

Abstract
Zebrafish serves as a promising transgenic animal model that can be used to study cardiac Ca2+ regulation. However, mechanisms of sarcoplasmic reticulum (SR) Ca2+ handling in zebrafish heart have not been systematically explored. We found that in zebrafish ventricular myocytes the action potential (AP)-induced Ca2+ transient is mainly (80%) mediated by Ca2+ influx via L-type Ca2+ channels (LTCC) and only 20% by Ca2+ released from the SR. This small contribution of the SR to the Ca2+ transient was not the result of depleted SR Ca2+ load. We found that the ryanodine receptor (RyR) expression level in zebrafish myocytes was 78% lower compared to rabbit myocytes. In permeabilized myocytes, increasing cytosolic [Ca2+] from 100 to 350 nM did not trigger SR Ca2+ release. However, an application of a low dose of caffeine activated Ca2+ sparks. These results show that the zebrafish cardiac RyR is not sensitive to the mechanism of Ca2+-induced Ca2+ release. Activation of protein kinase A by forskolin increased phosphorylation of the RyR in zebrafish myocardium. In half of the studied cells, an increased Ca2+ transient by forskolin was entirely mediated by augmentation of LTCC current. In the remaining myocytes, the forskolin action was associated with an increase of both LTCC and SR Ca2+ release. These results indicate that the mechanism of excitation-contraction coupling in zebrafish myocytes differs from the mammalian one mainly because of the small contribution of SR Ca2+ release to the Ca2+ transient. This difference is due to a low sensitivity of RyRs to cytosolic [Ca2+].
Keywords: Zebrafish, Sarcoplasmic reticulum, Ca2+-induced Ca2+ release, Ryanodine Receptor, Phosphorylation, Heart
INTRODUCTION
Contraction of an adult mammalian cardiomyocyte vitally relies on well-regulated intracellular Ca2+ homeostasis. During an action potential (AP), a small inward Ca2+ current via L-type Ca2+ channels (LTCC) triggers global Ca2+ release from the sarcoplasmic reticulum (SR) by activating ryanodine receptor (RyR; type 2) Ca2+ release channels. This mechanism of cardiac excitation-contraction coupling (ECC) is known as Ca2+-induced Ca2+ release (CICR) [13]. For relaxation to occur, cytosolic Ca2+ has to be pumped back into the SR by the Ca2+-ATPase (SERCA) and extruded through the sarcolemma out of the cell mainly by the Na+-Ca2+ exchanger (NCX). Abnormalities in Ca2+ homeostasis can lead to contractile dysfunctions and arrhythmias under different pathological conditions including heart failure (HF). Thus, insight into the molecular mechanisms that underlie such abnormalities is important for devising effective therapeutic strategies to improve heart function during HF.
In the last few decades, a significant effort has been made in understanding how alterations of RyR function during different pathological conditions can lead to SR Ca2+ mishandling. The cardiac RyR is a giant multimolecular complex composed of many different accessory proteins including several protein kinases and phosphatases [5;15;19]. A proper arrangement of this complex structure is critical for normal SR Ca2+ regulation. RyR function is also regulated by various post-translational modifications such as phosphorylation and redox modification. These post-translational modifications have potential to impair normal binding of accessory proteins thereby causing RyR dysfunction. For example, it has been suggested that hyper-phosphorylation of the RyR either by protein kinase A (PKA) [20] or Ca2+/Calmodulin-dependent kinase type II (CaMKII) [1] destabilizes SR Ca2+ homeostasis leading to generation of Ca2+-mediated cardiac arrhythmias in HF. Studying transgenic mouse models with specific RyR mutations relevant to human cardiomyopathies provides particularly important insight into molecular mechanisms that may cause abnormalities in Ca2+ regulation in cardiac diseases [17;28]. However, progress in this field is limited due to high cost and long time required for the generation of a transgenic mouse. Thus, using alternative cost-efficient transgenic animal models with short lifecycle would significantly contribute to the understanding of the molecular mechanisms of RyR regulation during health and diseases.
In recent years, the zebrafish has emerged as a promising experimental model for a variety of genetic studies due to the ease by which transgenic animals can be generated. In contrast to mouse, zebrafish have shorter lifecycles, larger hatch sizes and are easy to maintain. Similar to mammals, the zebrafish ventricular myocyte possesses the major components of ECC [22;25;30]. Furthermore, electrophysiological properties of zebrafish cardiac myocyte (e.g. shape of AP) closely resemble human [10], making this animal model suitable for study of ion channel mutations relevant to human pathologies. However, little information is available regarding zebrafish cardiac SR Ca2+ handling and RyR regulation. The aim of this study is to characterize mechanisms of SR Ca2+ regulation in ventricular myocytes isolated from zebrafish hearts. We found that the SR of zebrafish ventricular myocytes contains functional RyRs, however, with very low sensitivity to cytosolic [Ca2+]. RyR activity can be increased with a low dose of caffeine and by RyR phosphorylation. Because RyRs in zebrafish myocytes are almost inactive under basal conditions, zebrafish could be a useful animal model to study human RyR mutations in cellular surroundings that are not contaminated by “noise” from endogenous RyRs.
MATERIALS AND METHODS
Myocyte Isolation
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Loyola University, and complies with US regulations on animal experimentation. Zebrafish (Danio rerio) were anesthetized using tricaine (0.4 mg/L) and euthanized by immersion in ice-cold water for 5 min. Single cardiac myocytes were isolated by enzymatic digestion, modified from [10;30]. Briefly, zebrafish hearts were quickly excised and perfused (0.5 ml/min) for 30 min at room temperature with a solution containing (in mM): NaCl 100, KCl 10, MgSO4 4, KH2PO4 1.2, Taurine 50, glucose 20 and HEPES 10 (pH was adjusted to 6.9 with NaOH). Perfusion solution was supplemented with bovine serum albumin (BSA; 1 mg/ml), collagenase (type 2, Worthington; 0.2 mg/ml) and trypsin (type IX-S, Sigma; 0.12 mg/ml). 50 μM of EGTA and 37.5 μM of Ca2+ were also added to perfusion solution. At the end of perfusion, atrium and bulbous were removed and the heart was placed in 1 ml of the low Ca2+ solution with BSA (1 mg/ml) without enzymes. Next, the tissue was gently torn up into small pieces followed by 30 min rest at room temperature before adding Ca. Then [Ca2+] was increased gradually to 1 mM. The cell suspension was filtered through a nylon mesh and stored at room temperature up to 8–10 hours. Rabbit ventricular myocytes were isolated from New Zealand White rabbits (2–2.5 kg) according to the procedure described previously [12]. Rabbits were anaesthetized with sodium pentobarbital (50 mg/kg I.V.). Following thoracotomy hearts were quickly excised, mounted on a Langendorff apparatus, and retrogradely perfused with Liberase (Roche Applied Science, Indianapolis, IN) Blendzyme-containing solution at 37°C. Digested tissue was then minced, filtered, and washed in a MEM solution containing Ca2+ (50 μM) and bovine serum albumin (10 mg/ml). Chemicals and reagents were purchased from Sigma-Aldrich and Fisher Scientific unless otherwise stated. All experiments were performed at room temperature (20–24°C).
Confocal microscopy
Changes in [Ca2+]i, were measured using laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK and LSM 410, Zeiss, Germany) equipped with a ×40 oil-immersion objective lens (N.A=1.3).
Measurements of [Ca2+]i in intact myocytes
To record [Ca2+]i we used the high affinity Ca2+ indicator Fluo-4 (Molecular Probes/Invitrogen, Carlsbad, USA). To load the cytosol with the Ca2+ dye, cells were incubated at room temperature with 10 μM Fluo-4/AM for 15 minutes followed by a 20 minute wash. Fluo-4 was excited with the 488 nm line of an argon ion laser and fluorescence was measured at 515±15 nm. Action potentials were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med. Inc., USA) set at a voltage ~50% above the threshold. Fluo-4 images were acquired in line-scan mode (3 ms per scan; pixel size 0.12 μm). The scan line was positioned along a region of the cell where cell motion is minimal during contraction. Rabbit ventricular myocytes were studied in Tyrode solution containing (in mM): NaCl 140; KCl 4; CaCl2 2; MgCl2 1; glucose 10; HEPES 10; pH 7.4. Fish myocytes were studied in Tyrode solution containing (in mM): NaCl 132; KCl 2.5; MgCl2 1.6; NaH2PO4 0.33; NaHCO3 4; CaCl2 2; glucose 5; Na pyruvate 5 and HEPES 10; pH 7.5.
Measurements of [Ca2+]i in permeabilized myocytes
Myocytes were permeabilized with saponin [32]. The saponin free internal solution was composed of (in mM): potassium aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.12; MgCl2 0.75; phosphocreatine 10; Hepes 10; Rhod-2 tripotassium salt 0.04; creatine phosphokinase 5 U ml−1; dextran (MW: 40,000) 8%; pH 7.2 (KOH). Free [Ca2+] and [Ca2+] of this solution were 150 nM and 1 mM, respectively. Rhod-2 was excited with the 543 nm line of a He–Ne laser and fluorescence was measured at wavelengths >600 nm. Images were acquired in line-scan mode (3 ms per scan; pixel size 0.12 μm). Ca2+ sparks were detected and analyzed using SparkMaster [23].
Labeling of the surface membrane
The density of the t-tubular system was measured after staining the surface membrane of rabbit and zebrafish myocytes with the voltage-sensitive fluorescent dye Di-8-ANEPPS. Cells were incubated with extracellular solution containing 5 μM of Di-8-ANEPPS for 15 min. The Di-8-ANEPPS was excited at 488 nm and the fluorescence was recorded at >600 nm.
Western blot analysis of RyR and RyR phosphorylation
To prepare tissue samples, zebrafish hearts (from 6 fish) and pieces of rabbit ventricle (from 3 rabbits) were homogenized in saline buffer containing a protein inhibitors cocktail (Complete Mini, Roche, USA). Total protein concentration was quantified using Coomassie Plus reaction (Thermoscientific, USA). 20 μg of each sample was subjected to 4–15% SDS-PAGE and transferred onto nitrocellulose membrane using Turbo-transfer (Bio-Rad, USA). The RyR expression level was assessed with the 34C primary antibody (DSHB, USA) and normalized to the total level of protein (Ponceau). RyR phosphorylation at the PKA site was measured in three different tissue samples (12 zebrafish hearts total): control, forskolin (5 μM) and forskolin (5 μM) plus the phosphatase inhibitor okadaic acid (500 nM). The last sample was used to quantify the maximum level of RyR phosphorylation. Afterwards each sample was homogenized in Laemmli buffer and the same amount of total homogenate was subjected to 4–15% SDS-PAGE and western blot. RyR phosphorylation at the PKA site (Ser 2715) was detected using a phospho-specific antibody RyR-Ser 2809 (Badrilla; UK). The signal was normalized to the total RyR level measured with the 34C antibody. The secondary antibodies were conjugated with horseradish peroxidase (HRP). The signals obtained with the HRP substrate (Luminata forte, Millipore, USA) were quantified using the UVP EpiChemi imaging system and ImageJ software.
Statistics
Data are presented as mean ± SEM of n measured cells. Statistical comparisons between groups were performed by the Student’s t test. Differences were considered statistically significant at P<0.05.
RESULTS
Consistent with previous works [10;30], zebrafish ventricular myocytes had an elongated shape (width to length ratio is 0.09) compared to rabbit myocytes (ratio is 0.26). Furthermore, staining of myocytes with Di-8-ANEPPS revealed that zebrafish ventricular myocytes lack t-tubular organization (Fig. 1).
Figure 1. Morphology of zebrafish and rabbit ventricular myocytes.
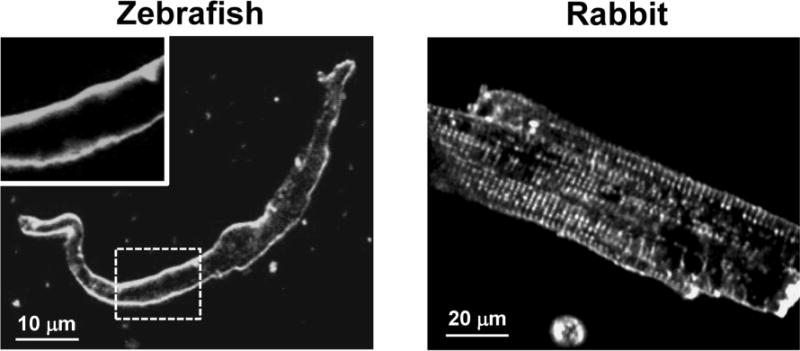
Confocal 2-D images of ventricular myocytes from zebrafish (left panel) and rabbit (right panel) labeled with the voltage-sensitive fluorescent dye Di-8-ANEPPS. The left-top panel is an enlarged area marked with the dashed white line, to highlight the absence of the t-tubular network in the zebrafish ventricular myocytes.
SR Ca2+ release during ECC in zebrafish ventricular myocytes
In the following experiments, we compared the contribution of RyR and LTCC to the global Ca2+ transient during ECC in zebrafish and rabbit ventricular myocytes. Figs 2A and B show line-scan images of [Ca2+]i and corresponding profiles of Ca2+ transients from zebrafish and rabbit ventricular myocytes. Ca2+ transients were evoked by electrical field stimulation (0.75 Hz) to induce an AP or by caffeine (10 mM) application to release Ca2+ stored in the SR. Despite a relatively similar SR Ca2+ content in both species, the AP-induced Ca2+ transient amplitude was smaller in zebrafish myocytes compared to rabbit myocytes. Since the SR is entirely depleted of Ca2+ in the presence of high doses of caffeine, the first AP-induced Ca2+ transient after caffeine application is entirely mediated by LTCC Ca2+ current. Indeed, a selective LTCC inhibitor verapamil (10 μM) applied immediately after the caffeine application prevented any increase in [Ca2+]i in myocytes from both species (data not shown). AP-induced Ca2+ transients measured in the presence of the SERCA inhibitor thapsigargin (TG; 5 μM) revealed similar amplitude compared to those recorded immediately following caffeine application (Fig 2, C). These results indicate that contribution of SR Ca2+ release to the first AP-induced Ca2+ transient following caffeine application is negligible. By analyzing Ca2+ transients before and after caffeine application, we found that in zebrafish myocytes 84.1±3 % (n=10) of the global Ca2+ transient during ECC was mediated by Ca2+ entry via LTCC and only 15.9±3 % (n=10) was due to Ca2+ release from the SR (Fig 3, A). In agreement with previous works [2;3], we found that in rabbit myocytes the contribution of LTCCs and RyRs to the AP-induced Ca2+ transient is 44±5 % (n=10) and 55±4 % (n=10), respectively (Fig 3, A). These data indicate that the contribution of LTCC and RyR to the amplitude of the Ca2+ transient during ECC is significantly different between zebrafish and rabbit myocytes.
Figure 2. Ca2+ transients during ECC in zebrafish and rabbit ventricular myocytes.
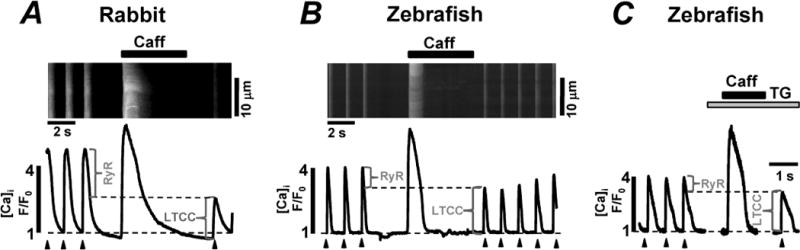
Confocal line-scan images of Fluo-4 fluorescence and corresponding F/F0 profiles recorded in rabbit (A) and zebrafish (B) ventricular myocytes. The F/F0 profiles were obtained by averaging the fluorescence throughout the entire image. The myocytes were electrically stimulated at constant rate (0.75 Hz; marked by black arrowheads). Contributions of LTCC and RyR to the AP-induced Ca2+ transients are indicated by the brackets. C, caffeine-induced and the AP-induced Ca2+ transients recorded in zebrafish ventricular myocytes in control conditions and after inhibition of SERCA with thapsigargin (TG; 10 μM).
Figure 3. Contribution of LTCC and RyR to Ca2+ transients during ECC in zebrafish and rabbit ventricular myocytes.
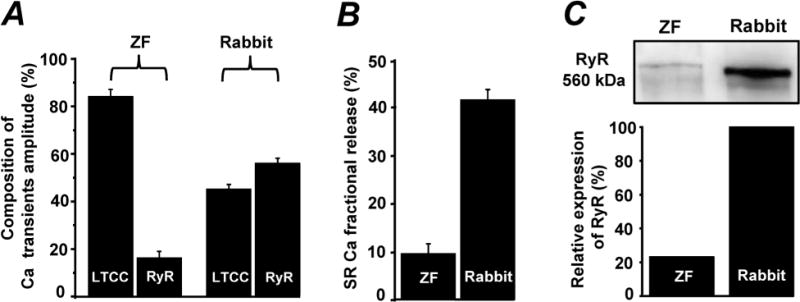
A, components of the AP-induced Ca2+ transient in zebrafish (left part) and in rabbit (right part) ventricular myocytes. B, fractional SR Ca release in zebrafish and rabbit ventricular myocytes. C, representative western blots (top) and correspondent analysis (bottom) of the RyR expression level in zebrafish and rabbit hearts. The signal corresponding to the RyR level in zebrafish was normalized to the signal obtained in rabbit hearts.
Next, we analyzed the fractional SR Ca2+ release during the AP to estimate the sensitivity of RyR to the trigger (i.e. LTCC current) in zebrafish myocytes. In order to accurately determine the fractional release, the amplitude of the LTCC-mediated Ca2+ transient (measured immediately following caffeine application) was first subtracted from the amplitude of the AP-induced Ca2+ transient (measured before caffeine application). The fractional SR Ca2+ release was then calculated as the ratio between the obtained value and the amplitude of the caffeine-induced Ca2+ transient. According to this analysis, in zebrafish myocytes only ~9% of the SR Ca2+ load is released into the cytosol during the AP, whereas in rabbit this value is ~40% (Fig 3, B). These results indicate that in zebrafish myocytes only a small fraction of Ca2+ stored in the SR contributes to ECC. This can be explained either by low sensitivity of the RyR to trigger Ca2+ or by a low expression level of RyRs in zebrafish heart.
RyR expression level in zebrafish ventricular myocytes
In order to investigate the expression of RyRs in zebrafish and in rabbit hearts, western blot analysis with a monoclonal antibody against RyR (type 2) was performed. Fig 3, C shows that in zebrafish, this antibody could detect a band which corresponds to MW ~560 kDa with a signal density that is significantly lower (by ~78%) in comparison to rabbit hearts. However, it should be noted that this value could be overestimated due to a different immunoreactivity of the used antibody towards the zebrafish cardiac RyR.
The sensitivity of CICR in zebrafish ventricular myocytes
In the following experiments we studied SR Ca2+ release in permeabilized zebrafish ventricular myocytes. The advantage of the membrane permeabilization is that an experimental solution with known [Ca2+] can be easily introduced into the cytosol. We could not detect any Ca2+ spark activity at 100 nM [Ca2+]i. The increase of [Ca2+]i up to 350 nM, a concentration that in permeabilized rabbit myocytes produces Ca2+ waves [9], also did not induce spark activity (data not shown). However, application of caffeine (10 mM) at the end of each experiment produced a large Ca2+ transient, indicating that the absence of sparks was not due to depleted SR Ca2+ load, but rather to low RyR activity or distribution of RyRs on the SR as isolated channels.
In order to increase the RyR activity, we used different concentrations of caffeine. The application of 200 μM caffeine induced Ca2+ sparks at a frequency of 3.5 sparks×s−1×(100 μm)−1 (Fig. 4, A). Caffeine (1 mM) increased RyR activity to the point that all the SR Ca2+ release events were detected in the first 500 ms of the caffeine application (Fig 4, B). Further increase of the caffeine concentration to 5 mM led to more synchronized SR Ca2+ release: isolated Ca2+ sparks were no longer detectable because the simultaneous activation of RyRs produced a global Ca2+ transient (Fig 4, C). These results revealed that in zebrafish myocytes RyRs are organized in the form of release clusters, however, these release clusters exhibit a low sensitivity to [Ca2+]i.
Figure 4. Effect of caffeine on RyR-mediated Ca2+ release in zebrafish ventricular myocytes.
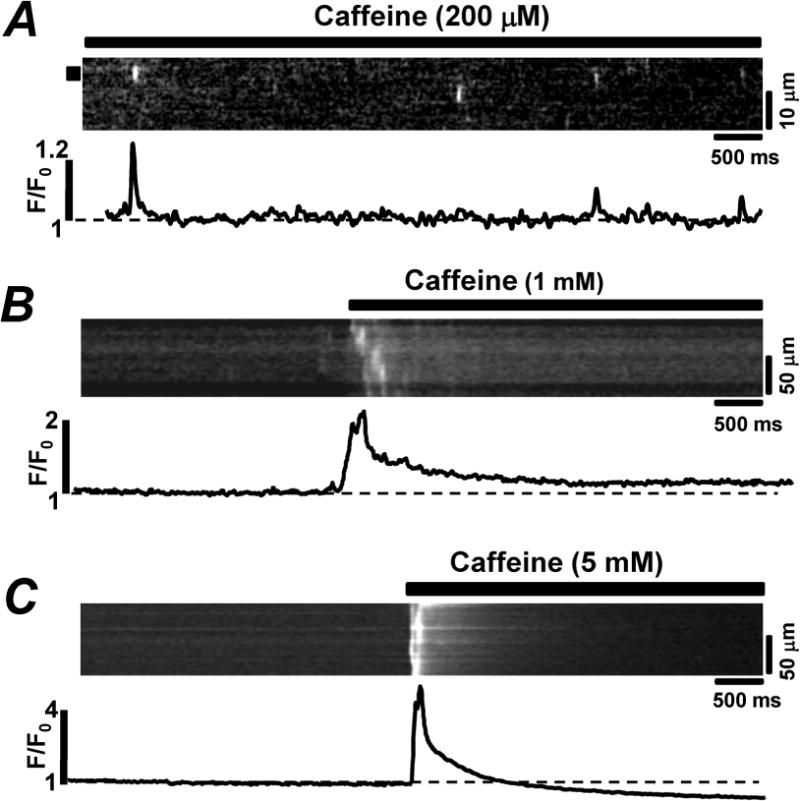
Confocal line-scan images of Rhod-2 fluorescence and corresponding F/F0 profiles recorded in permeabilized zebrafish ventricular myocytes in the presence of 200 μM (A), 1 mM (B) and 5 mM (C) caffeine. Profiles were obtained by averaging the fluorescence throughout the area labeled by the black box (A) or throughout the entire image (B and C).
Effect of RyR phosphorylation by PKA on SR Ca2+ release during ECC
RyR phosphorylation by PKA plays an important role in the regulation of SR Ca2+ release in the mammalian heart [20;21;27]. Thus, in the following experiments we investigated the susceptibility of the zebrafish RyR to PKA phosphorylation. First, we analyzed whether the zebrafish RyR amino acid sequence contained the consensus sites for PKA phosphorylation. Using “Blast P” software, we identified a PKA consensus site in the zebrafish RyR at Ser 2715 corresponding to the characterized Ser 2809 in the rabbit RyR. Using western blot analysis we measured the level of phosphorylation at Ser 2715. The basal level of phosphorylation at this site was ~38%. The treatment of zebrafish hearts with the adenylate cyclase activator forskolin (5 μM) almost doubled the RyR phosphorylation level (Fig 5, A). These values were obtained after normalization to the maximal level of phosphorylation achieved in the presence of both forskolin and the phosphatase inhibitor okadaic acid.
Figure 5. Effects of forskolin on RyR phosphorylation and Ca2+ transients in zebrafish ventricular myocytes.
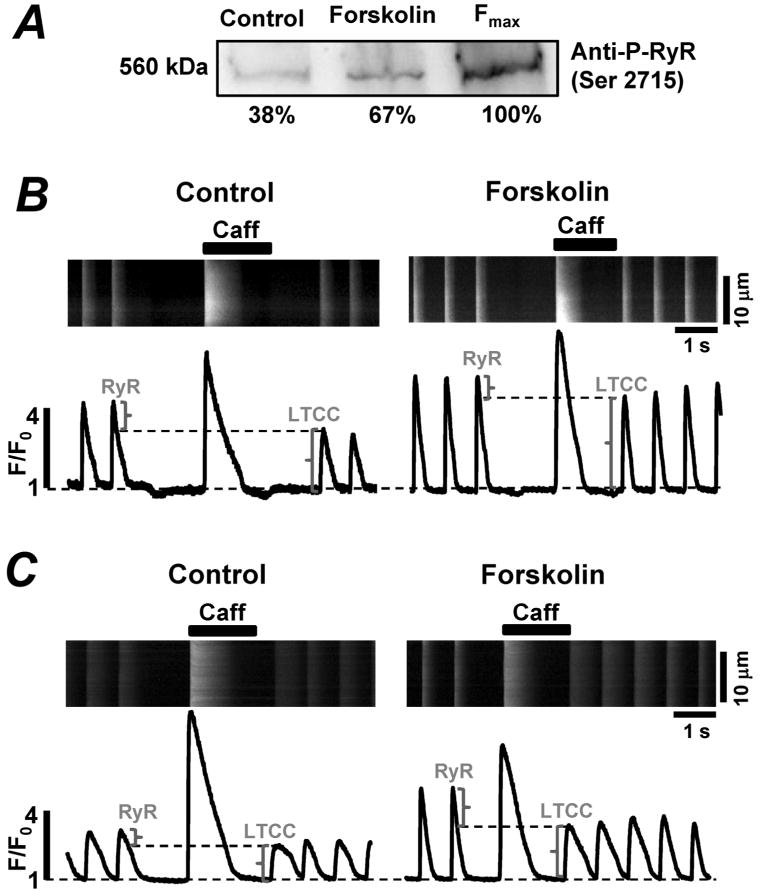
A, representative western blots with corresponding statistical analysis showing the effect of forskolin (5 μM) on RyR phosphorylation at the PKA specific site. The levels of phosphorylation were normalized to the maximal level of RyR phosphorylation at the PKA site (achieved after treatment of zebrafish hearts with forskolin and the phosphatase inhibitor okadaic acid). B, C, confocal line-scan images of Fluo-4 fluorescence and corresponding F/F0 profiles recorded in zebrafish ventricular myocytes in control conditions (left panels) and in the presence of forskolin (5 μM; right panels). The myocytes were electrically stimulated at 0.75 Hz. The brackets indicate the contribution of RyR and LTCC to the AP-induced Ca2+ transient.
We studied the effects of forskolin on Ca2+ release during ECC in intact zebrafish ventricular myocytes. In all myocytes studied, Ca2+ transient amplitude increased significantly upon application of forskolin (5 μM). The increase was mainly due to augmentation of LTCC activity (as can be seen from the increased LTCC-mediated Ca2+ transients in Figs 5 B and C). However, the effect of forskolin on the RyR activity was more complex. In half of the studied cells, forskolin increased SR Ca2+ load without a significant effect on RyR-mediated Ca2+ release (Fig 5, B). These results suggest that in these cells, PKA activation has a more profound effect on SERCA-mediated Ca2+ uptake than on RyR-mediated Ca2+ release. Furthermore, these myocytes commonly responded to forskolin by producing a second peak during the Ca2+ transient indicating early after depolarization (EAD) (Fig 6, A). In the other half of the cells, the application of forskolin enhanced RyR-mediated Ca2+ release and decreased SR Ca2+ load (Fig. 5, C). Thus, in these cells PKA activation had a more significant effect on RyR activity and diastolic SR Ca2+ leak, than on SERCA activity. In these myocytes, the increased diastolic Ca2+ leak was commonly associated with Ca2+ spark activity (Fig 6, B). However, none of these cells showed Ca2+ waves or delayed after depolarization (DAD).
Figure 6. Effect of forskolin on Ca2+ transients in zebrafish ventricular myocytes.
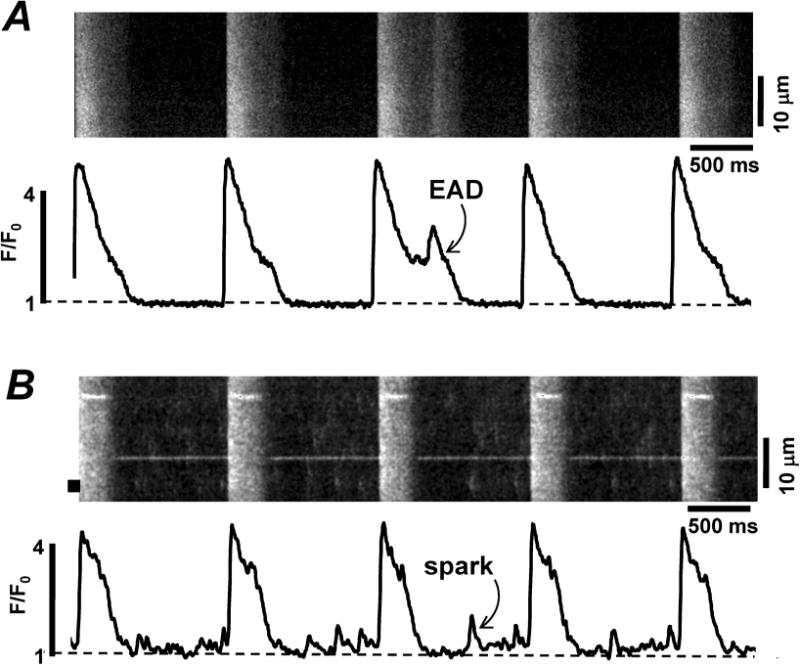
Confocal line-scan images of Fluo-4 fluorescence and corresponding F/F0 profiles recorded in electrically stimulated zebrafish myocytes during the application of forskolin (5 μM). A, forskolin induced a second peak during the Ca2+ transient indicating early after depolarization (EAD). B, forskolin produced diastolic Ca2+ sparks.
Effect of RyR phosphorylation by PKA on spontaneous SR Ca2+ release
We also studied the effect of PKA activation on SR Ca2+ release in permeabilized zebrafish ventricular myocytes. As described above, zebrafish myocytes did not exhibit any detectable SR Ca2+ release events at high [Ca2+]i. To maximally increase RyR phosphorylation at the PKA site, the internal solution containing cAMP (20 μM), the PKA catalytic subunit (5 U/ml) and the phosphatase inhibitor okadaic acid (100 nM) were introduced into the cytosol. The application of this “phosphorylation cocktail” caused a rapid rise in Ca2+ spark frequency in half of the studied cells. After reaching a peak within 1 min, Ca2+ spark frequency began to gradually decline and after 3 min Ca2+ sparks could not be detected (Fig 7, B). Spontaneous Ca2+ release events recorded in zebrafish myocytes had an average amplitude of 0.35±0.07 ΔF/F0 (n=10).
Figure 7. Effect of PKA activation on RyR-mediated Ca2+ release in zebrafish myocytes.
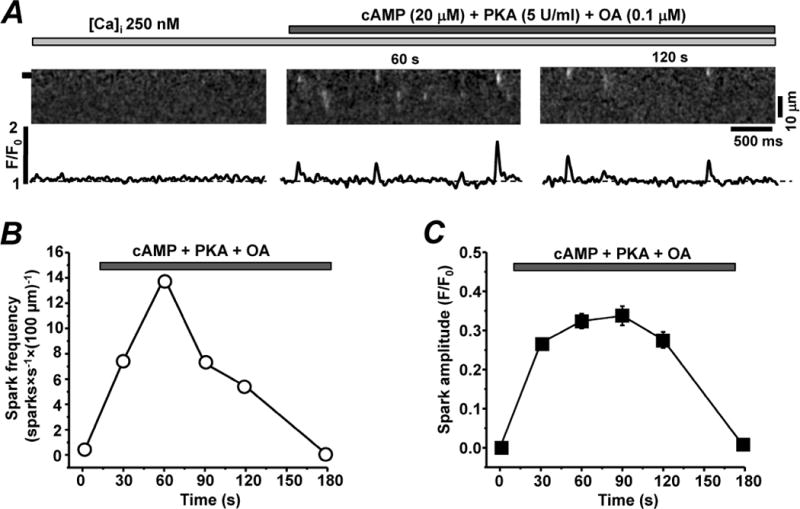
A, confocal line-scan images of Rhod-2 fluorescence and corresponding F/F0 profiles recorded in permeabilized zebrafish ventricular myocytes in the presence of 250 nM [Ca2+]i (left panel) and after a subsequent application of cAMP (20 μM), the PKA catalytic subunit (5 U/ml) and the phosphatase inhibitor okadaic acid (OA; 100 nM; central and right panel). Profiles were obtained by averaging the fluorescence throughout the area labeled by the black box. Effects of PKA activation on Ca spark frequency (B) and spark amplitude (C).
DISCUSSION
Results in this study revealed that in zebrafish ventricular myocytes the cytosolic Ca2+ transient induced by the AP is mainly mediated by Ca2+ influx via LTCCs, whereas only a small fraction of Ca2+ (<20%) is released from the SR via RyRs. Such composition of Ca2+ transients is considerably different from the one reported in adult mammalian cardiomyocytes (e.g. rabbit, human) where SR Ca2+ release contributes to nearly 70% of the Ca2+ transient during ECC (Fig 3 and also see [4]). Despite a small contribution of the SR to the Ca2+ transient, the SR of zebrafish myocytes is able to retain a significant amount of Ca2+ as demonstrated from the caffeine-induced Ca2+ release (Fig 2). Thus, the diminished SR Ca2+ release during ECC in zebrafish myocytes is likely to be the result of lower activity of RyRs. The spatial separation of triggering LTCCs and RyRs due to the lack of the t-tubular system (Fig 1) could reduce the efficacy of CICR contributing to the decreased SR Ca2+ release. It seems, however, that the lack of the t-tubular system in zebrafish ventricular myocytes is compensated by their small volume and elongated shape as in mammalian atrial cells [7]. Therefore, Ca2+ that enters the cytosol via LTCCs propagates throughout the cell without any detectable delay (Fig 2; Ca2+ transients mediated by LTTCs). Therefore, other factors may play a role in the diminished SR Ca2+ release in zebrafish myocytes, including low expression level of the RyR. Our data show that in the zebrafish heart the density of the band corresponding to the RyR is at least 78% lower than that found in rabbit myocardium. However, this difference might be an underestimation of the RyR expression level in zebrafish hearts due to the poor immunoreactivity of the antibody towards the fish cardiac RyR isoform.
The entire amino acid sequence of the zebrafish cardiac RyR (type 2) has not been completely determined. The characterized sequences of the RyR from zebrafish and rabbit share ~85% similarity. Therefore, the unknown domains might be profoundly different from the rabbit RyR. Moreover, differences in the amino acid sequences between these two species might be located within some important regulatory sites of the channel. For instance, activation of the mammalian RyR is mediated by the high affinity cytosolic Ca2+ binding site [14] composed of the glutamates at position 3885 of each RyR monomer [11]. Because this region of the zebrafish RyR has not been determined, there might be crucial amino acid differences that decrease the channel sensitivity to [Ca2+]i. This would lead to a smaller SR Ca2+ release during the AP in zebrafish myocytes. It can also explain the absence of spontaneous Ca2+ sparks at 350 nM [Ca2+]i, a concentration that triggers Ca2+ waves in rabbit myocytes [9]. Clearly our spark data suggest that RyRs in zebrafish myocytes are significantly less sensitive to [Ca2+]i than the cardiac RyR in mammals. Low activity of the RyR has also been reported in trout ventricular myocytes [25]. It has been suggested that in trout myocytes the absence of Ca2+ sparks at high [Ca2+]i is a result of the lack of continuity in the RyR cluster organization. In zebrafish myocytes, however, it was not the case, since the application of 200 μM caffeine in permeabilized myocytes triggered Ca2+ sparks with spatio-temporal characteristics similar to a typical mammalian cardiac spark. It is well established that the propensity of spontaneous Ca2+ release from the SR directly depends on SR Ca2+ load [18;24;31]. We found that the SR of zebrafish myocytes contains a significant amount of Ca2+ at rest (Fig 4). However, the role of this SR Ca2+ content that is not fully used on a beat to beat basis remains elusive. The fact that the SR of zebrafish myocytes is equipped with Ca2+ release and uptake machineries suggests that this Ca2+ load could be used to increase heart contraction during stimulation of certain signaling pathways that sensitize the RyR to [Ca2+]i (similar to the caffeine effect).
In mammalian myocytes, the RyR-mediated Ca2+ release can be significantly increased by β-adrenergic receptor (β-AR) stimulation [4]. The stimulation of β-ARs leads to activation of the adenylate cyclase-PKA pathway with following phosphorylation of several important proteins involved in ECC, including the RyR [8;16;20;29]. Although controversial [6;26], it has been shown that phosphorylation of mammalian RyR at Ser 2808 by PKA increases RyR activity and SR Ca2+ release [20;27;29]. In zebrafish ventricular myocytes, the role of PKA phosphorylation as a possible mechanism that can “unlock” the RyR remains obscure. We found that in zebrafish myocytes activation of adenylate cyclase by forskolin application increased phosphorylation of the RyR at the PKA site (Fig 5A). We also found that in intact myocytes forskolin increased the amplitude of AP-induced Ca2+ transients (Figs 5 B and C). However, the contributions of LTCC and RyR to the global [Ca2+]i increase varied significantly between cells. That is, in ~50% of the myocytes the inotropic effect of forskolin was mainly mediated via augmentation of Ca2+ current via LTCCs, without any significant effect on SR Ca2+ release. In contrast, in the other myocytes the forskolin inotropic action was associated with an increase of both LTCC-mediated Ca2+ current and RyR-mediated Ca2+ release. In rabbit myocytes increased RyR sensitivity during β-AR stimulation causes SR Ca2+ leak in the form of sparks and waves [8]. In zebrafish myocytes that are characterized by increased RyR activity, the forskolin application also increased SR Ca2+ leak in the form of diastolic Ca2+ sparks (Fig 6). As a result, the increased SR Ca2+ leak partially depleted SR Ca2+ load. In agreement with the intact cell results, we found that treatment of permeabilized myocytes with PKA triggered Ca2+ sparks only in half of the studied cells (Fig 7). We do not know the reason why only in some of zebrafish myocytes PKA activation increased SR Ca2+ release. It is possible that high protein phosphatase activity in some cells prevents RyR phosphorylation from reaching a sufficient level that is required for the RyR activity.
The presented results highlight two important aspects of the zebrafish SR Ca2+ handling that might be essential for considering this animal model to study human cardiac diseases associated with RyR mutations. The mechanism of ECC in zebrafish myocytes differs from that in mammalian myocardium mainly by the small contribution of SR Ca2+ release to the global Ca2+ transient. This difference could be a result of the zebrafish RyR structure that translates into a low sensitivity of the channel to [Ca2+]i. On the other hand, RyR phosphorylation by PKA appears to affect RyR function similarly in both mammalian and zebrafish myocytes. Further investigations of SR Ca2+ release in zebrafish myocytes can provide important information how alterations of the RyR structure translate into altered function. Furthermore, the fact that RyRs in zebrafish myocytes are almost inactive under basal conditions provides an advantage for the generation of transgenic animals expressing human RyR mutations. In such transgenic models, the activity of the expressed RyR will be easy to study because it would not be contaminated by “noise” from endogenous zebrafish RyRs. Thus, the zebrafish may constitute an ideal model for investigation of human pathologies associated with RyR malfunction such as in heart failure.
Acknowledgments
We would like to thank Olga Alekhina for excellent technical assistance. This work was supported by the National Institutes of Health Grants HL62426 and HL75494 to P.d.T., The Research Career Development Award from The Schweppe Foundation to A.V.Z. and American Heart Association Fellowship Grant 14510047 to A.V.D.
REFERENCE LIST
- 1.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 2.Bassani JW, Bassani RA, Bers DM. Twitch-dependent SR Ca accumulation and release in rabbit ventricular myocytes. Am J Physiol. 1993;265:C533–C540. doi: 10.1152/ajpcell.1993.265.2.C533. [DOI] [PubMed] [Google Scholar]
- 3.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- 5.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 6.Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 7.Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, Lipsius SL. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol. 2003;546:19–31. doi: 10.1113/jphysiol.2002.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bovo E, Mazurek SR, Blatter LA, Zima AV. Regulation of sarcoplasmic reticulum Ca2+ leak by cytosolic Ca2+ in rabbit ventricular myocytes. J Physiol. 2011;589:6039–6050. doi: 10.1113/jphysiol.2011.214171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brette F, Luxan G, Cros C, Dixey H, Wilson C, Shiels HA. Characterization of isolated ventricular myocytes from adult zebrafish (Danio rerio) Biochem Biophys Res Commun. 2008;374:143–146. doi: 10.1016/j.bbrc.2008.06.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SR, Ebisawa K, Li X, Zhang L. Molecular identification of the ryanodine receptor Ca2+ sensor. J Biol Chem. 1998;273:14675–14678. doi: 10.1074/jbc.273.24.14675. [DOI] [PubMed] [Google Scholar]
- 12.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 14.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 16.Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lompre AM, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ, Marks AR. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation. 2010;121:822–830. doi: 10.1161/CIRCULATIONAHA.109.890954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukyanenko V, Gyorke I, Gyorke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflugers Arch. 1996;432:1047–1054. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- 19.Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- 20.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 21.Morimoto S, Uchi J, Kawai M, Hoshina T, Kusakari Y, Komukai K, Sasaki H, Hongo K, Kurihara S. Protein kinase A-dependent phosphorylation of ryanodine receptors increases Ca2+ leak in mouse heart. Biochem Biophys Res Commun. 2009;390:87–92. doi: 10.1016/j.bbrc.2009.09.071. [DOI] [PubMed] [Google Scholar]
- 22.Nemtsas P, Wettwer E, Christ T, Weidinger G, Ravens U. Adult zebrafish heart as a model for human heart? An electrophysiological study. J Mol Cell Cardiol. 2010;48:161–171. doi: 10.1016/j.yjmcc.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 23.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 24.Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol. 1997;272:H657–H668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- 25.Shiels HA, White E. Temporal and spatial properties of cellular Ca2+ flux in trout ventricular myocytes. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1756–R1766. doi: 10.1152/ajpregu.00510.2004. [DOI] [PubMed] [Google Scholar]
- 26.Valdivia HH. Ryanodine receptor phosphorylation and heart failure: phasing out S2808 and “criminalizing” S2814. Circ Res. 2012;110:1398–1402. doi: 10.1161/CIRCRESAHA.112.270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yano M. Ryanodine receptor as a new therapeutic target of heart failure and lethal arrhythmia. Circ J. 2008;72:509–514. doi: 10.1253/circj.72.509. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida A, Takahashi M, Imagawa T, Shigekawa M, Takisawa H, Nakamura T. Phosphorylation of ryanodine receptors in rat myocytes during beta-adrenergic stimulation. J Biochem. 1992;111:186–190. doi: 10.1093/oxfordjournals.jbchem.a123735. [DOI] [PubMed] [Google Scholar]
- 30.Zhang PC, Llach A, Sheng XY, Hove-Madsen L, Tibbits GF. Calcium handling in zebrafish ventricular myocytes. Am J Physiol Regul Integr Comp Physiol. 2011;300:R56–R66. doi: 10.1152/ajpregu.00377.2010. [DOI] [PubMed] [Google Scholar]
- 31.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of ca release. Biophys J. 2008;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]