

The development of solar water-splitting systems provides a route to renewable H2.[1] A prerequisite for an efficient water-splitting process is the availability of highly efficient, inexpensive, and stable catalysts for H2 and O2 production. Despite much progress by synthetic and materials chemists,[2] it is still the natural enzymes that set the benchmark efficiency in the reduction and oxidation of water.[3] [FeFe] and [NiFe] hydrogenases convert protons and electrons into H2 with remarkably high rates at a low overpotential.[4] Photocatalytic H2 generation has previously been reported with hydrogenases in a heterogeneous scheme with the enzyme attached directly to semiconducting particles or electrodes.[5] Homogeneous systems with a dye and hydrogenase in solution are well established, but these multicomponent systems require a soluble redox mediator to transport the electron from the light absorber to the catalyst.[6]
An important requirement for water splitting is that the catalyst for H2 evolution tolerates at least small levels of O2, which will enter the system either through leakage of atmospheric O2 into the photoreactor and/or from the in situ formation of O2 during the water-splitting process.[7] Although many hydrogenases, in particular [FeFe] hydrogenase, are highly sensitive to O2, much progress has recently been reported in identifying and understanding the factors leading to O2 tolerance in [NiFe] hydrogenases.[8] In addition, a photocatalytic H2 evolution system without a redox mediator is desirable, because a chemically reduced mediator is easily quenched by O2, which can result in drastically reduced photoactivity for the system in the presence of O2.
Herein we report on a photocatalytic H2 evolution system consisting of a Desulfomicrobium baculatum (Db) [NiFeSe] hydrogenase and an organic dye, Eosin Y (EY), which evolves H2 photocatalytically under high levels of O2 (Figure 1). Db [NiFeSe] hydrogenase was selected as the H2 evolution catalyst because it displays unique properties among hydrogenases.[9] This enzyme has been reported to be biased towards H2 evolution, showing electrocatalytic H2 production activity in the presence of as much as 1 % O2 and displaying little product inhibition.[10] This hydrogenase has previously been adsorbed on ruthenium-dye-sensitized TiO2 nanoparticles for the photocatalytic generation of H2, but the formation of reactive oxygen species (ROS) on TiO2 during irradiation prevented its use in the presence of O2.[5a, 11] In this study, we removed the necessity for TiO2 by replacing dye-sensitized TiO2 with soluble EY, which allows for photo-induced direct electron transfer to a catalyst.[7,12] Elimination of radical-forming TiO2 allowed us to produce H2 photocatalytically in our hydrogenase-based system under remarkably high levels of O2.
Figure 1.
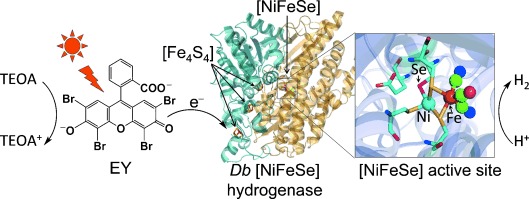
Schematic representation of photocatalytic H2 generation with EY and an O2-tolerant Db [NiFeSe] hydrogenase (EY–hydrogenase) system in the presence of TEOA in pH-neutral aqueous solution. Photo-induced electron transfer occurs directly from the EY to the [Fe4S4] cluster relay[3a, 6c, 8g] and then to the H2-evolving [NiFeSe]-active site of the enzyme (see text).
First, the photocatalytic activity was studied of a homogeneous aqueous solution of Db [NiFeSe] hydrogenase and EY in the presence of the electron donor triethanolamine (TEOA) under an inert atmosphere. Optimized conditions were obtained by varying the amount of hydrogenase, EY, and TEOA and the pH of the solution (Figures S1–S3). The EY–hydrogenase system worked efficiently in the absence of any soluble redox mediator when a stirred solution of hydrogenase (10 pmol) and EY (disodium salt, 1 μmol) in TEOA (2.25 mL, 150 mm) at pH 7 and 25 °C was exposed to visible light (solar light simulator; AM 1.5 G, 100 mW cm−2, λ>420 nm). The photoreactor was purged prior to the experiment with N2 containing 2 % CH4 (internal standard for gas chromatography (GC) measurements, see the Supporting Information).
The EY–hydrogenase system photogenerated (0.50±0.03) μmol of H2 per hour and almost linear H2 evolution rates up to 15 hours (Figure S4). This result corresponds to a hydrogenase-based turnover frequency (TOFhydrogenase) of (13.9±0.7) (mol H2)(mol hydrogenase)−1 s−1. The photoactivity of the system is lost after 24 h, whereupon (5.0±0.3) μmol of H2 had accumulated, corresponding to a TONhydrogenase of (5.0±0.3)×105 and an EY-based TON (TONEY) of (5.0±0.3) (mol H2)(mol EY)−1. No H2 was detected in the dark or in the absence of EY or hydrogenase. At 5 °C and 45 °C TOFhydrogenase values of (7.5±0.5) s−1 and (19±2) s−1 were obtained, respectively. The system exhibits an optimum activity at pH 7.0 and its performance is decreased by more than 50 % at pH 6.0 and 8.0 (Figure S2). This can be explained by the lower intrinsic activity of the hydrogenase under basic conditions[10] and an increased amount of protonated TEOA donor under an acidic environment.[5a, 12a] The EY–hydrogenase system operates with high photoactivity in the absence of any soluble redox mediator and electrons are transfered directly from the dye to the hydrogenase. The per-active-site performance of the EY–hydrogenase system is on the same order of magnitude as that of a previously reported system with Db [NiFeSe] hydrogenase on ruthenium-dye-sensitized TiO2 (TOFhydrogenase up to 50 s−1)[5a] and much higher than a photocatalytic system with EY and a synthetic Co catalyst (TOFCo=0.02 s−1)[7] in an aqueous pH-neutral TEOA solution.
Variation of the light intensity of monochromatic LED light (525 nm; pH 7.0 and 25 °C) from 1.5 to 5 and finally 18 mW cm−2 resulted in external quantum efficiencies (EQE) of (1.50±0.08), (0.49±0.03), and (0.18±0.01) % with a corresponding TOFhydrogenase of (16±1), (18±1), and (24±1) (mol H2)(mol hydrogenase)−1 s−1, respectively. The EQE increases with decreasing light intensity, whereas the TOFhydrogenase changes only marginally. The TOFhydrogenase also remained almost constant when the light intensity of visible light was increased from 50 to 100 mW cm−2 (Figure S5). At 100 mW cm−2 visible-light irradiation, an increasing amount of EY from 1 to 3 μmol did not result in the photogeneration of higher amounts of H2. Furthermore, when the amount of hydrogenase was increased from 10 to 50 pmol the amount of H2 photogenerated in the system more than doubled (Table S1, Figures S1 and S3). These experiments demonstrate that the hydrogenase limits the EY–hydrogenase system, which is an important requirement for studying the effect of inhibitors on the enzyme in the photocatalytic system.
Subsequently, the photocatalytic H2 production activity of the EY–hydrogenase system was investigated in the presence of varying concentrations of O2. Previously, protein film electrochemistry with Db [NiFeSe] hydrogenase on a pyrolytic graphite edge electrode demonstrated that this enzyme evolves H2 in the presence of 1 % O2 at an applied potential of −0.45 V versus the normal hydrogen electrode (NHE) in an aqueous electrolyte solution at pH 6.0.[10] Thus, H2 evolution under O2 should be possible if the photoexcited EY dye can efficiently transfer electrons directly to the hydrogenase.
To test this hypothesis, the EY–hydrogenase system (10 pmol of hydrogenase and 1 μmol of EY in 2.25 mL of aqueous 150 mm TEOA solution at pH 7 and 25 °C) was irradiated for one hour under N2 atmosphere (with 2 % CH4) to verify its activity under inert atmosphere. The photo-reactor was purged with 2 % CH4/N2 and different amounts of O2 were injected into the headspace after 1 hour with subsequent irradiation (Table 1 and Figure 2). In all cases, the photoactivity of the EY–hydrogenase system decreased with an increasing O2 concentration in the headspace. Remarkably, in the presence of 21 % O2 some photoactivity still remained ((11±3) % of the photoactivity under anaerobic conditions; Figure 2, insert).
Table 1.
Visible-light-driven (100 mW cm−2, AM 1.5 G, λ>420 nm) H2 production with EY–hydrogenase in an aqueous TEOA solution (2.25 mL, 150 mm) at pH 7 and 25 °C.
| Conditions | TOFhydrogenase[a] [s−1] | H2[a] [μmol H2 h−1] |
|---|---|---|
| EY (1 μmol) and hydrogenase (10 pmol) | ||
| 0 % O2 | 13.9±0.7 | 0.50±0.03 |
| 5 % O2 | 11.5±1.0 | 0.41±0.04 |
| 10 % O2 | 7.3±0.4 | 0.26±0.01 |
| 15 % O2 | 4.5±0.5 | 0.16±0.02 |
| 21 % O2 | 1.5±0.3 | 0.05±0.01 |
| 2 % CO | <0.4 | <0.02 |
| [Ru(bipy)3]2+ (1 μmol), hydrogenase (10 pmol), and MV (1 μmol) | ||
| 0 % O2 | 27±2 | 0.98±0.08 |
| 5 % O2 | 3.3±0.5 | 0.12±0.02 |
Calculated based on the amount of H2 produced in the first 0.5 h of irradiation; standard deviation for at least three experiments.
Figure 2.
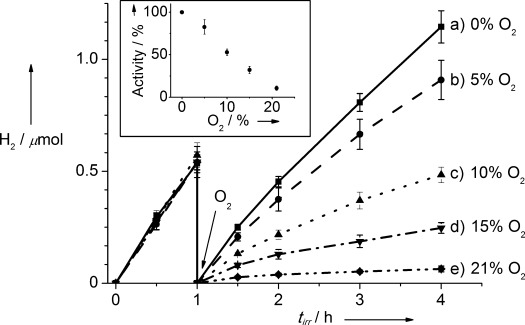
Amount of H2 generated with the EY–hydrogenase system in an aqueous TEOA solution (150 mm, pH 7.0) during visible-light irradiation (tirr, 100 mW cm−2, AM 1.5 G, λ>420 nm) at 25 °C. The EY–hydrogenase system was exposed to different O2 headspace concentrations after 1 h of irradiation under 2 % CH4/N2. Insert: Relative photocatalytic H2 evolution activity compared to that under anaerobic conditions (based on photoactivity within the first 0.5 h of irradiation).
The concentration of O2 was also measured in the solution for experiments with various concentrations of O2 in the headspace (Table S2). Irradiation of the EY–hydrogenase system under an atmosphere of 21 % O2 resulted in decrease of the concentration of dissolved O2 from (6.0±1.0) ppm to below 0.2 ppm within 1 min of irradiation, thereby creating conditions conducive for the O2-tolerant hydrogenase. The same behavior was observed in the absence of hydrogenase and can be ascribed to the known reaction of photo-excited EY with O2, resulting in the formation of singlet O2.[13] EY was used in excess in the system and the quenching of photo-excited EY with O2 explains the rapid decrease in the amount of dissolved O2 and the slow depletion of headspace O2 after several hours (Figure S6). Irradiation of the solution under a high concentration of O2 in the headspace presumably also resulted in the formation of increased amounts of singlet O2, causing the decreased lifetime of the photo-H2 evolution system (Figure 2).
The EY–hydrogenase system is fully photoactive after three hours under anaerobic conditions, but it is inactivated after three hours of irradiation under a 21 % O2 atmosphere. This endurance is remarkable when one considers the complete photo-decomposition of the same hydrogenase under air on dye-sensitized TiO2 within 2 min, which is presumably due to the decomposition of the enzyme by ROS formed upon reduction of O2 by conduction band electrons.[5a, 11] Notable differences between the dye–TiO2–hydrogenase and EY–hydrogenase systems are the generation of radical species in close proximity to the hydrogenase in the former case, whereas singlet O2 is mainly produced remote from the enzyme in the latter system.
In related work, a photocatalytic H2 evolution system consisting of a ruthenium dye covalently linked to [NiFe] hydrogenase from Thiocapsa roseopersicina in the presence of the soluble redox mediator methyl viologen (MV) was exposed to O2 in a closed photoreactor.[6b] Initial irradiation did not show formation of H2, but resulted in the depletion of O2 in the system, whereupon an anoxic environment in the system allowed for reactivation of the enzyme and formation of H2.[6b]
Therefore, we also tested a homogeneous system comprising [Ru(2,2′-bipyridine)3]Cl2 (1 μmol), [NiFeSe] hydrogenase (10 pmol), and MV (1 μmol) for comparison with the EY–hydrogenase system. The multicomponent Ru–MV–hydrogenase system is only photo-active in the presence of MV because electron transfer does not occur from photoexcited Ru directly to the hydrogenase.[5a, 6a,b] Under anaerobic conditions, the Ru–MV–hydrogenase system evolves (0.98±0.08) μmol H2 h−1 with a TOFhydrogenase of (27±2) mol H2 (mol hydrogenase)−1 s−1 during visible-light irradiation. In the presence of 5 % headspace O2, the photoactivity decreased dramatically to (3.3±0.5) s−1 and (0.12±0.02) μmol H2 h−1 (Table 1) and no significant amounts of H2 were observed under 21 % O2. Photoexcited [Ru(2,2′-bipyridine)3]2+ and reduced MV react with O2,[14] resulting in almost complete inactivation of the system.
In order to investigate the reversibility of the inhibitory effect of O2 in the EY–hydrogenase system, reactivation of hydrogenase under inert conditions after exposure to atmospheric O2 (21 %) was also examined. The EY–hydrogenase system was exposed to aerobic conditions for different periods of time at continuous white-light irradiation. After repurging the photoreactor with 2 % CH4/N2, we measured the photocatalytic H2 production. The resulting TOF dropped to (81±4), (65±6), and (31±3) % of the initial value after 30, 60, and 120 min of continuous exposure to air and light, respectively (Table S3 and Figure S7). Control experiments with EY–hydrogenase exposed to air in the dark for the same duration has led to a negligible inactivation of the hydrogenase (Table S3 and Figure S7).
Thus, the hydrogenase displays a good robustness in the presence of air and can be partially reactivated under inert conditions following light exposure with EY. Exposure of [NiFeSe] hydrogenases to O2 results in the oxidation of sulfur and/or selenium in cysteine and selenocysteine ligands at the active site.[9b–d] These inactive states can be reactivated through reduction and recovery of the amino acids. Exposing the EY–hydrogenase system to light results in photoexcitation of EY and formation of a long-lived triplet state.[12] Low-potential electrons are transferred to the hydrogenase, thereby reactivating O2-inactive states in the hydrogenase.[10] This fast reactivation of any O2-inactivated hydrogenase can be explained by the negative excited state reduction potential of EY (*EY=−0.91 V vs. NHE).[12a]
The effect of carbon monoxide, a well-known inhibitor of hydrogenases,[10] on the photocatalytic formation of H2 was also tested with EY–hydrogenase. Introduction of 2 % CO in N2 resulted in complete inactivation of the EY–hydrogenase system (Figure S8) and only a negligible amount of H2 was detectable by GC. Inhibition by CO was at least partially reversible and purging the inactive system with N2 containing 2 % CH4 resulted in (52±3) % of the initial TOFhydrogenase activity. The reversibility of CO inhibition is in agreement with the previously reported electrochemical studies.[10]
In conclusion, a photocatalytic H2 evolution system with an O2-tolerant Db [NiFeSe] hydrogenase and EY was assembled, which contains solely earth-abundant materials and maintains photoactivity under remarkably high levels of headspace O2. H2 evolution is driven efficiently by photoinduced direct electron transfer from EY to hydrogenase, making a soluble redox mediator unnecessary and thereby allowing for remarkable photostability under high O2 levels. This work demonstrates an unprecedented robustness of a hydrogenase towards O2 and paves the way to the exploitation of hydrogenases in full water splitting.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Artero V, Chavarot-Kerlidou M, Fontecave M. Angew. Chem. 2011;123:7376–7405. doi: 10.1002/anie.201007987. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2011;50:7238–7266. doi: 10.1002/anie.201007987. [DOI] [PubMed] [Google Scholar]
- 1b.Reece SY, Hamel JA, Sung K, Jarvi TD, Esswein AJ, Pijpers JJH, Nocera DG. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 1c.Barber J, Tran PD. J. R. Soc. Interface. 2013;10:20120984. doi: 10.1098/rsif.2012.0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d.Barroso M, Mesa CA, Pendlebury SR, Cowan AJ, Hisatomi T, Sivula K, Grätzel M, Klug DR, Durrant JR. Proc. Natl. Acad. Sci. USA. 2012;109:15640–15645. doi: 10.1073/pnas.1118326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Duan L, Bozoglian F, Mandal S, Stewart B, Privalov T, Llobet A, Sun L. Nat. Chem. 2012;4:418–423. doi: 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- 2b.Helm ML, Stewart MP, Bullock RM, DuBois MR, DuBois DL. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 2c.Han Z, Qiu F, Eisenberg R, Holland PL, Krauss TD. Science. 2012;338:1321–1324. doi: 10.1126/science.1227775. [DOI] [PubMed] [Google Scholar]
- 2d.Brillet J, Yum J-H, Cornuz M, Hisatomi T, Solarska R, Augustynski J, Graetzel M, Sivula K. Nat. Photonics. 2012;6:824–828. [Google Scholar]
- 2e.Cobo S, Heidkamp J, Jacques P-A, Fize J, Fourmond V, Guetaz L, Jousselme B, Ivanova V, Dau H, Palacin S, Fontecave M, Artero V. Nat. Mater. 2012;11:802–807. doi: 10.1038/nmat3385. [DOI] [PubMed] [Google Scholar]
- 2f.Vrubel H, Hu X. Angew. Chem. 2012;124:12875–12878. [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:12703–12706. doi: 10.1002/anie.201207111. [DOI] [PubMed] [Google Scholar]
- 2g.Smith RDL, Prévot MS, Fagan RD, Zhang Z, Sedach PA, Siu MKJ, Trudel S, Berlinguette CP. Science. 2013;340:60–63. doi: 10.1126/science.1233638. [DOI] [PubMed] [Google Scholar]
- 3a.Armstrong FA, Hirst J. Proc. Natl. Acad. Sci. USA. 2011;108:14049–14054. doi: 10.1073/pnas.1103697108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.Rapatskiy L, Cox N, Savitsky A, Ames WM, Sander J, Nowaczyk MM, Rögner M, Boussac A, Neese F, Messinger J, Lubitz W. J. Am. Chem. Soc. 2012;134:16619–16634. doi: 10.1021/ja3053267. [DOI] [PubMed] [Google Scholar]
- 3c.Kato M, Cardona T, Rutherford AW, Reisner E. J. Am. Chem. Soc. 2012;134:8332–8335. doi: 10.1021/ja301488d. [DOI] [PubMed] [Google Scholar]
- 4a.Abou Hamdan A, Dementin S, Liebgott P-P, Gutierrez-Sanz O, Richaud P, De Lacey AL, Rousset M, Bertrand P, Cournac L, Léger C. J. Am. Chem. Soc. 2012;134:8368–8371. doi: 10.1021/ja301802r. [DOI] [PubMed] [Google Scholar]
- 4b.McIntosh CL, Germer F, Schulz R, Appel J, Jones AK. J. Am. Chem. Soc. 2011;133:11308–11319. doi: 10.1021/ja203376y. [DOI] [PubMed] [Google Scholar]
- 4c.Ciaccafava A, Infossi P, Ilbert M, Guiral M, Lecomte S, Giudici-Orticoni MT, Lojou E. Angew. Chem. 2012;124:977–980. doi: 10.1002/anie.201107053. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:953–956. doi: 10.1002/anie.201107053. [DOI] [PubMed] [Google Scholar]
- 5a.Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. J. Am. Chem. Soc. 2009;131:18457–18466. doi: 10.1021/ja907923r. [DOI] [PubMed] [Google Scholar]
- 5b.Hambourger M, Gervaldo M, Svedruzic D, King PW, Gust D, Ghirardi M, Moore AL, Moore TA. J. Am. Chem. Soc. 2008;130:2015–2022. doi: 10.1021/ja077691k. [DOI] [PubMed] [Google Scholar]
- 5c.Greene BL, Joseph CA, Maroney MJ, Dyer RB. J. Am. Chem. Soc. 2012;134:11108–11111. doi: 10.1021/ja3042367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Okura I. Coord. Chem. Rev. 1985;68:53–99. [Google Scholar]
- 6b.Zadvornyy OA, Lucon JE, Gerlach R, Zorin NA, Douglas T, Elgren TE, Peters JW. J. Inorg. Biochem. 2012;106:151–155. doi: 10.1016/j.jinorgbio.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 6c.Reisner E. Eur. J. Inorg. Chem. 2011:1005–1016. [Google Scholar]
- 7.Lakadamyali F, Kato M, Muresan NM, Reisner E. Angew. Chem. 2012;124:9515–9518. doi: 10.1002/anie.201204180. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:9381–9384. doi: 10.1002/anie.201204180. [DOI] [PubMed] [Google Scholar]
- 8a.Goris T, Wait AF, Saggu M, Fritsch J, Heidary N, Stein M, Zebger I, Lendzian F, Armstrong FA, Friedrich B, Lenz O. Nat. Chem. Biol. 2011;7:310–318. doi: 10.1038/nchembio.555. [DOI] [PubMed] [Google Scholar]
- 8b.Fritsch J, Scheerer P, Frielingsdorf S, Kroschinsky S, Friedrich B, Lenz O, Spahn CMT. Nature. 2011;479:249–252. doi: 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- 8c.Abou Hamdan A, Burlat B, Gutiérrez-Sanz O, Liebgott P-P, Baffert C, De Lacey AL, Rousset M, Guigliarelli B, Léger C, Dementin S. Nat. Chem. Biol. 2013;9:15–17. doi: 10.1038/nchembio.1110. [DOI] [PubMed] [Google Scholar]
- 8d.Mouesca J-M, Fontecilla-Camps JC, Amara P. Angew. Chem. 2013;125:2056–2060. doi: 10.1002/anie.201209063. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2013;52:2002–2006. [Google Scholar]
- 8e.Evans RM, Parkin A, Roessler MM, Murphy BJ, Adamson H, Lukey MJ, Sargent F, Volbeda A, Fontecilla-Camps JC, Armstrong FA. J. Am. Chem. Soc. 2013;135:2694–2707. doi: 10.1021/ja311055d. [DOI] [PubMed] [Google Scholar]
- 8f.Fritsch J, Lenz O, Friedrich B. Nat. Rev. Microbiol. 2013;11:106–114. doi: 10.1038/nrmicro2940. [DOI] [PubMed] [Google Scholar]
- 8g.Armstrong FA, Belsey NA, Cracknell JA, Goldet G, Parkin A, Reisner E, Vincent KA, Wait AF. Chem. Soc. Rev. 2009;38:36–51. doi: 10.1039/b801144n. [DOI] [PubMed] [Google Scholar]
- 9a.Garcin E, Vernede X, Hatchikian EC, Volbeda A, Frey M, Fontecilla-Camps JC. Structure. 1999;7:557–566. doi: 10.1016/s0969-2126(99)80072-0. [DOI] [PubMed] [Google Scholar]
- 9b.Volbeda A, Amara P, Iannello M, De Lacey AL, Cavazza C, Fontecilla-Camps JC. Chem. Commun. 2013;49:7061–7063. doi: 10.1039/c3cc43619e. [DOI] [PubMed] [Google Scholar]
- 9c.Baltazar CSA, Marques MC, Soares CM, DeLacey AM, Pereira IAC, Matias PM. Eur. J. Inorg. Chem. 2011:948–962. [Google Scholar]
- 9d.Marques MC, Coelho R, De Lacey AL, Pereira IAC, Matias PM. J. Mol. Biol. 2010;396:893–907. doi: 10.1016/j.jmb.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Parkin A, Goldet G, Cavazza C, Fontecilla-Camps JC, Armstrong FA. J. Am. Chem. Soc. 2008;130:13410–13416. doi: 10.1021/ja803657d. [DOI] [PubMed] [Google Scholar]
- 11.Li Y-F, Selloni A. J. Am. Chem. Soc. 2013;135:9195–9199. doi: 10.1021/ja404044t. [DOI] [PubMed] [Google Scholar]
- 12a.Lazarides T, McCormick T, Du P, Luo G, Lindley B, Eisenberg R. J. Am. Chem. Soc. 2009;131:9192–9194. doi: 10.1021/ja903044n. [DOI] [PubMed] [Google Scholar]
- 12b.Li X, Wang M, Chen L, Wang X, Dong J, Sun L. ChemSusChem. 2012;5:913–919. doi: 10.1002/cssc.201100490. [DOI] [PubMed] [Google Scholar]
- 12c.Hashimoto K, Kawai T, Sakata T. Nouv. J. Chim. 1984;8:693–700. [Google Scholar]
- 12d.Shimidzu T, Iyoda T, Koide Y. J. Am. Chem. Soc. 1985;107:35–41. [Google Scholar]
- 13a.Gerola AP, Semensato J, Pellosi DS, Batistela VR, Rabello BR, Hioka N, Caetano W. J. Photochem. Photobiol. A. 2012;232:14–21. [Google Scholar]
- 13b.Knox JP, Dodge AD. Planta. 1985;164:22–29. doi: 10.1007/BF00391021. [DOI] [PubMed] [Google Scholar]
- 13c.Amat-Guerri F, López-González MMC, Martínez-Utrilla R, Sastre R. J. Photochem. Photobiol. A. 1990;53:199–210. [Google Scholar]
- 14a.Kong C, Qin L, Liu J, Zhong X, Zhu L, Long Y-T. Anal. Methods. 2010;2:1056–1062. [Google Scholar]
- 14b.Morris KJ, Roach MS, Xu W, Demas JN, DeGraff BA. Anal. Chem. 2007;79:9310–9314. doi: 10.1021/ac0712796. [DOI] [PubMed] [Google Scholar]
- 14c.Leventis N, Rawashdeh A-MM, Elder IA, Yang JH, Dass A, Sotiriou-Leventis C. Chem. Mater. 2004;16:1493–1506. [Google Scholar]
- 14d.Lin Q, Li Q, Batchelor-McAuley C, Compton RG. Phys. Chem. Chem. Phys. 2013;15:7760–7767. doi: 10.1039/c3cp50873k. [DOI] [PubMed] [Google Scholar]
- 14e.Reynolds EW, Demas JN, DeGraff BA. J. Fluoresc. 2013;23:237–241. doi: 10.1007/s10895-012-1139-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information