

Aryl, heteroaryl, and alkenyl sulfones play a prominent role in organic and medicinal chemistry. Not only are they versatile intermediates in organic synthesis[1], but they are also of particular pharmaceutical relevance, and exhibit an extensive and broad range of biological activities (Scheme 1).[2] In addition, sulfone-containing polymers display novel properties as materials.[3] Although there are a variety of methods known for the preparation of sulfones, the majority of these processes feature associated limitations. For example, oxidation of the corresponding sulfide relies on functional-group compatibility with oxidizing agents and the availability of catalysts to promote selectivity for sulfone formation over sulfoxide formation; in addition, the preparation of the required sulfide substrate often involves the use of foul-smelling thiols.[4,5] Friedel–Crafts-type sulfonylation of arenes is limited not only by the generally harsh reaction conditions, but also by the inherent regioselective bias imposed by the electronic and steric properties of the arene substrate.[5,6]
Scheme 1.

Pharmaceuticals featuring an aryl sulfone unit.
In response to these limitations, the use of transition-metal-catalyzed cross-coupling has been exploited as a route to sulfones.[7] The most utilized has been copper catalysis, in which sodium sulfinates couple with aryl, heteroaryl, or alkenyl halides,[8] or boronic acids.[9] However, the requirement for stoichiometric phase-transfer additives, high temperatures, and the use of undesirable solvents or ionic liquids, are significant drawbacks. An analogous palladium-catalyzed approach has also been developed using (pseudo)halides as coupling partners, although it too features the use of stoichiometric additives and high temperatures, and consequently suffers from limited scope.[10,11] In addition to these problems, the variety of sulfones prepared by these metal-catalyzed methods is poor; only a small number of sodium sulfinates (typically methyl, phenyl, and p-tolyl) have been used to demonstrate the scope. This reflects the very limited commercial availability of sodium sulfinates, perhaps as a result of impractical or limited synthetic routes. Organometallic addition to sulfur dioxide gas[12] (followed by cation exchange) and reduction of the sulfonyl chloride[12,13] are two such examples, but both have shortcomings such as the harsh conditions associated with sulfonyl chloride formation or the use of toxic gaseous sulfur dioxide. Taken together, these limitations present a clear mandate for the development of a more general, modular synthesis of sulfones, with the versatility to allow ready variation of both groups attached to the SO2 linker.[14]
We have recently developed DABSO (1), which is a solid, bench-stable complex formed between 1,4-diazabicyclo[2.2.2]octane (DABCO) and two sulfur dioxide molecules,[15] as an easy-to-handle surrogate for sulfur dioxide gas (Scheme 2). We have demonstrated its use in known reactions of sulfur dioxide,[16] as well as in a novel palladium-catalyzed aminosulfonylation of aryl, heteroaryl, and alkenyl halides to yield medicinally important sulfonamides.[17] Herein, we report on a three-component convergent synthesis of a broad range of aryl, heteroaryl, and alkenyl sulfones, employing palladium catalysis and DABSO (Scheme 2).
Scheme 2.

A convergent three-component sulfone synthesis exploiting DABSO, an easy-to-handle solid replacement for sulfur dioxide gas.
Given our previous demonstration of the efficient addition of Grignard reagents to DABSO to form magnesium halide sulfinate salts,[16] we envisioned that these would couple with (pseudo)halides to form sulfones in a similar fashion to the reported sodium salt examples using transition metal catalysis. Unfortunately, after testing a variety of catalytic conditions, we were not able to achieve this. Suspecting that this was due to the difference in the nature of the counter ions, we elected to study the addition of organolithiums to form the more closely related lithium sulfinates. We chose to explore palladium rather than copper catalysis because the reported conditions for sodium salts were the most amenable to our organolithium proposal.[10,11] We were mindful, however, of the precedent for certain metal sulfinates to undergo palladium-catalyzed desulfonylative cross-couplings, whereby extrusion of SO2 from the palladium-bound sulfinate ultimately leads to a biaryl product.[18]
We began our investigations with the addition of PhLi solution to a suspension of DABSO in THF at −40 °C. Pleasingly, HPLC analysis revealed that quantitative conversion to the lithium sulfinate 2 was achieved; only traces of benzene from deleterious protonation were detected. This crude sulfinate suspension was then combined with [Pd2(dba)3] (dba=dibenzylideneacetone), XantPhos, Cs2CO3, nBu4NCl, and p-tolyl iodide; the conditions reported for sodium sulfinate coupling.[10] Analysis of the crude 1H NMR spectrum indicated the presence of the desired sulfone 3 a, albeit at a low conversion (see the Supporting Information). However, by switching to Pd(OAc)2, conversion increased to 65 % and it was also found that the nBu4NCl additive was effectively redundant. Postulating that the incomplete conversion was a result of the lower temperature used relative to that for the sodium salt, the solvent was switched from THF to 1,4-dioxane, and the reaction heated at 85 °C. Gratifyingly, a quantitative conversion was achieved (Scheme 3).
Scheme 3.
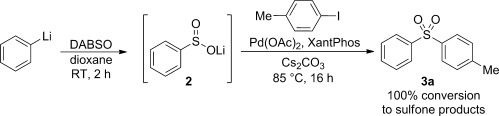
Initial investigation of the generation and Pd-catalyzed coupling of lithium sulfinate 2.
Unfortunately, upon purification, two different sulfone products were isolated. Desired sulfone 3 a was present in 70 % yield together with 23 % of diphenylsulfone. From the observation that a phenyl group was always incorporated in place of the aryl halide fragment upon varying the organolithium component, we deduced that this side product was a result of aryl–phenyl exchange with the XantPhos ligand. Such exchange processes are known in arylation of electron-poor nucleophiles and in Suzuki couplings.[19–22] Conditions for the palladium coupling step were fully explored, with the choice of base, additive, solvent, palladium source, and temperature being investigated. However, no improvement in selectivity whilst maintaining good conversion of starting material was observed.[22] Finally, variation of the phosphine ligand was undertaken. We found that, in agreement with the work of Cacchi et al., the XantPhos backbone was key to reactivity;[10] many other ligands, including those with similar structures, such as DPEPhos, as well as alkyl phosphines (which cannot undergo this exchange process), demonstrated little or no activity.[22] With the XantPhos architecture established as optimal, we next explored electronic and steric variations of this unique backbone.[23] These ligands were evaluated under the cross-coupling conditions previously established (Table 1). Alkyl (entries 3–5) and sterically hindering aryl substituents (entries 3 and 7) on the XantPhos backbone led only to recovered starting material. As expected, a more electron-rich phosphine gave more aryl–phenyl transfer product (entry 6).[21] Electron-poor phosphines, which have been reported to suppress transfer for the arylation of ureas,[20] gave similar or a small increase in selectivity as hoped, but for the more selective reaction, the conversion was poor (entries 8-10). However, by switching to the aryl bromide substrate in place of the iodide substrate, and increasing the temperature of the reaction, full conversion was achieved with high selectivity when using the 3,5-bis(CF3)-substituted XantPhos 5 f (entry 13). Using these optimized conditions, the scope of the aryl bromide coupling partner was next investigated (Scheme 4).
Table 1.
Evaluation of XantPhos-type ligands to improve selectivity through the suppression of aryl–aryl exchange[a]
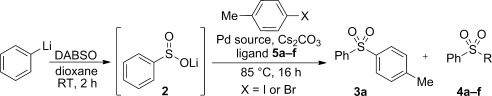
| Entry | Ligand | Pd source | I/Br | Conversion [%][d] | 3 a:4 a–f[d] |
|---|---|---|---|---|---|
| 1 | XantPhos | Pd(OAc)2 | I | 100 | 3:1 |
| 2[b] | XantPhos | [Pd2(dba)3] | I | 100 | 2:1 |
| 3 | 5 a | Pd(OAc)2 | I | – | – |
| 4 | 5 b | Pd(OAc)2 | I | – | – |
| 5[b] | 5 b | [Pd2(dba)3] | I | – | – |
| 6 | 5 c | Pd(OAc)2 | I | 40 | 1:1 |
| 7 | 5 d | Pd(OAc)2 | I | trace | – |
| 8 | 5 e | Pd(OAc)2 | I | 95 | 3:1 |
| 9 | 5 f | Pd(OAc)2 | I | – | – |
| 10[b] | 5 f | [Pd2(dba)3] | I | 50 | 5:1 |
| 11[c] | XantPhos | Pd(OAc)2 | Br | 100 | 7:2 |
| 12[b,c] | 5 f | [Pd2(dba)3] | Br | 100 | 4:1 |
| 13[c] | 5 f | Pd(OAc)2 | Br | 100 | 15:1 |
Reaction conditions: PhLi (1.4 equiv), DABSO (0.75 equiv), 1,4-dioxane (0.18 m) then p-tolyl halide (1 equiv, 0.35 mmol), palladium source (10 mol % Pd), ligand (10 mol %), Cs2CO3 (1.5 equiv).
With nBu4NCl additive (1.5 equiv).
Performed at 110 °C. [d] Determined by 1H NMR spectrum integration to the nearest 5 %. 
Scheme 4.
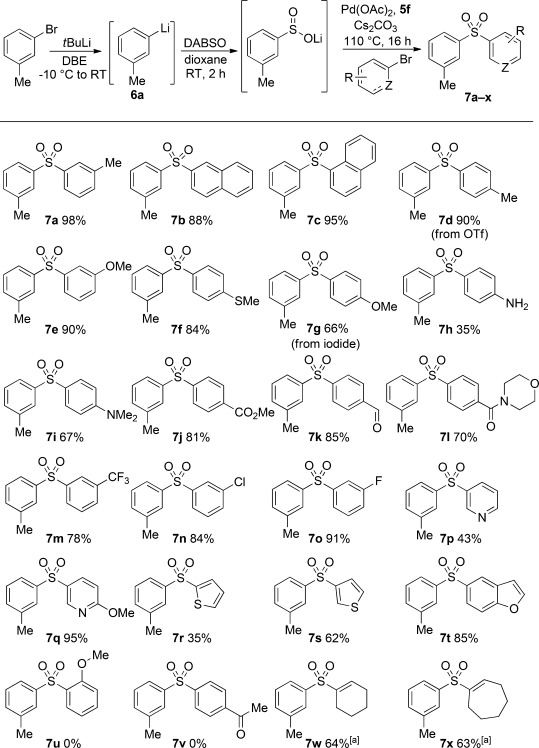
Scope of the aryl halide component in the Pd-catalyzed preparation of sulfones through coupling with lithium sulfinates. Coupling conditions as described in Table 1, entry 13. [a] When using the alkenyl tosylate, K3PO4 (1.5 equiv) employed in the place of Cs2CO3.
m-Tolyllithium 6 a was chosen to demonstrate the synthesis of sulfones that have not been prepared through sodium sulfinate coupling. A stock solution was readily synthesized by using lithium–halogen exchange between butyllithium and 3-bromotoluene in dibutylether, which is a Lewis-basic solvent in which lithium–halogen exchange is kinetically facile and yet aryl lithiums are more stable to storage than in Et2O or THF.[24] A wide range of aryl and heteroaryl bromides coupled in good to excellent yields (Scheme 2). Electron-rich substrates performed well, with C-, O-, S-, and N-based functionality all tolerated. Electron-poor aryl bromides also proved to be compatible, with ester, amide, aldehyde, chloro, and trifluoromethyl functional groups retained. The use of unprotected anilines, aryl fluoride, and napthyl derivatives demonstrated further scope. We established additional versatility by using heteroaromatic pyridyl, thiophenyl, and benzofuryl bromides, as well as using iodide or triflate substrates in place of bromide. Unfortunately, ketones and arenes bearing ortho substituents led to no reaction. Provided that K3PO4 was used in place of Cs2CO3, alkenyl tosylates could be successfully employed as the electrophilic coupling partner and allowed access to the corresponding aryl–alkenyl sulfone products.[11]
Next, we investigated the scope of the organolithium species (Scheme 5). Using 3-bromoanisole as the standard coupling partner, a range of aryl lithiums were successfully subjected to the optimized conditions. Pleasingly, all the organolithiums prepared added, as expected, to DABSO to form the corresponding lithium sulfinate, and the subsequent palladium-catalyzed coupling worked well for a broad range of aryl sulfinates, including those with electron-donating, neutral, and electron-withdrawing functionality; trifluoromethyl, chloro, and silane functional groups were all successfully tolerated. Unfortunately, heterocyclic lithium 2-thienylsulfinate led to biaryl formation from desulfonylative coupling through SO2 extrusion,[18] while ortho-substituted lithium 2-anisyl sulfinate resulted in the recovery of aryl bromide starting material.
Scheme 5.

Scope of the aryl lithium component in the Pd-catalyzed preparation of sulfones through the coupling of lithium sulfinates with aryl halides. Coupling conditions as described in Table 1, entry 13. [a] Organolithium used as commercial solution. [b] From the corresponding alkenyl tosylate. [c] Biaryl formation observed.
A limitation of our presented methodology was the inability to include functionality positioned ortho to the sulfone moiety. To address this issue, we exploited the capacity of the sulfone functional group to direct ortho metalation.[25] We demonstrated this through two regioselective ortho functionalizations of sulfone 8 d by using the co-operative effect of meta-disposed directing groups to give 1,2,3-trisubsituted aryl sulfones 9 and 10 in good yields (not optimized, Scheme 6).
Scheme 6.

Exploiting sulfonyl groups to direct ortho functionalization.
As a proof of the versatility and applicability of the presented methodology, we sought to demonstrate the synthesis of a significant target molecule. GSK-742457 (11) is a quinoline-based drug molecule that is currently under development for treating Alzheimer’s disease.[26] We envisaged disconnection to the known bis-halogenated quinoline derivative 12.[27] In the forward direction, palladium-catalyzed sulfonylation of 12 would generate chloro derivative 13, and a subsequent palladium-catalyzed Buchwald–Hartwig amination would produce the target medicinal agent (Scheme 7). The proposed sequence was successfully achieved, with the sulfonylation being realized in 70 % yield, and the amination in 68 % yield (both not optimized). This is an attractive route to the target system via the bis-halogenated quinoline 12, which provides a suitable branch point for the rapid preparation of analogues.
Scheme 7.

Synthesis of GSK-742457 using the combination of a Pd-catalyzed aryl halide–lithium sulfinate coupling, and a Pd-catalyzed aryl halide amination. Conditions: a) Sulfonylation with aryl lithium (see Table 1, entry 13). b) Pd(OAc)2, RuPhos, NaOtBu, piperazine, 1,4-dioxane, 90 °C, 16 h. RuPhos=2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl.
In summary, we have demonstrated the synthesis of a broad range of aryl, heteroaryl, and alkenyl sulfones through a convergent three-component palladium-catalyzed coupling approach. This method allows the straightforward production of varied sulfones through the union of two readily available coupling partners; an aryl lithium species[28] and aryl, heteroaryl, or alkenyl (pseudo)halides; with SO2 provided by the easy-to-handle bench-stable solid surrogate DABSO. The use of an electron-poor XantPhos-type ligand offers much improved yields over XantPhos itself. In addition, ortho functionality can be introduced by exploiting the ability of the sulfone group to direct ortho metalation. A demonstration of the utility of the methodology was achieved through the synthesis of a medicinal agent that is currently in development, utilizing a versatile bis-halogenated quinoline as the starting material. Finally, evidence for the nontrivial nature of the preparation of aryl–aryl (and related) sulfones is found in the observation that of the 36 sulfone products reported herein, more than 90 % are novel compounds.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Simpkins NS. Sulfones in Organic Synthesis. Oxford: Pergamon Press; 1993. [Google Scholar]
- 2a.Prasit P, Wang Z, Brideau C, Chan CC, Charleson S, Cromlish W, Ethier D, Evans JF, Ford-Hutchinson AW, Gauthier JY, et al. Bioorg. Med. Chem. Lett. 1999;9:1773. doi: 10.1016/s0960-894x(99)00288-7. [DOI] [PubMed] [Google Scholar]
- 2b.Sturino CF, O’Neill G, Lachance N, Boyd M, Berthelette C, Labelle M, Li L, Roy B, Scheigetz J, Tsou N, et al. J. Med. Chem. 2007;50:794. doi: 10.1021/jm0603668. [DOI] [PubMed] [Google Scholar]
- 2c.Wolf WM. J. Mol. Struct. 1999;474:113. [Google Scholar]
- Murár M, Addová G, Boháč A. Beilstein J. Org. Chem. 2013;9:173. doi: 10.3762/bjoc.9.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d.Neamati N, Mazumder A, Zhao H, Sunder S, Burke TR, Schultz RJ, Pommier Y. Antimicrob. Agents Chemother. 1997;41:385. doi: 10.1128/aac.41.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e.Ettari R, Nizi E, Di Francesco ME, Dude M-A, Pradel G, Vicik R, Schirmeister T, Micale N, Grasso S, Zappala M. J. Med. Chem. 2008;51:988. doi: 10.1021/jm701141u. [DOI] [PubMed] [Google Scholar]
- 2f.Brot ECA, Keefe DKJ, Haney BP, Metcalfe N, Palmer GJ, Isbester PK. 2008. (CeNeRx BioPharma Inc.), US2008009542 (A1)
- 2g.Zhu YI, Stiller MJ. J. Am. Acad. Dermatol. 2001;45:420. doi: 10.1067/mjd.2001.114733. [DOI] [PubMed] [Google Scholar]
- 2h.Schellhammer PF, Sharifi R, Block NL, Soloway MS, Venner PM, Patterson AL, Sarosdy MF, Vogelzang NJ, Schellenger JJ, Kolvenbag GJ. Urology. 1997;50:330. doi: 10.1016/s0090-4295(97)00279-3. [DOI] [PubMed] [Google Scholar]
- 3.El-Hibri MJ, Weinberg SA. Encyclopedia of Polymer Science and Technology. New York: Wiley; 2002. “Polysulfones”: [Google Scholar]
- 4a.Jereb M. Green Chem. 2012;14:3047. [Google Scholar]
- Bahrami K, Khodaei MM, Sheikh Arabi M. J. Org. Chem. 2010;75:6208. doi: 10.1021/jo1011784. [DOI] [PubMed] [Google Scholar]
- 4b.Schank K. The Chemistry of Sulfones and Sulfoxides. New York: Wiley; 1988. Chapter 7. [Google Scholar]
- 5.Oae S. Organic Sulfur Chemistry. Boca Raton, FL: CRC; 1992. [Google Scholar]
- 6a.Ueda M, Uchiyama K, Kano T. Synthesis. 1984:323. [Google Scholar]
- 6b.Singh DU, Singh PR, Samant SD. Tetrahedron Lett. 2004;45:9079. [Google Scholar]
- 6c.Graybill BM. J. Org. Chem. 1967;32:2931. [Google Scholar]
- 6d.Nara SJ, Harjani JR, Salunkhe MM. J. Org. Chem. 2001;66:8616. doi: 10.1021/jo016126b. [DOI] [PubMed] [Google Scholar]
- 6e.Frost CG, Hartley JP, Whittle AJ. Synlett. 2001:830. [Google Scholar]
- Bandgar BP, Kasture SP. Synth. Commun. 2001;31:1065. [Google Scholar]
- 6f.Marquié J, Laporterie A, Dubac J, Roques N, Desmurs J-R. J. Org. Chem. 2001;66:421. doi: 10.1021/jo0010173. [DOI] [PubMed] [Google Scholar]
- 7.Bandgar BP, Bettigeri SV, Phopase J. Org. Lett. 2004;6:2105. doi: 10.1021/ol049692c. [DOI] [PubMed] [Google Scholar]
- 8a.Baskin JM, Wang ZY. Org. Lett. 2002;4:4423. doi: 10.1021/ol0269190. [DOI] [PubMed] [Google Scholar]
- 8b.Zhu W, Ma DW. J. Org. Chem. 2005;70:2696. doi: 10.1021/jo047758b. [DOI] [PubMed] [Google Scholar]
- 8c.Bian M, Xu F, Ma C. Synthesis. 2007:2951. [Google Scholar]
- 9a.Kantam ML, Neelima B, Sreedhar B, Chakravarti R. Synlett. 2008:1455. [Google Scholar]
- 9b.Huang F, Batey RA. Tetrahedron. 2007;63:7667. [Google Scholar]
- 9c.Kar A, Sayyed IA, Lo WF, Kaiser HM, Beller M, Tse MK. Org. Lett. 2007;9:3405. doi: 10.1021/ol071396n. [DOI] [PubMed] [Google Scholar]
- 9d.Yang H, Li Y, Jiang M, Wang J, Fu H. Chem. Eur. J. 2011;17:5652. doi: 10.1002/chem.201003711. [DOI] [PubMed] [Google Scholar]
- 10.Cacchi S, Fabrizi G, Goggiamani A, Parisi LM, Bernini R. J. Org. Chem. 2004;69:5608. doi: 10.1021/jo0493469. [DOI] [PubMed] [Google Scholar]
- 11.Reeves DC, Rodriguez S, Lee H, Haddad N, Krishnamurthy D, Senanayake CH. Tetrahedron Lett. 2009;50:2870. [Google Scholar]
- 12.Schubart R. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH; 2000. “Sulfinic Acids and Derivatives”: [Google Scholar]
- 13a.Kao Liu L, Chi Y, Jen K-Y. J. Org. Chem. 1980;45:406. [Google Scholar]
- 13b.Umierski N, Manolikakes G. Org. Lett. 2013;15:188. doi: 10.1021/ol303248h. [DOI] [PubMed] [Google Scholar]
- 14. During the preparation of our manuscript, Manolikakes and Umierski published on the metal-free coupling of a broader range sodium sulfinates with diaryliodonium salts (Ref. [13b]). Whilst the progress towards metal-free couplings is attractive, the use of poorly atom-economical aryl iodonium salts and their associated problems with versatility towards using nontrivial aryl groups represents a significant constraint.
- 15.Santos PS, Mello MTS. J. Mol. Struct. 1988;178:121. [Google Scholar]
- 16.Woolven H, Gonzalez-Rodriguez C, Marco I, Thompson AL, Willis MC. Org. Lett. 2011;13:4876. doi: 10.1021/ol201957n. [DOI] [PubMed] [Google Scholar]
- 17a.Emmett EJ, Richards-Taylor CS, Nguyen B, Garcia-Rubia A, Hayter BR, Willis MC. Org. Biomol. Chem. 2012;10:4007. doi: 10.1039/c2ob07034k. [DOI] [PubMed] [Google Scholar]
- 17b.Nguyen B, Emmett EJ, Willis MC. J. Am. Chem. Soc. 2010;132:16372. doi: 10.1021/ja1081124. [DOI] [PubMed] [Google Scholar]
- 18a.Sévigny S, Forgione P. Chem. Eur. J. 2013;19:2256. doi: 10.1002/chem.201204201. [DOI] [PubMed] [Google Scholar]
- Ortgies DH, Barthelme A, Aly S, Desharnais B, Rioux S, Forgione P. Synthesis. 2013:694. [Google Scholar]
- 18b.Zhou C, Liu Q, Li Y, Zhang R, Fu X, Duan C. J. Org. Chem. 2012;77:10468. doi: 10.1021/jo302005s. [DOI] [PubMed] [Google Scholar]
- 19a.Kong KC, Cheng CH. J. Am. Chem. Soc. 1991;113:6313. [Google Scholar]
- 19b.Goodson FE, Wallow TI, Novak BM. Org. Synth. 1998;75:61. [Google Scholar]
- 20.Sergeev AG, Artamkina GA, Beletskaya IP. Tetrahedron Lett. 2003;44:4719. [Google Scholar]
- 21.Hamann BC, Hartwig JF. J. Am. Chem. Soc. 1998;120:3694. [Google Scholar]
- 22. See the Supporting Information for proposed mechanism, and associated conditions screened.
- 23. These were all prepared through double ortho lithiation of 9,9-dimethylxanthene, followed by trapping with the appropriate chlorophosphine, according to reported procedures (see the Supporting Information)
- 24.Schwindeman JA, Sutton DE, Morrison RC, Stryker SS. 1996. (FMC Corporation), US 587813.
- 25a.Iwao M, Iihama T, Mahalanabis KK, Perrier H, Snieckus V. J. Org. Chem. 1989;54:24. [Google Scholar]
- 25b.Clayden J, Senior J. Synlett. 2009:2769. [Google Scholar]
- 26.Gladwin AE. 2005. (Glaxo Group Ltd.), WO2005040124 (A1)
- 27.This bis-halogenated quinoline was synthesized from commercially available 8-chloroquinoline through an NIS iodination (see Ref. [26]
- 28.Giannerini M, Fañanás-Mastral M, Feringa BL. Nat. Chem. 2013;5:667. doi: 10.1038/nchem.1678. For the recent use of aryl lithium species in efficient palladium-catalyzed cross-couplings with aryl halides, see. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information