

Abstract
Glioblastoma is the most common and most aggressive diffuse glioma, associated with short survival and uniformly fatal outcome irrespective of treatment. It is characterized by morphologic, genetic, and gene-expression heterogeneity. The current standard of treatment is maximal surgical resection, followed by radiation, with concurrent and adjuvant chemotherapy. Due to the heterogeneity most tumors develop resistance to treatment and shortly recur. Following recurrence glioblastoma is quickly fatal in the majority of cases. Recent genetic molecular advances have contributed to a better understanding of glioblastoma pathophysiology and disease stratification. In this paper we review the basic glioblastoma pathophysiology with emphasis on clinically relevant genetic molecular alterations and potential targets for further drug development.
Keywords: glioblastoma, pathogenesis, IDH, EGFR, MGMT, FGFR, TACC, 1p/19q, proneural, mesenchymal
Introduction
Glioblastoma is a uniformly fatal extremely heterogeneous disease. Tumor heterogeneity translates into histological patterns [1], genetic alterations, and gene expression profiles [2–4]. The current treatment for glioblastoma is maximal surgical resection, followed by radiation therapy with concurrent and adjuvant temozolomide (the Stupp regimen) [5]. Recent genetic molecular advances have contributed to a better understanding of glioblastoma pathophysiology. In this paper we review the basic glioblastoma pathophysiology emphasizing the clinically relevant genetic molecular alterations and potential targets for further drug development.
Epidemiology
Glioblastoma is the most common intrinsic primary brain tumor in adults, and represents the most common diffuse glioma. According to the latest Central Brain Tumor Registry of the United States (CBTRUS) statistical report, the age-adjusted incidence rate for glioblastoma is 3.19 per 100,000. The incidence of glioblastoma increases with age and peaks between 75 and 84 years old, being more common in white males. It is uncommon in children, approximating 3% of all primary brain and CNS tumors. The five-years survival rate is approximately 12% in children and less than 5% in adults [6].
Definitions and histopathological diagnosis
Glioblastoma is the most aggressive diffuse glioma of astrocytic lineage and corresponds to grade IV according to the latest WHO Classification of Tumors of the CNS (4th ed., 2007). It may involve any neuroanatomical level or structure, but is most common in the cerebral hemispheres [1].
The defining biological feature of all diffuse gliomas is tumor cell infiltration. The neoplastic cells have the ability to travel long distances from the place of origin by tracking neuropil structures. They may travel in the opposite hemisphere and may give rise to additional tumor foci (i.e multifocal glioblastoma). Due to this biological behavior, a complete microscopic resection can never be achieved, and remnant neoplastic cells are frequently the source for disease recurrence (Fig. 1). Compared to other diffuse gliomas, glioblastoma has an extremely rapid infiltrative growth. Histologically, glioblastoma is composed of pleomorphic cells, mitotic activity, intravascular microthrombi, necrosis with or without cellular pseudopalisading, and/or microvascular proliferation (MVP) (Fig. 2). Either one of the latter two features are the sine qua non criteria for diagnosis. Called in the past “glioblastoma multiforme”, this highly aggressive tumor encloses under its umbrella multiple distinct patterns (i.e. small cell, giant cell, gliosarcoma etc). Irrespective of the histological pattern the clinical outcome remains the same [1].
Fig. 1.

Left upper: H&E stained cortical section of glioblastoma. Right upper: IDH1 R132H immunostain highlights the neoplastic cells. Lower images: Numerous IDH1 R132H positive neoplastic cells infiltrate the neuropil at a distance from the main tumor focus.
Fig. 2.
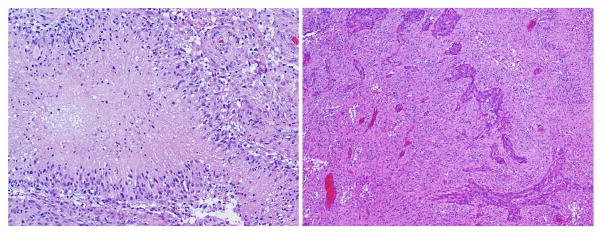
Histopathology of glioblastoma: H&E, intermediate power – proliferating atypical cells with mitotic activity, necrosis with pseudopalisading (left), and microvascular proliferation (right).
Glioblastoma may be divided into two identical morphological subtypes based on the presence or absence of a precursor lesion. Primary glioblastoma is the most common type (~90%); it arises de novo, without evidence of a precursor lesion and is common in older adults (over 50 years old). Secondary glioblastoma represents progression from a pre-existent, lower grade astrocytoma (WHO grades II or III) [7]. The time to progression from diffuse astrocytoma (WHO grade II) to glioblastoma is longer (~ 5 years) than the time to progression from anaplastic astrocytoma (WHO grade III) (~ 2 years) [8]. However, this is not always the rule. In our experience we have seen instances of protracted progression of diffuse astrocytoma to glioblastoma of 10 or more years or anaplastic astrocytomas progressing extremely fast to glioblastoma, in less than a year time frame [9].
Overview of pathogenesis
The vaso-occlusive model of glioblastoma progression
Genetic alterations in cancer result in cell proliferation, tumor growth, and angiogenesis [10,11]. In glioblastoma genetic abnormalities translate into morphological changes (infiltration, necrosis with pseudopalisading, and MVP) responsible for its accelerated tumor growth and aggressive behavior. At the center of these morphological changes sit prothrombotic events that shape a vaso-occlusive model for glioblastoma progression [12]. The neoplastic cells secrete procoagulant proteins (i.e. tissue factor and plasminogen activator inhibitor-1) responsible for endothelial injury and intravascular thrombosis. Intravascular thrombosis causes hypoxia and perivascular tumor necrosis with tumor cell pseudopalisading. Cellular pseudopalisading represents an adaptation to hypoxia. Compared to the rest of the tumor cells pseudopalisades overexpress Hypoxia inducible factor - 1α (HIF-1α). By binding to its promoter, HIF-1α upregulates the transcription of Vascular endothelial growth factor (VEGF) [13]. Under hypoxic and prothrombotic conditions pseudopalisades express and activate proteins (matrix metalloproteinases, components of the plasminogen activator system, protease activated receptors) that modulate glioblastoma cell migration away from the injured vessels [12,14]. As the tumor cells retract, they secrete proangiogenic factors (i.e. VEGF, IL-8) that stimulate MVP and outward expansion of tumor towards the newly formed vessels [12].
Understanding the modulation of angiogenesis in cancer was followed by the development of anti-angiogenic drugs. Bevacizumab is a FDA approved anti-VEGF humanized monoclonal antibody that interacts with VEGF receptor binding on the surface of tumor endothelial cells (VEGFR-1 and -2), and reduces angiogenesis. In subgroups of patients the drug improved progression free survival but failed to improve overall survival while a significant percentage of patients did not respond at all [15–18]. More importantly after bevacizumab treatment tumors recur, are more infiltrative [19,20], and develop drug resistance [18,21]. Angiogenesis in glioblastoma is complex and multiple theories may explain drug resistance. Besides VEGF, there are other pro-angiogenic factors secreted by other cell types like bone marrow derived cells (macrophages – M2 subtype, fibroblasts). Recent studies have shown that multipotent cancer stem cells give rise to a genetically abnormal tumor endothelium in glioblastoma [22–24]. Cancer stem cell derived endothelial cells might have different response to VEGFR blockade as compared to normal tissue derived endothelial cells (by sprouting angiogenesis, vasculogenesis or intussusception) [25]. Recently Piao et al. demonstrated differentiation from the proneural to the more aggressive mesenchymal glioblastoma phenotype [2,3] (see below) in developed cell lines with acquired resistance to bevacizumab [26]. The proneural to mesenchymal transition has been studied before and several transcription factors have been implicated (STAT3, C/EBP, TAZ) [27–29] in pathophysiology, however an exact mechanism and causality have not been accurately described. The explanation for this transition might reside at least in part in the surrounding tumor microenvironment. Our group demonstrated similar transitions in glioma sphere cultures in a TNFα/NFkB dependent manner, with associated increased CD44 positive cell fraction. CD44 is a cell surface protein expressed by cancer stem cells with possible roles in cell adhesion and migration [30]. The proneural to mesenchymal transition, CD44 expression, and NFkB activation correlated with lower rates of response to radiation therapy and with shorter median overall survival [31]. As we describe below, proinflammatory cytokines can activate NFkB transcription mediated signaling pathways that ultimately stimulate tumor growth. Inflammatory cells secrete cytokines and therefore the tumor infiltrating macrophages or residing microglia might be possible culprits for glioblastoma plasticity and aggressive phenotype transformation.
The role of cytokines
Extracellular modulators have also been implicated in glioblastoma growth/progression. Inflammatory cytokines, chemokines, and growth factors modulate tumor proliferation, infiltration, and angiogenesis and ultimately tumor growth. However, glioblastoma does not seem to be an inflammation-induced cancer, like are some cancers that develop secondary to microbial infections (Helicobacter Pylori in gastric carcinoma, HCV in hepatocellular carcinoma, etc.) or inflammatory conditions (colorectal carcinoma in inflammatory bowel disease) etc. [32]. The inflammation in glioblastoma is likely intrinsic in origin, driven by tumor cell genetic alterations, changes in the tumor microenvironment, or by responses to therapy (necrosis following chemotherapy or radiotherapy accompanied by release of cytokines) [33].
The glioblastoma microenvironment has increased levels of IL-1β, IL-6 and IL-8. High levels of IL-1β have been demonstrated in glioblastoma cell lines [34] and tumor tissues [35,36]. Increased IL-6 expression has been shown in glioblastoma [35,37] and was associated with decreased survival [38]. Inda et al has recently demonstrated a direct interaction between EGFR and gp130, the signal transducing receptor protein that forms a complex with IL-6, in tumor cells that express ΔEGFR (see details on EGFR below). Tumor cells harboring the ΔEGFR mutation express an increased level of cytokines (IL-6 and leukemia inhibitory factor (LIF)) that activate gp130, which in turn activates wtEGFR in adjacent cells in a paracrine fashion, leading to cell proliferation [39]. Same group later showed that ΔEGFR is associated with increased levels of IL-8 mediated by NFkB, AP-1, and C/EBP regulated transcription. IL-8 does not seem to have an autocrine effect on tumor cells as transfection of glioblastoma cell lines with IL-8 siRNA did not significantly alter proliferation. Growth effects are likely related to interactions with the microenvironment and to the process of angiogenesis. Moreover they showed that IL-8 signaling in ΔEGFR cells is independent of Phosphatidylinositol-3 kinase (PI3K), Akt, or Mitogen activated protein kinase (MAPK) and depended on c-Jun N-terminal kinase (JNK) [40].
Briefly, IL-1β, IL-6 and IL-8 bind to their respective receptors (IL-1R, IL-6R, and IL-8R) and activate signaling pathways (p38α MAPK, JAK2, and JNK2) that ultimately activate a series of oncogenic transcription factors (STAT3, AP-1, NFkB) or post-transcriptional regulatory molecules (HuR, MnK1/2) leading to tumor growth, impaired protein translation and mRNA stability [33].
Cancer stem cells in GBM
Several studies have demonstrated the presence of a small fraction of cancer stem cells in glioblastoma [41–44]. Pallini et al showed the impact these cells have on survival: the patients whose tumors had an increased number of proliferating cancer stem cells had decreased overall and progression-free survival [45]. Cancer stem cells are characterized by self-renewal, multipotency, stem cell marker expression (i.e. CD133, nestin, etc), and in vivo tumor growth even under critical conditions (i.e. serial cell transplantation) [41,46]. It is not yet clear what is the cancer stem cells precursor. They may arise following acquisition of genetic alterations either from normal neural stem cells [43,47] or from mature, differentiated neural cells (i.e. astrocytes) that acquire a more primitive phenotype [44]. As shown above, besides genetic alterations, the tumor microenvironment has an important role in gliomagenesis and signaling, potentially regulating the cancer stem cell phenotype [48]. As cancer stem cells are resistant to current treatment modalities [49–53], it is likely that these cells are at least partially responsible for tumor recurrence [54].
Gong et al showed that newer drugs (bortezomib, a proteasome inhibitor and erlotinib) decreased the fraction of cancer stem cells while minimally affecting normal neural stem cells in high-grade glioma cell lines. At the same time, ‘older’ drugs (temozolomide and cisplatin) primarily decreased the normal neural stem cell population while minimally affecting cancer stem cells [53].
Molecular Classification of Glioblastoma
Although morphologically identical, different glioblastoma tumors translate into different clinical outcomes. It has been shown that different clinical behaviors can be partially explained by different tumor molecular fingerprints [7,9,55–57]. The molecular alterations in glioblastoma are extremely complex.
Numerous tumor suppressor genes and oncogenes are inactivated and activated, respectively. The most commonly altered signaling pathways are RTK/RAS/MAPK/PI3KA; the p53 pathway - TP53, p14ARF, MDM2, MDM4; and the RB pathway - CDKN2A/CDKN2B, CDK4/CDK6, RB1 [58]. Primary glioblastomas are characterized by EGFR amplification, PTEN mutation, and absence of IDH mutations, while secondary glioblastomas are characterized by TP53 mutations, IDH mutations, and lack of EGFR amplification [59]. Recently Jiao et al described ATRX (Alpha thalassemia/mental retardation syndrome X-linked) mutations in diffuse glioma, associated with an alternative lengthening of telomeres. They seem to cluster with TP53 mutations within the secondary glioblastoma subcategory [60].
Gene expression profiling of glioblastoma identified several transcriptomal subgroups (Fig. 3). Two of the subgroups seem to replicate similar profiles among different studies and sit at opposite ends: proneural and mesenchymal. Proneural glioblastoma is common in young adults, corresponds to the secondary glioblastoma subtype, has neuronal differentiation, and is associated with better outcome. Is characterized by IDH/TP53 mutations/positivity for the glioma-CpG island methylator phenotype (G-CIMP) and normal EGFR/PTEN/Notch signaling. The G-CIMP phenotype, like IDH mutation, appears to be a general feature of lower grade gliomas, and practically speaking, provides a molecular definition of secondary glioblastoma. The mesenchymal glioblastoma is common in older adults, has mesenchymal differentiation, is associated with worse outcome, and is characterized by abnormal EGFR amplification/PTEN loss/NF1 mutations/Akt signaling [2–4,61–63].
Fig. 3.

Gene expression profiling defines three major tumor subsets: proneural (PN), mesenchymal (Mes), and proliferative (Prolif) – from Philips et al [2] 2006 with permission.
Singh et al. added to the glioblastoma pathogenesis by describing the FGFR-TACC fusion in a small fraction of glioblastoma (3/97, 3.1%). Oncogenic translocations cause in-frame fusion of the tyrosine kinase domains of FGFR (Fibroblast growth factor receptor) and TACC (transforming acidic coiled-coil). The resulting abnormal protein localizes to the mitotic spindle and induces chromosomal instability and aneuploidy promoting oncogenesis [64]. The clinical significance of this finding remains to be described.
Pediatric glioblastoma is a distinct disease from the molecular standpoint. Are characterized by frequent PDGFRA amplification, lack of IDH mutations, frequent gains of chromosome 1q, and lower frequencies of chromosome 7 gain and 10q loss [65]. Here we will consider the most clinically relevant genetic alterations and discuss treatment options in glioblastoma focusing on adult disease.
Isocitrate dehydrogenase (IDH) mutations
Isocitrate dehydrogenases (IDH1-3) are tricarboxylic acid cycle enzymes that catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate, with subsequent NADPH/NADH release [66]. Mutations involving the encoding genes (IDH1, IDH2) have been described in diffuse gliomas of which approximately 90% involve IDH1 [59,67,68]. IDH1-R132H (G395A) is most common mutation (~ 90%), followed at a distance by IDH1-R132S (C394A), IDH1-R132C (C394T), IDH1-R132G (C394G), IDH1-R132L (G395T), IDH1-R132V (C394G G395T) (0.5–5%). Codon 172 is involved in IDH2 mutations, most common amino acid substitution being R172K (G515A) [59,67–71]. The resulting mutated protein undergoes a conformational change with subsequent loss and gain of function effects and 2-hydroxyglutarate accumulation. The latter acts as a competitive inhibitor of α-ketoglutarate-dependent dioxygenases, a family of enzymes responsible for epigenetic deregulation (hypermethylation/G-CIMP phenotype [63]–see below) and gliomagenesis [66,72]. An antibody against the most common mutated protein, IDH1-R132H, is commercially available for formalin-fixed paraffin embedded tissue use and shows excellent correlation with the mutation [68].
IDH mutations are common in younger adults and adolescents [56,57,73] and in secondary glioblastoma (~ 80%), while only about 5% of primary glioblastoma have IDH mutations [56,59]. The mutations have prognostic significance in high-grade gliomas [69,74–78] that might extend beyond WHO histological grade (IDH mutated glioblastoma may have better outcomes than IDH wild-type anaplastic astrocytoma) [74]. However this is not absolute, as Jiao et al recently pointed to a small subgroup of patients with IDH mutated gliomas across all grades and an extremely dismal prognosis [60]. The underlying mechanism is not yet understood; possible additional genetic alterations yet to be identified might be responsible for this phenotype This information should not be ignored, especially in patient counseling. The rate of IDH mutations in infratentorial (spinal cord, brainstem, and cerebellum) glioblastoma is not known. Inferring from the low levels of IDH-R132H protein expression in WHO grade II and III diffuse glioma of similar location [79], it is probably low; however more evidence is needed.
IDH mutations seem to be early events in gliomagenesis, followed by acquisition of TP53 mutations and are the markers of secondary glioblastoma [55–57]. IDH and TP53 mutations tend to cluster with ATRX mutations [60] in grade II and III astrocytomas and secondary glioblastoma suggesting that together they might be biomarkers of astrocytic lineage. ATRX mutations are not frequent in primary and pediatric glioblastoma, feature shared by IDH mutations.
As shown by our group, absence of IDH1-R132H protein expression in nonenhancing diffuse glioma of older adults (> 50 years of age) predicted rapid progression (in less than a year) to glioblastoma. Interestingly, this rapidly progressing type of secondary glioblastoma implied molecular features associated with primary glioblastoma; however molecular genetic analysis for other IDH mutations has not been performed at the time of the study [9]. At our institution we now routinely perform IDH-R132H immunohistochemistry on all WHO grade II and III diffuse gliomas of all ages and glioblastoma in patients younger than 50 years of age. Antibody-negative gliomas are interrogated at IDH1132 and IDH2172 by DNA sequencing.
The mutated IDH-R132H protein might be a potential target for drug development. Recently Rohle et al., using high-throughput screening, developed a selective IDH-R132H mutant enzyme inhibitor - AGI-5198. Following observations on the inhibitory effect AGI-5198 had on IDH-R132H mutant human glioma cell line colony formation, the effects of its oral administration on human glioma xenografts were investigated. AGI-5198 blocked the production of 2-hydroxyglutarate and induced histone demethylation in a dose-dependent manner, however without significant CIMP phenotype alteration. Interestingly at high doses the compound induced expression of genes associated with glial differentiation. Tumor growth was inhibited independent of dosage while the drug was tolerated well without signs of toxicity. The authors observed that the lack of correlation between changes in DNA methylation and RNA expression suggests that IDH1-R132H mutated gliomas might promote gliomagenesis through other mechanisms besides epigenetic regulation. This therapeutic approach shows promise directed at a tumor-specific lesion in a subset of gliomas. [80].
1p/19q co-deletion
Oligodendroglioma is a diffuse glioma with monotonous cellular morphology (resembling oligodendroglia) and “fried-egg” formalin fixation cellular artifact. It was divided into two WHO grades (II and III) of prognostic significance. By definition, anaplastic oligodendroglioma (mitotic activity, MVP with or without necrosis) is the most aggressive subtype in this morphological category, but has better prognosis compared to patients with glioblastoma (grade IV). The WHO describes a subtype of glioblastoma called ‘with oligodendroglioma component’ (GBM-o) defined as glioblastoma with areas that resemble oligodendroglioma that might have a better prognosis compared to standard glioblastoma. A mixed anaplastic oligoastrocytoma is defined as a diffuse glioma with both astrocytic and oligodendroglial morphology and mitotic activity (MVP might be present, but without necrosis) or an anaplastic oligodendroglioma with ‘significant’ astrocytic component. According to the WHO, anaplastic oligoastrocytoma with necrosis (with or without MVP) should be classified as GBM-o [1]. Obviously there is a lot of room for confusion and molecular subtyping might offers the solution in these situations.
Combined loss of heterozygosity (LOH) on 1p and 19q is strongly correlated with classic oligodendroglioma morphology and associated with IDH mutations [69,81,82], increased MGMT promoter methylation [83,84], G-CIMP phenotype (see below) [85,86], a proneural profile [2–4], and is predictive of response to chemotherapy [87,88]. Co-deletion of 1p/19q was reported to be mutually exclusive with TP53 mutations [57,89] and recently was shown to be associated with CIC and FUBP1 mutations mainly seen in oligodendroglioma and rarely in mixed glioma [60]. On the other hand ATRX mutations were mainly seen in astrocytoma and mixed glioma (WHO grade II and III) and were mutually exclusive with CIC/FUBP1 suggesting a ‘pure’ astrocytic phenotype for mixed glioma [60].
Studies on GBM-o reported low 1p/19q co-deletion rates (~ 30% or less) and genetic heterogeneity [90–95]. Interrogating ATRX/CIC/FUBP1 would serve as a good start to further characterize these tumors. According to the NCCN treatment guidelines, 1p/19q co-deletion is the only molecular biomarker used for therapeutic stratification in anaplastic oligodendroglioma, mixed glioma, and GBM-o at this point [96]. Until these or other molecular alterations are described to better classify this entity, 1p/19q co-deletion status should be used for clinical guidance.
Epidermal growth factor receptor (EGFR)
Approximately 40% to 50% of glioblastomas have EGFR amplification [7,8,97–100]. Although EGFR amplification was described in both primary and secondary glioblastomas, it is more common in primary glioblastomas and seems to be encountered in patients over 35 years of age [7]. EGFR amplification is a characteristic signature of the TCGA classical glioblastoma subtype [3] and of the Phillips proliferative and mesenchymal glioblastoma subtypes [2].
Among tumors with EGFR amplification, other genetic alterations have been described. The loss of the coding sequence for amino acids 6–273 due to an in-frame deletion of exons 2–7 (801 bp) or EGFRvIII (variant III or ΔEGFR) is the most common mutation [101]. It has been described in up to approximately 60–70% of EGFR amplified glioblastomas [101–103]. Although in approximately 50% of cases, ΔEGFR is not accompanied by other mutations, multiple EGFR transcript alterations are present in about 30% of glioblastomas with EGFR amplification [101].
EGFR is located on the short arm of chromosome 7 (7p12) [98] and encodes a cell-surface receptor tyrosine kinase (EGFR or Erb-1), a member of the ErbB family of receptors [104]. Upon growth factor ligand binding to its extracellular domain and phosphorylation of its intracellular tyrosine kinase domain, EGFR becomes activated [104] and initiates signal transduction cascades (Ras/MAPK and PI3K/Akt) leading to increased DNA transcription, antiapoptosis, angiogenesis, and cellular proliferation [104]. ΔEGFR on the other hand does not bind growth factor ligands, but is constitutively phosphorylated. The receptor is truncated with a short extracellular domain. This structural abnormality seems to mimic the effect of ligand binding and induce receptor conformational changes followed by increased intracellular signaling and cell proliferation [105].
As mentioned above, ΔEGFR maintains tumor heterogeneity through cytokine-mediated paracrine effects on the surrounding, EGFR wild-type cells, independent of MAPK and PI3K/Akt and dependent on JNK [39,40]. Therefore, by targeting ΔEGFR a possible better tumor growth control might be achieved.
Soeda et al, on glioblastoma cancer stem cell lines showed that EGF, but not other growth factors, promoted cancer stem cell proliferation. Moreover, in the presence of gefitinib, the apoptosis rate was increased, the fraction of cancer stem cells decreased from 6–10% to 0–0.05% and the cancer stem cell colony formations were suppressed by 90% [106].
The clinical implications of EGFR amplification or ΔEGFR in glioblastoma are not yet clear. Several studies have shown contradictory results, with a positive impact on survival in some studies [107–110], decreased survival and poor prognosis in others [110–114], or no impact on survival at all [99,115]. Recently, Hobbs et al showed that the degree of EGFR amplification might influence survival. They showed that glioblastoma patients with higher levels of EGFR amplification (EGFR:CEP7 ratio > 20 assessed by FISH) had longer median survival compared to patients whose glioblastomas were EGFR non-amplified and had low or moderate levels of EGFR amplification [116].
Most studies reported minimal drug activity or no improved survival with EGFR-tyrosine kinase inhibitors (erlotinib, gefitinib) in newly-diagnosed or recurrent glioblastoma [117–121]. Hegi et al showed that the tissue drug concentrations are increased and efficient EGFR dephosphorylation takes place both in vivo and in vitro, but the downstream pathway remains active [120]. Persistent EGFR downstream signaling has also been reported by Hasselbalch et al on glioblastoma cell lines treated with cetuximab, an anti-EGFR monoclonal antibody [122] and by Vollman et al using siRNAs [123]. These findings suggest that EGFR downstream signaling is independent of receptor activation and simultaneous downstream signals might co-exist or be initiated as a response to EGFR blockade. Solomón et al however, recently published results on a randomized, double blind, multicentric clinical trial conducted on anaplastic astrocytomas and glioblastomas treated with radiation with nimotuzumab or with placebo. There was significant increase in survival for patients in the nimotuzumab arm [124]. As mentioned above Gong et al obtained encouraging results on cancer stem cells from high-grade glioma cell lines [53].
O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation status
O(6)-alkylating agents are molecules commonly found in the environment or endogenously formed during normal cellular metabolism, that have also been used in cancer therapy (i.e. temozolomide [125]). Their function is to transfer alkyl groups (CnH2n+1-) to guanine bases causing DNA damage and cellular death. MGMT is a suicidal DNA repair protein that removes alkyl groups in a stoichiometric fashion [126].
The MGMT promoter lacks the constitutive regulatory elements known as the TATA box and the CAT box, and contains a CpG island rich in 97 CG dinucleotides or CpG sites, located in the 5′ region of MGMT. Methylated CpG sites bind specific proteins, this complex causing altered chromatin structure and loss of transcription (MGMT silencing), ultimately interfering with DNA repair [127]. Although correlation has been reported [85], MGMT promoter methylation may not be directly related to the G-CIMP profile. G-CIMP is characterized by increased DNA methylation at an increased number of CpG sites and seems to be conditioned by IDH mutations [72]. This phenotype is more common in lower grade gliomas compared to glioblastoma, is associated with younger age, with a proneural phenotype, and improved prognosis [63] (Fig. 4, 5).
Fig. 4.

Hierarchical unsupervised clustering of TCGA Glioblastoma by methylation status. Each row represents a probe; each column represents a sample. The level of DNA methylation is represented with a color scale as shown in the key; white indicates missing data. Legend: M.SssI - CpG Methyltransferase treated DNA; WGA – whole genome amplified DNA. From Noushmehr et al, 2010 [63] with permission.
Fig. 5.
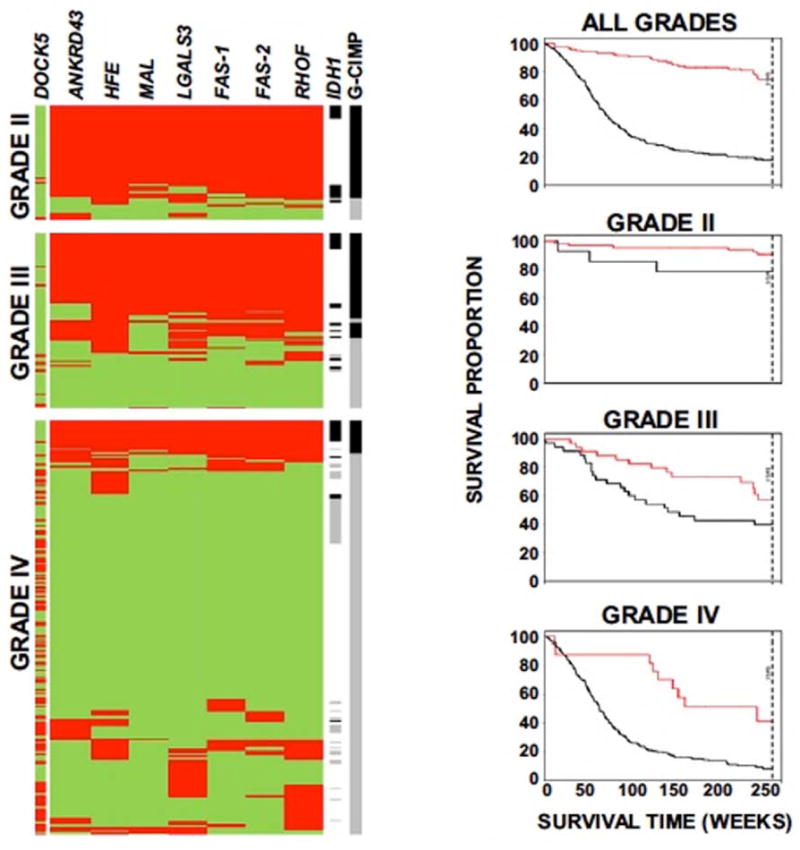
G-CIMP prevalence and outcome in diffuse glioma: left – Methylation profiling shows an association of CIMP status with tumor grade (red = methylated; green = unmethylated; black line = G-CIMP/IDH1 positive; gray line = G-CIMP/IDH1 negative; white lines = unknown IDH status); right – Kaplan-Meier survival curves plotting patient outcome by G-CIMP status (red lines = G-CIMP positive; black lines = G-CIMP negative). From Noushmehr et al, 2010 [63] with permission.
The MGMT promoter is methylated in approximately 50% of newly diagnosed glioblastomas overall [84,128–133] and more commonly in secondary glioblastoma [84,128,129,132] (reviewed by Weller et al [134]). MGMT promoter methylation has prognostic and predictive significance in patients with glioblastoma [128,130,131,135] with better overall survival irrespective of treatment choices [84,131]. Besides response to temozolomide [130,131], MGMT promoter methylation in glioblastoma was associated with response to radiotherapy [135,136]. A combined treatment approach (temozolomide with radiotherapy) seems to have better outcomes with improved progression-free and overall survival compared to either treatment modality alone [131,137]. Similar MGMT promoter methylation frequencies and clinical outcomes have been described in older patients (> 70 years of age) [138] and in children (< 18 years of age) with glioblastoma [139,140].
Methylation specific PCR or pyrosequencing are the tests of choice to determine MGMT promoter methylation. Immunohistochemistry for MGMT protein expression is not recommended as several studies showed discordant correlations with the MGMT status [133,141].
Following treatment with temozolomide the phenomenon of pseudoprogression is common. Pseudoprogression is defined by an increase in contrast-enhancement accompanied or not by clinical symptomatology, but with subsequent improvement and stabilization. Its underlying mechanism could be induced by a local inflammatory reaction, with abnormal vessel permeability and edema [142]. If an MGMT-methylated tumor under treatment with temozolomide develops pseudoprogression on imaging, chemotherapy should not therefore be stopped [143]. On the other hand, if clinical and radiological features suggestive of pseudoprogression are present in temozolomide-treated non MGMT-methylated glioblastoma, they likely represent bona fide tumor progression, and a change in therapy should be considered [144,145].
In glioblastoma MGMT methylation status does not correlate with the IDH, TP53 mutation [128] or the EGFR amplification status [146] as happens in grade II and III diffuse glioma for IDH mutations and 1p/19q co-deletion [128,146,147].
The predictive value of MGMT in recurrent glioblastoma is not yet established. Some tumors might change their methylation status following therapy while overall survival might be influenced only by methylation interrogation of pre-treatment tissue [148,149]. A post-treatment hypermutated tumor phenotype with acquisition of mutations in non–CpG regions has been associated with MGMT promoter methylation in primary glioblastoma. Because these mutations are not present in CpG regions they cannot be cleared by MGMT[58]. This might explain the overwhelming treatment resistance and poor outcome in treated recurrent glioblastoma.
Conclusions
The first step towards efficient treatment development, of maximum benefit and minimal toxicity, is to understand the pathophysiology of disease. Glioblastoma is an extremely complex entity encompassing a series of tightly interconnected cell signaling networks. Despite recent progress in understanding the molecular biology of glioblastoma and numerous targeted drug therapies available, the progress is extremely slow. Giving the complex network of mechanisms and molecules involved, one approach would be multi-targeted therapy, simultaneously aimed at different pathway constituents. However, evidence from clinical trials is growing very slowly and drug toxicity is a limiting factor that cannot be ignored. On the other hand treatment-induced changes following chemo- and radiotherapy is likely to push tumor cells in a vicious circle by actually promoting tumor growth. One might wonder, is ordo ad chaos even possible? In theory it seems that the best approach would be to target the initiating tumor cells and cut the tumor at its roots. Such modality is maybe more optimistic as cancer stem cells seem to be responding to certain drugs while normal stem cells are spared in vitro. The list of targeted drug strategies developed so far is long and awaiting clinical trial testing.
The key to successful treatment of glioblastoma will no doubt lie in the realization that this clinicopathologic entity is in biologic terms, more than one disease, and it is likely that specific targeted therapies will be efficacious in molecularly defined subsets. In this way, the molecular classification of these tumors will of necessity be defined in clinically relevant terms based of the identification of markers that define subsets and are predictive of response to promising agents. Additional investigation is necessary to identify new biomarkers and potential therapeutic targets will help to better define the clinical and biologic subtypes of glioblastoma and an improved disease control.
Acknowledgments
Funding provided by SPORE grant P50CA127001 from NIH/NCI. Dr. Adriana Olar is supported by the training grant 5T32CA163185 from NIH/NCI.
Footnotes
Conflict of interest: The authors report no conflict of interest.
Author contributions: Both authors contributed equally to the writing and editing of the manuscript.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of tumors of the central nervous system. 4. IARC; Lyon (France): 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng S, Chheda MG, Verhaak RG. Studying a complex tumor: potential and pitfalls. Cancer J. 2012;18:107–114. doi: 10.1097/PPO.0b013e3182431c57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 6.Dolecek TA, Propp JM, Stroup NE, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2005–2009. Neuro-Oncol. 2012;14:v1–v49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 9.Olar A, Raghunathan A, Albarracin CT, et al. Absence of IDH1-R132H mutation predicts rapid progression of nonenhancing diffuse glioma in older adults. Ann Diagn Pathol. 2011 doi: 10.1016/j.anndiagpath.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Rong Y, Post DE, Pieper RO, et al. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005;65:1406–1413. doi: 10.1158/0008-5472.CAN-04-3376. [DOI] [PubMed] [Google Scholar]
- 11.Rak J, Milsom C, May L, et al. Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin Thromb Hemost. 2006;32:54–70. doi: 10.1055/s-2006-933341. [DOI] [PubMed] [Google Scholar]
- 12.Rong Y, Durden DL, Van Meir EG, et al. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529–539. doi: 10.1097/00005072-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brat DJ, Castellano-Sanchez AA, Hunter SB, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- 15.Lai A, Tran A, Nghiemphu PL, et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2011;29:142–148. doi: 10.1200/JCO.2010.30.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinot O, Wick W, Mason W, et al. Phase III Trial of Bevacizumab added to standard radiotherapy and temozolomide for newly-diagnosed glioblastoma: Mature progression-free survival and preliminary overall survival results in Avaglio. Neuro-oncology. 2012;14:vi101–vi105. doi:110.1093/neuonc/nos1232. [Google Scholar]
- 17.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 18.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 19.de Groot JF, Fuller G, Kumar AJ, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-oncology. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayana A, Kunnakkat S, Chacko-Mathew J, et al. Bevacizumab in recurrent high-grade pediatric gliomas. Neuro-oncology. 2010;12:985–990. doi: 10.1093/neuonc/noq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott B, Quant E, McNamara M, et al. Bevacizumab salvage therapy following progression in high-grade glioma patients treated with VEGF receptor tyrosine kinase inhibitors. Neuro-oncology. 2010;12:603–607. doi: 10.1093/neuonc/nop073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Chadalavada K, Wilshire J, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 24.Soda Y, Marumoto T, Friedmann-Morvinski D, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;108:4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takano S. Glioblastoma angiogenesis: VEGF resistance solutions and new strategies based on molecular mechanisms of tumor vessel formation. Brain Tumor Pathol. 2012;29:73–86. doi: 10.1007/s10014-011-0077-6. [DOI] [PubMed] [Google Scholar]
- 26.Piao Y, Liang J, Holmes LS, et al. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- 27.Bhat KP, Salazar KL, Balasubramaniyan V, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011;25:2594–2609. doi: 10.1101/gad.176800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper LA, Gutman DA, Chisolm C, et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol. 2012;180:2108–2119. doi: 10.1016/j.ajpath.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nature reviews Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 31.Bhat KPL, Veerakumar B, Vaillant B, et al. Mesenchymal Differentiation Mediated by NF-kB Promotes Radiation Resistance in Glioblastoma. Cancer cell. 2013 doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 33.Yeung Y, McDonald K, Grewal T, et al. Interleukins in glioblastoma pathophysiology: implications for therapy. Br J Pharmacol. 2013;168:591–606. doi: 10.1111/bph.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu T, Tian L, Han Y, et al. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104:4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeung YT, Bryce NS, Adams S, et al. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J Neurooncol. 2012;109:35–44. doi: 10.1007/s11060-012-0875-7. [DOI] [PubMed] [Google Scholar]
- 36.Sharma V, Dixit D, Koul N, et al. Ras regulates interleukin-1beta-induced HIF-1alpha transcriptional activity in glioblastoma. J Mol Med (Berl) 2011;89:123–136. doi: 10.1007/s00109-010-0683-5. [DOI] [PubMed] [Google Scholar]
- 37.Tchirkov A, Rolhion C, Bertrand S, et al. IL-6 gene amplification and expression in human glioblastomas. Br J Cancer. 2001;85:518–522. doi: 10.1054/bjoc.2001.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tchirkov A, Khalil T, Chautard E, et al. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer. 2007;96:474–476. doi: 10.1038/sj.bjc.6603586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inda M-d-M, Bonavia R, Mukasa A, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonavia R, Inda MM, Vandenberg S, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene. 2012;31:4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 42.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 43.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 44.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 45.Pallini R, Ricci-Vitiani L, Banna GL, et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:8205–8212. doi: 10.1158/1078-0432.CCR-08-0644. [DOI] [PubMed] [Google Scholar]
- 46.Lathia JD, Gallagher J, Myers JT, et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PloS one. 2011;6:e24807. doi: 10.1371/journal.pone.0024807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alcantara Llaguno S, Chen J, Kwon CH, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lathia JD, Heddleston JM, Venere M, et al. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell stem cell. 2011;8:482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou BB, Zhang H, Damelin M, et al. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nature reviews Drug discovery. 2009;8:806–823. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 50.Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Molecular cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 52.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong X, Schwartz PH, Linskey ME, et al. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology. 2011;76:1126–1134. doi: 10.1212/WNL.0b013e318212a89f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Li Y, Yu T-S, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohgaki H, Kleihues P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol. 2011;28:177–183. doi: 10.1007/s10014-011-0029-1. [DOI] [PubMed] [Google Scholar]
- 56.Nobusawa S, Watanabe T, Kleihues P, et al. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15:6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe T, Nobusawa S, Kleihues P, et al. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cancer, Genome, Atlas et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cooper LA, Gutman DA, Long Q, et al. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PloS one. 2010;5:e12548. doi: 10.1371/journal.pone.0012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- 63.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paugh BS, Qu C, Jones C, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012 doi: 10.1007/s10014-012-0090-4. [DOI] [PubMed] [Google Scholar]
- 67.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 68.Horbinski C, Kofler J, Kelly LM, et al. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68:1319–1325. doi: 10.1097/NEN.0b013e3181c391be. [DOI] [PubMed] [Google Scholar]
- 69.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 71.Felsberg J, Wolter M, Seul H, et al. Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol. 2010;119:501–507. doi: 10.1007/s00401-010-0647-4. [DOI] [PubMed] [Google Scholar]
- 72.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pollack IF, Hamilton RL, Sobol RW, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010;120:707–718. doi: 10.1007/s00401-010-0781-z. [DOI] [PubMed] [Google Scholar]
- 75.van den Bent MJ, Dubbink HJ, Marie Y, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16:1597–1604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- 76.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27:5874–5880. doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 77.Weller M, Felsberg J, Hartmann C, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol. 2009;27:5743–5750. doi: 10.1200/JCO.2009.23.0805. [DOI] [PubMed] [Google Scholar]
- 78.Sonoda Y, Kumabe T, Nakamura T, et al. Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer science. 2009;100:1996–1998. doi: 10.1111/j.1349-7006.2009.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ellezam B, Theeler BJ, Walbert T, et al. Low rate of R132H IDH1 mutation in infratentorial and spinal cord grade II and III diffuse gliomas. Acta Neuropathol. 2012;124:449–451. doi: 10.1007/s00401-012-1011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Labussiere M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74:1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- 83.Xiong J, Liu Y, Wang Y, et al. Chromosome 1p/19q status combined with expression of p53 protein improves the diagnostic and prognostic evaluation of oligodendrogliomas. Chin Med J (Engl) 2010;123:3566–3573. [PubMed] [Google Scholar]
- 84.Eoli M, Menghi F, Bruzzone MG, et al. Methylation of O6-methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin Cancer Res. 2007;13:2606–2613. doi: 10.1158/1078-0432.CCR-06-2184. [DOI] [PubMed] [Google Scholar]
- 85.van den Bent MJ, Gravendeel LA, Gorlia T, et al. A hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. Clin Cancer Res. 2011;17:7148–7155. doi: 10.1158/1078-0432.CCR-11-1274. [DOI] [PubMed] [Google Scholar]
- 86.Mur P, Mollejo M, Ruano Y, et al. Codeletion of 1p and 19q determines distinct gene methylation and expression profiles in IDH-mutated oligodendroglial tumors. Acta Neuropathol. 2013;126:277–289. doi: 10.1007/s00401-013-1130-9. [DOI] [PubMed] [Google Scholar]
- 87.Kaloshi G, Benouaich-Amiel A, Diakite F, et al. Temozolomide for low-grade gliomas: predictive impact of 1p/19q loss on response and outcome. Neurology. 2007;68:1831–1836. doi: 10.1212/01.wnl.0000262034.26310.a2. [DOI] [PubMed] [Google Scholar]
- 88.Cairncross G, Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer J. 2008;14:352–357. doi: 10.1097/PPO.0b013e31818d8178. [DOI] [PubMed] [Google Scholar]
- 89.Ichimura K, Pearson DM, Kocialkowski S, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncology. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Appin CL, Gao J, Chisolm C, et al. Glioblastoma with Oligodendroglioma Component (GBM-O): Molecular Genetic and Clinical Characteristics. Brain Pathol. 2013;23:454–461. doi: 10.1111/bpa.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ha SY, Kang SY, Do IG, et al. Glioblastoma with oligodendroglial component represents a subgroup of glioblastoma with high prevalence of IDH1 mutation and association with younger age. J Neurooncol. 2013;112:439–448. doi: 10.1007/s11060-013-1073-y. [DOI] [PubMed] [Google Scholar]
- 92.Hegi ME, Janzer RC, Lambiv WL, et al. Presence of an oligodendroglioma-like component in newly diagnosed glioblastoma identifies a pathogenetically heterogeneous subgroup and lacks prognostic value: central pathology review of the EORTC_26981/NCIC_CE. 3 trial. Acta Neuropathol. 2012;123:841–852. doi: 10.1007/s00401-011-0938-4. [DOI] [PubMed] [Google Scholar]
- 93.Wang Y, Li S, Chen L, et al. Glioblastoma with an oligodendroglioma component: distinct clinical behavior, genetic alterations, and outcome. Neuro-oncology. 2012;14:518–525. doi: 10.1093/neuonc/nor232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Klink B, Schlingelhof B, Klink M, et al. Glioblastomas with oligodendroglial component - common origin of the different histological parts and genetic subclassification. Anal Cell Pathol (Amst) 2010;33:37–54. doi: 10.3233/ACP-CLO-2010-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Franco-Hernandez C, Martinez-Glez V, de Campos JM, et al. Allelic status of 1p and 19q in oligodendrogliomas and glioblastomas: multiplex ligation-dependent probe amplification versus loss of heterozygosity. Cancer Genet Cytogenet. 2009;190:93–96. doi: 10.1016/j.cancergencyto.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 96.NCCN. [Accessed 7/31/2013 2013];Clinical Practice Guidelines in Oncology. Central Nervous System Cancers. Version 2. Available from: http://www.nccn.org.
- 97.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, Ichimura K, Pettersson EH, et al. Chromosome 7 rearrangements in glioblastomas; loci adjacent to EGFR are independently amplified. J Neuropathol Exp Neurol. 1998;57:1138–1145. doi: 10.1097/00005072-199812000-00005. [DOI] [PubMed] [Google Scholar]
- 99.Benito R, Gil-Benso R, Quilis V, et al. Primary glioblastomas with and without EGFR amplification: relationship to genetic alterations and clinicopathological features. Neuropathology : official journal of the Japanese Society of Neuropathology. 2010;30:392–400. doi: 10.1111/j.1440-1789.2009.01081.x. [DOI] [PubMed] [Google Scholar]
- 100.Ekstrand AJ, James CD, Cavenee WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164–2172. [PubMed] [Google Scholar]
- 101.Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 102.Schwechheimer K, Huang S, Cavenee WK. EGFR gene amplification--rearrangement in human glioblastomas. Int J Cancer. 1995;62:145–148. doi: 10.1002/ijc.2910620206. [DOI] [PubMed] [Google Scholar]
- 103.Sugawa N, Ekstrand AJ, James CD, et al. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Soeda A, Inagaki A, Oka N, et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283:10958–10966. doi: 10.1074/jbc.M704205200. [DOI] [PubMed] [Google Scholar]
- 107.Houillier C, Lejeune J, Benouaich-Amiel A, et al. Prognostic impact of molecular markers in a series of 220 primary glioblastomas. Cancer. 2006;106:2218–2223. doi: 10.1002/cncr.21819. [DOI] [PubMed] [Google Scholar]
- 108.Smith JS, Tachibana I, Passe SM, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 109.Montano N, Cenci T, Martini M, et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia. 2011;13:1113–1121. doi: 10.1593/neo.111338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bienkowski M, Piaskowski S, Stoczynska-Fidelus E, et al. Screening for EGFR Amplifications with a Novel Method and Their Significance for the Outcome of Glioblastoma Patients. PloS one. 2013;8:e65444. doi: 10.1371/journal.pone.0065444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Korshunov A, Sycheva R, Golanov A. The prognostic relevance of molecular alterations in glioblastomas for patients age < 50 years. Cancer. 2005;104:825–832. doi: 10.1002/cncr.21221. [DOI] [PubMed] [Google Scholar]
- 112.Simmons ML, Lamborn KR, Takahashi M, et al. Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res. 2001;61:1122–1128. [PubMed] [Google Scholar]
- 113.Feldkamp MM, Lala P, Lau N, et al. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery. 1999;45:1442–1453. doi: 10.1097/00006123-199912000-00034. [DOI] [PubMed] [Google Scholar]
- 114.Shinojima N, Tada K, Shiraishi S, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- 115.Batchelor TT, Betensky RA, Esposito JM, et al. Age-dependent prognostic effects of genetic alterations in glioblastoma. Clin Cancer Res. 2004;10:228–233. doi: 10.1158/1078-0432.ccr-0841-3. [DOI] [PubMed] [Google Scholar]
- 116.Hobbs J, Nikiforova MN, Fardo DW, et al. Paradoxical relationship between the degree of EGFR amplification and outcome in glioblastomas. Am J Surg Pathol. 2012;36:1186–1193. doi: 10.1097/PAS.0b013e3182518e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Uhm JH, Ballman KV, Wu W, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2011;80:347–353. doi: 10.1016/j.ijrobp.2010.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brown PD, Krishnan S, Sarkaria JN, et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008;26:5603–5609. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hegi ME, Diserens AC, Bady P, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Molecular cancer therapeutics. 2011;10:1102–1112. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 121.Raizer JJ, Abrey LE, Lassman AB, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-oncology. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hasselbalch B, Lassen U, Poulsen HS, et al. Cetuximab insufficiently inhibits glioma cell growth due to persistent EGFR downstream signaling. Cancer Invest. 2010;28:775–787. doi: 10.3109/07357907.2010.483506. [DOI] [PubMed] [Google Scholar]
- 123.Vollmann A, Vornlocher HP, Stempfl T, et al. Effective silencing of EGFR with RNAi demonstrates non-EGFR dependent proliferation of glioma cells. Int J Oncol. 2006;28:1531–1542. [PubMed] [Google Scholar]
- 124.Solomon MT, Selva JC, Figueredo J, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC cancer. 2013;13:299. doi: 10.1186/1471-2407-13-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wesolowski JR, Rajdev P, Mukherji SK. Temozolomide (Temodar) AJNR Am J Neuroradiol. 2010;31:1383–1384. doi: 10.3174/ajnr.A2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaina B, Christmann M, Naumann S, et al. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA repair. 2007;6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 127.Nakagawachi T, Soejima H, Urano T, et al. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene. 2003;22:8835–8844. doi: 10.1038/sj.onc.1207183. [DOI] [PubMed] [Google Scholar]
- 128.Mellai M, Monzeglio O, Piazzi A, et al. MGMT promoter hypermethylation and its associations with genetic alterations in a series of 350 brain tumors. J Neurooncol. 2012;107:617–631. doi: 10.1007/s11060-011-0787-y. [DOI] [PubMed] [Google Scholar]
- 129.Nakamura M, Watanabe T, Yonekawa Y, et al. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C --> A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis. 2001;22:1715–1719. doi: 10.1093/carcin/22.10.1715. [DOI] [PubMed] [Google Scholar]
- 130.Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 131.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 132.Ohka F, Natsume A, Motomura K, et al. The global DNA methylation surrogate LINE-1 methylation is correlated with MGMT promoter methylation and is a better prognostic factor for glioma. PloS one. 2011;6:e23332. doi: 10.1371/journal.pone.0023332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Preusser M, Charles Janzer R, Felsberg J, et al. Anti-O6-methylguanine-methyltransferase (MGMT) immunohistochemistry in glioblastoma multiforme: observer variability and lack of association with patient survival impede its use as clinical biomarker. Brain Pathol. 2008;18:520–532. doi: 10.1111/j.1750-3639.2008.00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nature reviews Neurology. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 135.Olson RA, Brastianos PK, Palma DA. Prognostic and predictive value of epigenetic silencing of MGMT in patients with high grade gliomas: a systematic review and meta-analysis. J Neurooncol. 2011;105:325–335. doi: 10.1007/s11060-011-0594-5. [DOI] [PubMed] [Google Scholar]
- 136.Rivera AL, Pelloski CE, Gilbert MR, et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro-oncology. 2010;12:116–121. doi: 10.1093/neuonc/nop020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The lancet oncology. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 138.Reifenberger G, Hentschel B, Felsberg J, et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer. 2011 doi: 10.1002/ijc.27385. [DOI] [PubMed] [Google Scholar]
- 139.Donson AM, Addo-Yobo SO, Handler MH, et al. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatric blood & cancer. 2007;48:403–407. doi: 10.1002/pbc.20803. [DOI] [PubMed] [Google Scholar]
- 140.Srivastava A, Jain A, Jha P, et al. MGMT gene promoter methylation in pediatric glioblastomas. Childs Nerv Syst. 2010;26:1613–1618. doi: 10.1007/s00381-010-1214-y. [DOI] [PubMed] [Google Scholar]
- 141.Rodriguez FJ, Thibodeau SN, Jenkins RB, et al. MGMT immunohistochemical expression and promoter methylation in human glioblastoma. Applied immunohistochemistry & molecular morphology : AIMM/official publication of the Society for Applied Immunohistochemistry. 2008;16:59–65. doi: 10.1097/PAI.0b013e31802fac2f. [DOI] [PubMed] [Google Scholar]
- 142.Hygino da Cruz LC, Jr, Rodriguez I, Domingues RC, et al. Pseudoprogression and pseudoresponse: imaging challenges in the assessment of posttreatment glioma. AJNR Am J Neuroradiol. 2011;32:1978–1985. doi: 10.3174/ajnr.A2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jansen M, Yip S, Louis DN. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet neurology. 2010;9:717–726. doi: 10.1016/S1474-4422(10)70105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26:2192–2197. doi: 10.1200/JCO.2007.14.8163. [DOI] [PubMed] [Google Scholar]
- 145.Fabi A, Russillo M, Metro G, et al. Pseudoprogression and MGMT status in glioblastoma patients: implications in clinical practice. Anticancer Res. 2009;29:2607–2610. [PubMed] [Google Scholar]
- 146.Jha P, Suri V, Jain A, et al. O6-methylguanine DNA methyltransferase gene promoter methylation status in gliomas and its correlation with other molecular alterations: first Indian report with review of challenges for use in customized treatment. Neurosurgery. 2010;67:1681–1691. doi: 10.1227/NEU.0b013e3181f743f5. [DOI] [PubMed] [Google Scholar]
- 147.Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 148.Brandes AA, Franceschi E, Tosoni A, et al. O(6)-methylguanine DNA-methyltransferase methylation status can change between first surgery for newly diagnosed glioblastoma and second surgery for recurrence: clinical implications. Neuro-oncology. 2010;12:283–288. doi: 10.1093/neuonc/nop050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Felsberg J, Thon N, Eigenbrod S, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659–670. doi: 10.1002/ijc.26083. [DOI] [PubMed] [Google Scholar]