

Abstract
Background:
Medullary thyroid carcinoma (MTC) is a tumor arising from the para follicular (C) cells of the thyroid gland and can occur either sporadically or as part of an inherited syndrome. A proportion of these cases carry an autosomal dominant mutation in the RET (REarranged during Transfection) proto-oncogene. Screening for these mutations in the affected patients and the carriers “at risk” which includes the first-degree relatives is of utmost importance for early detection and prompt treatment including prophylactic thyroidectomy in cases that harbor these mutations.
Results:
This report presents details of screening and subsequent follow-up of a large Indian family, where the index case was found to carry p. Cys634Ser mutation involving exon 11 of the RET gene. These data are of value considering the paucity of information within the region in context of screening large families affected by these mutations.
Keywords: MTC, medullary thyroid carcinoma, mutations, RET gene
INTRODUCTION
Medullary thyroid carcinoma (MTC) is a tumor arising from the para follicular (C) cells of the thyroid gland. These tumors produce calcitonin a 32-amino acid peptide.[1] MTC can occur either sporadically or as part of an inherited syndrome. Hereditary MTC can be subdivided into three clinical subtypes: Familial MTC (FMTC), multiple endocrine neoplasia type 2A (MEN 2A) and type 2B (MEN 2B). MEN 2A is characterized by the presence of the following MTC, hyperparathyroidism, and pheochromocytoma. MEN 2B is characterized by the presence of MTC, pheochromocytoma, mucosal neuromas, and marfanoid body habitus.[2,3]
FMTC is of interest because a proportion of these cases carry an autosomal dominant gain-of-function mutation in the RET (REarranged during Transfection) proto-oncogene. We screened 11 members of a large Indian family with FMTC for mutations in the RET oncogene. Six of them carried a mutation in codon p.Cys634Ser involving exon 11 of the RET gene. Five of them were clinically affected and one underwent prophylactic thyroidectomy.
PATIENT DETAILS
The index case [A] was a 56-year-old lady who presented to us in June 1997 for evaluation of a thyroid swelling. She was diagnosed to have MTC [Figure 1 and Table 1]. She underwent total thyroidectomy with central neck dissection and external beam radiation therapy; she subsequently received I-131 (42 mci) therapy in October 1997. Her last medical checkup was in 2010. At this stage there was no palpable tumor, her serum calcitonin level was elevated (755 pg/mL), her iodine-131-meta-iodobenzylguanidine (MIBG) scan was, however, negative.
Figure 1.
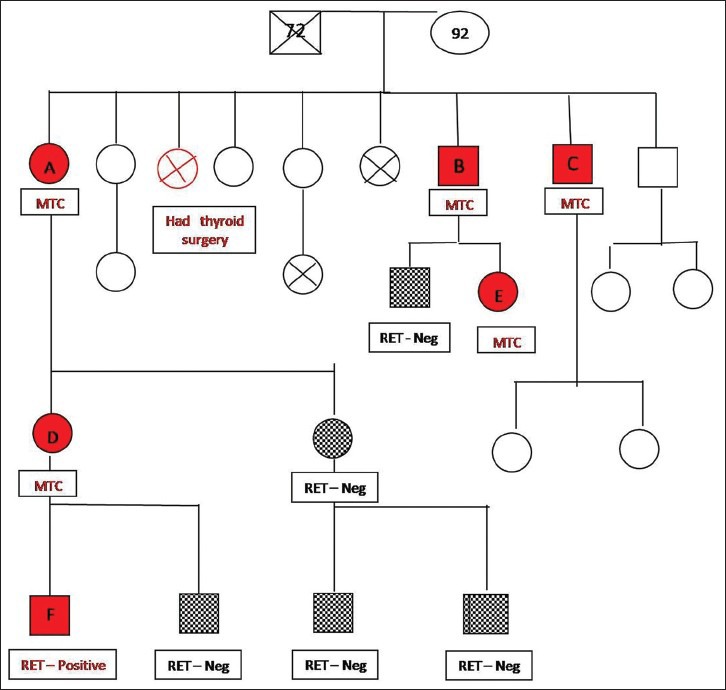
Family pedigree showing six affected members, five of whom were diagnosed to have medullary thyroid carcinoma, one underwent prophylactic thyroidectomy. The RET mutational analysis is incomplete because some of the family members could not be contacted and the others refused testing
Table 1.
Characteristics of the affected family members
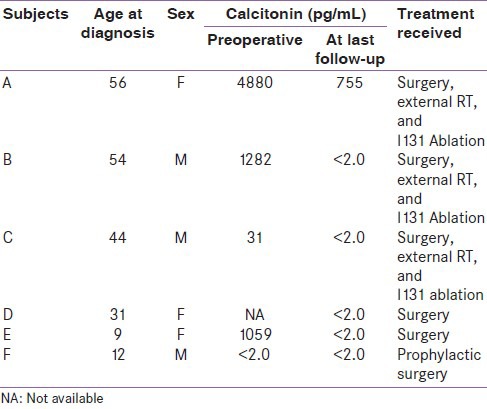
Following surgery, two of her younger brothers who had long-standing thyroid swellings came for further evaluation. Both of them were also diagnosed to have MTC. One of them [C] was 44 years old when he was diagnosed. He underwent total thyroidectomy and external beam radiation therapy in 1998. This was followed by I-131 ablation in January 1999. He was last reviewed in 2010. He has remained disease free and his serum calcitonin level had normalized (< 2 pg/mL).
His elder brother [B] was 54 years old when he underwent surgery in May 2002. After surgery, he received external beam radiation therapy. He was last reviewed in January 2012. There was no palpable tumor recurrence. His serum calcitonin level was normal (< 2 pg/mL). While screening his family members, his 9-year-old daughter [E] was diagnosed to have MTC. She underwent total thyroidectomy in June 2002 and was last reviewed by us in January 2012. She has remained disease-free. Her serum calcitonin level has normalized (<2 pg/mL).
The index case A has two daughters. Her eldest daughter [D] was diagnosed to have MTC when she was 31 years old. She underwent surgery at another center. Her son [F] who also carried the same mutation underwent prophylactic thyroidectomy at 12 years of age.
None of the affected family members had the other features associated with MEN II A or II B. All of them carried p.Cys634Ser mutation involving exon 11 of the RET gene [Figure 2].
Figure 2.
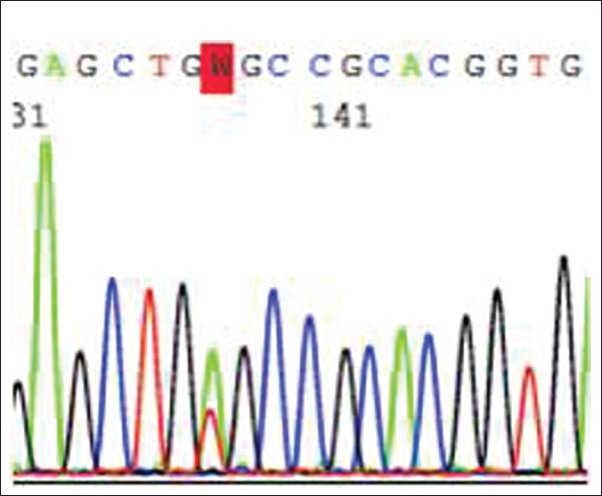
Electropherogram showing the mutation involving Codon 634 (p.Cys634Ser) on exon 11
GENETIC TESTING
A total of 200 μL of blood was used for extraction of genomic deoxyribonucleic acid (DNA) using the QIAamp DNA blood minikit (QIAGEN, Hilden, Germany). The extract was quantitated using the Nanodrop (Nanodrop technologies, USA) and 50 ng of DNA was used for polymerase chain reaction (PCR) amplification of the target regions. A total of 20 picomoles of primers were used for amplification of exons 10, 11, 13, 14, 15, and 16 in a 25 μL volume containing 1 unit of amplitaq gold (Applied Bio systems, USA) and were amplified in a Veriti thermal cycler (Applied Bio systems, USA).[4] The PCR products were detected using a 1.5% agarose gel and both the sense and antisense strands for all six exons were sequenced using the ABI PRISM 310 genetic analyzer with the ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Bio systems, USA).
DISCUSSION
FMTC is diagnosed when either MTC or a RET gene mutation is found in two or more generations of a family.[5] About 5%−15% of all cases of MTC are familial,[6] emphasizing the need to screen all family members at risk of developing disease. If identified early, these tumors can be cured by surgery.
FMTC represents a less aggressive form of hereditary MTC and has an older age at onset, often between 20 and 40 years, as compared with MEN 2A and MEN 2B. In the current series, the mean age at presentation of these tumors was 38.8 years, similar to earlier reports from India.[4,7] The index case and her siblings presented in the 5th and 6th decade of life, while their offsprings were affected much earlier, as documented in other studies on MTC.[7,8,9,10] Data from the West indicate that most patients present in the 4th or 5th decade of life, though a distinct age-related progression of familial MTC in carriers is also known to depend on the type of RET mutation.[2,11] Several studies have also reported female predominance of these tumors.[9,10] In the present series, males and females were affected equally (3:3).
Most of our patients presented with a neck swelling, some were identified during screening. The usual clinical presentation of MTC is a painless thyroid swelling (65%) or isolated cervical lymphadenopathy (15%).[12] These tumors secrete calcitonin, calcitonin gene-related peptide, prostaglandins, and vasoactive intestinal polypeptide, which can cause diarrhea and flushing.[13]
Calcitonin is a sensitive tumor marker and preoperative calcitonin levels correlate well with the tumor size and stage of the disease.[14] The positive predictive value of basal calcitonin in the preoperative diagnosis of MTC are as follows: for levels 20-50 pg/mL, for levels 50-100 pg/mL, and for levels >100 pg/mL they are 8.3%, 25%, and 100%, respectively.[15] In our series, three of the four patients who underwent surgery had calcitonin levels >1000 pg/mL and one (Subject C) had a calcitonin level of 31 pg/mL. Calcitonin levels 150-400 pg/mL are seen in those with distant metastasis and extrathyroid extension.[16]
Three of our patients with high calcitonin levels had extrathyroid extension of tumor and they received external beam radiation therapy and also underwent I131 ablation. The sensitivity of FNAC in detecting MTC is 63% as compared with 98% for measured serum calcitonin.[11] The typical cytological features seen in these tumors include a dispersed cell pattern, polygonal appearance of the cells, and binucleated cells with amyloid deposits. Carcino embryonic antigen (CEA) is another important marker for MTC. CEA levels > 30 ng/mL are less likely to be associated with a surgical remission and CEA levels > 100 ng/mL are associated with contralateral nodal disease and distant metastasis.[17]
Genetic testing
MTC has nearly a 100% penetrance in familial and MEN-2 syndromes.[18] The current standard of care for MTC includes genetic testing of all potentially affected family members. Germ line mutations in the RET proto oncogene have been identified in patients with hereditary MTC and somatic RET mutations in a proportion of sporadic MTC.[19] RET mutations can be detected in about 85% of families with MTC.[20,21]
The RET gene encodes a receptor tyrosine kinase that is expressed in neural crest derived cell lineages. The RET receptor plays a crucial role in regulating cell proliferation, migration, differentiation, and survival during embryo genesis. Mutations in the intracellular tyrosine kinase residues are generally found in MEN 2B and FMTC; these activate tyrosine, leading to aberrant phosphorylation of substrate and activation of downstream signaling pathways.[18] Patients who present with MTC should undergo DNA analysis for detection of mutations in the RET proto oncogene, because the likelihood of a germ line RET mutation is relatively high (9%) even in apparently sporadic cases.[4]
Clinically relevant mutations are located on exon 10, 11, 13, 14, 15, and 16 and these are to be analyzed.[4,18] The majority of RET mutations in FMTC are germ line mutations found on exon 10 and 11, specifically those involving codon 609, 611, 618, 620, and 634.[3,19] Mutation involving codon 634 is the most frequently reported mutation from this region,[9] including from our series of patients with MTC, which was reported earlier.[4] A total of 6 of 11 members of this large Indian family carried RET mutation involving codon p.Cys634Ser on exon 11.
Identification of mutations in asymptomatic carriers, as seen in this study, adds value to the existing data from India especially since there is paucity of information in this area.[9] Relatives with RET mutations should undergo prophylactic total thyroidectomy at an appropriate age depending on the type of mutation.[11] For couples carrying this mutation, we would suggest that they screen their children for RET mutations as early as possible. Prophylactic thyroidectomy should definitely be carried prior to 5 years of age, since certain mutations are associated with aggressive disease. In our series, one subject (F) underwent prophylactic thyroidectomy. The RET mutational analysis of the entire family is incomplete, because some members could not be contacted, while the others refused testing. The initial treatment will include total thyroidectomy with central compartment dissection. Three of our patients who had extensive local disease also required local radiation therapy and I131 ablation following surgery.
The sensitivity and specificity of 18F DOPA positron emission tomography/computed tomography is 100% when the serum calcitonin level is > 150 pg/mL.[22] Liver metastases are usually multiple and are usually not amenable to surgical treatment.[11,23] I-131 MIBG and 90Yttrium DOTATOC have been used for the treatment of metastatic disease.[12,24]
Current recommendation is to recheck serum calcitonin 2-3 months after surgery in view of variable time taken for normalization of calcitonin levels.[11] In our series, clinical and biochemical remission was achieved in five of six patients, while our index case continued to have elevated calcitonin levels even after 13 years, she has otherwise been keeping well.
Calcitonin doubling time is a predictor of survival in MTC.[11] In a retrospective series, when the calcitonin doubling time was < 6 months, the 5- and 10-year survival rates were 25% and 8%, respectively; when the doubling time was 6 − 24 months, the 5- and 10-year survival rates were 92% and 37%, respectively; and in those with doubling time > 2 years, survival was 100% at 10 years.[25]
Biochemical cure predicts a survival rate of 97.7% at 10 years. All our patients were followed-up for 10 years, while our index case was followed-up for 13 years. Persistent hypercalcitoninemia does not always indicate poor prognosis as seen in our index case. When there is no anatomic evidence of disease on imaging despite detectable serum calcitonin levels it would be most appropriate to keep the patient on follow-up.[11] Postoperatively, all our patients were on replacement doses of levothyroxine, as MTC is not a TSH dependent tumor.[11]
SUMMARY
We report the occurrence of MTC in six members of a family spread over three generations all of whom carried p.Cys634Ser mutation on exon 11 of the RET gene. The index case presented to us when she was 56 years old, while the youngest member in this family was 9 years old when she was detected to have MTC. This emphasizes the need for RET mutational screening of all the asymptomatic family members of patients with MTC.
ACKNOWLEDGEMENT
This work was supported by the start-up funds provided by the institutional review board of the Christian Medical College, Vellore, India.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Alsanea O, Clark OH. Familial thyroid cancer. Curr Opin Oncol. 2001;13:44–51. doi: 10.1097/00001622-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Gagel RF, Hoff AO, Cote GJ. Medullary thyroid carcinoma. In: Braverman LE, Utiger RD, editors. The Thyroid. Philadelphia: Lippincott Williams and Wilkins; 2005. pp. 967–88. [Google Scholar]
- 3.Kouvaraki MA, Shapiro SE, Perrier ND, Cote GJ, Gagel RF, Hoff AO, et al. RET proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumours. Thyroid. 2005;15:531–44. doi: 10.1089/thy.2005.15.531. [DOI] [PubMed] [Google Scholar]
- 4.Pai R, Nehru GA, Samuel P, Paul MJ, Thomas N, Premkumar AJ, et al. Mutational analysis of RET proto-oncogene among patients with medullary thyroid carcinoma and ‘at risk’ carriers from India. Clin Endocrinol (oxf) 2011;75:571–2. doi: 10.1111/j.1365-2265.2011.04069.x. [DOI] [PubMed] [Google Scholar]
- 5.Desai SS, Sarkar S, Borges AM. A study of histopathological features of medullary carcinoma of the thyroid: Cases from a single institute in India. Indian J Cancer. 2005;42:25–9. doi: 10.4103/0019-509x.15096. [DOI] [PubMed] [Google Scholar]
- 6.Jung J, Uchino S, Lee Y, Park H. A Korean family of familial medullary thyroid cancer with Cys618Ser RET germline mutation. J Korean Med Sci. 2010;25:226–9. doi: 10.3346/jkms.2010.25.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma BP, Saranath D. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J Biosci. 2011;36:603–11. doi: 10.1007/s12038-011-9095-0. [DOI] [PubMed] [Google Scholar]
- 8.Machens A, Dralle H. Genotype-phenotype based surgical concept of hereditary medullary thyroid carcinoma. World J Surg. 2007;31:957–68. doi: 10.1007/s00268-006-0769-y. [DOI] [PubMed] [Google Scholar]
- 9.Gilliland FD, Hunt WC, Morris DM, Key CR. Prognostic factors for thyroid carcinoma. A population based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program 1973-1991. Cancer. 1997;79:564–73. doi: 10.1002/(sici)1097-0142(19970201)79:3<564::aid-cncr20>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Raue F. German medullary thyroid carcinoma/multiple endocrine neoplasia registry. German MTC/MEN Study Group. Medullary thyroid carcinoma/multiple endocrine neoplasia type 2. Langenbecks Arch Surg. 1998;383:334–6. doi: 10.1007/s004230050143. [DOI] [PubMed] [Google Scholar]
- 11.American Thyroid Association Guidelines Task Force. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, et al. Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid. 2009;19:565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 12.Finny P, Jacob JJ, Thomas N, Phillip J, Rajaratnam S, Oommen R, et al. Medullary thyroid carcinoma: A 20-year experience from a center in South India. ANZ J Surg. 2007;77:130–4. doi: 10.1111/j.1445-2197.2006.03992.x. [DOI] [PubMed] [Google Scholar]
- 13.Genden EM, Brett E. Contemporary management of thyroid carcinoma. Cancer Ther. 2009;7:7–20. [Google Scholar]
- 14.Cohen R, Campos R, Salaun C, Heshmati HM, Kraimps JL, Proye C, et al. Preoperative calcitonin levels are predictive of tumor size and postoperative calcitonin normalization in medullary thyroid carcinoma. Groupe d’Etudes des Tumeurs a Calcitonine (GETC) J Clin Endocrinol Metab. 2000;85:919–22. doi: 10.1210/jcem.85.2.6556. [DOI] [PubMed] [Google Scholar]
- 15.Costante G, Meringolo D, Durante C, Bianchi D, Nocera M, Tumino S, et al. Predictive value of serum calcitonin levels for preoperative diagnosis of medullary thyroid carcinoma in a cohort of 5817 consecutive patients with thyroid nodules. J Clin Endocrinol Metab. 2007;92:450–5. doi: 10.1210/jc.2006-1590. [DOI] [PubMed] [Google Scholar]
- 16.Machens A, Schneyer U, Holzhausen HJ, Dralle H. Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab. 2005;90:2029–34. doi: 10.1210/jc.2004-1836. [DOI] [PubMed] [Google Scholar]
- 17.Machens A, Ukkat J, Hauptmann S, Dralle H. Abnormal carcinoembryonic antigen levels and medullary thyroid cancer progression: A Multivariate analysis. Arch Surg. 2007;142:289–93. doi: 10.1001/archsurg.142.3.289. [DOI] [PubMed] [Google Scholar]
- 18.de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;27:535–60. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- 19.Ball DW. Medullary thyroid cancer: Monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36:823–37. doi: 10.1016/j.ecl.2007.04.001. viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponder BA, Ponder MA, Coffey R, Pembrey ME, Gagel RF, Telenius-Berg M, et al. Risk estimation and screening in families of patients with medullary thyroid carcinoma. Lancet. 1988;1:397–401. doi: 10.1016/s0140-6736(88)91191-9. [DOI] [PubMed] [Google Scholar]
- 21.Mulligan LM, Eng C, Healey CS, Ponder MA, Feldman GL, Li P, et al. A de novo mutation of the RET proto-oncogene in a patient with MEN 2A. Hum Mol Genet. 1994;3:1007–8. doi: 10.1093/hmg/3.6.1007. [DOI] [PubMed] [Google Scholar]
- 22.Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H, et al. Clinical value of 18-fluorine-fluorodihydroxyphenylalanine positron emission totomography/computed tomography in the follow-up of medullary thyroid carcinoma. Thyroid. 2010;20:527–33. doi: 10.1089/thy.2009.0342. [DOI] [PubMed] [Google Scholar]
- 23.Isozaki T, Kiba T, Numata K, Saito S, Shimamura T, Kitamura T, et al. Medullary thyroid carcinoma with multiple hepatic metastases: Treatment with transcatheter arterial embolization and percutaneous ethanol injection. Intern Med. 1999;38:17–21. doi: 10.2169/internalmedicine.38.17. [DOI] [PubMed] [Google Scholar]
- 24.Iten F, Müller B, Schindler C, Rochlitz C, Oertli D, Mäcke HR, et al. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: A phase II clinical trial. Clin Cancer Res. 2007;13:6696–702. doi: 10.1158/1078-0432.CCR-07-0935. [DOI] [PubMed] [Google Scholar]
- 25.Barbet J, Campion L, Kraeber-Bodéré F, Chatal JF GTE Study Group. Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2005;90:6077–84. doi: 10.1210/jc.2005-0044. [DOI] [PubMed] [Google Scholar]