

Abstract
Clinical symptoms in MOG-induced EAE mice significantly exacerbated following hondroitin sulfate A (CS-A) injection, whereas administration of a degraded product, CSPG-DS, caused dramatic inhibition of EAE development. Also, administration of CSPG-DS but not CS-A, after the onset of clinical symptoms of EAE, was able to suppress the disease. Further studies demonstrated that CS-A up-regulated STAT4 expression and thus, induced IFN-γ production and Th1 CD4 T cell differenttiation. CS-A also up-regulated STAT3 and IL-23 expression and thus increased IL-17 producing T cells. CSPG-DS treatment both in vivo and in vitro decreased TNFα production from splenocytes. In vitro and in vivo studies indicated that CSPG-DS treatment in EAE mice significantly blocked migration of lymphocytes, whereas CS-A treatment increased lymphocyte infiltration in the brain.
Keywords: Chondroitin sulfate, Immune modulation, Cytokines, Cell migration, EAE/MS
1. Introduction
Multiple sclerosis (MS) is viewed as an autoimmune disease, however, neither the etiology nor the mechanism of disease is fully understood, and current treatments have only modest effect. The conceptual understanding of MS rests on the animal model of experimental autoimmune encephalomyelitis (EAE). It is widely believed that MS is an inflammatory attack on myelin and neurons orchestrated by myelin specific T cells (Holmoy, 2007).
Chondroitin sulfate (CS) is a major component of the extracellular matrix of many connective tissues, including cartilage, bone, skin, ligaments and tendons. It is a sulfated glycosaminoglycan (GAG) composed of a long unbranched polysaccharide chain with a repeating disaccharide structure of N-acetylgalactosamine and glucuronic acid. CS is an ingredient found commonly in dietary supplements used as complementary and alternative medicine (CAM) to treat osteoarthritis (OA). Researchers conducted a 4-year study known as the Glucosamine/chondroitin Arthritis Intervention Trial at 16 sites across the United States and found that the popular dietary supplement combination of glucosamine plus chondroitin sulfate did not provide significant relief from osteoarthritis pain among all participants. However, a smaller subgroup of study participants with moderate-to-severe pain showed significant relief with the combined supplements (Clegg et al., 2006). A recent report also demonstrates that CS has a slight to moderate efficacy in the symptomatic treatment of OA, with an excellent safety profile (Monfort et al., 2008a). The benefit of chondroitin sulfate in patients with OA is likely the result of a number of effects including its anti-inflammatory activity (Monfort et al., 2008b). Such findings suggest that CS may play an important role as a CAM that could potentially benefit patients suffering from other autoimmune diseases such as MS.
Chondroitin sulfate proteoglycan (CSPG), a matrix protein that occurs naturally in the CNS, is considered to be a major inhibitor of axonal regeneration and is known to participate in activation of the inflammatory response. The disaccharide degradation of CSPG by a specific enzyme, chondroitinase ABC, promotes repair (Rolls et al., 2004). Thus it has been postulated that a disaccharidic degradation product of chondroitin sulfate proteoglycan (CSPG-DS), participates in the modulation of the inflammatory responses and can, therefore, promote recovery in immune-induced neuropathologies of the CNS, such as experimental autoimmune uveitis (EAU) and EAE (Rolls et al., 2006). In these disease models, the dramatic increase in T cells infiltrating the CNS is far in excess of the numbers needed for regular maintenance. It has been reported that CSPG-DS markedly alleviated the clinical symptoms of EAE and protected against the neuronal loss in EAU (Rolls et al., 2006)
Although the functions of chondroitin sulfate in OA have been extensively studies Uebelhart, 2008; Uebelhart et al., 2006; Vangsness et al., 2009; Volpi, 2004), the role of chondroitin sulfate in EAE is still unknown. Studies on the immune modulation by chondroitin sulfate and its degraded disaccharide in EAE will provide a novel effective therapeutic target for multiple sclerosis in the patients and have great implications in the elucidation of mechanisms underlying multiple sclerosis. In the current study, we provide evidence for the first time that CS-A, which is naturally present in the central nervous system, may play a role in enhancing the clinical symptoms of EAE, whereas its degraded product, CSPG-DS, inhibits EAE development. Thus, CSPG-DS may constitute a novel drug candidate for suppressing inflammation and clinical disease in MS patients.
2. Materials and Methods
2.1 Mice
C57BL/6 mice were obtained from the National Institutes of Health (Frederick, MD). Female mice, 8–10 weeks of age, were used throughout these studies. Animals were housed in specific pathogen-free conditions, and all experiments were approved by Institutional Animal Care and Use Committee.
2.2 EAE induction
Myelin oligodendrocyte glycoprotein MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK) was diluted in PBS to a final concentration of 3 mg/ml. Equal volumes of MOG/PBS were mixed with complete Freund's adjuvant (CFA, Sigma-Aldrich, St. Louis, MO) with 4-6 mg/mL killed Mycobacterium tuberculosis H37 Ra thoroughly to form a thick peptide/CFA emulsion and left overnight on ice in the refrigerator. On the following day (day 0), the emulsion was centrifuged gently and loaded slowly into 1-ml syringe using an 18 gauge needle. The18 gauge needle was replaced with a 27 gauge needle for subcutaneous injection of 100 μl of MOG/CFA emulsion into two sites at mouse flank, 50 μ1 per site. On the same day, 200 ng or 300 ng (100 μl) of Bordetella Pertussis toxin (Sigma-Aldrich, St. Louis, MO) was injected intraperitoneally. On day 2, mice received an additional intraperitoneal injection of Pertussis toxin. Then, mice were monitored daily for signs of clinical disease. The severity of disease was recorded according to the following scale: 0, no symptoms; 1, partial loss of tail tonicity/inability to curl the distal end of tail; 2, complete loss of tail tone; 3, hind limb weakness/partial paralysis; 4, complete hind limb paralysis/fore limb weakness; 5, tetraplegia/moribund; 6, death. Data will be presented as mean clinical scores for each group of 10 mice. When necessary, food was provided on the cage floor and access to drinking water was made available for the paralyzed mice. C57BL/6 mice, which did not receive MOG35-55 immunization and Pertussis toxin, were used as control mice.
2.3 Administration of CS-A and CSPG-DS
CS-A and CSPG-DS were purchased from Sigma-Aldrich (St. Louis, MO). After EAE induction, mice received intravenous injection of CS-A (50 mg/kg of body weight) or CSPG-DS (5 or 50 mg/kg of body weight) in 100 μl of PBS on day 3, 5, 7, 9, 11 and 13. Then mice were sacrificed at day 7, 14, 21 and 28 for histological and immunological analysis. To investigate if CS-A and CSPG-DS would be effective after the onset of clinical symptoms, EAE was induced in mice as described above, and such mice received CS-A or CSPG-DS injection on days 13, 15, 17, 19, 21, 23, 25 and 27.
2.4 Examination of cell infiltration in brain
Brain tissues from control, CS-A or CSPG-DS treated mice were collected 14 days after EAE induction. The brain tissues were fixed, paraffin blocks were prepared, microtome sections were generated, and tissue sections were stained using hematoxylin and eosin. The sections were examined for inflammatory cell infiltrates under Nikon Optiphot Epifluorescence system.
2.5 Preparation and flow cytometric analysis of brain-infiltrating cells
At day 14 after EAE induction, the brain tissues were removed surgically from control, CS-A or CSPG-DS treated mice. The brain tissues were smashed in a Stomacher 80 Biomaster lab blender (Metrohm USA, Riverview, FL), lysed with Red Blood Lysing Buffer (Sigma-Aldrich, St. Louis, MO) to remove red blood cells, washed with PBS, filtered and then stained with FITC-conjugated anti-CD8 antibody and PE-conjugated anti-CD4 antibody for flow cytometric analysis. The brain cell suspension was also stained with FITC-conjugated anti-CD4 antibody and PE-conjugated CD27, CD44 or CD62L for flow cytometric analysis of lymphocyte phenotypic markers. Flow cytometric analysis was carried out using Cytomics FC500 flow cytometer (Beckman Coulter, Fullerton, CA). All antibodies were purchased from Biolegend (San Diego, CA).
2.6 Intracellular staining of cytokine production
At day 14 after EAE induction, the spleens were obtained from control, CS-A or CSPG DS treated mice. Splenocytes were obtained after the spleens were crushed in a Stomacher 80 Biomaster lab blender (Metrohm USA, Riverview, FL), lysed with Red Blood Lysing Buffer (Sigma-Aldrich, St. Louis, MO) to remove red blood cells, washed with PBS and filtered through sterile mesh (70 μm). Then, splenocytes were fixed and permeabilized using Cytofix/Cytoperm (BD, Franklin Lakes, NJ) and stained with PE-conjugated anti-IL-6, IL-17 or IFN-γ antibody. IL-6, IL-17 and IFN-γ producing splenocytes were determined by flow cytometric analysis using Cytomics FC500 flow cytometer (Beckman Coulter, Fullerton, CA). All antibodies were purchased from Biolegend (San Diego, CA).
2.7 In vitro stimulation of splenocytes and brain-infiltrating cells
Splenocytes (5×106) were stimulated in 2 mL of X-VIVO 15 medium (Lonza, Walkersville, MD) with 150 μg/mL MOG peptide supplemented with 100 μg/mL CS-A or CSPG-DS on 24-well plates in a humidified 5% CO2 incubator at 37°C. After 48 h, the splenocytes were stimulated with MOG and CS-A again. After another 48 h, the culture supernatants were collected for IFN- γ and TNFα release assay by ELISA and the splenocytes were used for intracellular staining of cytokine production and FACS analysis.
The brain-infiltrating lymphocytes were also isolated from brain cell suspensions in 33% Percoll under 2000rpm centrifugation for 15 min. 1×106 lymphocytes were used in 2 mL of X-VIVO 15 medium (Lonza, Walkersville, MD) in the presence of 150μg/mL MOG35–55 peptide for in vitro stimulation. After culture for one day, the culture supernatants were collected for IFN- γ release assay by ELISA.
2.8 ELISA analysis of IFN- γ and TNFα production
ELISA kits for IFN- γ and TNFα production analysis were purchased from PeproTech (Rocky Hill, NY). ELISA assay was performed according to the manufacturer's instructions. The culture supernatants from in vitro stimulation of splenocytes or sera obtained from mice after EAE induction were used for IFN- γ and TNFα ELISA assay.
2.9 Detection of IL-23, STAT3, STAT4 and TNFα expression by RT-PCR
Total RNAs from the splenocytes harvested from mice after EAE induction and CS-A or CSPG-DS treatment were prepared using the RNeasy minikit (QIAGEN, Valencia, CA). First-strand cDNA synthesis was performed in a 20μl-reaction mix containing 500 ng total RNA using ThermoScript™ RT-PCR system and following the protocol of the manufacturer (Invitrogen, Carlsbad, CA). After first-strand synthesis, 2 μl (10% of the reaction volume) was used as a template for PCR amplification. To detect the expression of IL-23, forward (5′- TGTTGCCCTGGGTCACTCA-3′) and reverse (5′-CCAGGCTAGCATGCAGAGATT-3′) primers specific to mouse IL-23 were used. To detect the expression of STAT3, forward (5′- CAATACCATTGACCTGCCGAT-3′) and reverse (5′-GAGCGACTCAAACTGCCCT 3′) primers specific to mouse STAT3 were used. To detect the expression of STAT4, forward (5′- TGGCAACAATTCTGCTTCAAAAC-3′) and reverse (5′ AGGTCCCTGGATAGGCATGT- 3′) primers specific to mouse STAT4 were used. To detect the expression of TNFα, forward (5′- TCTCATTCCTGCTTGTGGC-3′) and reverse (5′-GCTGGCACCACTAGTTGGTT-3′) primers specific to mouse TNFα were used. PCR was performed by the initial cycle of 2 min at 94°C, 35 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 68°C, and the final cycle of 10 min at 68°C using Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA) in a PTC-100 Peltier Thermal Cycler (MJ Research, Waltham, MA). The PCR products, generated from mouse IL-23, STAT3, STA4 and TNFα primer pairs, were normalized against PCR products generated from β-actin forward (5′-GTCCCTTCCAGCAGATGT-3′) and reverse (5′-AGCTCAGTAACAGTCCGCCTAGA-3′) primers after electrophoresis on 1.2 % agarose gel and visualization with UV light. The band intensity of PCR products was determined using Bio-Rad image analysis system (Bio-Rad Laboratories).
2.10. T cell migration assay in vitro
Splenocytes (5×105) isolated from naïve and EAE-induced mice were seeded into the upper well with 1 mL of RPMI 1640 serum-free medium in the presence of vehicle control, 100 μg/mL CSPG-DS or 100 μg/mL CS-A in BioCoat™ Matrigel™ Invasion Chamber (BD, San Jose, CA). The bottom well contained 1 mL of RPMI 1640 serum-free medium (control medium) or 1 mL of X-VIVO 15 medium (Lonza, Walkersville, MD) supplemented with 10% FCS and 50CU/mL IL- 2. After incubation at 37°C with 5% CO2 for 20 h, all T cells in the bottom wells were collected and counted after trypan blue exclusion staining. The number of migrating T cells in six wells was calculated and the mean ± SEM values were depicted.
2.11. Statistical Analysis
Each experiment was repeated at three times with consistent results. Data were expressed as mean ± SEM. Statistical analyses were performed using Student's t test or two-factor NOVA as appropriate, with a P value of <0.05 considered to be statistically significant. For EAE, significant difference between control and experimental groups was determined using the Mann-Whitney U test, as described in our previous studies (Singh et al., 2007).
3. Results
3.1. Modulation of EAE development by CS-A and CSPG-DS
Chondroitin sulfate is a GAG that is widely present in animal tissues, including the CNS and when externally administered has been shown to have anti-inflammatory and chondroprotective properties (Akiyama et al., 2004). We were interested in delineating the role of chondroitin sulfate A and its degraded disaccharide product on the lymphocytic infiltration in the CNS and development of experimental autoimmune encephalomyelitis in mice. Mice received subcutaneous injection of myelin oligodendrocyte glycoprotein peptide (MOG35–55) emulsified in the complete Freund's adjuvant containing killed Mycobacterium tuberculosis on day 0 and intraperitoneal injection of Bordetella Pertussis toxin on day 0 and 2 for EAE induction. After EAE induction, mice received intravenous injection of CS-A or CSPG-DS on day 3, 5, 7, 9, 11 and 13. Our data showed that chondroitin sulfate compounds dramatically affected EAE development in mice. Clinical symptoms in MOG-induced EAE mice significantly enhanced following CS-A injection, whereas CSPG-DS injection completely inhibited EAE development in mice (Fig.1A). Also, when a clinically more severe form of EAE was induced by injection of higher doses of Pertussis toxin, CS-A treatment increased the severity of the clinical disease particularly between 18-25 days post-MOG immunization, whereas CSPG-DS significantly delayed EAE development as well as decreased the severity of the clinical disease (Fig. 1B). In addition, we also tested whether treatment with CS-A or CSPG-DS was effective after the onset of clinical symptoms. To this end, EAE was induced in mice as described above, and such mice received CS-A or CSPG-DS injection on days 13, 15, 17, 19, 21, 23, 25 and 27 (Fig 1C). Mann-Whitney U test showed that there were no significant differences in clinical scores between EAE induced mice and the EAE mice that received CSPG-DS (p = 0.9577) or CS-A (p = 0.9584), prior to day 13 before CS-A or CSPG-DS injection. There was also no significant difference of clinical scores between EAE control mice and the EAE mice that received CS-A treatment (p = 0.2497) after day 13, however, EAE mice treated with CSPG-DS showed a significant decrease in clinical symptoms (p < 0.005) (Fig. 1C). These data together suggested that CSPG-DS but not CS-A may not only have the ability to prevent the development of EAE but also exhibit a therapeutic efficacy in disease control.
Fig. 1.
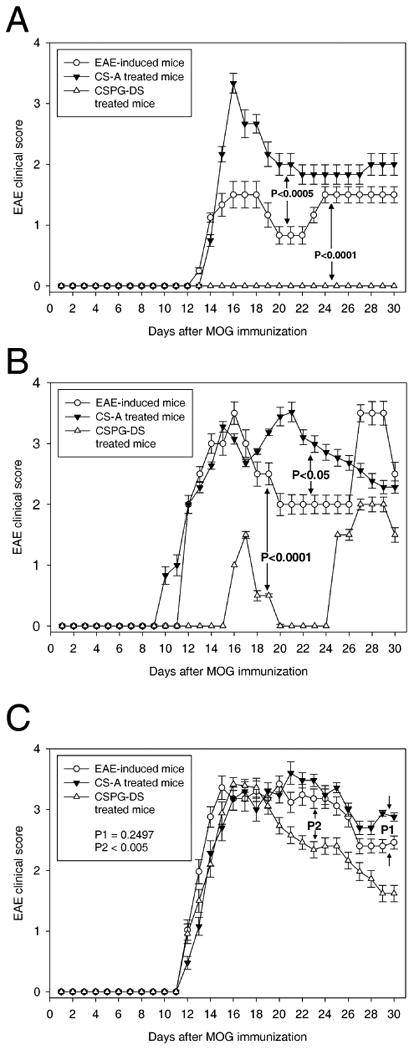
Effect of CS-A and CSPG-DS on clinical disease in EAE-induced mice. (A) Modulation of less severe form of EAE by CS-A and CSPG-DS. EAE was induced in C57BL/6 mice by MOG35-55 immunization and each mouse received i.p. injection of 200 ng Pertussis toxin on days 0 and 2. Then, the mice received i.v. injection of CS-A (50 mg/kg of body weight) or CSPG-DS (5 mg/kg of body weight) in 100 μl PBS on days 3, 5, 7, 9, 11 and 13. (B) Modulation of more severe form of EAE by CS-A and CSPG-DS. EAE induction and treatment with CS-A or CSPG-DS was identical to the procedure described above except that the mice received higher doses of Pertussis toxin (300 ng) to induce increased clinical score. (C) Therapeutic efficacy of CSPG-DS in the treatment of EAE. After EAE induction by MOG35-55 immunization and Pertussis toxin, the mice received CS-A (50 mg/kg of body weight) or CSPG-DS (50 mg/kg of body weight) in 100 μl PBS on days 13, 15, 17, 19, 21, 23, 25 and 27. Mann-Whitney U test was used to compare clinical scores between CS-A treated and untreated EAE mice (P1) as well as CSPG-DS treated and untreated EAE mice (P2) after day 13. EAE vs. EAE treated with CS-A (p = 0.2497); EAE vs. EAE treated with CSPG-DS (p < 0.005). Data were presented as mean ± SEM of clinical scores from 10 mice each group.
3.2. Modulation of cell infiltration in the brain by CS-A and CSPG-DS during clinical EAE
It has also been reported that infiltration of the central nervous system by CD4+ Th1 cells precedes onset and relapses of experimental autoimmune encephalomyelitis (Skundric et al., 2005). Our studies showed that EAE induction triggered significant lymphocytic infiltration in the mouse brain tissue (Fig. 2B) when compared to control mice (Fig. 2A) and CS-A treatment further increased cell infiltration (Fig. 2D), however, CSPG-DS treatment dramatically inhibited cell infiltration in the brain tissues (Fig. 2C). Flow cytometric analysis demonstrated that the increased proportions of lymphocytes, predominantly CD4+ T cells, were present in the brain tissues of mice with EAE disease (Fig. 3B) when compared with control mice (Fig. 3A), CS-A treatment further significantly increased the percentage of CD4+ T cell infiltration in the brain and in contrast, CSPG-DS significantly decreased the percentage of CD4+ T cell infiltration (Fig. 3C). These data correlated with the clinical efficacy of CS-A and CSPG-DS. Further studies using flow cytometry revealed that EAE induction significantly increased percentage of cells bearing the phenotype of CD4+CD27+, CD4+CD44+ and CD4+CD62L+ cells. CS-A treatment further augmented while CSPG-DS considerably decreased those cell populations (Fig. 3D, 3E and 3F). CD27, CD44 and CD62L expression is usually used in defining memory T cells (Appay et al., 2002; Sallusto et al., 2004; Sprent and Surh, 2002). Thus, these results indicated that memory-phenotypic CD4+ T cells may play an important role in the progression of EAE disease.
Fig. 2.
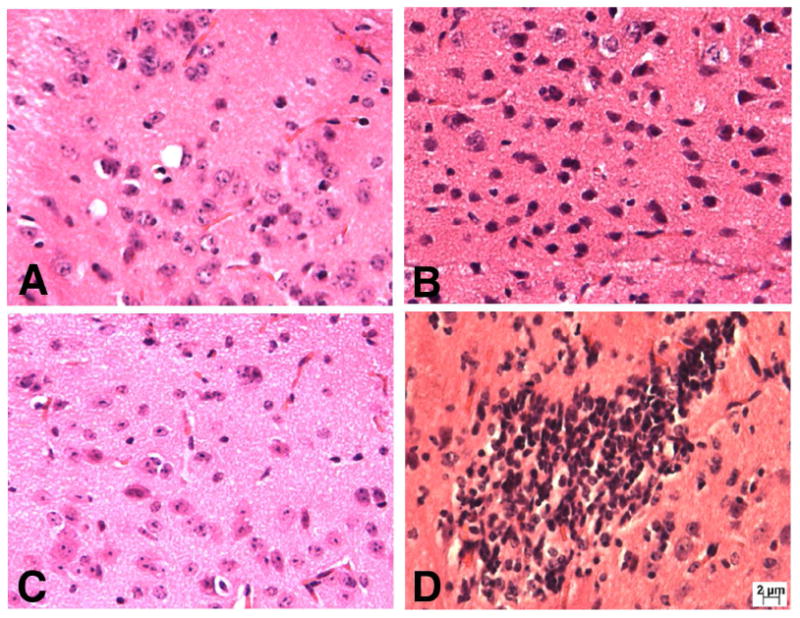
Effect of CS-A and CSPG-DS on cell infiltration in mouse brain tissues after EAE induction. A representative brain section has been shown from: (A) control mice, which did not receive MOG35-55 immunization and Pertussis toxin; (B) EAE-induced mice; (C) CSPG-DS treated mice after EAE induction; and (D) CS-A treated mice after EAE induction. EAE induction and treatment with CS-A or CSPG-DS was carried out as described in Fig. 1. On day 14, brain tissues were examined following Haematoxylin & Eosin staining.
Fig. 3.
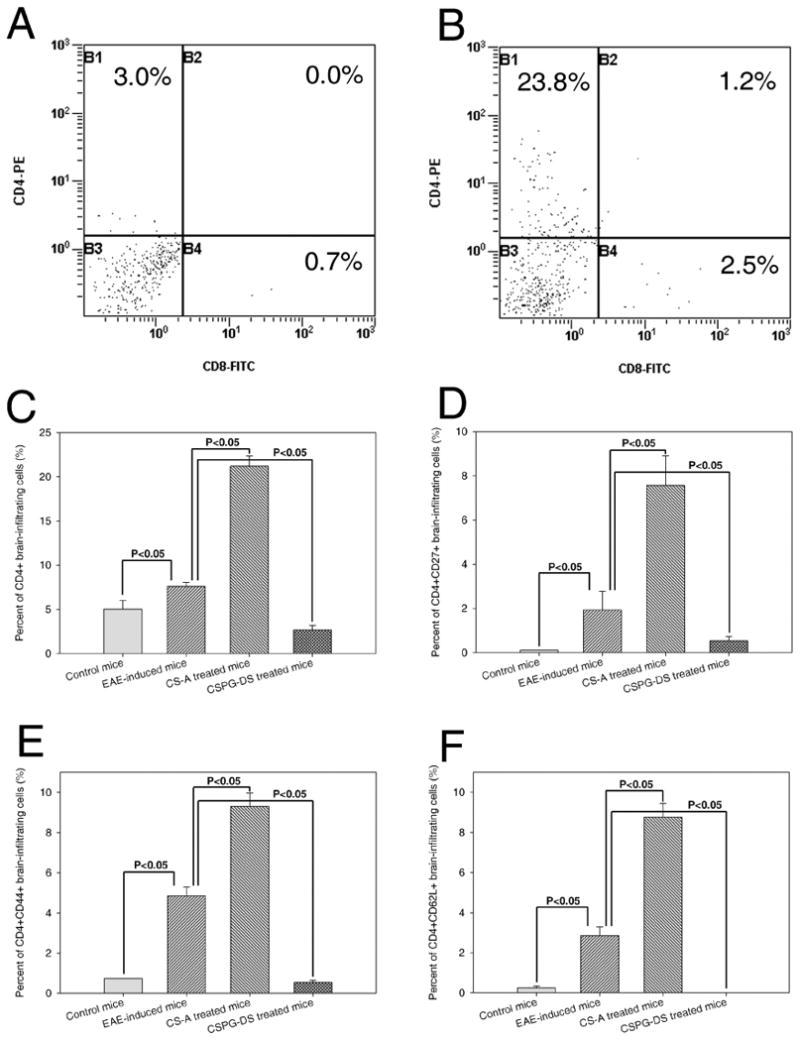
Phenotypic characterization of infiltrating cells in the brains of EAE mice after CS-A and CSPG-DS treatment. (A) A representative dot plot of flow cytometric analysis of CD4+ and CD8+T cell subpopulations found in the brain from control mice, which did not receive MOG35-55 immunization and Pertussis toxin. (B) A representative dot plot of flow cytometric analysis of T cell population infiltrated in the brain from EAE-induced mice. Percentage of CD4+ T cells (C), CD4+CD27+ T cells (D), CD4+CD44+ T cells (E) and CD4+CD62L+ T cells (F) in EAE mice with or without treatment with CS-A and CSPG-DS from multiple experiments was depicted as mean ± SEM (error bars) from three mice/group.
3.3. Modulation of Cytokine production by CS-A and CSPG-DS
Recent studies have indicated that Th17 cells and IFN-γ-secreting Th1 cells play an important role in the clinical pathogenesis in EAE/MS disease (Komiyama et al., 2006; Nagelkerken, 1998; Segal, 2003; Skarica et al., 2009; Yura et al., 2001). In addition, proinflammatory cytokines such as TNFα and IL-6 are also considered important for induction and pathogenesis involving inflammation and neuroglial damage (Penkowa and Hidalgo, 2001; Villarroya et al., 1997; Wiemann et al., 1998). To study the cytokine production, splenocytes from MOG-immunized mice after CS-A or CSPG-DS treatment were washed, fixed and permeabilized. Then the splenocytes were stained with PE-conjugated anti-IL-6, IL-17 or IFN-γ antibody. IL-6, IL-17 and IFN-γ producing splenocytes were determined by flow cytometric analysis. Our analysis confirmed that EAE induction in mice by MOG35–55 peptide significantly increased percentage of spleen cells expressing IL-6, IL-17 and IFN-γ (Fig. 4A, 4B and 4C). CS-A caused further increase of IL-6, IL-17 and IFN-γ producing cells (Fig. 4A, 4B and 4C), while CSPG-DS caused a significant decrease of cells expressing IFN- γ (Fig. 4C). Accordingly, the cytokine IFN-γ production in the brain-infiltrating lymphocytes was significantly decreased after CSPG-DS treatment, whereas it was dramatically increased after CS-A treatment (Fig. 4D).
Fig. 4.
Effect of CS-A and CSPG-DS on IL-6, IL-17 and IFN-γ production in splenocytes and brain-infiltrating cells from EAE mice. EAE induction and treatment with CS-A or CSPG-DS were carried out as described in Fig 1. Without re-stimulation in vitro, splenocytes from control mice, which did not receive MOG35-55 immunization and Pertussis toxin, and EAE mice untreated or treated with CS-A or CSPG-DS, on day 14, were stained for intracellular expression of: (A) IL-6; (B) IL-17; and (C) IFN-γ. IFN-γ production in the brain-infiltrating cells after in vitro stimulation was also determined by ELISA from EAE mice untreated or treated with CS-A or CSPG-DS on day 14 (D). Data were presented as mean ± SEM (error bars) of multiple experiments from three mice/group.
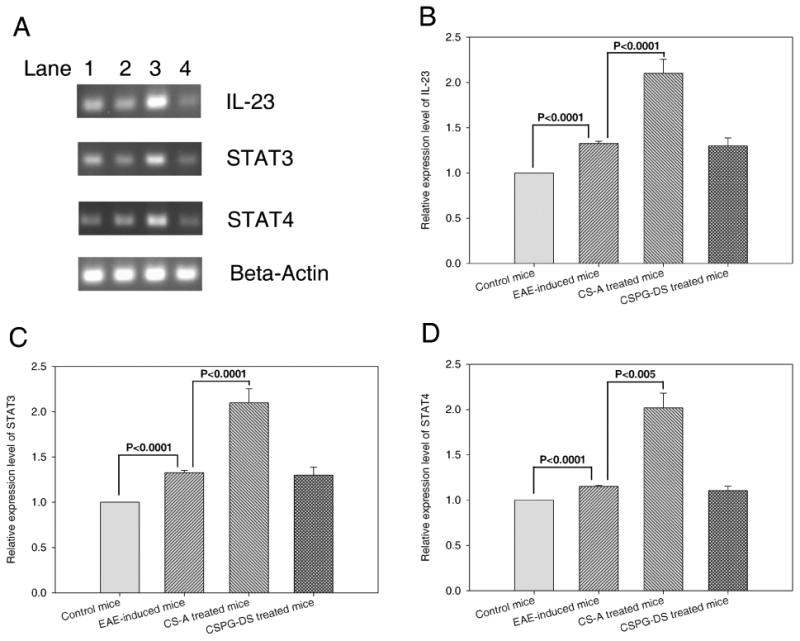
There is evidence to suggest that IL-23 stimulated CD4+ T cells express various genes, including IL-17, and that IL-23-stimulated T cells are important in the pathogenesis of EAE (Langrish et al., 2005). Also, it has become clear that IL-23 is not the direct initiator of Th17 production (Veldhoen et al., 2006), but rather is an important factor for expanding and maintaining Th17 cells in vivo (McGeachy et al., 2007). The differentiation of Th17 cells is actually initiated by signal transducer and activator of transcription 3 (STAT3), downstream of IL-6- and IL-21-induced signaling. Activation of STAT3 induced the expression of retinoic-acid-receptor-related orphan receptor-α (RORα) and RORγt. These two factors establish the Th17-cell-associated gene-expression program, leading to the production of IL-17, IL-17F and IL-22 (Dong, 2008). In addition, STAT4 is an essential element in the early events of Th1 differentiation (Nishikomori et al., 2002). Thus, we examined the effects of EAE induction and chondroitin sulfate treatment on gene expression of IL-23, STAT3 and STAT4. The results showed that EAE induction in mice enhanced the gene expression of IL-23, STAT3 and STAT4 in splenocytes and CS-A treatment in vivo further up-regulated the expression of these genes while CSPG-DS failed to cause significant alterations (Fig. 5). The results suggested that CS-A may promote Th17 and Th1 T cell development while CSPG-D may not influence the induction of these cells.
Fig. 5.
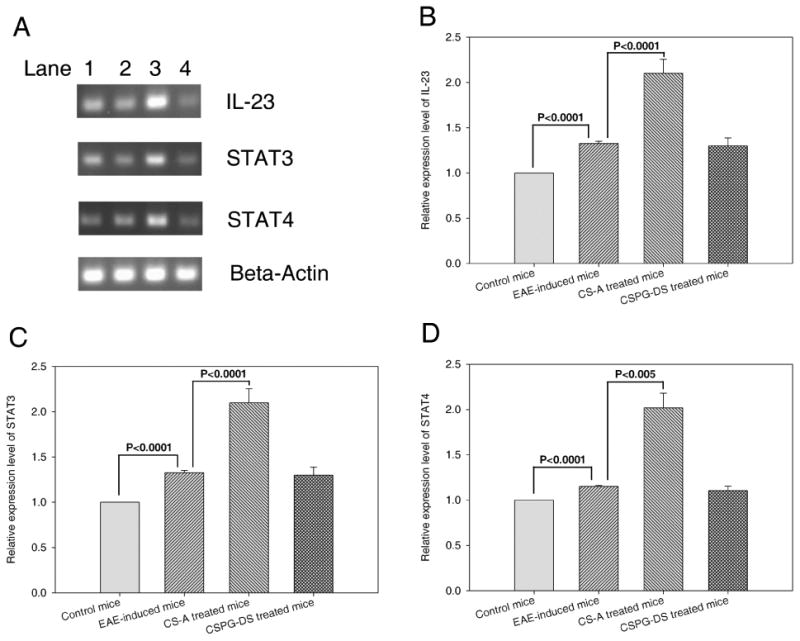
Effect of CS-A and CSPG-DS on IL-23, STAT3 and STAT4 expression in splenocytes. EAE was induced in mice that were untreated or treated with CS-A and CSPG-DS as described in Fig 1. (A) A representative gel electrophoresis profile of IL-23, STAT3, STAT4 and β-actin RT-PCR products from mouse splenocytes on day 7 after EAE induction. Data from normal control mice that did not receive MOG35-55 immunization and Pertussis toxin (Lane 1), untreated EAE mice (Lane 2), EAE mice treated with CS-A (Lane 3) and EAE mice treated with CSPG-DS (Lane 4) are shown. In panels (B), (C) and (D), data from multiple experiments has been plotted indicating the relative expression levels when compared to controls, which were presented as mean ± SEM (error bas) of multiple experiments from three mice/group.
We also investigated the induction of TNFα during EAE and its modulation following CS-A and CSPG-DS treatment. To this end, we measured TNFα in the serum (Fig. 6A), mRNA levels in splenocytes (Fig 6B) and following in vitro culture of splenocytes with MOG (Fig 6C). EAE induction in mice by MOG35–55 peptide significantly augmented TNFα production (Fig. 6). However, CSPG-DS treatment caused a significant decrease in TNFα whereas CS-A failed to block TNFα induction.
Fig. 6.
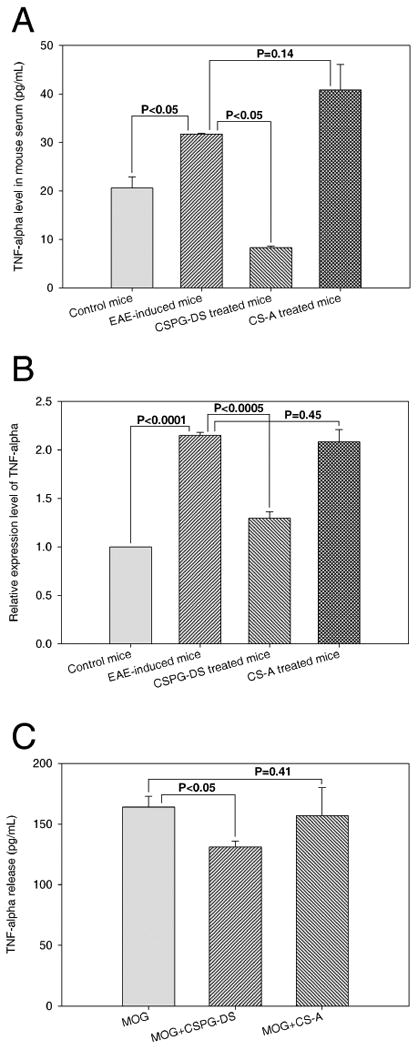
Effect of CS-A and CSPG-DS on TNFα production in splenocytes. (A) TNFα level in the sera of control mice that did not receive MOG35-55 immunization and Pertussis toxin or EAE mice, on day 7, treated with or without CSPG-DS and CS-A. (B) TNFα expression at mRNA level in splenocytes from control or EAE mice, on day 7, with or without CSPG-DS or CS-A treatment. (C) Detection of TNFα release from splenocytes after in vitro stimulation with MOG35-55 in the absence or presence of CSPG-DS or CS-A for four days. Data were presented as mean ± SEM (error bas) of multiple experiments from three mice/group.
3.4. Modulation of T cell migration by CS-A and CSPG-DS
Next, we tested if the decreased inflammation seen in the CNS of EAE mice following treatment with CS-A and CSPG-DS resulted from altered T cell migration into the CNS. To test if CSA and CSPG-DS would alter cell migration, transwell migration assays were performed. In this assay, mouse splenocytes were seeded into the upper well with serum-free medium in the presence of vehicle control, CSPG-DS or CS-A in a 24-well chamber. The bottom well contained the complete medium supplemented with 10% FCS and IL-2 as T cell migration chemoattractant. After incubation, the number of migrating T cells in the bottom wells were collected and counted. The assay demonstrated that the migration of splenocytes from EAE mice was markedly blocked by CSPG-DS while CS-A did not significantly inhibit the migration (Fig 7). These data are consistent with the observation that CSPG-DS injection into the mice dramatically blocked the cell infiltration in the brain tissues (Figs. 2C and 3C).
Fig. 7.
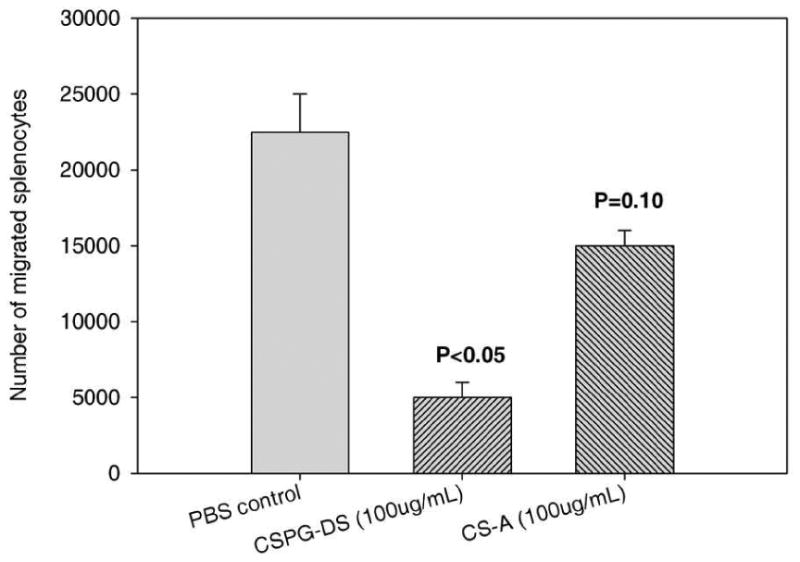
Effect of CSPG-DS and CS-A on T cell migration in vitro. Splenocytes were harvested from mice on day 30 after EAE induction. Migration assay of splenocytes was performed in BD Matrigel Invasion Chamber in the presence of IL-2 in the lower well and chondroitin sulfate compounds in the upper well. After 20h culture, the number of splenocytes migrated from the upper well into the lower well were determined. Data represented Mean ± SEM of three independent experiments. Three mice/group were used in each experiment.
4. Discussion
Chondroitin sulfate (CS) is a sulfated GAG composed of a chain of alternating sugars (N-acetylgalactosamine and glucuronic acid). It is usually found attached to proteins as part of a proteoglycan. CS is an important structural component of cartilage and provides much of its resistance to compression (Baeurlea, 2009). Along with glucosamine, chondroitin sulfate is a widely used dietary supplement for treatment of OA. The use of CS is somewhat controversial and recent research has questioned its efficacy in treating OA (Morris and Smith, 2009). It has been reported that after administration of glucosamine 500 mg 3 times daily, CS 400 mg 3 times daily, or a combination of glucosamine and CS for 2 years, there was no clinically important difference in joint space width (JSW) loss when compared with placebo (Sawitzke et al., 2008), however, patients taking chondroitin sulfate were noted to have a statistically significant improvement in knee joint swelling (Hochberg and Clegg, 2008). On the other hand, enough clinical data are available supporting the view that oral CS is a valuable and safe symptomatic treatment for OA disease (Uebelhart, 2008). The benefit of chondroitin sulfate in patients with OA is likely the result of a number of effects including its anti-inflammatory activity (Monfort et al., 2008b) and a structure-modifying effect of glucosamine and chondroitin sulfate (Bruyere and Reginster, 2007). It should be noted that injection of GAGs such as hyaluronic acid, heparin, and chondroitin sulfates A, B, and C can also induce rheumatoid arthritis (RA) in mice, which is a chronic, systemic, and inflammatory disease of connective tissue with unknown etiology (Wang and Roehrl, 2002). Increased level of chondroitin sulfate was observed in blood serum of RA patients (Merkur'eva and Bukhtoiarova, 1976). Consistent with such findings, our studies suggested that CS- A significantly increased the immune response and thus promoted EAE development when administered prior to disease onset (Figs. 1-5). However, it did not alter the clinical outcome administered after the disease (Fig 1C). Thus, chondroitin sulfate may represent a natural immune modulator, which plays a stimulatory role in autoimmune diseases such as RA and EAE.
The persistence of human autoimmune diseases is thought to be mediated predominantly by memory T cells (Elyaman et al., 2008). Murine memory cells are high in CD44 and low in the expression of activation markers such as CD25. Two subsets of memory T cells, called central-memory (TCM) and effector-memory (TEM) T cells, are described based on their anatomical location, expression of cell surface markers, and effector functions (Dutton et al., 1998). TCM cells express molecules such as CD62L and CCR7, which allow efficient homing to lymph nodes, whereas TEM cells lack expression of these lymph node homing receptors and are located in nonlymphoid tissues. However, both T cell subsets are present in the blood and spleen. The phenotype of T cells that infiltrate the spinal cord has been studied in EAE. It has been shown that in EAE in mice, T cells are mainly activated/memory (CD44high/LFA-1high/ICAM-1high/CD45RBlow) (Engelhardt et al., 1998). It has also been reported that memory CD4+ T cells (CD4+CD44hi T cells, isolated from C57BL/6 mice after recovery from acute disease, greater than 100 days after immunization with MOG35-55) induce more severe disease than effector CD4+ T cells caused by a preferential differentiation of memory T cells into the Th1 phenotype and a differential expression of chemokine receptors and costimulatory molecules leading to severe CNS inflammation in mice after EAE induction (Elyaman et al., 2008). However, the function of memory T cells in EAE has not been fully understood. Our studies indicated that EAE induction significantly increased cell populations of CD4+CD27+, CD4+CD44+ and CD4+CD62L+ infiltrated in the brain and CS-A treatment further augmented those cell populations whereas CSPG-DS considerably decreased those cell populations (Fig. 3D, 3E and 3F). These results provided the evidence that chondroitin sulfate dramatically increases memory phenotypic CD4 T cells in mouse brain, which may play an important role in promoting EAE pathogenesis.
It was largely accepted that Th1 cells driven by IL-12 are pathogenic T cells in human MS and mouse EAE. The signal transducer and activator of transcription, STAT4, is required for most IL-12-stimulated functions and for development of IFN-γ-secreting Th1 cells from naïve CD4+ T cells (Kaplan et al., 1996; Thierfelder et al., 1996). Recent data have also established that IL- 17-producing CD4+ T cells, driven by IL-23 and referred to as Th17 cells, play a pivotal role in the pathogenesis of EAE (Aranami and Yamamura, 2008). Activation of STAT3 by transforming growth factor beta-1 (TGFβ-1), IL-6, IL-21 and IL-23 as well as the expression of RORγt lead to the differentiation of Th17 cells in mice. In conjunction with transforming growth factor beta-1, IL-6, IL-21 and IL-23, which activate STAT3, the expression of RORγt leads to the differentiation of Th17 cells in mice (Takatori et al., 2008). Our investigations showed that EAE induction and CS-A treatment increased STAT4 gene expression (Fig. 5D) and IFN-γ-producing cell population (Fig. 4C), suggesting that chondroitin sulfate may promote Th1 CD4 T cell differentiation and thus enhance EAE disease. Meanwhile, EAE induction and CS-A treatment also enhanced STAT3 and IL-23 gene expression (Fig. 5), IL-6 and IL-17 producing cells (Fig. 4), indicating that chondroitin sulfate may stimulate Th17 cell differentiation and thus augment EAE development. It should be noted that administration of CS-A before but not after the onset of clinical symptoms, augmented EAE. This suggested that the presence of CS-A during the induction phase of the immune response may promote Th1 and Th17 differentiation whereas, the presence of CS-A during the effector phase of the immune response may have limited role in further modulating the T cell response.
Proinflammatory cytokines such as TNFα and IL-6 are considered important for induction and pathogenesis of EAE/MS disease, which is characterized by significant inflammation and neuroglial damage (Penkowa and Hidalgo, 2001; Villarroya et al., 1997; Wiemann et al., 1998). It has also been reported that CSPG-DS markedly alleviated the clinical symptoms of EAE and protected against the neuronal loss in EAU (Rolls et al., 2006). This effect was associated with a reduction in the numbers of infiltrating T cells and marked microglia activation. This is further supported by in vitro results indicating that CSPG-DS attenuated T cell motility and decreased secretion of the cytokines IFN-γ and TNFα (Rolls et al., 2006). It has been reported that CSPG-DS attenuates the delayed-type hypersensitivity response in mice (Rolls et al., 2006). Attenuation of the delayed-type hypersensitivity response may be attributed to the decreased presence of T cells in an irritated region, a manifestation of reduced T cell migration (Rolls et al., 2006). Our studies demonstrated that CSPG-DS effectively inhibited EAE development (Fig. 1), which was associated with the dramatic reduction of cell migration of splenocytes from EAE-induced mice (Fig. 7) and cell infiltration in mouse brain (Figs. 2 and 3) as well as the significant decrease of TNFα and IFN-γ production (Figs. 6 and 4C). CS-A, however, did not significantly affect TNFα production (Fig. 6) and cell migration of splenocytes from both EAE-induced mice (Fig. 7). These results showed that CS-A and CSPG-DS may have different roles in the modulation of immune responses associated with autoimmune diseases such as EAE and MS. CD44 is an adhesion molecule, a cell surface transmembrane glycoprotein, encoded by a single gene. CD44 is known to be important in T-cell signaling and a variety of immune cell functions. CD44 has a well documented role in tumor metastasis (Marhaba and Zoller, 2004). Our recent studies demonstrated that CD44 plays a role in the differentiation of Th1 and Th2 cells and CD44-deficiency enhances the development of Th2 effectors in response to antigen stimulation (Guan et al., 2009). CD44 was also essential for the generation of memory Th1 cells by the phosphoinositide 3-kinase-Akt kinase signaling pathway that regulates cell survival (Baaten et al., 2010). Because CS-A readily interacts with proteins such as CD44 in the extracellular matrix due to its negative charges and these interactions are important for regulating a diverse array of cellular activities (Gotte and Yip, 2006), it is possible that CS-A may bind to CD44 and induce CD44-trigerred signaling to promote Th1 CD4 T cell differentiation (Figs. 4 and 5). CD44 has been demonstrated to play a critical role in cell migration (Marhaba and Zoller, 2004), and thus the degraded disaccharide product of CS-A, CSPG-DS, may interact with CD44 to interfere CD44-cytoskeleton signaling (Pure and Assoian, 2009) and hamper lymphocyte cell migration as shown in the current study (Fig. 7). However, the exact signaling pathways induced by CS-A and CSPG-DS still need to be further elucidated.
In summary, our results indicate that administration of CS-A, which is naturally present in the central nervous system, before but not after the onset of clinical symptoms, may play a role in the promotion of EAE. In contrast, its degraded product, CSPG-DS, inhibits EAE development when administered both before and after the disease onset. Thus, CSPG-DS may act as a negative regulator by suppressing the inflammation in the CNS. Together, the current study suggests that CSPG-DS may represent a novel drug candidate for suppressing inflammation and clinical disease in MS patients.
Acknowledgments
This work was supported by NIH grants R01ES09098, R01DA016545 and P01AT003961 (P. Nagarkatti); NIH grants R01AI053703, R01AI058300, and R01HL058641 (M. Nagarkatti).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Sakai S, Linhardt RJ, Goda Y, Toida T, Maitani T. Chondroitin sulphate structure affects its immunological activities on murine splenocytes sensitized with ovalbumin. Biochem J. 2004;382:269–278. doi: 10.1042/BJ20031851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, Gruener N, Pape G, Waters A, Easterbrook P, Salio M, Cerundolo V, McMichael AJ, Rowland-Jones SL. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- Aranami T, Yamamura T. Th17 Cells and autoimmune encephalomyelitis (EAE/MS) Allergol Int. 2008;57:115–120. doi: 10.2332/allergolint.R-07-159. [DOI] [PubMed] [Google Scholar]
- Baaten BJ, Li CR, Deiro MF, Lin MM, Linton PJ, Bradley LM. CD44 Regulates Survival and Memory Development in Th1 Cells. Immunity. 2010;32:104–115. doi: 10.1016/j.immuni.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeurlea SA, Kiselevb MG, Makarovab ES, Nogovitsinb EA. Effect of the counterion behavior on the frictional–compressive properties of chondroitin sulfate solutions Polymer. 2009;50:1805–1813. [Google Scholar]
- Bruyere O, Reginster JY. Glucosamine and chondroitin sulfate as therapeutic agents for knee and hip osteoarthritis. Drugs Aging. 2007;24:573–580. doi: 10.2165/00002512-200724070-00005. [DOI] [PubMed] [Google Scholar]
- Clegg DO, Reda DJ, Harris CL, Klein MA, O'Dell JR, Hooper MM, Bradley JD, Bingham CO, Weisman MH, 3rd, Jackson CG, Lane NE, Cush JJ, Moreland LW, Schumacher HR, Jr, Oddis CV, Wolfe F, Molitor JA, Yocum DE, Schnitzer TJ, Furst DE, Sawitzke AD, Shi H, Brandt KD, Moskowitz RW, Williams HJ. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795–808. doi: 10.1056/NEJMoa052771. [DOI] [PubMed] [Google Scholar]
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- Elyaman W, Kivisakk P, Reddy J, Chitnis T, Raddassi K, Imitola J, Bradshaw E, Kuchroo VK, Yagita H, Sayegh MH, Khoury SJ. Distinct functions of autoreactive memory and effector CD4+ T cells in experimental autoimmune encephalomyelitis. Am J Pathol. 2008;173:411–422. doi: 10.2353/ajpath.2008.080142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Martin-Simonet MT, Rott LS, Butcher EC, Michie SA. Adhesion molecule phenotype of T lymphocytes in inflamed CNS. J Neuroimmunol. 1998;84:92–104. doi: 10.1016/s0165-5728(97)00237-3. [DOI] [PubMed] [Google Scholar]
- Gotte M, Yip GW. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. Cancer Res. 2006;66:10233–10237. doi: 10.1158/0008-5472.CAN-06-1464. [DOI] [PubMed] [Google Scholar]
- Guan H, Nagarkatti PS, Nagarkatti M. Role of CD44 in the differentiation of Th1 and Th2 cells: CD44-deficiency enhances the development of Th2 effectors in response to sheep RBC and chicken ovalbumin. J Immunol. 2009;183:172–180. doi: 10.4049/jimmunol.0802325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg MC, Clegg DO. Potential effects of chondroitin sulfate on joint swelling: a GAIT report. Osteoarthritis Cartilage 16 Suppl. 2008;3:S22–24. doi: 10.1016/j.joca.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmoy T. Immunopathogenesis of multiple sclerosis: concepts and controversies. Acta Neurol Scand Suppl. 2007;187:39–45. doi: 10.1111/j.1600-0404.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhaba R, Zoller M. CD44 in cancer progression: adhesion, migration and growth regulation. J Mol Histol. 2004;35:211–231. doi: 10.1023/b:hijo.0000032354.94213.69. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Merkur'eva RV, Bukhtoiarova FG. Glycosaminoglycans in blood serum of patients with rheumatoid arthritis. Vopr Med Khim. 1976;22:291–296. [PubMed] [Google Scholar]
- Monfort J, Martel-Pelletier J, Pelletier JP. Chondroitin sulphate for symptomatic osteoarthritis: critical appraisal of meta-analyses. Curr Med Res Opin. 2008a;24:1303–1308. doi: 10.1185/030079908x297231. [DOI] [PubMed] [Google Scholar]
- Monfort J, Pelletier JP, Garcia-Giralt N, Martel-Pelletier J. Biochemical basis of the effect of chondroitin sulphate on osteoarthritis articular tissues. Ann Rheum Dis. 2008b;67:735–740. doi: 10.1136/ard.2006.068882. [DOI] [PubMed] [Google Scholar]
- Morris JD, Smith KM. Chondroitin sulfate in osteoarthritis therapy. Orthopedics. 2009;32 doi: 10.3928/01477447-20090401-04. [DOI] [PubMed] [Google Scholar]
- Nagelkerken L. Role of Th1 and Th2 cells in autoimmune demyelinating disease. Braz J Med Biol Res. 1998;31:55–60. doi: 10.1590/s0100-879x1998000100007. [DOI] [PubMed] [Google Scholar]
- Nishikomori R, Usui T, Wu CY, Morinobu A, O'Shea JJ, Strober W. Activated STAT4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of IL-12R beta 2 chain expression and signaling. J Immunol. 2002;169:4388–4398. doi: 10.4049/jimmunol.169.8.4388. [DOI] [PubMed] [Google Scholar]
- Penkowa M, Hidalgo J. Metallothionein treatment reduces proinflammatory cytokines IL-6 and TNF-alpha and apoptotic cell death during experimental autoimmune encephalomyelitis (EAE) Exp Neurol. 2001;170:1–14. doi: 10.1006/exnr.2001.7675. [DOI] [PubMed] [Google Scholar]
- Pure E, Assoian RK. Rheostatic signaling by CD44 and hyaluronan. Cell Signal. 2009;21:651–655. doi: 10.1016/j.cellsig.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Avidan H, Cahalon L, Schori H, Bakalash S, Litvak V, Lev S, Lider O, Schwartz M. A disaccharide derived from chondroitin sulphate proteoglycan promotes central nervous system repair in rats and mice. Eur J Neurosci. 2004;20:1973–1983. doi: 10.1111/j.1460-9568.2004.03676.x. [DOI] [PubMed] [Google Scholar]
- Rolls A, Cahalon L, Bakalash S, Avidan H, Lider O, Schwartz M. A sulfated disaccharide derived from chondroitin sulfate proteoglycan protects against inflammation-associated neurodegeneration. Faseb J. 2006;20:547–549. doi: 10.1096/fj.05-4540fje. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- Sawitzke AD, Shi H, Finco MF, Dunlop DD, Bingham CO, 3rd, Harris CL, Singer NG, Bradley JD, Silver D, Jackson CG, Lane NE, Oddis CV, Wolfe F, Lisse J, Furst DE, Reda DJ, Moskowitz RW, Williams HJ, Clegg DO. The effect of glucosamine and/or chondroitin sulfate on the progression of knee osteoarthritis: a report from the glucosamine/chondroitin arthritis intervention trial. Arthritis Rheum. 2008;58:3183–3191. doi: 10.1002/art.23973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal BM. Experimental autoimmune encephalomyelitis: cytokines, effector T cells, and antigen-presenting cells in a prototypical Th1-mediated autoimmune disease. Curr Allergy Asthma Rep. 2003;3:86–93. doi: 10.1007/s11882-003-0017-6. [DOI] [PubMed] [Google Scholar]
- Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4′-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72:1508–1521. doi: 10.1124/mol.107.038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarica M, Wang T, McCadden E, Kardian D, Calabresi PA, Small D, Whartenby KA. Signal transduction inhibition of APCs diminishes th17 and Th1 responses in experimental autoimmune encephalomyelitis. J Immunol. 2009;182:4192–4199. doi: 10.4049/jimmunol.0803631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skundric DS, Dai R, Zakarian VL, Bessert D, Skoff RP, Cruikshank WW, Kurjakovic Z. Anti-IL-16 therapy reduces CD4+ T-cell infiltration and improves paralysis and histopathology of relapsing EAE. J Neurosci Res. 2005;79:680–693. doi: 10.1002/jnr.20377. [DOI] [PubMed] [Google Scholar]
- Sprent J, Surh CD. T cell memory. Annu Rev Immunol. 2002;20:551–579. doi: 10.1146/annurev.immunol.20.100101.151926. [DOI] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Chen Z, O'Shea JJ. New complexities in helper T cell fate determination and the implications for autoimmune diseases. Mod Rheumatol. 2008;18:533–541. doi: 10.1007/s10165-008-0099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Uebelhart D. Clinical review of chondroitin sulfate in osteoarthritis. Osteoarthritis Cartilage 16 Suppl. 2008;3:S19–21. doi: 10.1016/j.joca.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Uebelhart D, Knols R, de Bruin ED, Verbruggen G. Treatment of knee osteoarthritis with oral chondroitin sulfate. Adv Pharmacol. 2006;53:523–539. doi: 10.1016/S1054-3589(05)53027-8. [DOI] [PubMed] [Google Scholar]
- Vangsness CT, Jr, Spiker W, Erickson J. A review of evidence-based medicine for glucosamine and chondroitin sulfate use in knee osteoarthritis. Arthroscopy. 2009;25:86–94. doi: 10.1016/j.arthro.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17- producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Villarroya H, Marie Y, Ouallet JC, Le Saux F, Tchelingerian JL, Baumann N. Expression of TNF alpha in central neurons of Lewis rat spinal cord after EAE induction. J Neurosci Res. 1997;49:592–599. doi: 10.1002/(SICI)1097-4547(19970901)49:5<592::AID-JNR9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Volpi N. The pathobiology of osteoarthritis and the rationale for using the chondroitin sulfate for its treatment. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:119–127. doi: 10.2174/1568008043339929. [DOI] [PubMed] [Google Scholar]
- Wang JY, Roehrl MH. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc Natl Acad Sci U S A. 2002;99:14362–14367. doi: 10.1073/pnas.222536599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann B, Van GY, Danilenko DM, Yan Q, Matheson C, Munyakazi L, Ogenstad S, Starnes CO. Combined treatment of acute EAE in Lewis rats with TNF-binding protein and interleukin-1 receptor antagonist. Exp Neurol. 1998;149:455–463. doi: 10.1006/exnr.1997.6723. [DOI] [PubMed] [Google Scholar]
- Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, Kiyono H. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE) J Autoimmun. 2001;17:17–25. doi: 10.1006/jaut.2001.0520. [DOI] [PubMed] [Google Scholar]