

Abstract
Many preclinical studies in critical care medicine and related disciplines rely on hypothesis-driven research in mice. The underlying premise posits that mice sufficiently emulate numerous pathophysiological alterations produced by trauma/sepsis and can serve as an experimental platform for answering clinically relevant questions. Recently the lay press severely criticized the translational relevance of mouse models in critical care medicine. A series of provocative editorials were elicited by a highly-publicized research report in the Proceedings of the National Academy of Sciences (PNAS; February 2013), which identified an unrecognized gene expression profile mismatch between human and murine leukocytes following burn/trauma/endotoxemia. Based on their data, the authors concluded that mouse models of trauma/inflammation are unsuitable for studying corresponding human conditions. We believe this conclusion was not justified. In conjunction with resulting negative commentary in the popular press, it can seriously jeopardize future basic research in critical care medicine. We will address some limitations of that PNAS report to provide a framework for discussing its conclusions and attempt to present a balanced summary of strengths/weaknesses of use of mouse models. While many investigators agree that animal research is a central component for improved patient outcomes, it is important to acknowledge known limitations in clinical translation from mouse to man. The scientific community is responsible to discuss valid limitations without over-interpretation. Hopefully a balanced view of the strengths/weaknesses of using animals for trauma/endotoxemia/critical care research will not result in hasty discount of the clear need for using animals to advance treatment of critically ill patients.
Keywords: mouse models of critical illness, trauma, endotoxemia, sepsis, burn
Introduction
In the USA (1) and European Union countries (2), approximately 15 and 7 million laboratory rodents, respectively, are used annually for research and testing. While it is difficult to precisely define the exact number of mice used in the field of critical care illnesses (such as trauma, burn, infections, endotoxemia, and sepsis), it is not an overstatement that the majority of preclinical studies rely on this species. As of the end of October 2013, a search of PubMed produced between approximately 4200 and 24300 publications in response to a “mouse and burn/sepsis/shock/trauma” query (queries ordered from the lowest to highest number of hits). One of these studies, the collaborative report by Seok et al. published in the February 26, 2013 issue of the Proceedings of the National Academy of Sciences (PNAS), identified a potentially serious mismatch in the translational utility of the mouse-to-human data in the area of critical care medicine (3). The original data stem from the Inflammation and Host Response to Injury, Large Scale Collaborative Research Program Project (under the GLUE GRANT scientific consortium; www.gluegrant.org/index.htm). The aim of the GLUE GRANT project was to compare the genetic responses of humans and mice following burn, trauma and endotoxemia by analyzing approximately 5000 human genes and their mouse orthologs. Based on their interpretation of these results, the authors concluded that “genomic responses in mouse models poorly mimic human inflammatory diseases” and claimed the mouse gene profile response appeared random when compared to the human gene response to burn, trauma, or lipopolysaccharide (LPS) challenge. To those investigators familiar with how the immune system reacts to these innate stimuli, this broad generalization of their analysis appears, at least partly, unwarranted. Nevertheless, this labor-intensive study (3) is among a growing list of publications (4-6) challenging the merits of using mice and other animal models in basic and preclinical research and thus should be thoroughly discussed.
The Ripple Effect
Given the surprising and controversial nature of the data concerning mouse-to-human (in)compatibilities in tested inflammatory disease models, the findings of the PNAS paper were quickly publicized in the lay press. The initial account of the research in the New York Times entitled “Mice Fall Short as Test Subjects for Some of Humans’ Deadly Ills” (7) led to a subsequent ripple effect in the form of several alarming follow-up editorials, posts and/or blogs (8-11). Their collective conclusion was clear and implied that decades of mouse-based research culminated in few scientific advances, wasted precious research opportunities, and was a poor use of taxpayers’ money. Consequently, given that “mouse models of inflammation are basically worthless” (10), “it seems that researchers have tortured mice in vain for decades in the search for drugs to help humans recover from certain traumas, like severe burns, blunt force, and sepsis” (11). There is concern that the sensational tone of those communications will be damaging to preclinical mouse-based research programs. As a result, public perception, research progress and funding support for basic discovery and hypothesis-driven research for many medical disciplines may be impeded. As the authors of the original PNAS paper were mostly directing their criticisms towards inflammation, trauma, shock, and sepsis research, we felt compelled, following the recent comments by others (12-16), to collectively address their controversial conclusions. By discussing its main limitations, we aim to delineate the boundaries within which the work of Seok at al. should be viewed and evaluated. Importantly, we also provide objective information demonstrating that animal research using mice has led to ground-breaking studies that have improved patient care and outcomes.
Lost in Translation: What Does the PNAS Study Really Say?
Seok and colleagues report that the genomic response to trauma, burns, or endotoxin challenge shows an extremely low correlation between mice and humans, while these different types of injury responses showed high similarity among humans. The authors state in the first paragraph that “Among genes changed significantly in humans, the murine orthologs are close to random in matching their human counterparts (e.g., R2 between 0.0 and 0.1).” We contend that the authors have over-interpreted their data due to the many limitations of their study design and analysis, some of which they have failed to acknowledge. Furthermore, it remains uncertain whether and/or to what extent the results of gene expression profiling should be used to judge the biological validity of animal models for human disease. Although we disagree with the overall conclusion and interpretation of this PNAS report, the intention of this manuscript is not to lessen the value of the authors’ work but to analyze conclusions of the study in an appropriate evidence-based framework. The following is a partial list of limitations and issues that were identified subsequent to the publication of the PNAS paper (17-19) and that were featured in a debate session at the 2013 annual Shock Society meeting in San Diego, CA, USA (20).
1) Comparing strain, sex and age
Gene profiles of a highly heterogeneous (outbred) population of burn/trauma/endotoxemic male and female patients were compared to inbred, genetically identical C57BL/6J male mice at the approximate age of 2 months. Using inbred mice that are genetically identical for such a comparative analysis is equivalent to comparing a single individual burn or trauma patient to 167 trauma or 244 burn patients. Furthermore, comparing individual responses to these injuries in inbred versus outbred subjects represents an important study limitation because of the differential immune system response among inbred mice to various parasitic (21;22), viral (23;24) and bacterial (25;26) infections. The most recent review by Fink (27) offers a deeper insight into the limitations of the inbred strains in modeling of sepsis (also in the context of the PNAS article). Summarizing, testing a single strain of mice does not justify the assertion that all mouse models poorly mimic human inflammatory diseases given that this or any other strain fails to represent the genetic diversity of the entire mouse population. Furthermore, comparison of a single mouse sex to the mixed male/female population of human patients demands further caution regarding data interpretation given that sex modifies leukocyte responses after trauma-hemorrhage (28-30), bacterial infection (31) or LPS (32;33). Additionally, it cannot be assumed that 8-week-old mice (used in the PNAS study) constitute a reliable surrogate for older patients. In the C57BL/6J strain, the age of 8 weeks corresponds approximately to the prescent human age of 8 years (34). This contrasts sharply with the average age of 34 years in trauma patients (35), 30 years in healthy control subjects and 18-40 years in the eight volunteers injected with endotoxin (3;35). It remains to be established how strongly (if at all) this particular age disparity can influence the studied responses in mice and humans. However, the role of age should not be minimized as differential age-dependent outcomes and responses in the immuno-inflammatory compartment were demonstrated in pediatric versus adult patients with trauma (36; 37), burns (38;39), endotoxemia (40) and infections (41;42) as well as in corresponding mouse models (43-46). Of note, although not directly pertinent to data analyzed in the study by Seok and colleagues (3) (as leukocytes from septic patients were not analyzed), age is a major mismatch in the mouse-to-human comparison of data from sepsis syndromes; while a majority of human septic patients are old, preclinical sepsis models (including the most relevant) typically rely on young mice (47).
2) Gene expression analysis using unfractionated blood leukocytes
The composition of circulating leukocytes differs markedly between humans and mice (i.e., 60% versus 15% neutrophils and 30% versus 70% lymphocytes, respectively) (6) and a recent study by Shay et al. demonstrated that human versus mouse granulocytes and lymphocytes have distinct gene expression profiles (48). Because the comparison was made on only unfractionated whole blood leukocytes in the study, this translates into the profiling of a neutrophil-rich versus lymphocyte-rich sample, which undoubtedly skewed the final results. It must be noted that an oral communication by one of the PNAS co-authors indicated that reanalysis of genomic responses in a more narrow PMN population did not markedly improve the overall correlation (20), although a peer-reviewed publication of such a reanalysis has not yet been reported. The risks of imprecise comparison of genomic responses in circulating white blood cells (WBC) are not solely restricted to mouse versus human studies; earlier GLUE GRANT-based reports that analyzed various inflammation/injury scenarios using only mouse models, voiced identical concerns (49;50). Analyses of gene expression in discrete leukocyte subsets are certainly warranted as they provide more precise information regarding individual activation patterns in injury and/or infection.
3) Model and severity mismatch
The influence of this potential limitation was suggested by the authors in the original paper. For example, mice may have higher resistance to inflammation/trauma/infection because they are housed in a controlled environment and most often lack predisposing conditions present in the human population. This injury severity mismatch may translate into a dissimilar response both on the genomic and protein levels. Though animal models are designed to produce a pronounced, clinically relevant post-traumatic physiological effect, a near-death severity threshold is often needed since milder severity only recapitulates some features of trauma or sepsis response (51-53). It is also noteworthy that due to their innate high resistance to trauma and inflammation, mouse models cannot adequately recapitulate a full pallet of the most severe responses that occur in patients. This may be partly due to yet another mismatch: it is generally appreciated that any patient admitted to the ICU and survives would have likely died without intervention. Our inability to replicate this aggravating phenomenon may be another influential limitation in animal modeling.
To approximate sepsis, the authors used LPS delivery into human volunteers and mice for their gene expression profile comparison. Although this is a commonly used model in humans that supposedly mimics some clinical features of the sepsis syndrome, it is not a true model for sepsis. Sepsis and sepsis syndrome are complex responses and most of the existing experimental models fail to reproduce the entire spectrum of sepsis syndromes diagnosed in patients, especially the bolus injection of LPS. In other words, it is inappropriate to compare aged patients suffering from monobacterial pneumonia induced bacteremia with the cecal ligation and puncture (CLP) model of polymicrobial peritonitis. Other mismatched secondary comparisons were made based on the Gene Expression Omnibus database (54); data from each of the existing specific sepsis models should only be compared to the corresponding clinical condition (47). Another debatable point, recently raised in a letter by Cauwels et al. (12), is that the relative dose of LPS differed markedly between mice and human volunteers. It is also important to consider that besides genetic background, environment and underlying diseases can markedly affect the host response to infection. In such a scenario mice can be rendered either hyporesponsive or hyperresponsive to LPS with major differences in inflammatory responses that in turn alter susceptibility to infection. For example, mice with chronic kidney disease exhibit an increase in morbidity when subjected to sepsis (55). Increased inflammatory responses may be induced by previous Propionibacterium acnes infection, hepatotoxic agents (e.g., D-Galactosamine) and growing Lewis lung carcinoma, while exposure to minute amounts of LPS renders the animals tolerant to LPS (56). This modulation is likely to occur in humans as well, in whom underlying diseases such as diabetes mellitus and end stage renal dysfunction are known to impair host response to infection (47). Thus, diverse underlying conditions or exposure to pathogens may further underscore differences between experimental studies with healthy animals and studies in humans in clinical settings.
4) Analysis of responses in a single source
Many critically ill patients trigger activation of virtually all body systems so that the total amount of secreted cytokines comes from multiple cells and organs (Fig. 1). Due to technical and/or diagnostic ease, blood, with its cellular and soluble components, is the most frequently used source of information in critical conditions (57-60). Although the blood-based diagnostics have proven utility in many facets of ICU monitoring, blood leukocytes represent a relatively narrow source considering the entire pool of inflammatory mediators released to the systemic circulation after trauma and during severe infections (61;62). Given that virtually all inflammatory mediators detected in the blood are produced by various sources (Fig. 1), it cannot be ignored that the interspecies compatibility is better when other sites of cytokine synthesis are compared (e.g., hepatocytes). Yet, despite rapid technological advancements in the ICU, such interspecies comparisons are currently either beyond reach or very difficult to perform. While this caveat is not a shortcoming of the paper, it does highlight the fact that the systemic or organ-specific contribution to the inflammatory response needs to be taken into consideration when interpreting results.
Fig. 1. Sources of inflammatory mediators produced in response to a critical condition.
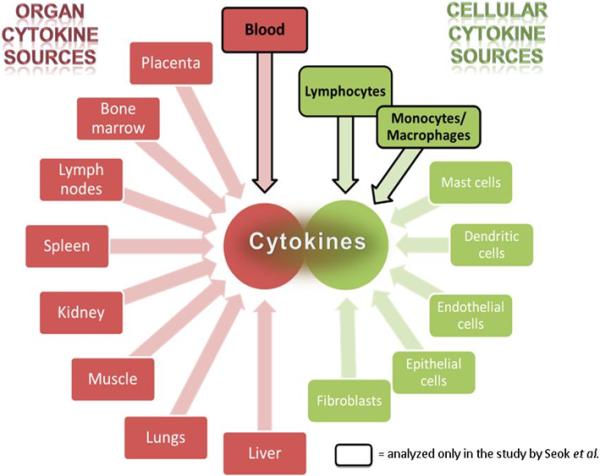
A typical critical illness, regardless whether of purely traumatic or infectious origin, triggers a broad inflammatory response that involves virtually all organ and cellular systems. The sum of inflammatory mediators circulating in the blood, the most expedient source of diagnostic information, is composed of proteins synthetized at multiple sites and sources. The simplified (given the overlap of cells and organs) schematic displays the most important and recognized sources of cytokine production. Black framing indicates the organ/cell populations which were used in the comparative genomic response study by Seok et al. (3). CNS: central nervous system.
5) Compartmentalization of the immune-inflammatory response
The multi-source synthesis of inflammatory mediators has yet another important aspect – the compartmentalization of the immuno-inflammatory response. It has been suggested that in systemic inflammatory response syndrome (SIRS) and/or infection, compartmentalized synthesis and release of inflammatory cytokines are equally important (63-65) and that cells other than leukocytes may be responsible for morbidity and mortality in trauma (61), inflammatory shock (66), endotoxemia (67-69) and/or infection (70). Studies have also shown that peripheral blood cells fail to reflect what occurs in the tissue fixed cells within the same or different organs (30;49;50;71;72). Furthermore, the concept of compartmentalization pertains to coexistence of differential (and often contrasting) responses that depend on the specific location of the immune-competent machinery and this notion has been supported by numerous preclinical studies. For example, in mice, hemorrhage resulted in a contrasting cytotoxic capacity with reduction in peritoneal and splenic macrophages and enhancement in Kupffer cells (73). The disparity in the post-hemorrhage response also extends to the cytokine compartment: transcriptional activity of TNF-α, IL-1ß and TGF-β (but not IL-6) as well as the release capacity of TNF-α and IL-1 were shown to be enhanced in Kupffer cells but diminished in peritoneal and splenic macrophages (73;74). Remarkably, compartmentalization of the gene and protein expression response after trauma-hemorrhage (30;75), infection (71) and no challenge (76) was also reported to occur in different cell types of the same organ (e.g., liver) and in cells of the same origin (e.g. mononuclear cells) but different locations (i.e., blood, spleen, peritoneum). Interestingly, data reported by the two aforementioned GLUE GRANT studies that compared genomic WBC and splenic responses in the mouse injury/infection model, further substantiate the notion of compartmentalization given that responses in those two compartments were highly dissimilar both at the early (50) and late phase after challenge (49). Collectively, the existing evidence strongly suggests that changes occurring in the blood compartment are not tantamount to the alterations arising in tissues/organs. Thus, any changes in treatment protocols based on changes occurring in circulating leukocytes only, regardless of whether on the genomic or protein level, and without considering the role of less diagnostically accessible compartments, could be very detrimental to patients.
6) Genome-to-protein gap
The poor correlation between mouse and man of the genomic responses reported in the original PNAS publication was not verified on the protein level. While fully understandable because of the labor-intensive nature of genomic screening, the wide gap between the genomic activation and the final protein product should not be summarily discounted. We speculate, for example, that correlation of circulating cytokines between patients and corresponding mouse models, had it been performed, would have shown better correlation than the changes in mRNA content in leukocytes. Prior work by the GLUE GRANT investigators (77) showed that using the same type of endotoxin in mice and humans produced nearly identical kinetics of cytokine production. Additionally, the concentrations of many of the cytokines or cytokine inhibitors that were produced were relatively close. Also, both mice and humans showed equivalent lymphopenia after endotoxin injection. Furthermore, the existing evidence demonstrates comparable temporal patterns for systemic inflammatory responses after both injury (78;79) and/or systemic infection (47), especially when patients are matched with the corresponding (and appropriate) mouse models. Collectively, these data suggest the total concentration of circulating cytokines is either substantially enriched by non-leukocyte sources (Fig. 1) or strongly influenced by various post-transcriptional/translational modifications (Fig. 2). Regarding the latter point, the strong relationship between mRNA and protein abundance levels is not self-evident and cancer research demonstrates that the correlation greatly varies. For example, comparison of mRNA and protein expression of three genes/protein pairs (MMP-2, MMP-9 and TIMP-1) showed no significant relationship in prostate cancer patients (80), while in human lung adenocarcinomas, only 20% of the 98 total screened genes demonstrated statistically significant correlation with their respective products (81). In human bladder cancers (82), gene-to-protein correlation was highly significant in some targets and low to negligible in others. A very similar outcome (i.e., high correlation variability between protein and mRNA expression levels) was demonstrated in circulating monocytes from healthy females (83) and human livers (84) suggesting that the use of mRNA expression to predict protein expression levels even in healthy organisms appears to be burdened with a relatively high uncertainty. The presence of a severe condition such as trauma or sepsis may further influence the gene-to-protein conduit. Overall, our current understanding of the dynamic chain of events occurring between genomic activation and arrival of the final product in diseased organisms remains poor and a straightforward gene-to-protein relationship should not be reflexively assumed in any biological system (85).
Fig. 2. From DNA to the final product - Regulation of gene expression.
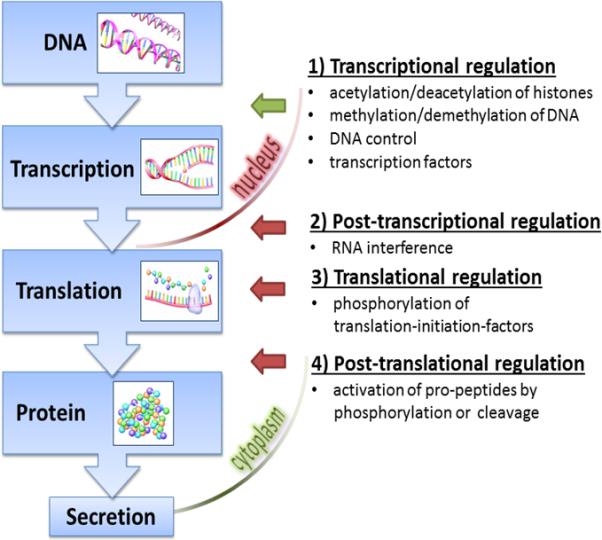
The path from the genomic activation to the final protein is interrupted by a series of complex regulatory steps that may either completely halt and/or markedly modify the abundance of the final product. The simplified schematic lists the most important regulatory mechanisms between DNA activation and synthesis of the ready protein. 1) To initiate transcription, demethylated DNA must be bound to acetylated histones; 2) RNA-polymerase and transcription factors bind on DNA to start transcription; 3) mRNA leaves the nucleus and is translated into protein; 4) Some proteins are produced in a dormant state and require subsequent activation. Green arrow indicates the first regulatory step between genomic activation and generation of the corresponding mRNA. Red arrows indicate remaining regulatory steps prior to the emergence of the final protein coded by the mRNA.
7) (Mis)matching the temporal response patterns
The analysis of the genomic data included comparisons of temporal expression patterns between human and mouse models. This is a commendable approach given the rapid fluctuation produced by inflammatory responses in trauma (blood loss and/or burn injury) and endotoxemia (47;86;87). Yet, execution of such a longitudinal comparison with large databases is particularly prone to both type I and II errors. One example of such a potential inaccuracy was discussed in the most recent commentary by Perlman et al. (14): the authors suggested that temporal dynamics of mouse and human critical conditions should not be matched 1:1 on the time scale given that compared to patients, murine disease models evolve over a shorter time course. A similar concern regarding the human-to-mouse time mismatch was echoed by Osterburg et al. (15). Furthermore, preliminary re-assessment of the same GLUE GRANT data, with a specific focus on the temporal match, by another independent group (ImmGen Consortium) was presented at the recent Shock Society Pro and Con Debate (20). This alternative analysis demonstrated markedly better human-to-mouse correlations (e.g., reaching r = 0.41 on day 7 post-burn injury by Fold-Change Quadrant Plot analysis) suggesting the original analysis of temporal responses might not have been sufficiently rigorous. The complete and detailed findings of the aforementioned reassessment will be soon submitted for peer review and will be published in a separate report to allow impartial comparison of the two different analytical methods applied.
In summary, the above concerns warrant additional mouse versus man studies and verification by an independent investigative team to validate the analysis of the findings reported by Seok and coworkers. If confirmed, the asserted dissimilarity of the genomic responses in human versus mouse leukocytes will have important and long-term implications for preclinical animal research, for example, alerting against the inter-species translational incompatibility of data generated in preclinical (i.e., in the mouse) testing of potential therapeutic genetic manipulations on the level of circulating leukocytes. At present, however, due to the rapid character of interventions in critical medicine, the vast majority of inflammation-targeted therapies focus on circulating inflammatory mediators – irrespective of the site of their synthesis, and focus less on manipulations of gene expression.
The Good and the Bad: The Role of Mouse Models in Critical Illness
Compared to other protracted disease conditions such as cancer or atherosclerosis, critical care research faces more difficult hurdles. These challenges stem from the fact that critical illness typically progresses very quickly, triggers rapid reactions from all body systems, results in heterogeneous reactions and causes extreme fluctuations in the elicited responses. Consequently, the challenge of trying to develop clinically relevant mouse models to simultaneously mimic all components of human critical injury and illness likely represents a fool's errand. Paradoxically, the wide public uproar provoked by the work of Seok and colleagues may simultaneously be - regardless of the ultimate accuracy the study's findings - its great service to the scientific community as it has elevated the discussion on the adequacy of preclinical mouse modeling in the contemporary research realm to a deserving premiere spot. In doing so, it is likely to also stimulate the discussion of the fidelity of mouse models designed to mimic other human pathological conditions (e.g., heart disease, obesity, cancer, etc.).
The limitations of existing mouse models of critical care illnesses (and beyond) are not a new concern. Apart from the ones discussed here, previous papers have focused on the mouse-to-human mismatch, e.g., characterizing major differences in the immuno-inflammatory signature (6). Additionally, responses to inflammatory stimuli (88;89), biochemical functionality of homologous proteins (90), makeup of serum proteins (91) as well as the influence of so called “cold stress” in the laboratory setting (92) have been identified as important differences between mice and humans. However, some of these same arguments could be used to criticize the way in which clinical studies are conducted. For example, laboratory isolation approaches for blood leukocytes vary widely between laboratories. This could lead to contrasting results in experiments using patient samples in a manner similar to that of mouse studies. Also, most human studies are limited to using blood samples, while other tissue and organ compartments can be effectively studied in mice, which improves the scientific profundity of well-designed animal research studies. No less important for modeling/translational purposes are variations within populations of human patients themselves. Trauma/sepsis patients typically receive allogeneic blood transfusion (versus shed blood in animals), different doses of morphine-based products (versus lack of and/or set doses of non-/morphine substances), inotropic agents and others, a majority of which produce strong immune-inflammatory effects that are very difficult to account for and match experimentally (93). Thus, patient studies are fraught with numerous small and large differences in the manner in which clinical care and standard procedures are carried out at different institutions. Furthermore, apart from the biologically-based differences, preclinical testing is simultaneously influenced by no less important study design flaws such as: 1) an excessive focus on early (acute) events; 2) age mismatch; 3) a lack of comorbidities; 4) using pre-treatment versus post-treatment in an appropriate manner; 5) lack of broad spectrum antibiotic coverage for sepsis treatment studies; and 6) difficulty in reproducing specific ICU conditions in animal models (47). This is far from an exhaustive list of mouse-to-human mismatches and/or potential confounding factors - many new pieces of this puzzle are yet to be identified.
From a global perspective, however, these differences do not appear to supersede similarities as there are countless physiological and pathological traits shared by both species in response to critical illness. We have selected dozens of relevant examples demonstrating the striking similarities in the responses of mice and humans (Table 1). This list could serve as a starting point for designing animal models for answering clinically relevant questions. Ideally, the origins of a “perfect” animal model should be deeply rooted in the clinical problem-solving and the model itself should reproduce as closely as possible the entire spectrum of pathophysiological consequences and the mechanisms of the human condition it aims to duplicate. Such an ideal match may be challenging to achieve, not only with mice or rats but with larger species as well, even non-human primates. An obvious first step in responsible modeling is to identify and verify a murine system with an acceptable resemblance to the studied critical illness and in the specific context of the defined scientific question. For example, one should not set out to study septic acute lung injury in CD-1 mice as this strain does not typically develop this condition (94), or to expect that pre-treatment data generated in a burn model conducted in healthy 4-week-old male mice will be translatable to the entire spectrum of human patients. The initial selection of the model must be subsequently followed by a fine tuning of the study design and, finally, a realistic interpretation of the acquired data. The latter element requires careful consideration. Too frequently (the PNAS article notwithstanding) lax interpretations of animal-based results are published in the scientific literature creating confusing (if not outright misleading) “background noise” (95). Compulsory disclosure of relevant animal experimentation details in compliance with the ARRIVE Guidelines (http://www.nc3rs.org.uk; recently adapted by the Shock journal; Instructions for Authors. Shock 41(1), January 2014) will partly help to remedy this confusion. A more diligent peer-review process is even stronger medicine given the growing stock market-like competition in the area of scientific publishing (96).
Table 1.
Selected Mouse-to-Human Translational Examples
| # | Translational phenomenon/response | Specific comments - mouse: | Specific comments - human: |
|---|---|---|---|
| 1 | Antibodies to TNFa given indiscriminately fail to reduce sepsis mortality | BALB/c mice were pretreated with antibodies to TNF prior to CLP sepsis. The murine studies were published 3 years before the failed human trials (101;116) | Anti-TNF antibodies failed to be an effective treatment strategy in a general population of septic patients (117;118) |
| 2 | Pretreatment with an anti-TNF strategy prevents early systemic inflammatory response syndrome (SIRS) | Passive immunization with the antiserum to TNF-α in BALB/c mice protected them against the lethal hyperinflammation by Escherichia coli LPS (98) | Anti-TNFα therapy was effective in humans with louse-borne relapsing fever when given as a pre-treatment against Jarisch-Herxheimer reactions (119) |
| 3 | Low-dose steroid therapy is associated with decreased mortality in septic mice and humans | Demonstrated in C57BL/6 male mice subjected to CLP and treated with different corticoid concentrations; low but not high-dose steroids improved 21 day survival (120) | Early initiation of low-dose corticosteroid therapy decreased mortality in septic shock patients (121) |
| 4 | Regulation of chemotactic behavior of mouse and human neutrophils via purinergic signaling | Human and mouse neutrophils rely on same purinergic receptor subtypes (P2Y2, A3, and A2a receptors) for autocrine signaling (122;123;124) | Demonstrated in vitro and in vivo; mice are suitable to study chemotaxis in inflammation, trauma, and sepsis (122;123;124; NCT01180361b) |
| 5 | Human and mouse neutrophils rely on similar signaling mechanisms for their activation during bacteria-induced acute lung injury | Increased nuclear activation of NF-kB in pulmonary neutrophils of mice after in vivo administration with endotoxin (125;126) | Increased nuclear accumulation of NF-kB in peripheral or pulmonary neutrophils of human volunteers after in vitro or in vivo stimulation with endotoxin (127) or in peripheral neutrophils of patients with sepsis (128) |
| 6 | Sepsis always in MARS: simultaneous systemic release of both pro- and anti-inflammatory cytokines in sepsis | Demonstrated in ICR/CD-1 (outbred) female mice subjected to CLP sepsis (129;130) | Demonstrated in septic shock patients (131) and patients with post-operative abdominal sepsis (132) |
| 7 | IL-6 serves as a biomarker for sepsis mortality | IL-6 measured 6 hours after the onset of CLP sepsis in BALB/c (133) and CD-1 mice (129) accurately predicts survival | Patients with high levels of IL-6 are at increased risk of dying from sepsis (134;135) |
| 8 | Role of nicotinic receptors in inflammatory responses after endotoxemia is similar in mice and humans | Demonstrated in C57BL/6 mice and α7 nicotinic receptor deficient mice; endotoxin-induced response was abrogated via activation of anti-inflammatory cholinergic pathway (vagus nerve stimulation) (136) | Human volunteers were administered endotoxin and GTS-21 (α7nAChR agonist) or placebo to study anti-inflammatory effects of cholinergic pathway (137; NCT00783068b) |
| 9 | Similar mode of pathogen-associated molecular patterns detection via toll like receptors (TLR) in mice and humans | TLR-4 was identified as the receptor that senses LPS in experiments with congenic sensitive (C3H/HeN; C57BL/10ScSn) and resistant (C3H/HeJ and C57BL/10ScCr) mice (138); TLR-4 expression level determines the degree of LPS-susceptibility in mice (139) | Human volunteers administered with LPS demonstrated altered TLR-induced genes expression (140). TLR-signaling pathways are strongly modulated in septic patients (141) |
| 10 | Sepsis induces profound apoptosis of immune and gastrointestinal epithelial (GIE) cells | Demonstrated in CLP female ND4 mice (142) and Pseudomonas aeruginosa pneumonia-induced septic FVB/N mice (143); apoptosis in B and T lymphocytes and dendritic cells. GIE cell apoptosis in large and small intestine | Demonstrated in patients who died of sepsis and sepsis and MODS; Data obtained by retrospective (rapid autopsy) and prospective (tissue resection) examination (144;145;146) |
| 11 | Sepsis increases while septic shock decreases the rate of hepatic gluconeogenesis | Demonstrated in mechanically ventilated C57BL/6 (inbread) male mice subjected to CLP sepsis (147) | Demonstrated in “infected” patients after surgery or trauma (148) and “bacteremic”/“complicated bacteremic” burn patients (149) |
| 12 | Increased nitric oxide reduces systolic contractility but supports (adaptive) left ventricular diastolic relaxation | Demonstrated in mechanically ventilated male C57BL/6 mice subjected to CLP sepsis (150) | Demonstrated in patients with septic shock (151), rat cardiomyocytes exposed to serum from patients with septic shock (152), and patients with chest pain (153) |
| 13 | Identical pathomechanism of increased intestinal mucosal permeability during inflammatory conditions in mice and humans | Increased intestinal permeability is associated with an increase in IL-1β-induced NF-κB activation and MLCK expression (154). This action involves p38 kinase and ATF-2 activation in mice and humans (155) | Production of IL-1β in ulcerative and Crohn's colitis (156) increases intestinal epithelial tight junction permeability (157); these actions are mediated by an NF-κB-dependent increase in MLCK gene transcription (158) |
| 14 | Epithelial tight junction barrier failure in mice and humans | Demonstrated in anti-CD3 murine diarrhea (159;160) model, Card15/NOD2- (159) and NOD2-deficient (160) mice; MLCK activation is necessary for epithelial barrier dysfunction and mucosal permeability increase (159, 160); NOD2 gene regulates this response (161,162) | Demonstrated in human colonic epithelial cell line HT-29/B6 (163), Caco-2 cells (164,165) and intestinal epithelial cells (165); TNFα impairs mucosal barrier function of the epithelial tight junction (163) by NF-κB activation (164) and increasing gene/protein expression of MLCK (165) |
| 15 | Intrauterine group A streptococcal infections in mice and humans are similarly modulated by prostaglandin (PG) E2 | Demonstrated in a mouse (C57BL/6) GAS (Group A Streptococcus) infection model; PGE2 impaired the phagocytic ability of mouse peritoneal macrophages in vitro (166) | An in vitro human macrophage–GAS interaction model was used. In GAS infected human THP-1 (macrophage-like) cells, PGE2 impairs the phagocytic ability of THP-1 and of human placental macrophages (166) |
| 16 | LPS-tolerant cells present a reprogramming of gene expression and function following a second challenge with LPS | Differentially regulated genes were found in bone marrow- derived macrophages tolerant to LPS. The tolerizeable (T; proinflammatory action) genes were typically not reinduced in macrophages upon second LPS exposure, while the nontolerizeable (NT; antimicrobial action) genes were reinduced (167) | Demonstrated in human monocytes made tolerant to LPS (168,169); Class T genes encompass antigen presentation-related and pro-inflammatory cytokine genes; NT genes code for anti-inflammatory/microbial factors (168); T and NT genes modulate TLR signaling pathway (169) |
| 17 | Sepsis increases muscle protein degradation and proteasome activity | CLP-induced sepsis in B6 mice increased total and myofibrillar protein breakdown in EDL with evidence of increased proteasome activity (170) | Critically ill patients with sepsis show increased muscle proteolysis and proteasome proteolytic activity (171) |
| 18 | Sepsis decreases lean body mass and increases muscle wasting | CLP-induced sepsis in C57BL/6 male mice decreases body weight, lean body mass and muscle mass (172) | Patients with severe peritonitis sepsis show loss of body weight and muscle mass and protein (173) |
| 19 | Sepsis induces elevated thrombin–antithrombin complexes are a part of TF/FVIIa pathway activation of coagulation | Using a mouse model peritonitis was induced by an intraperitoneal injection of live Escherichia coli (174) | Demonstrated in patients with severe bacterial peritonitis (175) and in healthy volunteers injected with endotoxin (176) |
| 20 | Sepsis induces systemic elevation of matrix metalloproteinase (MMP)-8 as part of the inflammatory process in humans and mice | Increased activity of MMP-8 was observed in plasma of C57BL/6 mice. Generation of MMP-8 genetically deficient mice was used to understand the biological function of the enzyme in inflammation (177) | Increased mRNA expression and activity of MMP-8 was observed in blood of pediatric septic patients and correlated with disease severity and mortality (177) |
| 21 | Similar mechanisms of mitochondrial dysfunction and mitochondrial biogenesis during sepsis in mice and humans. | Mitochondrial dysfunction was demonstrated in CLP male BALB/c mice (178); mitochondrial biogenesis was associated with an increase in PGC-1α in peritonitis by fibrin clot in male C57BL/6 mice (179) | Mitochondrial dysfunction was demonstrated in muscle biopsies of septic shock patients (180). Septic survivors presented increased expression of the mitochondrial biogenesis-related PGC-1α (181) |
| 22 | Inflammation-induced autophagy in tissues | Demonstrated in C57BL/6 male mice subjected to CLP (182) and Atg16L1 deficient healthy mice (183) and mice infected with Salmonella enterica (184); autophagy regulated by Atg16L1 gene | Demonstrated in patients that died of sepsis (182) and in patients with Crohn's disease (183) |
| 23 | Similar dynamics of circulating plasminogen activator inhibitor (PAI)-1 in subjects surviving and dying from sepsis | Demonstrated in CD-1 female mice subjected to CLP (185) and post-traumatic sepsis (186); PAI-1 increase correlates with sepsis severity and/or outcome | Demonstrated in surviving and non-surviving patients with sepsis, severe sepsis and/or septic shock(187) |
| 24 | Reduced antioxidant defense and increased oxidative stress in spinal cord injury | Demonstrated in CD-1 male mice with spinal cord injury; increase in protein nitration and membrane lipid peroxidation and secondary damage (188;189). | Demonstrated in patients with spinal cord injury; strong and long-term reduction of circulating antioxidants and increase of oxidative stress (190) |
| 25 | Heat shock protein (HSP)-72 serves as a biomarker for early detection of acute kidney injury | Demonstrated in B6/129-j/F2 male iNOS knockout mice subjected to renal injury by ischemia/reperfusion; Increase of renal HSP-72 mRNA and protein correlated with the extent of renal injury (191) | Urinary HSP-72 was significantly increased in patients with clinical acute kidney injury prior to elevation of serum creatinine (192) |
| 26 | Plasma gelsolin is a potential prognostic biomarker for critically ill surgical patients | Exogenous gelsolin infusion reduces brain inflammation and apoptotic signaling and improves survival of mice following major burn injury (193) as well as attenuating burn-induced pulmonary microvascular dysfunction (194) | Low plasma gelsolin levels were associated with increased risk of death occurring in the ICU (195) and correlated with development of multiple organ dysfunction syndrome and fatal outcome in burn patients (196) |
Abbreviations that are not explained in the table: TNF – tumor necrosis factor; CLP – cecal ligation and puncture; NF-κB – nuclear factor kappa B; MARS – mixed anti-inflammatory response syndrome; IL – interleukin; MLCK – myosin L chain kinase; NOD – nucleotide oligomerization domain; LPS – lipopolysaccharide; EDL – endothelial cell-derived lipase; TF – tissue factor; PGC-1α – peroxisome proliferator-activated receptor γ coactivator 1-α; MODS – multi organ dysfunction syndrome; iNOS – inducible nitric oxide synthase; mRNA – messenger ribonucleic acid; ICU – intensive care unit
www.clinicaltrials.gov identifier (NCT number)
Achieving the goal of implementing the elements listed above will likely generate datasets that are much more precise and relevant to patients. Yet, old habits die hard and many misconceptions surrounding applicability of data from mouse models are likely to have long half-lives. The most recent exchange between Cauwels et al. (12) and the authors of the original paper (3) is perhaps a telling example: in the reply letter to Cauwels et al. (97), the fact that anti-TNF treatment was life-saving in mice administered a lethal dose of LPS (98) but failed in septic patients, supported their notion that mouse models are generally unfit to predict human inflammatory diseases such as sepsis. However, when anti-TNF antibodies were used in a clinically relevant model of sepsis (i.e. cecal ligation and puncture) (99;100), they also failed to have any protective effects against sepsis (101); a finding similar to the lack of efficacy seen in patients. Thus, it is humans, and not mice, with their incomplete understanding of sepsis pathophysiology coupled with the use of an inappropriate animal model, which are to be blamed for the spectacular collapse of anti-inflammatory sepsis trials. After “successfully” executing various treatments (i.e., against circulating cytokines or endotoxins) in the mouse (98), rat (102), rabbit (103), dog (104) and non-human primates (105), the striking failure of similar anti-inflammatory therapeutic protocols in septic patients has brought a painful realization that injecting mice and other species with a lethal dose of bacterial LPS is not a good predictive model for a typical human sepsis. It must be stressed, however, that a complete renouncement of anti-inflammatory treatments based on the failed trials would be equally short sighted and the most recent meta-analysis argues that application of anti-TNF agents in septic patients should be revisited (106). It has become clear that responses elicited in sepsis are highly mixed and the immunosuppressive component frequently exceeds hyperinflammation (107). Hence, in smaller cohorts of patients treated based on the similar type of acquired sepsis (e.g. meningococcal septicemia), their well-defined immuno-inflammatory makeup and/or predicted susceptibility toward the tailored treatment, even the “notorious” anti-TNF intervention may be lifesaving (108). Limited evidence in support of implementing such tactics is already available (109;110; www.clinicaltrials.gov identifier: NCT01046669).
Clearly, sepsis and septic shock are complex disease processes. We suggest multiple animal models should be considered for basic research and preclinical testing of therapeutics. The remarkable capability of contemporary science has opened many new investigative avenues such as emergence of humanized mice (111, 112, 113) and access to a growing selection of recombinant inbred mouse strains (with specific defined genetic traits) from the Collaborative Cross Project (114; 115). Yet, whereas using inbred mice for basic research remains important to advancing knowledge and may allow much more nuanced investigation of gene environment and gene-pathogen interactions, the efficacy testing of beneficial treatments should be tested in outbred mice as well. Attempts to test the efficacy of treatments with multiple types of infectious disease models (e.g., CLP, pneumonia, urosepsis ect.) should be considered and tested before moving forward with efficacy trials in large animals and then patients.
Final Conclusions
The current reality is that other than whole blood assays and isolated single organ cultures, animal models are the only viable and fully intact biological systems which allow examination of clinically relevant hypotheses and studies to decipher underlying mechanisms of biological phenomena. Mice have been in the forefront of these investigations precisely for the reason that they have served as an origin of many subsequent successes at the patient's bedside, including the area of critical care medicine. Yet, it is evident that any mouse study is merely a beginning in the long process that requires caution and series of subsequent verification steps. To continue with in vivo research in an ethically responsible way, the scientific community has an obligation to seek improvements and implement more fitting solutions in the currently used mouse model systems so they continuously adapt to the evolving understanding of the respective human critical illnesses and not vice versa. We must remain cognizant of the known limitations of the models, share newly discovered incompatibilities and be willing to abandon the erroneous models if necessary. In relation to this discussion on the relevance of mouse models in critical care medicine, the allegory of inflammatory response appears to be very fitting. Specifically, an exaggerated or excessively weak inflammatory reaction will typically lead to a poor outcome in an ICU patient. In a similar manner, excessive trust or hasty discounting the usefulness of mouse models for research will have a negative impact on preclinical critical care research and ultimately result in fewer discoveries which improve patient care and outcome.
Acknowledgements
We gratefully acknowledge Jean-Marc Cavaillon for his constructive input regarding the merits and balance of the manuscript. We thank Pia Rademann for her kind help with the Reference Manager and in drawing the Figures.
Footnotes
Competing interests: The authors declare that no competing interests exist.
“Extraordinary claims require extraordinary evidence.” Carl Sagan
Reference List
- 1.U.S. Congress, Office of Technology Assessment . Alternatives to Animal Use in Research, Testing, and Education. U.S. Government Printing Office; Washington, DC: Feb, 1986. OTA-BA-273 Available at: http://govinfo.library.unt.edu/ota/Ota_3/DATA/1986/8601. [Google Scholar]
- 2.Sixth Report on the Statistics on the Number of Animals used for Experimental and other Scientific Purposes in the Member States of the European Union. 2010 Available at: http://www.europarl.europa.eu/registre/docs_autres_institutions/commission_europeenne/sec/2010/1107/COM_SEC%282010%291107_EN.
- 3.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110(9):3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolker J. Model organisms: There's more to life than rats and flies. Nature. 2012;491(7422):31–33. doi: 10.1038/491031a. [DOI] [PubMed] [Google Scholar]
- 5.Handel AE, Lincoln MR, Ramagopalan SV. Of mice and men: experimental autoimmune encephalitis and multiple sclerosis. Eur J Clin Invest. 2011;41(11):1254–1258. doi: 10.1111/j.1365-2362.2011.02519.x. [DOI] [PubMed] [Google Scholar]
- 6.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 7.Kolata G. Mice Fall Short as Test Subjects for Humans’ Deadly Ills. 2013-02-12 ( nytimes.com): Available at: http://www.nytimes.com/2013/02/12/science/testing-of-some-deadly-diseases-on-mice-mislead-report-says.html?pagewanted=1&_r=3&ref=global-home&#comments.
- 8.In science.slashdot.org 2013-02-12 Available at: http://science.slashdot.org/story/13/02/12/1823238/drug-testing-in-mice-may-be-a-waste-of-time-researchers-warn.
- 9.Love SM. We are not mice. 2013-02-14 ( Huffingtonpost.com) Available at: http://www.huffingtonpost.com/susan-m-love/research-mice_b_2680201.html.
- 10.Derek Low. Mouse Models of Inflammation Are Basically Worthless. Now We Know. 2013-02-12 ( pipeline.corante.com) Available at: http://pipeline.corante.com/archives/2013/02/13/mouse_models_of_inflammation_a re_basically_worthless_now_we_know.php.
- 11.Chu E. This Is Why It's A Mistake To Cure Mice Instead Of Humans. 2012-12-20 (richarddawkins.net) Available at: http://www.popsci.com/science/article/2013-02/how-scientists-got-lost-curing-mice-instead-of-humans.
- 12.Cauwels A, Vandendriessche B. Brouckaert P: Of mice, men, and inflammation. Proc Natl Acad Sci USA. 2013;110(34):E3150. doi: 10.1073/pnas.1308333110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Souza N. Model organisms: Mouse models challenged. Nat Methods. 2013;10(4):288. doi: 10.1038/nmeth.2429. [DOI] [PubMed] [Google Scholar]
- 14.Perlman H, Budinger GR, Ward PA. Humanizing the mouse: in defense of murine models of critical illness. Am J Respir Crit Care Med. 2013;187(9):898–900. doi: 10.1164/rccm.201303-0489ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osterburg AR, Hexley P, Supp DM, Robinson CT, Noel G, Ogle C, Boyce ST, Aronow BJ, Babcock GF. Concerns over interspecies transcriptional comparisons in mice and humans after trauma. Proc Natl Acad Sci USA. 2013;110(36):E3370. doi: 10.1073/pnas.1306033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake AC. Of mice and men: what rodent models don't tell us. Cell Mol Immunol. 2013;10(4):284–285. doi: 10.1038/cmi.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cossin D. Do Mice Make Bad Models? The Scientist. 2013 Feb 11; Available at: http://www.the-scientist.com/?articles.view/articleNo/34346/title/Do-Mice-Make-Bad-Models-/
- 18.Barrington B. Are Mice Reliable Models for Human Disease Studies? Understand Nutrition. 2013 Feb 14; Available at: http://understandnutrition.com/2013/02/14/are-mice-reliable-models-for-human-disease-studies/
- 19.Wanner M. Why mice may succeed in research when a single mouse falls short? Genetics and Your Health Blog. 2013 Feb 13; Available at: http://community.jax.org/genetics_health/b/weblog/archive/2013/02/13/why-mice-may-succeed-in-research-when-a-single-mouse-falls-short.aspx.
- 20.Remick D. Use of animal models for the study of human disease-a shock society debate. Shock. 2013;40(4):345–346. doi: 10.1097/SHK.0b013e3182a2aee0. [DOI] [PubMed] [Google Scholar]
- 21.Sadarangani C, Skamene E, Kongshavn PA. Cellular basis for genetically determined enhanced resistance of certain mouse strains to listeriosis. Infect Immun. 1980;28(2):381–386. doi: 10.1128/iai.28.2.381-386.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B, Koo GC, Yap EH, Chua KL, Gan YH. Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect Immun. 2002;70(2):504–511. doi: 10.1128/IAI.70.2.504-511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brenner GJ, Cohen N, Moynihan JA. Similar immune response to nonlethal infection with herpes simplex virus-1 in sensitive (BALB/c) and resistant (C57BL/6) strains of mice. Cell Immunol. 1994;157(2):510–524. doi: 10.1006/cimm.1994.1246. [DOI] [PubMed] [Google Scholar]
- 24.Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol. 1995;69(12):7674–7681. doi: 10.1128/jvi.69.12.7674-7681.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Autenrieth IB, Beer M, Bohn E, Kaufmann SH, Heesemann J. Immune responses to Yersinia enterocolitica in susceptible BALB/c and resistant C57BL/6 mice: an essential role for gamma interferon. Infect Immun. 1994;62(6):2590–2599. doi: 10.1128/iai.62.6.2590-2599.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis. 1990;162(1):133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 27.Fink MP. Animal models of sepsis. Virulence. 2014;5(1):143–153. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angele MK, Schwacha MG, Ayala A, Chaudry IH. Effect of gender and sex hormones on immune responses following shock. Shock. 2000;14(2):81–90. doi: 10.1097/00024382-200014020-00001. [DOI] [PubMed] [Google Scholar]
- 29.Choudhry MA, Schwacha MG, Hubbard WJ, Kerby JD, Rue LW, Bland KI, Chaudry IH. Gender differences in acute response to trauma-hemorrhage. Shock. 2005;24(Suppl 1):101–106. doi: 10.1097/01.shk.0000191341.31530.5e. [DOI] [PubMed] [Google Scholar]
- 30.Schneider CP, Schwacha MG, Chaudry IH. Influence of gender and age on T-cell responses in a murine model of trauma-hemorrhage: differences between circulating and tissue-fixed cells. J Appl Physiol. 2006;100(3):826–833. doi: 10.1152/japplphysiol.00898.2005. 1985. [DOI] [PubMed] [Google Scholar]
- 31.Marriott I, Huet-Hudson YM. Sexual dimorphism in innate immune responses to infectious organisms. Immunol Res. 2006;34(3):177–192. doi: 10.1385/IR:34:3:177. [DOI] [PubMed] [Google Scholar]
- 32.Aoyama M, Kotani J, Usami M. Gender difference in granulocyte dynamics and apoptosis and the role of IL-18 during endotoxin-induced systemic inflammation. Shock. 2009;32(4):401–409. doi: 10.1097/SHK.0b013e31819c358a. [DOI] [PubMed] [Google Scholar]
- 33.Imahara SD, Jelacic S, Junker CE, O'Keefe GE. The influence of gender on human innate immunity. Surgery. 2005;138(2):275–282. doi: 10.1016/j.surg.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 34.Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS, Coopersmith CM, Buchman TG. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19(4):310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208(13):2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wood JH, Partrick DA, Johnston RB., Jr The inflammatory response to injury in children. Curr Opin Pediatr. 2010;22(3):315–320. doi: 10.1097/MOP.0b013e328338da48. [DOI] [PubMed] [Google Scholar]
- 37.Calkins CM, Bensard DD, Moore EE, McIntyre RC, Silliman CC, Biffl W, Harken AH, Partrick DA, Offner PJ. The injured child is resistant to multiple organ failure: a different inflammatory response? J Trauma. 2002;53(6):1058–1063. doi: 10.1097/00005373-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Finnerty CC, Jeschke MG, Herndon DN, Gamelli R, Gibran N, Klein M, Silver G, Arnoldo B, Remick D, Tompkins RG, et al. Temporal cytokine profiles in severely burned patients: a comparison of adults and children. Mol Med. 2008;14(9-10):553–560. doi: 10.2119/2007-00132.Finnerty. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.George RL, McGwin G, Jr, Schwacha MG, Metzger J, Cross JM, Chaudry IH, Rue LW., 3rd The association between sex and mortality among burn patients as modified by age. J Burn Care Rehabil. 2005;26(5):416–421. doi: 10.1097/01.bcr.0000176888.44949.87. [DOI] [PubMed] [Google Scholar]
- 40.Krabbe KS, Bruunsgaard H, Hansen CM, Møller K, Fonsmark L, Qvist J, Madsen PL, Kronborg G, Andersen HO, Skinhøj P, et al. Ageing is associated with a prolonged fever response in human endotoxemia. Clin Diagn Lab Immunol. 2001;8(2):333–338. doi: 10.1128/CDLI.8.2.333-338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raqib R, Qadri F, SarkEr P, Mia SM, Sansonnetti PJ, Albert MJ, Andersson J. Delayed and reduced adaptive humoral immune responses in children with shigellosis compared with in adults. Scand J Immunol. 2002;55(4):414–423. doi: 10.1046/j.1365-3083.2002.01079.x. [DOI] [PubMed] [Google Scholar]
- 42.Lundgren A, Bhuiyan TR, Novak D, Kaim J, Reske A, Lu YJ, Qadri F, Malley R. Characterization of Th17 responses to Streptococcus pneumoniae in humans: comparisons between adults and children in a developed and a developing country. Vaccine. 2012;30(26):3897–3907. doi: 10.1016/j.vaccine.2012.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider CP, Schwacha MG, Chaudry IH. Impact of sex and age on bone marrow immune responses in a murine model of trauma-hemorrhage. J Appl Physiol. 2007;102(1):113–121. doi: 10.1152/japplphysiol.00848.2006. 1985. [DOI] [PubMed] [Google Scholar]
- 44.Kang SC, Matsutani T, Choudhry MA, Schwacha MG, Rue LW, Bland KI, Chaudry IH. Are the immune responses different in middle-aged and young mice following bone fracture, tissue trauma and hemorrhage? Cytokine. 2004;26(5):223–230. doi: 10.1016/j.cyto.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Brubaker AL, Rendon JL, Ramirez L, Choudhry MA, Kovacs EJ. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol. 2013;190(4):1746–1757. doi: 10.4049/jimmunol.1201213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chorinchath BB, Kong LY, Mao L, McCallum RE. Age-associated differences in TNF-alpha and nitric oxide production in endotoxic mice. J Immunol. 1996;156(4):1525–1530. [PubMed] [Google Scholar]
- 47.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, Remick DG. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev. 2013;93(3):1247–1288. doi: 10.1152/physrev.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, Wakamatsu E, Benoist C, Koller D, Regev A, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci USA. 2013;110(8):2946–2951. doi: 10.1073/pnas.1222738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lederer JA, Brownstein BH, Lopez MC, Macmillan S, Delisle AJ, Macconmara MP, Choudhry MA, Xiao W, Lekousi S, Cobb JP, et al. Comparison of longitudinal leukocyte gene expression after burn injury or trauma-hemorrhage in mice. Physiol Genomics. 2008;32(3):299–310. doi: 10.1152/physiolgenomics.00086.2007. [DOI] [PubMed] [Google Scholar]
- 50.Brownstein BH, Logvinenko T, Lederer JA, Cobb JP, Hubbard WJ, Chaudry IH, Remick DG, Baker HV, Xiao W, Mannick JA. Commonality and differences in leukocyte gene expression patterns among three models of inflammation and injury. Physiol Genomics. 2006;24(3):298–309. doi: 10.1152/physiolgenomics.00213.2005. [DOI] [PubMed] [Google Scholar]
- 51.Wichmann MW, Zellweger R, DeMaso CM, Ayala A, Williams C, Chaudry IH. Immune function is more compromised after closed bone fracture and hemorrhagic shock than hemorrhage alone. Arch Surg. 1996;131(9):995–1000. doi: 10.1001/archsurg.1996.01430210093021. [DOI] [PubMed] [Google Scholar]
- 52.Wichmann MW, Ayala A, Chaudry IH. Severe depression of host immune functions following closed-bone fracture, soft-tissue trauma, and hemorrhagic shock. Crit Care Med. 1998;26(8):1372–1378. doi: 10.1097/00003246-199808000-00024. [DOI] [PubMed] [Google Scholar]
- 53.Gentile LF, Nacionales DC, Cuenca AG, Armbruster M, Ungaro RF, Abouhamze AS, Lopez C, Baker HV, Moore FA, Ang DN, et al. Identification and description of a novel murine model for polytrauma and shock. Crit Care Med. 2013;41(4):1075–1085. doi: 10.1097/CCM.0b013e318275d1f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leelahavanichkul A, Huang Y, Hu X, Zhou H, Tsuji T, Chen R, Kopp JB, Schnermann J, Yuen PS, Star RA. Chronic kidney disease worsens sepsis and sepsis-induced acute kidney injury by releasing High Mobility Group Box Protein-1. Kidney Int. 2011;80(11):1198–1211. doi: 10.1038/ki.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salomao R, Brunialti MK, Rapozo MM, Baggio-Zappia GL, Galanos C, Freudenberg M. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock. 2012;38(3):227–242. doi: 10.1097/SHK.0b013e318262c4b0. [DOI] [PubMed] [Google Scholar]
- 57.Samraj RS, Zingarelli B, Wong HR. Role of biomarkers in sepsis care. Shock. 2013;40(5):358–365. doi: 10.1097/SHK.0b013e3182a66bd6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schochl H, Voelckel W, Grassetto A, Schlimp CJ. Practical application of point-of-care coagulation testing to guide treatment decisions in trauma. J Trauma Acute Care Surg. 2013;74(6):1587–1598. doi: 10.1097/TA.0b013e31828c3171. [DOI] [PubMed] [Google Scholar]
- 59.Murray CK, Hoffmaster RM, Schmit DR, Hospenthal DR, Ward JA, Cancio LC, Wolf SE. Evaluation of white blood cell count, neutrophil percentage, and elevated temperature as predictors of bloodstream infection in burn patients. Arch Surg. 2007;142(7):639–642. doi: 10.1001/archsurg.142.7.639. [DOI] [PubMed] [Google Scholar]
- 60.Fan Y, Yu JL. Umbilical blood biomarkers for predicting early-onset neonatal sepsis. World J Pediatr. 2012;8(2):101–108. doi: 10.1007/s12519-012-0347-3. [DOI] [PubMed] [Google Scholar]
- 61.Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38(12):1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med. 2008;26(6):711–715. doi: 10.1016/j.ajem.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 63.Cavaillon JM, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. 2006;12(3):151–170. doi: 10.1179/096805106X102246. [DOI] [PubMed] [Google Scholar]
- 64.Cavaillon JM, Adib-Conquy M, Cloëz-Tayarani I, Fitting C. Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res. 2001;7(2):85–93. [PubMed] [Google Scholar]
- 65.Schwacha MG, Schneider CP, Chaudry IH. Differential expression and tissue compartmentalization of the inflammatory response following thermal injury. Cytokine. 2002;17(5):266–274. doi: 10.1006/cyto.2001.1003. [DOI] [PubMed] [Google Scholar]
- 66.Cauwels A. Nitric oxide in shock. Kidney Int. 2007;72(5):557–565. doi: 10.1038/sj.ki.5002340. [DOI] [PubMed] [Google Scholar]
- 67.Juskewitch JE, Knudsen BE, Platt JL, Nath KA, Knutson KL, Brunn GJ, Grande JP. LPS-induced murine systemic inflammation is driven by parenchymal cell activation and exclusively predicted by early MCP-1 plasma levels. Am J Pathol. 2012;180(1):32–40. doi: 10.1016/j.ajpath.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Juskewitch JE, Platt JL, Knudsen BE, Knutson KL, Brunn GJ, Grande JP. Disparate roles of marrow- and parenchymal cell-derived TLR4 signaling in murine LPS-induced systemic inflammation. Sci Rep. 2012;2:918. doi: 10.1038/srep00918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meisner M, Muller V, Khakpour Z, Toegel E, Redl H. Induction of procalcitonin and proinflammatory cytokines in an anhepatic baboon endotoxin shock model. Shock. 2003;19(2):187–190. doi: 10.1097/00024382-200302000-00017. [DOI] [PubMed] [Google Scholar]
- 70.Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95(1):55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kapetanovic R, Parlato M, Fitting C, Quesniaux V, Cavaillon JM, Adib-Conquy M. Mechanisms of TNF induction by heat-killed Staphylococcus aureus differ upon the origin of mononuclear phagocytes. Am J Physiol Cell Physiol. 2011;300(4):C850–C859. doi: 10.1152/ajpcell.00187.2010. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Schwacha MG, Chaudry IH. Tissue compartment-specific role of estrogen receptor subtypes in immune cell cytokine production following trauma-hemorrhage. J Appl Physiol. 2007;102(1):163–168. doi: 10.1152/japplphysiol.00964.2006. 1985. [DOI] [PubMed] [Google Scholar]
- 73.Ayala A, Perrin MM, Wang P, Ertel W, Chaudry IH. Hemorrhage induces enhanced Kupffer cell cytotoxicity while decreasing peritoneal or splenic macrophage capacity. Involvement of cell-associated tumor necrosis factor and reactive nitrogen. J Immunol. 1991;147(12):4147–4154. [PubMed] [Google Scholar]
- 74.Zhu XL, Zellweger R, Zhu XH, Ayala A, Chaudry IH. Cytokine gene expression in splenic macrophages and Kupffer cells following haemorrhage. Cytokine. 1995;7(1):8–14. doi: 10.1006/cyto.1995.1002. [DOI] [PubMed] [Google Scholar]
- 75.Wang P, Ba ZF, Morrison MH, Chaudry IH. Differential alterations in cyclic nucleotide levels in Kupffer cells and hepatocytes following trauma-hemorrhage and resuscitation. Shock. 1994;1(6):438–442. doi: 10.1097/00024382-199406000-00008. [DOI] [PubMed] [Google Scholar]
- 76.Gorgani NN, Ma Y, Clark HF. Gene signatures reflect the marked heterogeneity of tissue-resident macrophages. Immunol Cell Biol. 2008;86(3):246–254. doi: 10.1038/sj.icb.7100131. [DOI] [PubMed] [Google Scholar]
- 77.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick DG. Inflammation and Host Response to Injury Investigators: Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol. 2005;12(1):60–67. doi: 10.1128/CDLI.12.1.60-67.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Namas R, Ghuma A, Hermus L, Zamora R, Okonkwo DO, Billiar TR, Vodovotz Y. The acute inflammatory response in trauma / hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J Med. 2009;4(3):97–103. doi: 10.4176/090325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7(1):22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lichtinghagen R, Musholt PB, Lein M, Römer A, Rudolph B, Kristiansen G, Hauptmann S, Schnorr D, Loening SA, Jung K. Different mRNA and protein expression of matrix metalloproteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 in benign and malignant prostate tissue. Eur Urol. 2002;42(4):398–406. doi: 10.1016/s0302-2838(02)00324-x. [DOI] [PubMed] [Google Scholar]
- 81.Chen G, Gharib TG, Huang CC, Taylor JM, Misek DE, Kardia SL, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1(4):304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 82.Orntoft TF, Thykjaer T, Waldman FM, Wolf H, Celis JE. Genome-wide study of gene copy numbers, transcripts, and protein levels in pairs of non-invasive and invasive human transitional cell carcinomas. Mol Cell Proteomics. 2002;1(1):37–45. doi: 10.1074/mcp.m100019-mcp200. [DOI] [PubMed] [Google Scholar]
- 83.Guo Y, Xiao P, Lei S, Deng F, Xiao GG, Liu Y, Chen X, Li L, Wu S, Chen Y, et al. How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim Biophys Sin (Shanghai) 2008;40(5):426–436. doi: 10.1111/j.1745-7270.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- 84.Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 1997;18(3-4):533–537. doi: 10.1002/elps.1150180333. [DOI] [PubMed] [Google Scholar]
- 85.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4(9):117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stoecklein VM, Osuka A, Lederer JA. Trauma equals danger--damage control by the immune system. J Leukoc Biol. 2012;92(3):539–551. doi: 10.1189/jlb.0212072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith JW, Gamelli RL, Jones SB, Shankar R. Immunologic responses to critical injury and sepsis. J Intensive Care Med. 2006;21(3):160–172. doi: 10.1177/0885066605284330. [DOI] [PubMed] [Google Scholar]
- 88.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol. 2005;12(1):60–67. doi: 10.1128/CDLI.12.1.60-67.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, Hauschildt S. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31(5):379–446. doi: 10.1615/critrevimmunol.v31.i5.20. [DOI] [PubMed] [Google Scholar]
- 90.Nagae M, Nishi N, Nakamura-Tsuruta S, Hirabayashi J, Wakatsuki S, Kato R. Structural analysis of the human galectin-9 N-terminal carbohydrate recognition domain reveals unexpected properties that differ from the mouse orthologue. J Mol Biol. 2008;375(1):119–135. doi: 10.1016/j.jmb.2007.09.060. [DOI] [PubMed] [Google Scholar]
- 91.Warren HS, Fitting C, Hoff E, Adib-Conquy M, Beasley-Topliffe L, Tesini B, Liang X, Valentine C, Hellman J, Hayden D, et al. Resilience to bacterial infection: difference between species could be due to proteins in serum. J Infect Dis. 2010;201(2):223–232. doi: 10.1086/649557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karp CL. Unstressing intemperate models: how cold stress undermines mouse modeling. J Exp Med. 2012;209(6):1069–1074. doi: 10.1084/jem.20120988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chaudry IH. Sepsis: lessons learned in the last century and future directions. Arch Surg. 1999;134(9):922–929. doi: 10.1001/archsurg.134.9.922. [DOI] [PubMed] [Google Scholar]
- 94.Iskander KN, Craciun FL, Stepien DM, Duffy ER, Kim J, Moitra R, Vaickus LJ, Osuchowski MF, Remick DG. Cecal ligation and puncture-induced murine sepsis does not cause lung injury. Crit Care Med. 2013;41(1):159–170. doi: 10.1097/CCM.0b013e3182676322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hartung T. Look back in anger – what clinical studies tell us about preclinical work. ALTEX. 2013;30(3):275–291. doi: 10.14573/altex.2013.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bohannon J. Who's afraid of peer review? Science. 2013;342(6154):60–65. doi: 10.1126/science.2013.342.6154.342_60. [DOI] [PubMed] [Google Scholar]
- 97.Warren HS, Tompkins RG, Mindrinos MN, Xiao W, Davis RW. Reply to Cauwels et al.: Of men, not mice, and inflammation. Proc Natl Acad Sci USA. 2013;110(34):E3151. doi: 10.1073/pnas.1308943110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229(4716):869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 99.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock--a review of laboratory models and a proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 100.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 101.Remick D, Manohar P, Bolgos G, Rodriguez J, Moldawer L, Wollenberg G. Blockade of tumor necrosis factor reduces lipopolysaccharide lethality, but not the lethality of cecal ligation and puncture. Shock. 1995;4(2):89–95. doi: 10.1097/00024382-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 102.Goto M, Zeller WP, Hurley RM, Jong JS, Lee CH. Prophylaxis and treatment of newborn endotoxic shock with anti-lipid. A monoclonal antibodies. Circ Shock. 1991;35(1):60–64. [PubMed] [Google Scholar]
- 103.Ohlsson K, Bjork P, Bergenfeldt M, Hageman R, Thompson RC. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature. 1990;348(6301):550–552. doi: 10.1038/348550a0. [DOI] [PubMed] [Google Scholar]
- 104.Krausz MM, Utsunomiya T, Feuerstein G, Wolfe JH, Shepro D, Hechtman HB. Prostacyclin reversal of lethal endotoxemia in dogs. J Clin Invest. 1981;67(4):1118–1125. doi: 10.1172/JCI110125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330(6149):662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 106.Qiu P, Cui X, Sun J, Welsch J, Natanson C, Eichacker PQ, Hotchkiss RS, Monneret G, Payen D. Antitumor necrosis factor therapy is associated with improved survival in clinical sepsis trials – a meta-analysis. Crit Care Med. 2013;41(10):2419–2429. doi: 10.1097/CCM.0b013e3182982add. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Angus DS. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306(23):2614–2615. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- 109.Panacek EA, Marshall JC, Albertson TE, Johnson DH, Johnson S, MacArthur RD, Miller M, Barchuk WT, Fischkoff S, Kaul M, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32(11):2173–2182. doi: 10.1097/01.ccm.0000145229.59014.6c. [DOI] [PubMed] [Google Scholar]
- 110.Osuchowski MF, Connett J, Welch K, Granger J, Remick DG. Stratification is the key: inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit Care Med. 2009;37(5):1567–1573. doi: 10.1097/CCM.0b013e31819df06b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7(2):118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 112.Usinger J, McDonough JS, Schultz LD, Ferguson TA, Hotchkiss RS. Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized” mice. J Leukoc Biol. 2009;86(2):219–227. doi: 10.1189/jlb.1008615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ernst W, Zimara N, Hanses F, Männel DN, Seelbacj-Göbel B, Wege AK. Humanized mice, a new model to study the influence of drug treatment on neonatal sepsis. Infect Immun. 2013;81(5):1520–1531. doi: 10.1128/IAI.01235-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36(11):1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 115.Threadgill DW, Churchill GA. Ten years of the Collaborative Cross. Genetics. 2012;190(2):291–294. doi: 10.1534/genetics.111.138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eskandari MK, Bolgos G, Miller C, Nguyen DT, DeForge LE, Remick DG. Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J Immunol. 1992;148(9):2724–2730. [PubMed] [Google Scholar]
- 117.Dhainaut JF, Vincent JL, Richard C, Lejeune P, Martin C, Fierobe L, Stephens S, Ney UM, Sopwith M. CDP571, a humanized antibody to human tumor necrosis factor-alpha: safety, pharmacokinetics, immune response, and influence of the antibody on cytokine concentrations in patients with septic shock. CPD571 Sepsis Study Group. Crit Care Med. 1995;23(9):1461–1469. doi: 10.1097/00003246-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 118.Reinhart K, Wiegand-Löhnert C, Grimminger F, Kaul M, Withington S, Treacher D, Eckart J, Willatts S, Bouza C, Krausch D, et al. Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK 195F, in patients with sepsis and septic shock: a multicenter, randomized, placebo-controlled, dose-ranging study. Crit Care Med. 1996;24(5):733–742. doi: 10.1097/00003246-199605000-00003. [DOI] [PubMed] [Google Scholar]
- 119.Fekade D, Knox K, Hussein K, Melka A, Lalloo DG, Coxon RE, Warrell DA. Prevention of Jarisch-Herxheimer reactions by treatment with antibodies against tumor necrosis factor alpha. N Engl J Med. 1996;335(5):311–315. doi: 10.1056/NEJM199608013350503. [DOI] [PubMed] [Google Scholar]
- 120.van den Berg JW, van der Zee M, de Bruin RW, van Holten-Neelen C, Bastiaans J, Nagtzaam NM, IJzermans JN, Benner R, Dik WA. Mild versus strong anti-inflammatory therapy during early sepsis in mice: a matter of life and death. Crit Care Med. 2011;39(6):1275–1281. doi: 10.1097/CCM.0b013e31820edf75. [DOI] [PubMed] [Google Scholar]
- 121.Park HY, Suh GY, Song JU, Yoo H, Jo IJ, Shin TG, Lim SY, Woo S, Jeon K. Early initiation of low-dose corticosteroid therapy in the management of septic shock: a retrospective observational study. Crit Care. 2012;16(1):R3. doi: 10.1186/cc10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314(5806):1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 123.Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, Inoue Y, Woehrle T, Zhang Q, Hauser C, et al. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3(125):ra45. doi: 10.1126/scisignal.2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shenkar R, Abraham E. Mechanisms of lung neutrophil activation after hemorrhage or endotoxemia: roles of reactive oxygen intermediates, NF-kappa B, and cyclic AMP response element binding protein. J Immunol. 1999;163(2):954–962. [PubMed] [Google Scholar]
- 126.Abraham E, Arcaroli J, Shenkar R. Activation of extracellular signal-regulated kinases, NF-kappa B, and cyclic adenosine 5′-monophosphate response element-binding protein in lung neutrophils occurs by differing mechanisms after hemorrhage or endotoxemia. J Immunol. 2001;166(1):522–530. doi: 10.4049/jimmunol.166.1.522. [DOI] [PubMed] [Google Scholar]
- 127.Abraham E, Nick JA, Azam T, Kim SH, Mira JP, Svetkauskaite D, He Q, Zamora M, Murphy J, Park JS, et al. Peripheral blood neutrophil activation patterns are associated with pulmonary inflammatory responses to lipopolysaccharide in humans. J Immunol. 2006;176(12):7753–7760. doi: 10.4049/jimmunol.176.12.7753. [DOI] [PubMed] [Google Scholar]
- 128.Yang KY, Arcaroli JJ, Abraham E. Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am J Respir Crit Care. 2003;167(11):1567–1574. doi: 10.1164/rccm.200207-664OC. [DOI] [PubMed] [Google Scholar]
- 129.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177(3):1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 130.Osuchowski MF, Craciun F, Weixelbaumer KM, Duffy ER, Remick DG. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude, or phase of sepsis. J Immunol. 2012;189(9):4648–4656. doi: 10.4049/jimmunol.1201806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, Goncalves L, Gómez-Herreras JI, de Lejarazu RO, Bermejo-Martin JF. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw. 2011;22(2):82–87. doi: 10.1684/ecn.2011.0281. [DOI] [PubMed] [Google Scholar]
- 132.Novotny AR, Reim D, Assfalg V, Altmayr F, Friess HM, Emmanuel K, Holzmann B. Mixed antagonist response and sepsis severity-dependent dysbalance of pro- and anti-inflammatory responses at the onset of postoperative sepsis. Immunobiology. 2012;217(6):616–621. doi: 10.1016/j.imbio.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 133.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17(6):463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 134.Hack CE, De Groot ER, Felt-Bersma RJ, Nuijens JH, Strack Van Schijndel RJ, Eerenberg-Belmer AJ, Thijs LG, Aarden LA. Increased plasma levels of interleukin-6 in sepsis. Blood. 1989;74(5):1704–1710. [PubMed] [Google Scholar]
- 135.Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J Exp Med. 1989;169(1):333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2002;2003;421(6921):384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 137.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248(1):188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 139.Kalis C, Kanzler B, Lembo A, Poltorak A, Galanos C, Freudenberg MA. Toll-like receptor 4 expression levels determine the degree of LPS-susceptibility in mice. Eur J Immunol. 2003;33(3):798–805. doi: 10.1002/eji.200323431. [DOI] [PubMed] [Google Scholar]
- 140.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 141.Salomao R, Brunialti MK, Gomes NE, Mendes ME, Diaz RS, Komninakis S, Machado FR, da Silva ID, Rigato O. Toll-like receptor pathway signaling is differently regulated in neutrophils and peripheral mononuclear cells of patients with sepsis, severe sepsis, and septic shock. Crit Care Med. 2009;37(1):132–139. doi: 10.1097/CCM.0b013e318192fbaf. [DOI] [PubMed] [Google Scholar]
- 142.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25(8):1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 143.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM, 2nd, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287(13):1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 144.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 145.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166(11):6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 146.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, Karl IE. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. 2002;168(5):2493–2500. doi: 10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 147.Albuszies G, Radermacher P, Vogt J, Wachter U, Weber S, Schoaff M, Georgieff M, Barth E. Effect of increased cardiac output on hepatic and intestinal microcirculatory blood flow, oxygenation, and metabolism in hyperdynamic murine septic shock. Crit Care Med. 2005;33(10):2332–2338. doi: 10.1097/01.ccm.0000182817.20977.e9. [DOI] [PubMed] [Google Scholar]
- 148.Dahn MS, Mitchell RA, Lange MP, Smith S, Jacobs LA. Hepatic metabolic response to injury and sepsis. Surgery. 1995;117(5):520–530. doi: 10.1016/s0039-6060(05)80251-x. [DOI] [PubMed] [Google Scholar]
- 149.Wilmore DW, Goodwin CW, Aulick LH, Powanda MC, Mason AD, Jr, Pruitt BA., Jr Effect of injury and infection on visceral metabolism and circulation. Ann Surg. 1980;192(4):491–504. doi: 10.1097/00000658-198010000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barth E, Radermacher P, Thiemermann C, Weber S, Georgieff M, Albuszies G. Role of inducible nitric oxide synthase in the reduced responsiveness of the myocardium to catecholamines in a hyperdynamic, murine model of septic shock. Crit Care Med. 2006;34(2):307–313. doi: 10.1097/01.ccm.0000199070.46812.21. [DOI] [PubMed] [Google Scholar]
- 151.Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100(4):483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 152.Kumar A, Brar R, Wang P, Dee L, Skorupa G, Khadour F, Schulz R, Parrillo JE. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276(1 Pt 2):R265–R276. doi: 10.1152/ajpregu.1999.276.1.R265. [DOI] [PubMed] [Google Scholar]
- 153.Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89(5):2070–2070. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- 154.Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, Watterson Ma TY. Mechanism of interleukin-1β induced-increase in mouse intestinal permeability in vivo. J Interferon Cytokine Res. 2012;32(10):474–484. doi: 10.1089/jir.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Al-Sadi R, Guo S, Dokladny K, Alhmound T, Ereifej L, Said HM, Ma TY. Mechanism of interleukin-1β modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation. J Immunol. 2013;190(12):6596–6606. doi: 10.4049/jimmunol.1201876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease--enhanced production during active disease. Gut. 1990;31(6):686–689. doi: 10.1136/gut.31.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178(7):4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol. 2008;180(8):5653–5661. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116(10):2682–2694. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115(10):2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M, Foligne B, Ollendorff V, Heyman M, Bonacorsi S, et al. CARD15/NOD2 is required for Peyer's patches homeostasis in mice. PLoS One. 2007;2(6):e523. doi: 10.1371/journal.pone.0000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barreau F, Madre C, Meinzer U, Berrebi D, Dussaillant M, Merlin F, Eckmann L, Karin M, Sterkers G, Bonacorsi S. Nod2 regulates the host response towards microflora by modulating T cell function and epithelial permeability in mouse Peyer's patches. Gut. 2010;59(2):207–217. doi: 10.1136/gut.2008.171546. [DOI] [PubMed] [Google Scholar]
- 163.Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. 1999;112(1):137–146. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 164.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286(3):G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 165.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G422–6430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 166.Mason KL, Rogers LM, Soares EM, Bani-Hashemi T, Erb Downward J, Agnew D, Peters-Golden M, Weinberg JB, Crofford LJ, Aronoff DM. Intrauterine group A streptococcal infections are exacerbated by prostaglandin E2. J Immunol. 2013;191(5):2457–2465. doi: 10.4049/jimmunol.1300786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 168.del Fresno C, García-Rio F, Gómez-Piña V, Soares-Schanoski A, Fernández-Ruíz I, Jurado T, Kajiji T, Shu C, Marín E, Gutierrez del Arroyo A, et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol. 2009;182(10):6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 169.Mendes ME, Baggio-Zappia GL, Brunialti MK, Fernandes Mda L, Rapozo MM, Salomao R. Differential expression of toll-like receptor signaling cascades in LPS-tolerant human peripheral blood mononuclear cells. Immunobiology. 2011;216(3):285–295. doi: 10.1016/j.imbio.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 170.Williams A, Wang JJ, Wang L, Sun X, Fischer JE, Hasselgren PO. Sepsis in mice stimulates muscle proteolysis in the absence of IL-6. Am J Physiol. 1998;275(6 Pt 2):R1983–1991. doi: 10.1152/ajpregu.1998.275.6.R1983. [DOI] [PubMed] [Google Scholar]
- 171.Klaude M, Mori M, Tjäder I, Gustafsson T, Wernerman J, Rooyackers O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci (Lond) 2012;122(3):133–142. doi: 10.1042/CS20110233. [DOI] [PubMed] [Google Scholar]
- 172.Lang CH, Lynch CJ, Vary TC. BCATm deficiency ameliorates endotoxin-induced decrease in muscle protein synthesis and improves survival in septic mice. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R935–R944. doi: 10.1152/ajpregu.00297.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Plank LD, Connolly AB, Hill GL. Sequential changes in the metabolic response in severely septic patients during the first 23 days after the onset of peritonitis. Ann Surg. 1998;228(2):146–158. doi: 10.1097/00000658-199808000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Weijer S, Schoenmakers SH, Florquin S, Levi M, Vlasuk GP, Rote WE, Reitsma PH, Spek CA, van der Poll T. Inhibition of the tissue factor/factor VIIa pathway does not influence the inflammatory or antibacterial response to abdominal sepsis induced by Escherichia coli in mice. J Infect Dis. 2004;189(12):2308–2317. doi: 10.1086/421031. [DOI] [PubMed] [Google Scholar]
- 175.Aasen AO, Smith-Erichsen N, Amundsen E. Studies on pathological plasma proteolysis in patients with septicemia. Scand J Clin Lab Invest Suppl. 1985;178:37–45. [PubMed] [Google Scholar]
- 176.Franco RF, de Jonge E, Dekkers PE, Timmerman JJ, Spek CA, van Deventer SJ, van Deursen P, van Kerkhoff L, van Gemen B, ten Cate H, et al. The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood. 2000;96(2):554–559. [PubMed] [Google Scholar]
- 177.Solan PD, Dunsmore KE, Denenberg AG, Odoms K, Zingarelli B, Wong HR. A novel role for matrix metalloproteinase-8 in sepsis. Crit Care Med. 2012;40(2):379–387. doi: 10.1097/CCM.0b013e318232e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Crouser ED, Julian MW, Huff JE, Struck J, Cook CH. Carbamoyl phosphate synthase-1: a marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med. 2006;34(9):2439–2446. doi: 10.1097/01.CCM.0000230240.02216.21. [DOI] [PubMed] [Google Scholar]
- 179.Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2007;176(8):768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 181.Carré JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182(6):745–751. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE, Hotchkiss RS. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009;89(5):549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. Atg16l1 is Required for Autophagy in Intestinal Epithelial Cells and Protection of Mice From Salmonella Infection. Gastroenterology. 2013;145(6):1347–1357. doi: 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Raeven P, Feichtinger GA, Weixelbaumer KM, Atzenhofer S, Redl H, Van Griensven M, Bahrami S, Osuchowski MF. Compartment-specific expression of plasminogen activator inhibitor-1 correlates with severity/outcome of murine polymicrobial sepsis. Thromb Res. 2012;129(5):e238–245. doi: 10.1016/j.thromres.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 186.Raeven P, Feichtinger GA, Weixelbaumer KM, Atzenhofer S, Redl H, Van Griensven M, Bahrami S, Osuchowski MF. A non-lethal traumatic?hemorrhagic insult strongly modulates the compartment-specific PAI-1 response in the subsequent polymicrobial sepsis. PLoS One. 2013;8(2):e55467. doi: 10.1371/journal.pone.0055467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shapiro NI, Schuetz P, Yano K, Sorasaki M, Parikh SM, Jones AE, Trzeciak S, Ngo L, Aird WC. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care. 2010;14(5):R182. doi: 10.1186/cc9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Genovese T, Mazzon E, Esposito E, Di Paola R, Murthy K, Neville L, Bramanti P, Cuzzocrea S. Effects of a metalloporphyrinic peroxynitrite decomposition catalyst, ww-85, in a mouse model of spinal cord injury. Free Radic Res. 2009;43(7):631–645. doi: 10.1080/10715760902954126. [DOI] [PubMed] [Google Scholar]
- 189.Genovese T, Mazzon E, Esposito E, Muià C, Di Paola R, Di Bella P, Bramanti P, Cuzzocrea S. Role of endogenous glutathione in the secondary damage in experimental spinal cord injury in mice. Neurosci Lett. 2007;423(1):41–46. doi: 10.1016/j.neulet.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 190.Bastani NE, Kostovski E, Sakhi AK, Karlsen A, Carlsen MH, Hjeltnes N, Blomhoff R, Iversen PO. Reduced antioxidant defense and increased oxidative stress in spinal cord injured patients. Arch Phys Med Rehabil. 2012;93(12):2223–2228. doi: 10.1016/j.apmr.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 191.Ling H, Edelstein C, Gengaro P, Meng X, Lucia S, Knotek M, Wangsiripaisan A, Shi Y, Schrier R. Attenuation of renal ischemia-reperfusion injury in inducible nitric oxide synthase knockout mice. Am J Physiol. 1999;277(3 Pt 2):F383–390. doi: 10.1152/ajprenal.1999.277.3.F383. [DOI] [PubMed] [Google Scholar]
- 192.Barrera-Chimal J, Pérez-Villalva R, Cortés-González C, Ojeda-Cervantes M, Gamba G, Morales-Buenrostro LE, Bobadilla NA. Hsp72 is an early and sensitive biomarker to detect acute kidney injury. EMBO Mol Med. 2011;3(1):5–20. doi: 10.1002/emmm.201000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang QH, Chen Q, Kang JR, Liu C, Dong N, Zhu XM, Sheng ZY, Yao YM. Treatment with gelsolin reduces brain inflammation and apoptotic signaling in mice following thermal injury. J Neuroinflammation. 2011;8:118. doi: 10.1186/1742-2094-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rothenbach PA, Dahl B, Schwartz JJ, O'Keefe GE, Yamamoto M, Lee WM, Horton JW, Yin HL, Turnage RH. Recombinant plasma gelsolin infusion attenuates burn-induced pulmonary microvascular dysfunction. J Appl Physiol. 2003;96(1):25–31. doi: 10.1152/japplphysiol.01074.2002. 1985 2004. [DOI] [PubMed] [Google Scholar]
- 195.Lee PS, Drager LR, Stossel TP, Moore FD, Rogers SO. Relationship of plasma gelsolin levels to outcomes in critically ill surgical patients. Ann Surg. 2006;243(3):399–403. doi: 10.1097/01.sla.0000201798.77133.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang LF, Yao YM, Li JF, Dong N, Liu C, Yu Y, He LX, Sheng ZY. Reduction of plasma gelsolin levels correlates with development of multiple organ dysfunction syndrome and fatal outcome in burn patients. PLoS One. 2011;6(11):e25748. doi: 10.1371/journal.pone.0025748. [DOI] [PMC free article] [PubMed] [Google Scholar]