 10.1523/JNEUROSCI.5680-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gibbs J, Smith GP. Cholecystokinin and satiety in rats and rhesus monkeys. Am J Clin Nutr. 1977;30:758–761. doi: 10.1093/ajcn/30.5.758. [DOI] [PubMed] [Google Scholar]
- 83.Muurahainen N, Kissileff HR, Derogatis AJ, Pi-Sunyer FX. Effects of cholecystokinin-octapeptide (CCK-8) on food intake and gastric emptying in man. Physiol Behav. 1988;44:645–649. doi: 10.1016/0031-9384(88)90330-7. [DOI] [PubMed] [Google Scholar]
- 84.Melton PM, Kissileff HR, Pi-Sunyer FX. Cholecystokinin (CCK-8) affects gastric pressure and ratings of hunger and fullness in women. Am J Physiol. 1992;263:R452–R456. doi: 10.1152/ajpregu.1992.263.2.R452. [DOI] [PubMed] [Google Scholar]
- 85.Smith GP, Gibbs J. Satiating effect of cholecystokinin. Ann N Y Acad Sci. 1994;713:236–241. doi: 10.1111/j.1749-6632.1994.tb44071.x. [DOI] [PubMed] [Google Scholar]
- 86.van Dijk G, Thiele TE, Seeley RJ, Woods SC, Bernstein IL. Glucagon-like peptide-1 and satiety. Nature. 1997;385:214. doi: 10.1038/385214a0. [DOI] [PubMed] [Google Scholar]
- 87.Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Näslund E, Gutniak M, Skogar S, Rössner S, Hellström PM. Glucagon-like peptide 1 increases the period of postprandial satiety and slows gastric emptying in obese men. Am J Clin Nutr. 1998;68:525–530. doi: 10.1093/ajcn/68.3.525. [DOI] [PubMed] [Google Scholar]
- 89.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 90.Walker D, Smarandescu L, Wansink B. Half Full or Empty: Cues that lead wine drinkers to unintentionally overpour. Subst Use Misuse. 2013 doi: 10.3109/10826084.2013.832327. [DOI] [PubMed] [Google Scholar]
- 91.van Kleef E, Shimizu M, Wansink B. Serving bowl selection biases the amount of food served. J Nutr Educ Behav. 2012;44:66–70. doi: 10.1016/j.jneb.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 92.De Castro JM. Social facilitation of duration and size but not rate of the spontaneous meal intake of humans. Physiol Behav. 1990;47:1129–1135. doi: 10.1016/0031-9384(90)90363-9. [DOI] [PubMed] [Google Scholar]
- 93.De Castro JM. Social facilitation of food intake in humans. Appetite. 1995;24:260. doi: 10.1016/s0195-6663(95)99835-7. [DOI] [PubMed] [Google Scholar]
- 94.De Castro JM. Socio-cultural determinants of meal size and frequency. Br J Nutr. 1997;77 Suppl 1:S39–S54; discussion S54-S55. doi: 10.1079/bjn19970103. [DOI] [PubMed] [Google Scholar]
- 95.Lumeng JC, Hillman KH. Eating in larger groups increases food consumption. Arch Dis Child. 2007;92:384–387. doi: 10.1136/adc.2006.103259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pliner P, Bell R, Hirsch ES, Kinchla M. Meal duration mediates the effect of “social facilitation” on eating in humans. Appetite. 2006;46:189–198. doi: 10.1016/j.appet.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 97.Ferrari PF, Maiolini C, Addessi E, Fogassi L, Visalberghi E. The observation and hearing of eating actions activates motor programs related to eating in macaque monkeys. Behav Brain Res. 2005;161:95–101. doi: 10.1016/j.bbr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 98.James WT. Social facilitation of eating behavior in puppies after satiation. J Comp Physiol Psychol. 1953;46:427–428. doi: 10.1037/h0056028. [DOI] [PubMed] [Google Scholar]
- 99.Sweeting MP, Houpt CE, Houpt KA. Social facilitation of feeding and time budgets in stabled ponies. J Anim Sci. 1985;60:369–374. doi: 10.2527/jas1985.602369x. [DOI] [PubMed] [Google Scholar]
- 100.Strobel MG, Macdonald GE. Induction of eating in newly hatched chicks. J Comp Physiol Psychol. 1974;86:493–502. doi: 10.1037/h0036170. [DOI] [PubMed] [Google Scholar]
- 101.Levitsky DA, Youn T. The more food young adults are served, the more they overeat. J Nutr. 2004;134:2546–2549. doi: 10.1093/jn/134.10.2546. [DOI] [PubMed] [Google Scholar]
- 102.Levitsky DA, Halbmaier CA, Mrdjenovic G. The freshman weight gain: a model for the study of the epidemic of obesity. Int J Obes Relat Metab Disord. 2004;28:1435–1442. doi: 10.1038/sj.ijo.0802776. [DOI] [PubMed] [Google Scholar]
- 103.Levitsky DA, Iyer S, Pacanowski CR. Number of foods available at a meal determines the amount consumed. Eat Behav. 2012;13:183–187. doi: 10.1016/j.eatbeh.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 104.Blatt AD, Williams RA, Roe LS, Rolls BJ. Effects of energy content and energy density of pre-portioned entrées on energy intake. Obesity (Silver Spring) 2012;20:2010–2018. doi: 10.1038/oby.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Williams RA, Roe LS, Rolls BJ. Comparison of three methods to reduce energy density. Effects on daily energy intake. Appetite. 2013;66:75–83. doi: 10.1016/j.appet.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Williams RA, Roe LS, Rolls BJ. Assessment of satiety depends on the energy density and portion size of the test meal. Obesity (Silver Spring) 2014;22:318–324. doi: 10.1002/oby.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kendall A, Levitsky DA, Strupp BJ, Lissner L. Weight loss on a low-fat diet: consequence of the imprecision of the control of food intake in humans. Am J Clin Nutr. 1991;53:1124–1129. doi: 10.1093/ajcn/53.5.1124. [DOI] [PubMed] [Google Scholar]
- 108.Schindler K, Prager G, Ballaban T, Kretschmer S, Riener R, Buranyi B, Maier C, Luger A, Ludvik B. Impact of laparoscopic adjustable gastric banding on plasma ghrelin, eating behaviour and body weight. Eur J Clin Invest. 2004;34:549–554. doi: 10.1111/j.1365-2362.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 109.Sinha AC, Singh PM, Williams NW, Ochroch EA, Goudra BG. Aprepitant’s prophylactic efficacy in decreasing postoperative nausea and vomiting in morbidly obese patients undergoing bariatric surgery. Obes Surg. 2014;24:225–231. doi: 10.1007/s11695-013-1065-1. [DOI] [PubMed] [Google Scholar]
- 110.Mathus-Vliegen EM, Tytgat GN, Veldhuyzen-Offermans EA. Intragastric balloon in the treatment of super-morbid obesity. Double-blind, sham-controlled, crossover evaluation of 500-milliliter balloon. Gastroenterology. 1990;99:362–369. doi: 10.1016/0016-5085(90)91017-z. [DOI] [PubMed] [Google Scholar]
- 111.Mathus-Vliegen EM, Tytgat GN. Intragastric balloon for treatment-resistant obesity: safety, tolerance, and efficacy of 1-year balloon treatment followed by a 1-year balloon-free follow-up. Gastrointest Endosc. 2005;61:19–27. doi: 10.1016/s0016-5107(04)02406-x. [DOI] [PubMed] [Google Scholar]
- 112.Cannon WB, Washburn AL. An explanation of hunger. 1911. Obes Res. 1993;1:494–500. doi: 10.1002/j.1550-8528.1993.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 113.Berthoud HR, Lynn PA, Blackshaw LA. Vagal and spinal mechanosensors in the rat stomach and colon have multiple receptive fields. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1371–R1381. doi: 10.1152/ajpregu.2001.280.5.R1371. [DOI] [PubMed] [Google Scholar]
- 114.Phillips RJ, Powley TL. Tension and stretch receptors in gastrointestinal smooth muscle: re-evaluating vagal mechanoreceptor electrophysiology. Brain Res Brain Res Rev. 2000;34:1–26. doi: 10.1016/s0165-0173(00)00036-9. [DOI] [PubMed] [Google Scholar]
- 115.Powley TL, Phillips RJ. Vagal intramuscular array afferents form complexes with interstitial cells of Cajal in gastrointestinal smooth muscle: analogues of muscle spindle organs? Neuroscience. 2011;186:188–200. doi: 10.1016/j.neuroscience.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Phillips RJ, Powley TL. Gastric volume rather than nutrient content inhibits food intake. Am J Physiol. 1996;271:R766–R769. doi: 10.1152/ajpregu.1996.271.3.R766. [DOI] [PubMed] [Google Scholar]
- 117.Powley TL, Phillips RJ. Gastric satiation is volumetric, intestinal satiation is nutritive. Physiol Behav. 2004;82:69–74. doi: 10.1016/j.physbeh.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 118.Seimon RV, Lange K, Little TJ, Brennan IM, Pilichiewicz AN, Feltrin KL, Smeets AJ, Horowitz M, Feinle-Bisset C. Pooled-data analysis identifies pyloric pressures and plasma cholecystokinin concentrations as major determinants of acute energy intake in healthy, lean men. Am J Clin Nutr. 2010;92:61–68. doi: 10.3945/ajcn.2009.29015. [DOI] [PubMed] [Google Scholar]
- 119.Dethier VG, Solomon RL, Turner LH. Sensory input and central excitation and inhibition in the blowfly. J Comp Physiol Psychol. 1965;60:303–313. doi: 10.1037/h0022557. [DOI] [PubMed] [Google Scholar]
- 120.Dethier VG, Solomon RL, Turner LH. Central inhibition in the blowfly. J Comp Physiol Psychol. 1968;66:144–150. doi: 10.1037/h0025957. [DOI] [PubMed] [Google Scholar]
- 121.Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81:3419–3423. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- 122.Dubuc GR, Phinney SD, Stern JS, Havel PJ. Changes of serum leptin and endocrine and metabolic parameters after 7 days of energy restriction in men and women. Metabolism. 1998;47:429–434. doi: 10.1016/s0026-0495(98)90055-5. [DOI] [PubMed] [Google Scholar]
- 123.Kolaczynski JW, Considine RV, Ohannesian J, Marco C, Opentanova I, Nyce MR, Myint M, Caro JF. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes. 1996;45:1511–1515. doi: 10.2337/diab.45.11.1511. [DOI] [PubMed] [Google Scholar]
- 124.Schmitz O, Fisker S, Orskov L, Hove KY, Nyholm B, Møller N. Effects of hyperinsulinaemia and hypoglycaemia on circulating leptin levels in healthy lean males. Diabetes Metab. 1997;23:80–83. [PubMed] [Google Scholar]
- 125.Patel BK, Koenig JI, Kaplan LM, Hooi SC. Increase in plasma leptin and Lep mRNA concentrations by food intake is dependent on insulin. Metabolism. 1998;47:603–607. doi: 10.1016/s0026-0495(98)90247-5. [DOI] [PubMed] [Google Scholar]
- 126.Kolaczynski JW, Ohannesian JP, Considine RV, Marco CC, Caro JF. Response of leptin to short-term and prolonged overfeeding in humans. J Clin Endocrinol Metab. 1996;81:4162–4165. doi: 10.1210/jcem.81.11.8923877. [DOI] [PubMed] [Google Scholar]
- 127.Casabiell X, Piñeiro V, De la Cruz LF, Gualillo O, Folgar L, Diéguez C, Casanueva FF. Dual effect of insulin on in vitro leptin secretion by adipose tissue. Biochem Biophys Res Commun. 2000;276:477–482. doi: 10.1006/bbrc.2000.3506. [DOI] [PubMed] [Google Scholar]
- 128.Levy JR, Stevens W. The effects of insulin, glucose, and pyruvate on the kinetics of leptin secretion. Endocrinology. 2001;142:3558–3562. doi: 10.1210/endo.142.8.8313. [DOI] [PubMed] [Google Scholar]
- 129.Mueller WM, Gregoire FM, Stanhope KL, Mobbs CV, Mizuno TM, Warden CH, Stern JS, Havel PJ. Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology. 1998;139:551–558. doi: 10.1210/endo.139.2.5716. [DOI] [PubMed] [Google Scholar]
- 130.Malmström R, Taskinen MR, Karonen SL, Yki-Järvinen H. Insulin increases plasma leptin concentrations in normal subjects and patients with NIDDM. Diabetologia. 1996;39:993–996. doi: 10.1007/BF00403921. [DOI] [PubMed] [Google Scholar]
- 131.Rayner DV, Trayhurn P. Regulation of leptin production: sympathetic nervous system interactions. J Mol Med (Berl) 2001;79:8–20. doi: 10.1007/s001090100198. [DOI] [PubMed] [Google Scholar]
- 132.Tsai M, Asakawa A, Amitani H, Inui A. Stimulation of leptin secretion by insulin. Indian J Endocrinol Metab. 2012;16:S543–S548. doi: 10.4103/2230-8210.105570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wellhoener P, Fruehwald-Schultes B, Kern W, Dantz D, Kerner W, Born J, Fehm HL, Peters A. Glucose metabolism rather than insulin is a main determinant of leptin secretion in humans. J Clin Endocrinol Metab. 2000;85:1267–1271. doi: 10.1210/jcem.85.3.6483. [DOI] [PubMed] [Google Scholar]
- 134.Cammisotto PG, Bukowiecki LJ, Deshaies Y, Bendayan M. Leptin biosynthetic pathway in white adipocytes. Biochem Cell Biol. 2006;84:207–214. doi: 10.1139/o06-032. [DOI] [PubMed] [Google Scholar]
- 135.Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau JP, Bortoluzzi MN, Moizo L, Lehy T, Guerre-Millo M, Le Marchand-Brustel Y, et al. The stomach is a source of leptin. Nature. 1998;394:790–793. doi: 10.1038/29547. [DOI] [PubMed] [Google Scholar]
- 136.Mix H, Widjaja A, Jandl O, Cornberg M, Kaul A, Göke M, Beil W, Kuske M, Brabant G, Manns MP, et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut. 2000;47:481–486. doi: 10.1136/gut.47.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sobhani I, Bado A, Vissuzaine C, Buyse M, Kermorgant S, Laigneau JP, Attoub S, Lehy T, Henin D, Mignon M, et al. Leptin secretion and leptin receptor in the human stomach. Gut. 2000;47:178–183. doi: 10.1136/gut.47.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cammisotto PG, Renaud C, Gingras D, Delvin E, Levy E, Bendayan M. Endocrine and exocrine secretion of leptin by the gastric mucosa. J Histochem Cytochem. 2005;53:851–860. doi: 10.1369/jhc.5A6620.2005. [DOI] [PubMed] [Google Scholar]
- 139.Cammisotto PG, Gingras D, Bendayan M. Transcytosis of gastric leptin through the rat duodenal mucosa. Am J Physiol Gastrointest Liver Physiol. 2007;293:G773–G779. doi: 10.1152/ajpgi.00260.2007. [DOI] [PubMed] [Google Scholar]
- 140.Cammisotto PG, Bendayan M. Leptin secretion by white adipose tissue and gastric mucosa. Histol Histopathol. 2007;22:199–210. doi: 10.14670/HH-22.199. [DOI] [PubMed] [Google Scholar]
- 141.Cammisotto PG, Levy E, Bukowiecki LJ, Bendayan M. Cross-talk between adipose and gastric leptins for the control of food intake and energy metabolism. Prog Histochem Cytochem. 2010;45:143–200. doi: 10.1016/j.proghi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 142.Cammisotto P, Bendayan M. A review on gastric leptin: the exocrine secretion of a gastric hormone. Anat Cell Biol. 2012;45:1–16. doi: 10.5115/acb.2012.45.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yarandi SS, Hebbar G, Sauer CG, Cole CR, Ziegler TR. Diverse roles of leptin in the gastrointestinal tract: modulation of motility, absorption, growth, and inflammation. Nutrition. 2011;27:269–275. doi: 10.1016/j.nut.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hassink SG, de Lancey E, Sheslow DV, Smith-Kirwin SM, O’Connor DM, Considine RV, Opentanova I, Dostal K, Spear ML, Leef K, et al. Placental leptin: an important new growth factor in intrauterine and neonatal development? Pediatrics. 1997;100:E1. doi: 10.1542/peds.100.1.e1. [DOI] [PubMed] [Google Scholar]
- 145.Pico C, Jilkova ZM, Kus V, Palou A, Kopecky J. Perinatal programming of body weight control by leptin: putative roles of AMP kinase and muscle thermogenesis. Am J Clin Nutr. 2011;94:1830S–1837S. doi: 10.3945/ajcn.110.000752. [DOI] [PubMed] [Google Scholar]
- 146.Zheng D, Jones JP, Usala SJ, Dohm GL. Differential expression of ob mRNA in rat adipose tissues in response to insulin. Biochem Biophys Res Commun. 1996;218:434–437. doi: 10.1006/bbrc.1996.0077. [DOI] [PubMed] [Google Scholar]
- 147.Lee MJ, Fried SK. Multilevel regulation of leptin storage, turnover, and secretion by feeding and insulin in rat adipose tissue. J Lipid Res. 2006;47:1984–1993. doi: 10.1194/jlr.M600065-JLR200. [DOI] [PubMed] [Google Scholar]
- 148.Matson CA, Ritter RC. Long-term CCK-leptin synergy suggests a role for CCK in the regulation of body weight. Am J Physiol. 1999;276:R1038–R1045. doi: 10.1152/ajpregu.1999.276.4.R1038. [DOI] [PubMed] [Google Scholar]
- 149.Matson CA, Reid DF, Cannon TA, Ritter RC. Cholecystokinin and leptin act synergistically to reduce body weight. Am J Physiol Regul Integr Comp Physiol. 2000;278:R882–R890. doi: 10.1152/ajpregu.2000.278.4.R882. [DOI] [PubMed] [Google Scholar]
- 150.Williams DL, Baskin DG, Schwartz MW. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes. 2006;55:3387–3393. doi: 10.2337/db06-0558. [DOI] [PubMed] [Google Scholar]
- 151.Nowak A, Bojanowska E. Effects of peripheral or central GLP-1 receptor blockade on leptin-induced suppression of appetite. J Physiol Pharmacol. 2008;59:501–510. [PubMed] [Google Scholar]
- 152.Williams DL, Baskin DG, Schwartz MW. Hindbrain leptin receptor stimulation enhances the anorexic response to cholecystokinin. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1238–R1246. doi: 10.1152/ajpregu.00182.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li Y, Wu X, Zhou S, Owyang C. Low-affinity CCK-A receptors are coexpressed with leptin receptors in rat nodose ganglia: implications for leptin as a regulator of short-term satiety. Am J Physiol Gastrointest Liver Physiol. 2011;300:G217–G227. doi: 10.1152/ajpgi.00356.2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 154.Kentish SJ, O’Donnell TA, Isaacs NJ, Young RL, Li H, Harrington AM, Brierley SM, Wittert GA, Blackshaw LA, Page AJ. Gastric vagal afferent modulation by leptin is influenced by food intake status. J Physiol. 2013;591:1921–1934. doi: 10.1113/jphysiol.2012.247577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hargrave SL, Kinzig KP. Repeated gastric distension alters food intake and neuroendocrine profiles in rats. Physiol Behav. 2012;105:975–981. doi: 10.1016/j.physbeh.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sartor DM, Verberne AJ. Gastric leptin: a novel role in cardiovascular regulation. Am J Physiol Heart Circ Physiol. 2010;298:H406–H414. doi: 10.1152/ajpheart.00997.2009. [DOI] [PubMed] [Google Scholar]
- 157.How JM, Fam BC, Verberne AJ, Sartor DM. High-fat diet is associated with blunted splanchnic sympathoinhibitory responses to gastric leptin and cholecystokinin: implications for circulatory control. Am J Physiol Heart Circ Physiol. 2011;300:H961–H967. doi: 10.1152/ajpheart.01156.2010. [DOI] [PubMed] [Google Scholar]
- 158.Russell CD, Petersen RN, Rao SP, Ricci MR, Prasad A, Zhang Y, Brolin RE, Fried SK. Leptin expression in adipose tissue from obese humans: depot-specific regulation by insulin and dexamethasone. Am J Physiol. 1998;275:E507–E515. doi: 10.1152/ajpendo.1998.275.3.E507. [DOI] [PubMed] [Google Scholar]
- 159.Wabitsch M, Jensen PB, Blum WF, Christoffersen CT, Englaro P, Heinze E, Rascher W, Teller W, Tornqvist H, Hauner H. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes. 1996;45:1435–1438. doi: 10.2337/diab.45.10.1435. [DOI] [PubMed] [Google Scholar]
- 160.Bradley RL, Cheatham B. Regulation of ob gene expression and leptin secretion by insulin and dexamethasone in rat adipocytes. Diabetes. 1999;48:272–278. doi: 10.2337/diabetes.48.2.272. [DOI] [PubMed] [Google Scholar]
- 161.Norman D, Isidori AM, Frajese V, Caprio M, Chew SL, Grossman AB, Clark AJ, Michael Besser G, Fabbri A. ACTH and alpha-MSH inhibit leptin expression and secretion in 3T3-L1 adipocytes: model for a central-peripheral melanocortin-leptin pathway. Mol Cell Endocrinol. 2003;200:99–109. doi: 10.1016/s0303-7207(02)00410-0. [DOI] [PubMed] [Google Scholar]
- 162.Machinal F, Dieudonne MN, Leneveu MC, Pecquery R, Giudicelli Y. In vivo and in vitro ob gene expression and leptin secretion in rat adipocytes: evidence for a regional specific regulation by sex steroid hormones. Endocrinology. 1999;140:1567–1574. doi: 10.1210/endo.140.4.6617. [DOI] [PubMed] [Google Scholar]
- 163.Boden G, Chen X, Kolaczynski JW, Polansky M. Effects of prolonged hyperinsulinemia on serum leptin in normal human subjects. J Clin Invest. 1997;100:1107–1113. doi: 10.1172/JCI119621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Utriainen T, Malmström R, Mäkimattila S, Yki-Järvinen H. Supraphysiological hyperinsulinemia increases plasma leptin concentrations after 4 h in normal subjects. Diabetes. 1996;45:1364–1366. doi: 10.2337/diab.45.10.1364. [DOI] [PubMed] [Google Scholar]
- 165.Pratley RE, Nicolson M, Bogardus C, Ravussin E. Effects of acute hyperinsulinemia on plasma leptin concentrations in insulin-sensitive and insulin-resistant Pima Indians. J Clin Endocrinol Metab. 1996;81:4418–4421. doi: 10.1210/jcem.81.12.8954052. [DOI] [PubMed] [Google Scholar]
- 166.Muscelli E, Camastra S, Masoni A, Baldi S, Sironi AM, Natali A, Ferrannini E. Acute insulin administration does not affect plasma leptin levels in lean or obese subjects. Eur J Clin Invest. 1996;26:940–943. doi: 10.1111/j.1365-2362.1996.tb02142.x. [DOI] [PubMed] [Google Scholar]
- 167.Clapham JC, Smith SA, Moore GB, Hughes MG, Azam H, Scott A, Jung RT. Plasma leptin concentrations and OB gene expression in subcutaneous adipose tissue are not regulated acutely by physiological hyperinsulinaemia in lean and obese humans. Int J Obes Relat Metab Disord. 1997;21:179–183. doi: 10.1038/sj.ijo.0800384. [DOI] [PubMed] [Google Scholar]
- 168.Vidal H, Auboeuf D, De Vos P, Staels B, Riou JP, Auwerx J, Laville M. The expression of ob gene is not acutely regulated by insulin and fasting in human abdominal subcutaneous adipose tissue. J Clin Invest. 1996;98:251–255. doi: 10.1172/JCI118786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Carlson JJ, Turpin AA, Wiebke G, Hunt SC, Adams TD. Pre- and post- prandial appetite hormone levels in normal weight and severely obese women. Nutr Metab (Lond) 2009;6:32. doi: 10.1186/1743-7075-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dagogo-Jack S, Fanelli C, Paramore D, Brothers J, Landt M. Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes. 1996;45:695–698. doi: 10.2337/diab.45.5.695. [DOI] [PubMed] [Google Scholar]
- 171.Karhunen L, Haffner S, Lappalainen R, Turpeinen A, Miettinen H, Uusitupa M. Serum leptin and short-term regulation of eating in obese women. Clin Sci (Lond) 1997;92:573–578. doi: 10.1042/cs0920573. [DOI] [PubMed] [Google Scholar]
- 172.Ishida K, Murakami T, Mizuno A, Iida M, Kuwajima M, Shima K. leptin suppresses basal insulin secretion from rat pancreatic islets. Regul Pept. 1997;70:179–182. doi: 10.1016/s0167-0115(97)01002-1. [DOI] [PubMed] [Google Scholar]
- 173.Gray SL, Donald C, Jetha A, Covey SD, Kieffer TJ. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology. 2010;151:4178–4186. doi: 10.1210/en.2010-0102. [DOI] [PubMed] [Google Scholar]
- 174.Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes. 1997;46:1087–1093. doi: 10.2337/diab.46.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006;4:291–302. doi: 10.1016/j.cmet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 176.Levi J, Gray SL, Speck M, Huynh FK, Babich SL, Gibson WT, Kieffer TJ. Acute disruption of leptin signaling in vivo leads to increased insulin levels and insulin resistance. Endocrinology. 2011;152:3385–3395. doi: 10.1210/en.2011-0185. [DOI] [PubMed] [Google Scholar]
- 177.Harris RB, Zhou J, Redmann SM, Smagin GN, Smith SR, Rodgers E, Zachwieja JJ. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/endo.139.1.5675. [DOI] [PubMed] [Google Scholar]
- 178.Stich V, Berlan M. Physiological regulation of NEFA availability: lipolysis pathway. Proc Nutr Soc. 2004;63:369–374. doi: 10.1079/PNS2004350. [DOI] [PubMed] [Google Scholar]
- 179.McMurray RG, Hackney AC. Interactions of metabolic hormones, adipose tissue and exercise. Sports Med. 2005;35:393–412. doi: 10.2165/00007256-200535050-00003. [DOI] [PubMed] [Google Scholar]
- 180.Moinat M, Deng C, Muzzin P, Assimacopoulos-Jeannet F, Seydoux J, Dulloo AG, Giacobino JP. Modulation of obese gene expression in rat brown and white adipose tissues. FEBS Lett. 1995;373:131–134. doi: 10.1016/0014-5793(95)01030-i. [DOI] [PubMed] [Google Scholar]
- 181.Trayhurn P, Duncan JS, Rayner DV. Acute cold-induced suppression of ob (obese) gene expression in white adipose tissue of mice: mediation by the sympathetic system. Biochem J. 1995;311(Pt 3):729–733. doi: 10.1042/bj3110729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Trayhurn P, Duncan JS, Hoggard N, Rayner DV. Regulation of leptin production: a dominant role for the sympathetic nervous system? Proc Nutr Soc. 1998;57:413–419. doi: 10.1079/pns19980060. [DOI] [PubMed] [Google Scholar]
- 183.Zheng D, Wooter MH, Zhou Q, Dohm GL. The effect of exercise on ob gene expression. Biochem Biophys Res Commun. 1996;225:747–750. doi: 10.1006/bbrc.1996.1245. [DOI] [PubMed] [Google Scholar]
- 184.Pérusse L, Collier G, Gagnon J, Leon AS, Rao DC, Skinner JS, Wilmore JH, Nadeau A, Zimmet PZ, Bouchard C. Acute and chronic effects of exercise on leptin levels in humans. J Appl Physiol (1985) 1997;83:5–10. doi: 10.1152/jappl.1997.83.1.5. [DOI] [PubMed] [Google Scholar]
- 185.Pinkney JH, Coppack SW, Mohamed-Ali V. Effect of isoprenaline on plasma leptin and lipolysis in humans. Clin Endocrinol (Oxf) 1998;48:407–411. doi: 10.1046/j.1365-2265.1998.00480.x. [DOI] [PubMed] [Google Scholar]
- 186.Carulli L, Ferrari S, Bertolini M, Tagliafico E, Del Rio G. Regulation of ob gene expression: evidence for epinephrine-induced suppression in human obesity. J Clin Endocrinol Metab. 1999;84:3309–3312. doi: 10.1210/jcem.84.9.6007. [DOI] [PubMed] [Google Scholar]
- 187.Donahoo WT, Jensen DR, Yost TJ, Eckel RH. Isoproterenol and somatostatin decrease plasma leptin in humans: a novel mechanism regulating leptin secretion. J Clin Endocrinol Metab. 1997;82:4139–4143. doi: 10.1210/jcem.82.12.4434. [DOI] [PubMed] [Google Scholar]
- 188.Walder K, Filippis A, Clark S, Zimmet P, Collier GR. Leptin inhibits insulin binding in isolated rat adipocytes. J Endocrinol. 1997;155:R5–R7. doi: 10.1677/joe.0.155r005. [DOI] [PubMed] [Google Scholar]
- 189.Barzilai N, Wang J, Massilon D, Vuguin P, Hawkins M, Rossetti L. Leptin selectively decreases visceral adiposity and enhances insulin action. J Clin Invest. 1997;100:3105–3110. doi: 10.1172/JCI119865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shimabukuro M, Koyama K, Chen G, Wang MY, Trieu F, Lee Y, Newgard CB, Unger RH. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Barzilai N, She L, Liu L, Wang J, Hu M, Vuguin P, Rossetti L. Decreased visceral adiposity accounts for leptin effect on hepatic but not peripheral insulin action. Am J Physiol. 1999;277:E291–E298. doi: 10.1152/ajpendo.1999.277.2.E291. [DOI] [PubMed] [Google Scholar]
- 192.Huang W, Dedousis N, O’Doherty RM. Hepatic steatosis and plasma dyslipidemia induced by a high-sucrose diet are corrected by an acute leptin infusion. J Appl Physiol (1985) 2007;102:2260–2265. doi: 10.1152/japplphysiol.01449.2006. [DOI] [PubMed] [Google Scholar]
- 193.Kraegen EW, Saha AK, Preston E, Wilks D, Hoy AJ, Cooney GJ, Ruderman NB. Increased malonyl-CoA and diacylglycerol content and reduced AMPK activity accompany insulin resistance induced by glucose infusion in muscle and liver of rats. Am J Physiol Endocrinol Metab. 2006;290:E471–E479. doi: 10.1152/ajpendo.00316.2005. [DOI] [PubMed] [Google Scholar]
- 194.Ceci R, Sabatini S, Duranti G, Savini I, Avigliano L, Rossi A. Acute, but not chronic, leptin treatment induces acyl-CoA oxidase in C2C12 myotubes. Eur J Nutr. 2007;46:364–368. doi: 10.1007/s00394-007-0664-9. [DOI] [PubMed] [Google Scholar]
- 195.Janovská A, Hatzinikolas G, Staikopoulos V, McInerney J, Mano M, Wittert GA. AMPK and ACC phosphorylation: effect of leptin, muscle fibre type and obesity. Mol Cell Endocrinol. 2008;284:1–10. doi: 10.1016/j.mce.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 196.Linares C, Su D. Body mass index and health among Union Army veterans: 1891-1905. Econ Hum Biol. 2005;3:367–387. doi: 10.1016/j.ehb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 197.Todd MK, Yaspelkis BB, Turcotte LP. Short-term leptin treatment increases fatty acids uptake and oxidation in muscle of high fat-fed rats. Metabolism. 2005;54:1218–1224. doi: 10.1016/j.metabol.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 198.Steinberg GR, Dyck DJ. Development of leptin resistance in rat soleus muscle in response to high-fat diets. Am J Physiol Endocrinol Metab. 2000;279:E1374–E1382. doi: 10.1152/ajpendo.2000.279.6.E1374. [DOI] [PubMed] [Google Scholar]
- 199.Fishman S, Muzumdar RH, Atzmon G, Ma X, Yang X, Einstein FH, Barzilai N. Resistance to leptin action is the major determinant of hepatic triglyceride accumulation in vivo. FASEB J. 2007;21:53–60. doi: 10.1096/fj.06-6557com. [DOI] [PubMed] [Google Scholar]
- 200.Ceddia RB. Direct metabolic regulation in skeletal muscle and fat tissue by leptin: implications for glucose and fatty acids homeostasis. Int J Obes (Lond) 2005;29:1175–1183. doi: 10.1038/sj.ijo.0803025. [DOI] [PubMed] [Google Scholar]
- 201.Rouru J, Cusin I, Zakrzewska KE, Jeanrenaud B, Rohner-Jeanrenaud F. Effects of intravenously infused leptin on insulin sensitivity and on the expression of uncoupling proteins in brown adipose tissue. Endocrinology. 1999;140:3688–3692. doi: 10.1210/endo.140.8.6890. [DOI] [PubMed] [Google Scholar]
- 202.Suzuki R, Tobe K, Aoyama M, Sakamoto K, Ohsugi M, Kamei N, Nemoto S, Inoue A, Ito Y, Uchida S, et al. Expression of DGAT2 in white adipose tissue is regulated by central leptin action. J Biol Chem. 2005;280:3331–3337. doi: 10.1074/jbc.M410955200. [DOI] [PubMed] [Google Scholar]
- 203.Baldwin KM, Joanisse DR, Haddad F, Goldsmith RL, Gallagher D, Pavlovich KH, Shamoon EL, Leibel RL, Rosenbaum M. Effects of weight loss and leptin on skeletal muscle in human subjects. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1259–R1266. doi: 10.1152/ajpregu.00397.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Goldsmith R, Joanisse DR, Gallagher D, Pavlovich K, Shamoon E, Leibel RL, Rosenbaum M. Effects of experimental weight perturbation on skeletal muscle work efficiency, fuel utilization, and biochemistry in human subjects. Am J Physiol Regul Integr Comp Physiol. 2010;298:R79–R88. doi: 10.1152/ajpregu.00053.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jiang L, Wang Q, Yu Y, Zhao F, Huang P, Zeng R, Qi RZ, Li W, Liu Y. Leptin contributes to the adaptive responses of mice to high-fat diet intake through suppressing the lipogenic pathway. PLoS One. 2009;4:e6884. doi: 10.1371/journal.pone.0006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Steinberg GR, Parolin ML, Heigenhauser GJ, Dyck DJ. Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab. 2002;283:E187–E192. doi: 10.1152/ajpendo.00542.2001. [DOI] [PubMed] [Google Scholar]
- 208.Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, Su K, Cheng B, Li X, Harvey-White J, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med. 2008;14:667–675. doi: 10.1038/nm1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Boghossian S, Dube MG, Torto R, Kalra PS, Kalra SP. Hypothalamic clamp on insulin release by leptin-transgene expression. Peptides. 2006;27:3245–3254. doi: 10.1016/j.peptides.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 210.Pérez C, Fernández-Galaz C, Fernández-Agulló T, Arribas C, Andrés A, Ros M, Carrascosa JM. Leptin impairs insulin signaling in rat adipocytes. Diabetes. 2004;53:347–353. doi: 10.2337/diabetes.53.2.347. [DOI] [PubMed] [Google Scholar]
- 211.Tortoriello DV, McMinn JE, Chua SC. Increased expression of hypothalamic leptin receptor and adiponectin accompany resistance to dietary-induced obesity and infertility in female C57BL/6J mice. Int J Obes (Lond) 2007;31:395–402. doi: 10.1038/sj.ijo.0803392. [DOI] [PubMed] [Google Scholar]
- 212.Scarpace PJ, Matheny M, Pollock BH, Tümer N. Leptin increases uncoupling protein expression and energy expenditure. Am J Physiol. 1997;273:E226–E230. doi: 10.1152/ajpendo.1997.273.1.E226. [DOI] [PubMed] [Google Scholar]
- 213.Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol. 1998;275:E259–E264. doi: 10.1152/ajpendo.1998.275.2.E259. [DOI] [PubMed] [Google Scholar]
- 214.Casas AT, Hubsch AP, Doran JE. Effects of reconstituted high-density lipoprotein in persistent gram-negative bacteremia. Am Surg. 1996;62:350–355. [PubMed] [Google Scholar]
- 215.Zhou YT, Shimabukuro M, Koyama K, Lee Y, Wang MY, Trieu F, Newgard CB, Unger RH. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proc Natl Acad Sci USA. 1997;94:6386–6390. doi: 10.1073/pnas.94.12.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Scarpace PJ, Nicolson M, Matheny M. UCP2, UCP3 and leptin gene expression: modulation by food restriction and leptin. J Endocrinol. 1998;159:349–357. doi: 10.1677/joe.0.1590349. [DOI] [PubMed] [Google Scholar]
- 217.Liu Q, Bai C, Chen F, Wang R, MacDonald T, Gu M, Zhang Q, Morsy MA, Caskey CT. Uncoupling protein-3: a muscle-specific gene upregulated by leptin in ob/ob mice. Gene. 1998;207:1–7. doi: 10.1016/s0378-1119(97)00596-9. [DOI] [PubMed] [Google Scholar]
- 218.Reidy SP, Weber JM. Accelerated substrate cycling: a new energy-wasting role for leptin in vivo. Am J Physiol Endocrinol Metab. 2002;282:E312–E317. doi: 10.1152/ajpendo.00037.2001. [DOI] [PubMed] [Google Scholar]
- 219.Solinas G, Summermatter S, Mainieri D, Gubler M, Pirola L, Wymann MP, Rusconi S, Montani JP, Seydoux J, Dulloo AG. The direct effect of leptin on skeletal muscle thermogenesis is mediated by substrate cycling between de novo lipogenesis and lipid oxidation. FEBS Lett. 2004;577:539–544. doi: 10.1016/j.febslet.2004.10.066. [DOI] [PubMed] [Google Scholar]
- 220.Minaker KL, Rowe JW, Young JB, Sparrow D, Pallotta JA, Landsberg L. Effect of age on insulin stimulation of sympathetic nervous system activity in man. Metabolism. 1982;31:1181–1184. doi: 10.1016/0026-0495(82)90001-4. [DOI] [PubMed] [Google Scholar]
- 221.Woods SC, Ramsay DS. Food intake, metabolism and homeostasis. Physiol Behav. 2011;104:4–7. doi: 10.1016/j.physbeh.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Landsberg L, Saville ME, Young JB. Sympathoadrenal system and regulation of thermogenesis. Am J Physiol. 1984;247:E181–E189. doi: 10.1152/ajpendo.1984.247.2.E181. [DOI] [PubMed] [Google Scholar]
- 223.Olefsky JM, Kobayashi M. Mechanisms of fasting-induced increase in insulin binding to rat adipocytes. J Clin Invest. 1978;61:329–338. doi: 10.1172/JCI108943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fleig WE, Enderle D, Steudter S, Nöther-Fleig G, Ditschuneit H. Regulation of basal and insulin-stimulated glycogen synthesis in cultured hepatocytes. Inverse relationship to glycogen content. J Biol Chem. 1987;262:1155–1160. [PubMed] [Google Scholar]
- 225.Nielsen JN, Richter EA. Regulation of glycogen synthase in skeletal muscle during exercise. Acta Physiol Scand. 2003;178:309–319. doi: 10.1046/j.1365-201X.2003.01165.x. [DOI] [PubMed] [Google Scholar]
- 226.Barzilai N, She L, Liu BQ, Vuguin P, Cohen P, Wang J, Rossetti L. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes. 1999;48:94–98. doi: 10.2337/diabetes.48.1.94. [DOI] [PubMed] [Google Scholar]
- 227.Agren G, Wilander O, Jorpes E. Cyclic changes in the glycogen content of the liver and the muscles of rats and mice: Their bearing upon the sensitivity of the animals to insulin, and their influence on the urinary output of nitrogen. Biochem J. 1931;25:777–785. doi: 10.1042/bj0250777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Arner P. Adrenergic receptor function in fat cells. Am J Clin Nutr. 1992;55:228S–236S. doi: 10.1093/ajcn/55.1.228s. [DOI] [PubMed] [Google Scholar]
- 229.Assali AR, Ganor A, Beigel Y, Shafer Z, Hershcovici T, Fainaru M. Insulin resistance in obesity: body-weight or energy balance? J Endocrinol. 2001;171:293–298. doi: 10.1677/joe.0.1710293. [DOI] [PubMed] [Google Scholar]
- 230.Bergendahl M, Evans WS, Pastor C, Patel A, Iranmanesh A, Veldhuis JD. Short-term fasting suppresses leptin and (conversely) activates disorderly growth hormone secretion in midluteal phase women--a clinical research center study. J Clin Endocrinol Metab. 1999;84:883–894. doi: 10.1210/jcem.84.3.5536. [DOI] [PubMed] [Google Scholar]
- 231.Grinspoon SK, Askari H, Landt ML, Nathan DM, Schoenfeld DA, Hayden DL, Laposata M, Hubbard J, Klibanski A. Effects of fasting and glucose infusion on basal and overnight leptin concentrations in normal-weight women. Am J Clin Nutr. 1997;66:1352–1356. doi: 10.1093/ajcn/66.6.1352. [DOI] [PubMed] [Google Scholar]
- 232.Young JB, Rosa RM, Landsberg L. Dissociation of sympathetic nervous system and adrenal medullary responses. Am J Physiol. 1984;247:E35–E40. doi: 10.1152/ajpendo.1984.247.1.E35. [DOI] [PubMed] [Google Scholar]
- 233.Askari H, Tykodi G, Liu J, Dagogo-Jack S. Fasting plasma leptin level is a surrogate measure of insulin sensitivity. J Clin Endocrinol Metab. 2010;95:3836–3843. doi: 10.1210/jc.2010-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Owecki M, Nikisch E, Miczke A, Pupek-Musialik D, Sowiński J. Leptin, soluble leptin receptors, free leptin index, and their relationship with insulin resistance and BMI: high normal BMI is the threshold for serum leptin increase in humans. Horm Metab Res. 2010;42:585–589. doi: 10.1055/s-0030-1253422. [DOI] [PubMed] [Google Scholar]
- 235.Dube JJ, Bhatt BA, Dedousis N, Bonen A, O’Doherty RM. Leptin, skeletal muscle lipids, and lipid-induced insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;293:R642–R650. doi: 10.1152/ajpregu.00133.2007. [DOI] [PubMed] [Google Scholar]
- 236.Mizuno A, Murakami T, Doi T, Shima K. Effect of leptin on insulin sensitivity in the Otsuka Long-Evans Tokushima Fatty rat. Regul Pept. 2001;99:41–44. doi: 10.1016/s0167-0115(01)00215-4. [DOI] [PubMed] [Google Scholar]
- 237.Sáinz N, Rodríguez A, Catalán V, Becerril S, Ramírez B, Lancha A, Burgos-Ramos E, Gómez-Ambrosi J, Frühbeck G. Leptin reduces the expression and increases the phosphorylation of the negative regulators of GLUT4 traffic TBC1D1 and TBC1D4 in muscle of ob/ob mice. PLoS One. 2012;7:e29389. doi: 10.1371/journal.pone.0029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Roden M. Hepatic glucose production and insulin resistance. Wien Med Wochenschr. 2008;158:558–561. doi: 10.1007/s10354-008-0595-y. [DOI] [PubMed] [Google Scholar]
- 239.Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Obes Res. 1997;5:646–649. doi: 10.1002/j.1550-8528.1997.tb00590.x. [DOI] [PubMed] [Google Scholar]
- 240.Maclean PS, Bergouignan A, Cornier MA, Jackman MR. Biology’s response to dieting: the impetus for weight regain. Am J Physiol Regul Integr Comp Physiol. 2011;301:R581–R600. doi: 10.1152/ajpregu.00755.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.MacLean PS, Higgins JA, Wyatt HR, Melanson EL, Johnson GC, Jackman MR, Giles ED, Brown IE, Hill JO. Regular exercise attenuates the metabolic drive to regain weight after long-term weight loss. Am J Physiol Regul Integr Comp Physiol. 2009;297:R793–R802. doi: 10.1152/ajpregu.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Blundell JE, King NA. Effects of exercise on appetite control: loose coupling between energy expenditure and energy intake. Int J Obes Relat Metab Disord. 1998;22 Suppl 2:S22–S29. [PubMed] [Google Scholar]
- 243.Edholm OG, Fletcher JG, Widdowson EM, Mccance RA. The energy expenditure and food intake of individual men. Br J Nutr. 1955;9:286–300. doi: 10.1079/bjn19550040. [DOI] [PubMed] [Google Scholar]
- 244.Edholm OG, Adam JM, Healy MJ, Wolff HS, Goldsmith R, Best TW. Food intake and energy expenditure of army recruits. Br J Nutr. 1970;24:1091–1107. doi: 10.1079/bjn19700112. [DOI] [PubMed] [Google Scholar]
- 245.Mayer J, Roy P, Mitra KP. Relation between caloric intake, body weight, and physical work: studies in an industrial male population in West Bengal. Am J Clin Nutr. 1956;4:169–175. doi: 10.1093/ajcn/4.2.169. [DOI] [PubMed] [Google Scholar]
- 246.Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–2791. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- 247.Benomar Y, Wetzler S, Larue-Achagiotis C, Djiane J, Tomé D, Taouis M. In vivo leptin infusion impairs insulin and leptin signalling in liver and hypothalamus. Mol Cell Endocrinol. 2005;242:59–66. doi: 10.1016/j.mce.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 248.Jackman MR, Steig A, Higgins JA, Johnson GC, Fleming-Elder BK, Bessesen DH, MacLean PS. Weight regain after sustained weight reduction is accompanied by suppressed oxidation of dietary fat and adipocyte hyperplasia. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1117–R1129. doi: 10.1152/ajpregu.00808.2007. [DOI] [PubMed] [Google Scholar]
- 249.Gurr MI, Jung RT, Robinson MP, James WP. Adipose tissue cellularity in man: the relationship between fat cell size and number, the mass and distribution of body fat and the history of weight gain and loss. Int J Obes. 1982;6:419–436. [PubMed] [Google Scholar]
- 250.Löfgren P, Andersson I, Adolfsson B, Leijonhufvud BM, Hertel K, Hoffstedt J, Arner P. Long-term prospective and controlled studies demonstrate adipose tissue hypercellularity and relative leptin deficiency in the postobese state. J Clin Endocrinol Metab. 2005;90:6207–6213. doi: 10.1210/jc.2005-0596. [DOI] [PubMed] [Google Scholar]
- 251.Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab. 1976;5:299–311. doi: 10.1016/s0300-595x(76)80023-0. [DOI] [PubMed] [Google Scholar]
- 252.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Morrison CD, Huypens P, Stewart LK, Gettys TW. Implications of crosstalk between leptin and insulin signaling during the development of diet-induced obesity. Biochim Biophys Acta. 2009;1792:409–416. doi: 10.1016/j.bbadis.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ritchie IR, Gulli RA, Stefanyk LE, Harasim E, Chabowski A, Dyck DJ. Restoration of skeletal muscle leptin response does not precede the exercise-induced recovery of insulin-stimulated glucose uptake in high-fat-fed rats. Am J Physiol Regul Integr Comp Physiol. 2011;300:R492–R500. doi: 10.1152/ajpregu.00602.2010. [DOI] [PubMed] [Google Scholar]
- 255.Schwartz MW, Prigeon RL, Kahn SE, Nicolson M, Moore J, Morawiecki A, Boyko EJ, Porte D. Evidence that plasma leptin and insulin levels are associated with body adiposity via different mechanisms. Diabetes Care. 1997;20:1476–1481. doi: 10.2337/diacare.20.9.1476. [DOI] [PubMed] [Google Scholar]
- 256.Saltiel AR. Insulin resistance in the defense against obesity. Cell Metab. 2012;15:798–804. doi: 10.1016/j.cmet.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 257.Myers MG, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Seals DR, Bell C. Chronic sympathetic activation: consequence and cause of age-associated obesity? Diabetes. 2004;53:276–284. doi: 10.2337/diabetes.53.2.276. [DOI] [PubMed] [Google Scholar]
- 259.Ihara S, Shimamoto K, Watanabe H, Sakai R, Kawana M. An alpha1-receptor blocker reduces plasma leptin levels in hypertensive patients with obesity and hyperleptinemia. Hypertens Res. 2006;29:805–811. doi: 10.1291/hypres.29.805. [DOI] [PubMed] [Google Scholar]
- 260.Patel SB, Reams GP, Spear RM, Freeman RH, Villarreal D. Leptin: linking obesity, the metabolic syndrome, and cardiovascular disease. Curr Hypertens Rep. 2008;10:131–137. doi: 10.1007/s11906-008-0025-y. [DOI] [PubMed] [Google Scholar]
- 261.Ferrannini E, Cushman WC. Diabetes and hypertension: the bad companions. Lancet. 2012;380:601–610. doi: 10.1016/S0140-6736(12)60987-8. [DOI] [PubMed] [Google Scholar]
- 262.Cheung BM, Li C. Diabetes and hypertension: is there a common metabolic pathway? Curr Atheroscler Rep. 2012;14:160–166. doi: 10.1007/s11883-012-0227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 265.Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Figlewicz DP, Sipols AJ. Energy regulatory signals and food reward. Pharmacol Biochem Behav. 2010;97:15–24. doi: 10.1016/j.pbb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Davis JF, Choi DL, Schurdak JD, Fitzgerald MF, Clegg DJ, Lipton JW, Figlewicz DP, Benoit SC. Leptin regulates energy balance and motivation through action at distinct neural circuits. Biol Psychiatry. 2011;69:668–674. doi: 10.1016/j.biopsych.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hoebel BG, Teitelbaum P. Hypothalamic control of feeding and self-stimulation. Science. 1962;135:375–377. doi: 10.1126/science.135.3501.375. [DOI] [PubMed] [Google Scholar]
- 270.Berthoud HR, Münzberg H. The lateral hypothalamus as integrator of metabolic and environmental needs: from electrical self-stimulation to opto-genetics. Physiol Behav. 2011;104:29–39. doi: 10.1016/j.physbeh.2011.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Blum ID, Patterson Z, Khazall R, Lamont EW, Sleeman MW, Horvath TL, Abizaid A. Reduced anticipatory locomotor responses to scheduled meals in ghrelin receptor deficient mice. Neuroscience. 2009;164:351–359. doi: 10.1016/j.neuroscience.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Adams AC, Domouzoglou EM, Chee MJ, Segal-Lieberman G, Pissios P, Maratos-Flier E. Ablation of the hypothalamic neuropeptide melanin concentrating hormone is associated with behavioral abnormalities that reflect impaired olfactory integration. Behav Brain Res. 2011;224:195–200. doi: 10.1016/j.bbr.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Boutrel B, de Lecea L. Addiction and arousal: the hypocretin connection. Physiol Behav. 2008;93:947–951. doi: 10.1016/j.physbeh.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ludwig DS, Mountjoy KG, Tatro JB, Gillette JA, Frederich RC, Flier JS, Maratos-Flier E. Melanin-concentrating hormone: a functional melanocortin antagonist in the hypothalamus. Am J Physiol. 1998;274:E627–E633. doi: 10.1152/ajpendo.1998.274.4.E627. [DOI] [PubMed] [Google Scholar]
- 275.Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- 276.Tritos NA, Vicent D, Gillette J, Ludwig DS, Flier ES, Maratos-Flier E. Functional interactions between melanin-concentrating hormone, neuropeptide Y, and anorectic neuropeptides in the rat hypothalamus. Diabetes. 1998;47:1687–1692. doi: 10.2337/diabetes.47.11.1687. [DOI] [PubMed] [Google Scholar]
- 277.Segal-Lieberman G, Trombly DJ, Juthani V, Wang X, Maratos-Flier E. NPY ablation in C57BL/6 mice leads to mild obesity and to an impaired refeeding response to fasting. Am J Physiol Endocrinol Metab. 2003;284:E1131–E1139. doi: 10.1152/ajpendo.00491.2002. [DOI] [PubMed] [Google Scholar]
- 278.Mantzoros CS, Qu D, Frederich RC, Susulic VS, Lowell BB, Maratos-Flier E, Flier JS. Activation of beta(3) adrenergic receptors suppresses leptin expression and mediates a leptin-independent inhibition of food intake in mice. Diabetes. 1996;45:909–914. doi: 10.2337/diab.45.7.909. [DOI] [PubMed] [Google Scholar]
- 279.Kerman IA, Bernard R, Rosenthal D, Beals J, Akil H, Watson SJ. Distinct populations of presympathetic-premotor neurons express orexin or melanin-concentrating hormone in the rat lateral hypothalamus. J Comp Neurol. 2007;505:586–601. doi: 10.1002/cne.21511. [DOI] [PubMed] [Google Scholar]
- 280.España RA, Plahn S, Berridge CW. Circadian-dependent and circadian-independent behavioral actions of hypocretin/orexin. Brain Res. 2002;943:224–236. doi: 10.1016/s0006-8993(02)02653-7. [DOI] [PubMed] [Google Scholar]
- 281.Rodgers RJ, Halford JC, Nunes de Souza RL, Canto de Souza AL, Piper DC, Arch JR, Blundell JE. Dose-response effects of orexin-A on food intake and the behavioural satiety sequence in rats. Regul Pept. 2000;96:71–84. doi: 10.1016/s0167-0115(00)00203-2. [DOI] [PubMed] [Google Scholar]
- 282.Nakamura T, Uramura K, Nambu T, Yada T, Goto K, Yanagisawa M, Sakurai T. Orexin-induced hyperlocomotion and stereotypy are mediated by the dopaminergic system. Brain Res. 2000;873:181–187. doi: 10.1016/s0006-8993(00)02555-5. [DOI] [PubMed] [Google Scholar]
- 283.Adan RA, Hillebrand JJ, Danner UN, Cardona Cano S, Kas MJ, Verhagen LA. Neurobiology driving hyperactivity in activity-based anorexia. Curr Top Behav Neurosci. 2011;6:229–250. doi: 10.1007/7854_2010_77. [DOI] [PubMed] [Google Scholar]
- 284.Borer KT. Nonhomeostatic control of human appetite and physical activity in regulation of energy balance. Exerc Sport Sci Rev. 2010;38:114–121. doi: 10.1097/JES.0b013e3181e3728f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Rising R, Harper IT, Fontvielle AM, Ferraro RT, Spraul M, Ravussin E. Determinants of total daily energy expenditure: variability in physical activity. Am J Clin Nutr. 1994;59:800–804. doi: 10.1093/ajcn/59.4.800. [DOI] [PubMed] [Google Scholar]
- 286.Schulz LO, Schoeller DA. A compilation of total daily energy expenditures and body weights in healthy adults. Am J Clin Nutr. 1994;60:676–681. doi: 10.1093/ajcn/60.5.676. [DOI] [PubMed] [Google Scholar]
- 287.Vanhecke TE, Franklin BA, Miller WM, deJong AT, Coleman CJ, McCullough PA. Cardiorespiratory fitness and sedentary lifestyle in the morbidly obese. Clin Cardiol. 2009;32:121–124. doi: 10.1002/clc.20458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Pirke KM, Trimborn P, Platte P, Fichter M. Average total energy expenditure in anorexia nervosa, bulimia nervosa, and healthy young women. Biol Psychiatry. 1991;30:711–718. doi: 10.1016/0006-3223(91)90016-f. [DOI] [PubMed] [Google Scholar]
- 289.Casper RC. The ‘drive for activity’ and “restlessness” in anorexia nervosa: potential pathways. J Affect Disord. 2006;92:99–107. doi: 10.1016/j.jad.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 290.Borer KT, Kelch RP, White MP, Dolson L, Kuhns LR. The role of the septal area in the neuroendocrine control of growth in the adult golden hamster. Neuroendocrinology. 1977;23:133–150. doi: 10.1159/000122662. [DOI] [PubMed] [Google Scholar]
- 291.Borer KT, Kelch RP, Peugh J, Huseman C. Increased serum growth hormone and somatic growth in adult hamsters with hippocampal transections. Neuroendocrinology. 1979;29:22–33. doi: 10.1159/000122901. [DOI] [PubMed] [Google Scholar]
- 292.Borer KT, Peters NL, Kelch RP, Tsai AC, Holder S. Contribution of growth, fatness, and activity to weight disturbance after septohypothalamic cuts in adult hamsters. J Comp Physiol Psychol. 1979;93:907–918. doi: 10.1037/h0077624. [DOI] [PubMed] [Google Scholar]
- 293.Borer KT, Bonna R, Kielb M. Hippocampal serotonin mediates hypoactivity in dietarily obese hamsters: a possible manifestation of aging? Pharmacol Biochem Behav. 1988;31:885–892. doi: 10.1016/0091-3057(88)90400-5. [DOI] [PubMed] [Google Scholar]
- 294.Tou JC, Wade CE. Determinants affecting physical activity levels in animal models. Exp Biol Med (Maywood) 2002;227:587–600. doi: 10.1177/153537020222700806. [DOI] [PubMed] [Google Scholar]
- 295.Routtenberg A, Kuznesof AW. Self-starvation of rats living in activity wheels on a restricted feeding schedule. J Comp Physiol Psychol. 1967;64:414–421. doi: 10.1037/h0025205. [DOI] [PubMed] [Google Scholar]
- 296.Kavanau JL. Wheel-running preferences of mice. Z Tierpsychol. 1966;23:858–866. doi: 10.1111/j.1439-0310.1966.tb01715.x. [DOI] [PubMed] [Google Scholar]
- 297.Potter CD, Borer KT, Katz RJ. Opiate-receptor blockade reduces voluntary running but not self-stimulation in hamsters. Pharmacol Biochem Behav. 1983;18:217–223. doi: 10.1016/0091-3057(83)90366-0. [DOI] [PubMed] [Google Scholar]
- 298.Borer KT, Potter CD, Fileccia N. Basis for the hypoactivity that accompanies rapid weight gain in hamsters. Physiol Behav. 1983;30:389–397. doi: 10.1016/0031-9384(83)90142-7. [DOI] [PubMed] [Google Scholar]
- 299.Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol. 1991;36:343–362. doi: 10.1016/0301-0082(91)90015-s. [DOI] [PubMed] [Google Scholar]
- 300.Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.De Luca MA, Solinas M, Bimpisidis Z, Goldberg SR, Di Chiara G. Cannabinoid facilitation of behavioral and biochemical hedonic taste responses. Neuropharmacology. 2012;63:161–168. doi: 10.1016/j.neuropharm.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.El Khoury MA, Gorgievski V, Moutsimilli L, Giros B, Tzavara ET. Interactions between the cannabinoid and dopaminergic systems: evidence from animal studies. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:36–50. doi: 10.1016/j.pnpbp.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 303.Peciña S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci. 2005;25:11777–11786. doi: 10.1523/JNEUROSCI.2329-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Barbano MF, Cador M. Opioids for hedonic experience and dopamine to get ready for it. Psychopharmacology (Berl) 2007;191:497–506. doi: 10.1007/s00213-006-0521-1. [DOI] [PubMed] [Google Scholar]
- 305.Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E, Leibel RL. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115:3579–3586. doi: 10.1172/JCI25977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ehrlich S, Burghardt R, Schneider N, Broecker-Preuss M, Weiss D, Merle JV, Craciun EM, Pfeiffer E, Mann K, Lehmkuhl U, et al. The role of leptin and cortisol in hyperactivity in patients with acute and weight-recovered anorexia nervosa. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:658–662. doi: 10.1016/j.pnpbp.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 307.Exner C, Hebebrand J, Remschmidt H, Wewetzer C, Ziegler A, Herpertz S, Schweiger U, Blum WF, Preibisch G, Heldmaier G, et al. Leptin suppresses semi-starvation induced hyperactivity in rats: implications for anorexia nervosa. Mol Psychiatry. 2000;5:476–481. doi: 10.1038/sj.mp.4000771. [DOI] [PubMed] [Google Scholar]
- 308.Hebebrand J, Exner C, Hebebrand K, Holtkamp C, Casper RC, Remschmidt H, Herpertz-Dahlmann B, Klingenspor M. Hyperactivity in patients with anorexia nervosa and in semistarved rats: evidence for a pivotal role of hypoleptinemia. Physiol Behav. 2003;79:25–37. doi: 10.1016/s0031-9384(03)00102-1. [DOI] [PubMed] [Google Scholar]
- 309.Hebebrand J, Albayrak Ö. Leptin treatment of patients with anorexia nervosa? The urgent need for initiation of clinical studies. Eur Child Adolesc Psychiatry. 2012;21:63–66. doi: 10.1007/s00787-012-0243-3. [DOI] [PubMed] [Google Scholar]
- 310.Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes. 2000;49:1525–1533. doi: 10.2337/diabetes.49.9.1525. [DOI] [PubMed] [Google Scholar]
- 311.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 312.Chakravarthy MV, Booth FW. Eating, exercise, and “thrifty” genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Physiol (1985) 2004;96:3–10. doi: 10.1152/japplphysiol.00757.2003. [DOI] [PubMed] [Google Scholar]
- 313.Campbell BA, Fibiger HC. Effects of insulin on spontaneous activity during food deprivation. J Comp Physiol Psychol. 1970;71:341–346. doi: 10.1037/h0029132. [DOI] [PubMed] [Google Scholar]
- 314.Brooks CM, Lambert HF. A study of the effect of limitation of food intake and the method of feeding on the rate of weight gain during hypothalamic obesity in the albino rat. Am J Physiol. 1946;147:695–707. doi: 10.1152/ajplegacy.1946.147.4.695. [DOI] [PubMed] [Google Scholar]
- 315.Ferguson NB, Keesey RE. Comparison of ventromedial hypothalamic lesion effects upon feeding and lateral hypothalamic self-stimulation in the female rat. J Comp Physiol Psychol. 1971;74:263–271. doi: 10.1037/h0030315. [DOI] [PubMed] [Google Scholar]
- 316.Hoebel BG, Teitelbaum P. Weight regulation in normal and hypothalamic hyperphagic rats. J Comp Physiol Psychol. 1966;61:189–193. doi: 10.1037/h0023126. [DOI] [PubMed] [Google Scholar]
- 317.Jensen LB, Quaade F, Sørensen OH. Bone loss accompanying voluntary weight loss in obese humans. J Bone Miner Res. 1994;9:459–463. doi: 10.1002/jbmr.5650090404. [DOI] [PubMed] [Google Scholar]
- 318.Borer KT. Absence of weight regulation in exercising hamsters. Physiol Behav. 1974;12:589–597. doi: 10.1016/0031-9384(74)90207-8. [DOI] [PubMed] [Google Scholar]
- 319.Kennedy GC, Mitra J. Body weight and food intake as initiating factors for puberty in the rat. J Physiol. 1963;166:408–418. doi: 10.1113/jphysiol.1963.sp007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kennedy GC. The development with age of hypothalamic restraint upon the appetite of the rat. J Endocrinol. 1957;16:9–17. doi: 10.1677/joe.0.0160009. [DOI] [PubMed] [Google Scholar]
- 321.Kennedy GC, Mitra J. Hypothalamic control of energy balance and the reproductive cycle in the rat. J Physiol. 1963;166:395–407. doi: 10.1113/jphysiol.1963.sp007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Borer KT, Kooi AA. Regulatory defense of the exercise-induced weight elevation in hamsters. Behav Biol. 1975;13:301–310. doi: 10.1016/s0091-6773(75)91332-2. [DOI] [PubMed] [Google Scholar]
- 323.Borer KT, Kaplan LR. Exercise-induced growth in golden hamsters: effects of age, weight, and activity level. Physiol Behav. 1977;18:29–34. doi: 10.1016/0031-9384(77)90089-0. [DOI] [PubMed] [Google Scholar]
- 324.Browne SA, Borer KT. Basis for the exercise-induced hyperphagia in adult hamsters. Physiol Behav. 1978;20:553–557. doi: 10.1016/0031-9384(78)90246-9. [DOI] [PubMed] [Google Scholar]
- 325.Borer KT, Kelch RP. Increased serum growth hormone and somatic growth in exercising adult hamsters. Am J Physiol. 1978;234:E611–E616. doi: 10.1152/ajpendo.1978.234.6.E611. [DOI] [PubMed] [Google Scholar]
- 326.Borer KT, Nicoski DR, Owens V. Alteration of pulsatile growth hormone secretion by growth-inducing exercise: involvement of endogenous opiates and somatostatin. Endocrinology. 1986;118:844–850. doi: 10.1210/endo-118-2-844. [DOI] [PubMed] [Google Scholar]
- 327.Tomljenović Borer K, Kuhns LR. Radiographic evidence for acceleration of skeletal growth in adult hamsters by exercise. Growth. 1977;41:1–13. [PubMed] [Google Scholar]
- 328.Karsenty G, Oury F. The central regulation of bone mass, the first link between bone remodeling and energy metabolism. J Clin Endocrinol Metab. 2010;95:4795–4801. doi: 10.1210/jc.2010-1030. [DOI] [PubMed] [Google Scholar]