

Abstract
Background
Known linear knottins are unsuitable as scaffolds for oral peptide drug due to their gastrointestinal instability. Herein, a new subclass of knottin peptides from Porifera is structurally described and characterized regarding their potential for oral peptide drug development.
Methods
Asteropsins B–D (ASPB, ASPC, and ASPD) were isolated from the marine sponge Asteropus sp. The tertiary structures of ASPB and ASPC were determined by solution NMR spectroscopy and that of ASPD by homology modeling.
Results
The isolated asteropsins B–D, together with the previously reported asteropsin A (ASPA), compose a new subclass of knottins that share a highly conserved structural framework and remarkable stability against the enzymes in gastrointestinal tract (chymotrypsin, elastase, pepsin, and trypsin) and human plasma.
Conclusion
Asteropsins can be considered as promising peptide scaffolds for oral bioavailability.
General significance
The structural details of asteropsins provide essential information for the engineering of orally bioavailable peptides.
Keywords: Knottin, Marine sponge, Asteropus sp, NMR, Solution structure, Oral peptide drug delivery
1. Introduction
Peptides are attractive drug candidates because of their potent biological activities as well as high target specificities [1]. However, the short in vivo half-lives of peptide therapeutics remain a major issue. In particular, oral delivery, which provides improved patient compliance, remains a primary target of peptide drug development. Recently, head-to-tail cyclized knottins (cyclotides) have been placed in the spotlight as novel scaffolds for oral peptide drug administration because of their extraordinary proteolytic stability and relatively straightforward chemical and recombinant syntheses [2–5].
Knottin peptides share a rigid molecular disulfide arrangement (III–VI through I–IV, II–V), also called a ‘knot’, and a triple-stranded antiparallel β-sheet fold. In late 2004, the knottin, ω-conotoxin MVIIA (or ziconotide) from the cone snail Conus magus became the first fully approved drug obtained directly from the marine environment for the treatment of chronic pain [6]. Several other knottins are currently undergoing preclinical or phase I/II clinical trials for the treatment of pain related diseases [7]. In addition to their potential as drug candidates, the use of knottins, especially cyclotides, has been extended to peptide engineering for the development of protease-resistant ligand scaffolds [3,4,8–12]. The cyclotide kalata B1, which exhibits stability towards pepsin, trypsin, and chymotrypsin, is a promising scaffold for the development of orally effective peptide drugs; however, the oral bio-availability of kalata B1 was found to be dramatically reduced when its head-to-tail cyclization was not performed [3,4]. The linear knottins from squash (SE-EM and SE-EP), human agouti related protein (SE-AGAZ), and even hybrid recombinant knottins (SE-ET-TP-020 and SE-MC-TR-020) were found to be extensively degraded by chymotrypsin or trypsin [13–15]. The approved drug ziconotide, which is a linear knottin, is also unstable to trypsin, and thus, must be administered intrathecally [16,17]. In a recent study by Clark et al., it was found that the cyclic product obtained by cyclizing a 16-residue conotoxin with a hydrophobic linker consisting of six aliphatic amino acids was orally effective, whereas the linear native conotoxin remained orally ineffective [18]. Linear knottins reported so far are not suitable as scaffolds for oral delivery, although their syntheses are less complicated because they do not require an additional step for head-to-tail cyclization.
Knottins have been reported in many organisms, though most have been discovered in spider or cone snail venoms. Relatively few marine-derived knottins have been discovered in sources other than cone snails. In 2006, Fusetani et al. first identified asteropine A (APA; a bacterial sialidase inhibitor) in the marine sponge Asteropus simplex [19]; and later, we isolated asteropsin A (ASPA) from the same sponge genus and validated Porifera as a source of unusual knottin-like peptides [20]. The N-terminal modification, absence of basic residues, the cis conformation of prolines of ASPA and its unique bioactivity distinguish it from other reported knottins.
Herein we report three unusual knottin-like peptides, asteropsins B–D (ASPB, ASPC and ASPD) from the same sponge Asteropus sp. In addition to their moderate sequence homologies, NMR derived solution structures revealed that these sponge-derived peptides have highly conserved tertiary structures. Asteropsins A–D share several conserved residues at special locations that are likely to maintain structural stability. In addition, N-terminal blocking and the absence of basic residues make them inherently stable to aminopeptidases and trypsin. In our evaluation of their proteolytic resistances against major gastrointestinal proteases (chymotrypsin, elastase, pepsin, and trypsin) and human plasma, all four asteropsins exhibited notable resistance, which suggests that they can be utilized as novel linear knottin scaffolds for orally delivered peptide drugs.
2. Materials and methods
2.1. Animal material and peptide purification
The marine sponge Asteropus sp. (2.4 kg, wet weight) was collected by hand at a depth of 20 m in 2006 off the coast of Geoje Island, Korea, and stored at −20 °C until used. The frozen sponge (2.4 kg, wet weight) was extracted with MeOH at room temperature, and the extract (166 g) was then partitioned between water and CH2Cl2 (1:1, v/v). The aqueous layer was further partitioned with BuOH and water (1:1, v/v), and the organic layer (5.1 g) obtained was subjected to step-gradient MPLC (ODS-A, 120 Å, S-30/50 mesh) using 20–100% methanol as eluant. Asteropsins B–D (7.5, 68, and 2.7 mg, respectively) were purified by reversed-phase HPLC equipped with an RI detector (YMC ODS-H80 column 250 mm × 10 mm, i.d. 4 μm, 80 Å) using 60% MeOH + 0.2% HCOOH at a flow rate of 1 mL/min.
2.2. Reduction and alkylation of peptides
A portion of each peptide (100 μg) was dissolved in 100 μL of denaturation buffer (7 M guanidine hydrochloride in 0.4 M Tris-acetate-EDTA buffer, pH 8.3) and 10 μL of 45 mM dithiothreitol (DTT) was then added. The mixture was incubated at 60 °C for 90 min and then 20 μL of 100 mM iodoacetamide was added and incubated at room temperature for 45 min. The reaction mixture was purified by reversed-phase HPLC equipped with a UV detector (Waters ODS-2 column 250 mm × 4.6 mm, i.d. 5 μm; wavelength: 220 nm) using a linear gradient (30–80% solvent B; solvent A: H2O + 0.1% TFA, solvent B: 90% ACN + 0.1% TFA) to afford hexacarboxamidomethyl derivatives (m/z 4267.7 [M + Na]+, 4135.7 [M + Na]+, and 3774.3 [M + H]+ for ASPB, ASPC, and ASPD, respectively).
2.3. MALDI-TOF MS
Each peptide (0.1 mg) was dissolved in DMSO and then diluted tenfold with α-cyano-4-hydroxycinnamic acid matrix solution (7 mg/mL in 50% ACN, 0.1% TFA). Then, 1 μL of each sample/matrix solution was spotted onto a MALDI plate and inserted into a MALDI-TOF unit (Applied Biosystems 4700 proteomics analyzer, Framingham, MA).
2.4. N-terminal deblocking and sequence analysis
The hexacarboxamidomethyl derivatives (<30 μg) of asteropsins B–D were digested with 2 mU of pyroglutamate aminopeptidase (Takara Bio Inc., Shiga, Japan) dissolved in 100 μL of supplied buffer (50 mM sodium phosphate buffer containing 10 mM DTT and 1 mM EDTA; pH 7.0) containing 5% glycerol at 50 °C for 10 h. Digests were desalted with a sample preparation cartridge (Prosorb, Applied Biosystems, Foster City, CA), and subjected to automatic Edman degradation in a protein sequencing system (Procise 491; Applied Biosystems). Sequence analysis was performed at KBSI (Korea Basic Science Institute, Seoul). Sequence alignment was performed using the program ClustalX (ver. 2.0, http://www.clustal.org).
2.5. NMR spectroscopy
ASPB and ASPC were dissolved in DMSO-d6 and CD3OH (Sigma Aldrich Chemical Co., St. Louis, MO, USA), respectively, degassed, and topped with argon to minimize moisture absorption. Residual hydroxyl signal suppression was achieved using a standard presaturation method. Standard experimental parameters were used for the acquisition of HSQC (Adiabatic Decoupling), z-TOCSY (DIPSI, mixing time = 80 ms), DQF-COSY, and NOESY (mixing time = 100 and 300 ms) using Varian 900 and 600 MHz (13C = 150 MHz) INOVA at 298 K with reference to the internal lock signal. Processing was completed using NMRpipe (ver. 5.5, NIH) and chemical shift analysis using SPARKY (ver. 3.114, UCSF) [21], with spectra image rendering using MestReNova (ver. 6.2.0, MestreLab Research S.L.).
2.6. Structure calculations
Interproton distance constraints were obtained from the 100 ms and 300 ms mixing time NOESY spectra recorded in DMSO-d6 or CD3OH for ASPB and ASPC, respectively. NOESY spectra were analyzed with the program SPARKY [21]. Cross peaks were categorized into four classes by peak intensity (2.5, 3.0, 4.0, and 5.0 Å, which corresponded to strong, medium, weak, and very weak correlations). Pseudo-atoms were applied for methyl, non-stereospecifically assigned methylene, and aromatic protons according to a standard method [22]. Dihedral angle constraints were generated from 3JNH-Hα values > 8.0 Hz in DQF-COSY. Hydrogen bond restraints were collected by long-range NOE correlations together with hydrogen–deuterium exchange experiments using CD3OH and CD3OD as solvents. In addition, chemical shift indices (CSI) [23,24] were used to determine secondary structures.
Solution structural calculations were initially performed by CYANA 2.1 [25] using distance, dihedral angle, and hydrogen bond constraints, and further refined by simulated annealing within CNS 1.3 [26]. A final set of 200 structures was calculated within the CNS program, and the 20 structures with lowest energies and no residual restraint violations were used to represent the solution structures of the peptides. The calculated three-dimensional structures were analyzed with the program PYMOL [27] and MOLMOL [28]. Structure qualities were validated by the PSVS (protein structure validation software suite) server (http://psvs-1_4-dev.nesg.org/) [29]. The solution structures of ASPB and ASPC have been deposited in the RCSB Protein Data Bank (PDB ID: 2LZX and 2LZY, respectively).
2.7. Homology modeling of ASPD
Prediction including loop refinement of ASPD was performed in Prime (Schrödinger, LLC) using the solution structure of ASPC (PDB ID: 2LZY), its closest sequence homolog, as a template. The ASPD model obtained was further refined using ModRefiner [30], and energy minimization was performed by CHARMm force field. The resulting model was analyzed using PYMOL [27] and PSVS server (http://psvs-1_4-dev.nesg.org/) [29].
2.8. Gastrointestinal enzymatic stability of asteropsins A–D
Enzymatic degradations by chymotrypsin (Sigma C3142, 54 BTEE units/mg), elastase (Sigma E7885, 4 N-succinyl-L-Ala-Ala-Ala-p-nitroanilide units/mg), and trypsin (Sigma T4799, 1753 BAEE units/mg) were performed at protease concentrations typically found in human intestinal fluid [31]. Each peptide (100 μL of 1 mg/mL in 100 mM Tris–HCl buffer, pH 7.6) was added to: 200 μL of chymotrypsin (0.76 mg/mL in 100 mM Tris–HCl buffer containing 10 mM CaCl2, pH 7.6), 200 μL of elastase (0.0625 mg/mL in 100 mM Tris–HCl buffer containing 1% KCl, pH 7.6), or to 200 μL of trypsin (2.78 mg/mL in 100 mM Tris–HCl buffer, pH 7.6). For pepsin (Sigma P7000, 250 hemoglobin units/mg) degradation, peptide samples were prepared in 0.08 M HCl (the pH was adjusted to 2 with 1 M NaOH) containing 30% MeOH. Each peptide solution (100 μL) was added to 200 μL of pepsin (2.4 mg/mL in 0.08 M HCl, pH 2). The peptide–enzyme solutions were incubated at 37 °C with agitation (160 rpm). At predetermined time points (0, 5, 15, 30, 60, 120, and 240 min), aliquots (40 μL) were withdrawn and enzymatic reactions were stopped immediately by adding 0.1% TFA (40 μL) to intestinal protease solutions and by adding 0.1 M NaOH (40 μL) to pepsin solution. Insulin (1 mg/mL, prepared in 0.1 M NaHCO3 for intestinal proteases; and in 0.08 M HCl for pepsin) was used as a reference standard for enzyme activities. Reaction mixtures were analyzed by reversed-phase HPLC using a UV detector (YMC ODS column 250 mm × 4.6 mm, i.d. 5 μm; wavelength 220 nm) at a flow rate of 0.5 mL/min using linear gradient elution (30–80% solvent B; solvent A: H2O + 0.1% TFA, solvent B: 90% ACN + 0.1% TFA).
2.9. Plasma stability of asteropsins A–D
To 400 μL of human plasma (Sigma), 100 μL of peptide (1 mg/mL in 100 mM Tris–HCl buffer, pH 7.6) was added. The peptide–plasma solution was incubated at 37 °C while shaking (160 rpm) during the sampling period. At different time points (0, 30, 60, 120, and 240 min), 50 μL of reaction sample was taken out and quenched to precipitation by MeOH (50 μL). The precipitate was removed by centrifugation at 13500 rpm for 20 min. The supernatant was analyzed by reversed-phase HPLC using a UV detector (YMC ODS column 250 mm × 4.6 mm, i.d. 5 μm; wavelength: 220 nm) at a flow rate of 0.5 mL/min with a linear gradient elution (30–80% solvent B; solvent A: H2O + 0.1% TFA, solvent B: 90% ACN + 0.1% TFA).
3. Results
3.1. Sequence analysis
Asteropsins B–D (ASPB, ASPC, and ASPD) were isolated from the MeOH extract of the marine sponge Asteropus sp. by solvent partitioning followed by reversed-phase HPLC. The MALDI-TOF MS of asteropsins B–D exhibited monoisotopic pseudomolecular ion peaks at m/z 3919.5 [M + Na]+; 3787.5 [M + Na]+; and 3426.2 [M + H]+, respectively. Previously isolated pyroglutamyl dipeptides [32] and asteropsin A (ASPA) [20] from the same sponge aided confirmation of the presence of a pyroglutamyl ring NH (δH 7.82–7.84 ppm) based on signal shape, chemical shift, and HMBC (DMSO-d6) correlations. NMR evidence and difficulties with traditional sequence analysis indicated that the N-terminus of each peptide was blocked by a pyroglutamic acid (pGlu). Amino acid analyses and initial NMR spectral interpretations indicated that asteropsins B–D were composed of common amino acids with several disulfide bonds. Thus, before sequence analysis, pretreatments were required for disulfide bond reduction and N-terminal deblocking. Asteropsins B–D were reduced with dithiothreitol and alkylated with iodoacetamide to afford corresponding hexacarboxamidomethyl derivatives (m/z 4267.7 [M + Na]+, 4135.7 [M + Na]+, and 3774.3 [M + H]+, respectively), indicating the presence of three disulfide bonds per molecule. Derivatives were then digested with pyroglutamate aminopeptidase to remove N-terminal pGlu prior to Edman degradation sequence analysis. Sequence alignments of asteropsins B–D versus ASPA are shown in Fig. 1.
Fig. 1.
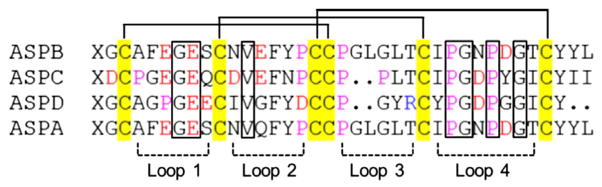
Sequence alignments of asteropsins A–D (ASPA, ASPB, ASPC, and ASPD). The disulfide bonds are connected by solid lines. Residues: red = acidic, blue = basic, yellow = Cys, purple = Pro. X = pyroglutamic acid. The seven conserved residues involved in structural maintenance are marked with boxes, and the loops indicate the residues comprising polypeptide backbones between two Cys residues. Sequence alignment was performed with ClustalX 2.0.
3.2. Secondary structures of ASPB and ASPC
As ASPB and ASPC were virtually insoluble in pure water or low pH buffers, DMSO-d6 and CD3OH were used to record 2D NMR spectra of ASPB (600 MHz) and ASPC (900 MHz), respectively. Sequence-specific resonance assignments were made using the standard method for small proteins [22] using SPARKY [21]. NOE distance constraints, 3JNH-Hα values, and chemical shift indices (CSI) [23,24] supported the assignment of three antiparallel β-sheets of ASPB (Ser9-Asn11, Leu23-Pro27, and Asp31-Tyr36), and ASPC (Glu9-Asp11, Thr22-Pro25, and Tyr29-Tyr33) (Fig. 2). Turns were identified by characteristic distances of backbone protons [22] knowing that the relationship between Cα(i) and Cα(i + 3) is <7 Å [33]. β-Turn types (turn I and turn II) were classified as described by Wilmot and Thornton [34]. As a result, short type I β-turns were identified in ASPB (Val12-Tyr15 and Gly28-Pro30) and ASPC (Gly26 and Asp27). As expected, the secondary structure of ASPB was consistent with that of previously isolated ASPA due to the 97% sequence homology between the two peptides.
Fig. 2.

Secondary structure elements of ASPC. (A) Summary of sequential and medium range NOE correlations (|i°–°j| < 5), hydrogen–deuterium exchange experiments, 3JNH-Hα (3JαN) values, and chemical shift indices (CSI). NOE intensities are grouped into 4 classes (2.5, 3.0, 4.0, and 5.0 Å) and are represented by the thicknesses of solid lines. Thicker lines indicate stronger NOE correlations. Open circles indicate the amide protons observed in 1H and TOCSY spectra recorded in CD3OD (500 MHz). Solid circles indicate the positions where 3JNH-Hα values are larger than 8 Hz. Consensus results of CSI derived from CαH, Cα, and Cβ chemical shifts of ASPC are indicated by a ternary index with values of −1, 0 and +1. The arrows at the top of the figure indicate the positions of β-sheets as determined by a combination of strong sequential dαN, weak dNN, non-exchanged amide protons, large 3JNH-Hα values, and a CSI value of +1. (B) Long-range NOE correlations (|i°–°j| ≥ 5) and H-bonds of the triple-stranded antiparallel β sheet region in ASPC. NOE correlations and H-bonds are indicated by solid arrows and dashed lines, respectively.
The disulfide arrangements of ASPB and ASPC were initially presumed to be identical to that of ASPA (I–IV, II–V, and III–VI) based on sequence framework similarities verified by experimental NOE correlations. NOE correlations between CβH-CβH and CαH-CβH indicated the presence of a disulfide bonding pattern (dββ < 4.0 Å, 95.7% of cysteines form a pair, dββ < 5.0 Å, 88.8%) [35]. ASPB exhibited CβH-CβH NOE correlations between Cys3/Cys18, and between Cys17/Cys34, and CαH-CβH NOEs of Cys3/Cys18 and Cys17/Cys34, which strongly suggested Cys3–Cys18 and Cys17–Cys34 cysteine pairs. Thus, by default, the third disulfide bond was located between Cys10 and Cys25. ASPC showed CβH-CβH NOEs of Cys3/Cys18 and Cys17/Cys32, and CαH-CβH NOEs of Cys3/Cys18, indicative of Cys3–Cys18, Cys10–Cys23, and Cys17–Cys32 disulfide pairings. A range of additional CαH-CβH NOEs between cysteines (ASPB: Cys34/Cys25; ASPC: Cys17/Cys3, Cys17/Cys18, Cys32/Cys23) that was inconsistent with typical disulfide connectivity were also observed. However, analysis of the possible disulfide bonding patterns by calculating the S–S distances (Å) from the structures without disulfide bond restraints further supported that both ASPB and ASPC conformed I–IV, II–V and III–VI disulfide pairings (Table S1).
The cis–trans conformation of the Pro residues was determined by Δβ γ(δ[13Cβ] – δ[13Cγ]) chemical shift differences [36] and NOE analysis [22]. ASPB and ASPC were found to have two highly conserved cis prolines (Pro27 and Pro30 in ASPB; and Pro25 and Pro28 in ASPC) located at the end of the second and before the third β-strands, respectively; this conformation was also conserved in ASPA [20].
3.3. Solution structures of ASPB and ASPC
The solution structures of ASPB and ASPC were initially calculated by CYANA 2.1 [25] using distance, dihedral angle, and hydrogen bond constraints, and further refined by simulated annealing within CNS 1.3 [26]. For each peptide, the 20 structures with lowest energies and no residual restraint violations were selected to represent solution structures (Fig. 3). Structure qualities were validated using the PSVS server (http://psvs-1_4-dev.nesg.org/) [29]; statistics are detailed in Table 1. The Ramachandran plot of ASPB from Richardson Lab’s Molprobity (integrated in PSVS) showed 91.3% in the most favored region and 8.7% in the allowed region. For ASPC, these values were 84.4% and 15.6%, respectively.
Fig. 3.

Solution NMR structures of ASPB, ASPC, and ASPA. (A) Overlapped 20 lowest-energy NMR structures representing solution structures of ASPB (left), ASPC (middle), and ASPA (right). The side chain of N-terminal pGlu (PCA) is modeled using sticks and labeled. The seven conserved residues involved in structural maintenance in each peptide are labeled and colored red; their side chains are modeled using sticks. Six Cys residues and disulfide bonds are colored yellow. The average root-mean-square deviations (RMSDs) calculated for residues 2–37 of ASPB were 0.02 Å for backbone atoms, and 0.47 Å for all heavy atoms (N, Cα, C). RMSDs calculated for residues 2–35 of ASPC were 0.13 Å for backbone atoms and 0.46 Å for all heavy atoms. (B) Lowest energy solution structures of ASPB [PDB ID: 2LZX], ASPC [PDB ID: 2LZY], and ASPA [PDB ID: 2LQA]. The three disulfide bonds and β-sheets are colored yellow and blue, respectively. The six Cys residues are labeled. The figure was drawn using PYMOL.
Table 1.
Structural statistics for the 20 lowest energy solution structures of ASPB and ASPC as determined by PSVS (protein structure validation suite).
| ASPBa | ASPCb | |
|---|---|---|
| Experimental constraints | ||
| Total NOE | 639 | 552 |
| Intra-residue [i = j] | 94 | 114 |
| Sequential [|i – j| = 1] | 199 | 197 |
| Medium range [1 < |i – j| < 5] | 91 | 56 |
| Long range [|i – j| ≥ 5] | 255 | 185 |
| Dihedral angles | 18 | 18 |
| Hydrogen bonds | 18 | 20 |
| Violations | ||
| Distance (>0.1 Å) | 0 | 0 |
| Dihedral angle (>1°) | 0 | 0 |
| Van der Waals (<1.6 Å) | 0 | 0 |
| RMSD from idealized geometryc | ||
| Bond lengths (Å) | 0.009 | 0.011 |
| Bond angles (°) | 0.6 | 0.8 |
| RMS of distance violation (Å) | 0.01 | 0.01 |
| RMS of dihedral angle violation (°) | 0 | 0.008 |
| Average pairwise RMSD values (Å)c | ||
| All backbone atoms | 0.02 | 0.13 |
| All heavy atoms | 0.47 | 0.46 |
| Ramachandran plot statistics from Richardson’s lab (%) | ||
| Most favored regions | 91.3 | 84.4 |
| Allowed regions | 8.7 | 15.6 |
| Disallowed regions | 0 | 0 |
Analyzed for residues 2 to 37.
Analyzed for residues 2 to 35.
RMSD values are mean values.
The average root-mean-square deviations (RMSDs) of ASPB (residues 2–37) were 0.02 Å for backbone atoms, and 0.47 Å for all heavy atoms (N, Cα, C). The solution structure of ASPB matched that previously reported for ASPA, with exception to slight structural differences, especially in the loop 3 region (Pro19-Gly22), which may have resulted from their acquisition in different solvent environments.
The RMSDs of the backbone and heavy atoms of ASPC (residues 2–35) were 0.13 Å and 0.46 Å, respectively. Although its ASPA sequence homology is relatively low compared to other asteropsin analogs, ASPC still exhibited tertiary structures similar to ASPA [backbone RMSDs: 0.88 Å for residues 3–17 (loops 1 and 2) and 0.48 Å for residues 21–33 (loop 4)], and ASPB [backbone RMSDs: 1.02 Å for residues 3–17 (loops 1 and 2) and 0.71 Å for residues 21–33 (loop 4)]. The solution structures of ASPB and ASPC have been deposited in the RCSB PDB (ID: 2LZX and 2LZY, respectively).
3.4. Homology modeling of ASPD
Because the amount of ASPD isolated was not sufficient for a NMR-based structure elucidation, its tertiary structure was built by homology modeling using the solution structure of ASPC (PDB ID: 2LZY) as a template, and further refined by ModRefiner [30]. According to the sequence alignment analysis (Fig. 1), ASPD displayed 57% sequence homology with ASPC, which provided sufficient identity to carry out homology modeling. The minimized total energy of the ASPD model refined by a CHARMm force field was −1388.09 kcal/mol. The final conformer was validated using a Ramachandran plot. This showed 93.3% in the most favored region with 6.7% in the allowed region. According to the homology model, ASPD forms three anti-parallel β-sheets composed of the residues Glu9-Ile11, Arg22-Pro25, and Gly29-Tyr33 (Fig. 4). The homology model of ASPD was highly comparable to ASPA [backbone RMSDs: 1.11 Å for residues 3–17 (loops 1 and 2) and 0.64 Å for residues 21–31 (loop 4)], ASPB [backbone RMSDs: 1.28 Å for residues 3–17 (loops 1 and 2) and 0.54 Å for residues 21–31 (loop 4)], and ASPC [backbone RMSD: 0.83 Å for residues 1–31].
Fig. 4.
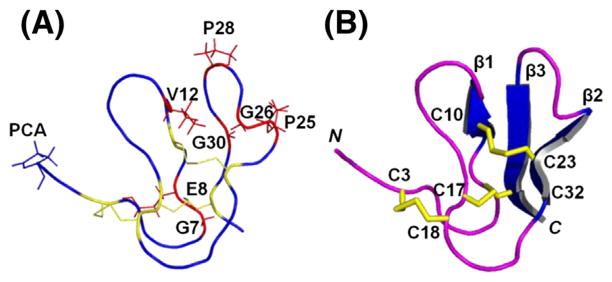
Homology model of ASPD using ASPC [PDB ID: 2LZY] as a template. (A) Ribbon representation of the homology model of ASPD. The N-terminal pGlu (PCA) side chain is modeled using sticks and labeled. The seven conserved residues involved in the structural maintenance are labeled and colored red; their side chains are modeled using sticks. The six Cys residues and disulfide bonds are colored yellow. (B) Cartoon representation of the refined homology model of ASPD. The three disulfide bonds and β-sheets are colored yellow and blue, respectively. The six Cys residues are labeled. The figure was drawn using PYMOL.
3.5. Description of tertiary structures
Several structural aspects are shared by asteropsins B–D. They have hemispherical tertiary structures. Their anti-parallel β-sheets are reinforced by two (III–VI and II–V) of the three disulfide bridges that lie at their centers with their third disulfide bridge closer to the surface near the N-terminus (Figs. 3 and 4).
In addition to N-terminal pGlu and six Cys, asteropsins A–D share several other conserved residues at specified locations. The first β-sheet is always located between GlyI-GluII and ValIII; the second β-sheet ends with a cis-ProIV followed by GlyV; the third β-sheet starts after another cis-ProVI and always contains a conserved GlyVII residue (Fig. 5A). According to the tertiary structures of asteropsins A–D, the distances of dHN-O(GlyI,CysVI), dHN-O(CysVI,GluII), dN-O(ValIII,ProVI), dHN-O(CysII,GlyVII), and dHN-O(GlyVI, CysII) were <3 Å, indicating the possible presence of backbone H-bonds. These seven conserved residues, which protect the secondary structures, are most likely to provide an H-bond network within the cystine knot and are a key structural support of the asteropsins.
Fig. 5.

(A) Conserved structure-maintaining sequence pattern of asteropsins A–D. The residues with superscript represent the conserved residues. The disulfide bonds are connected by solid lines, and three β-strands are shown with arrows. The loops indicate the residues comprising polypeptide backbones between two Cys residues. (B) Structure alignments of asteropsins A–D. The six Cys residues and seven structurally important residues are labeled, and colored yellow and red, respectively. The loop 3 region is indicated by the box. The backbone RMSD (residues 2–36) of ASPB with ASPA was 0.86 Å. The backbone RMSDs of ASPC with ASPA were 0.88 Å for residues 3–17 (loops 1 and 2) and 0.48 Å for residues 21–33 (loop 4); and those with ASPB were 1.02 Å for residues 3–17 (loops 1 and 2), and 0.71 Å for residues 21–33 (loop 4). The homology model of ASPD was highly comparable to ASPA [backbone RMSDs: 1.11 Å for residues 3–17 (loops 1 and 2), and 0.64 Å for residues 21–31 (loop 4)], ASPB [backbone RMSDs: 1.28 Å for residues 3–17 (loops 1 and 2), and 0.54 Å for residues 21–31 (loop 4)], and ASPC [backbone RMSD: 0.83 Å for residues 1–31]. The figure was drawn using PYMOL.
The N-terminal pGlu, Phe in loop 2, and Tyr near the C-terminus are highly exposed to the solvent, which are not considered necessary for the structural maintenance of asteropsins. The most significant structural differences among asteropsins A–D were observed within the region of loop 3 (Fig. 5B), thus, the conserved trans-Pro residue in this region was excluded in consideration for being the residue involved in the structural integrity of asteropsins. Residue variations at other locations do not substantially affect the structural framework of asteropsins, which is supported by highly conserved tertiary structures with average backbone RMSDs < 1 Å between each other (Fig. 5B). Thus, the conserved sequence pattern of – CI – GIEII(–CII–)β1VIII – CIIICIV – (–CV – PIV)β2GV – PVI(–GVII – CVI–)β3–, together with a tightly knotted disulfide core, is speculated to maintain the tertiary structural framework of asteropsins.
3.6. Enzymatic stabilities of asteropsins A–D
Considering the potential biological activity of asteropsins, and their utilities for the oral delivery of peptide drugs like kalata B1 [3,4], we examined the stabilities of asteropsins A–D in the presence of chymotrypsin, elastase, pepsin, or trypsin at concentrations present in human intestinal fluid [31]. Insulin was used as the reference standard to estimate enzyme activities. Asteropsins A–D were found to be stable in the presence of chymotrypsin, elastase, and pepsin for up to 4 h, whereas insulin was completely degraded in less than 5 min by chymotrypsin and pepsin, and degraded over 60% by elastase within 4 h (Fig. 6A–C). Because asteropsins A–C do not contain basic residues (Arg, His, or Lys), they do not have a trypsin cleavage site. Thus, only ASPD, which contains Arg, was tested. It was found to be unaffected by exposure to trypsin for 4 h (Fig. 6D).
Fig. 6.

Stabilities of asteropsins A–D against enzymatic degradation by (A) chymotrypsin, (B) elastase, (C) pepsin, and (D) trypsin. Insulin was used as a reference standard to estimate enzyme activities. Enzymatic degradation was performed at protease concentrations found in human intestinal fluid. Each point represents the means ± SDs of three experiments.
The stability of asteropsins in human plasma was also investigated, and asteropsins A–D showed remarkable stability up to 4 h (Fig. 7).
Fig. 7.

Stabilities of asteropsins A–D against the enzymes in human plasma. Each point represents the means ± SDs of three experiments.
4. Discussion
In the present study, we describe three new knottin-like peptides, which were isolated from the marine sponge Asteropus sp. The solution structures of ASPB and ASPC were determined using NMR techniques, whereas the tertiary structure of ASPD was derived by homology modeling. With the exception of cystine arrangements, asteropsins bear little sequence resemblance to other reported knottins. Asteropsins B–D and the previously reported ASPA compose a new knottin subfamily with unique features of N-terminal blocking, a highly acidic nature, conserved structurally important residues including two cis-prolines, and henceforth highly conserved tertiary structures. As is common for many knottins, the side chains of most residues in asteropsins appear to radiate out from the center of the structure where the disulfide bridges are located, which may increase their interaction with receptor proteins.
Regarding peptide sequences, the four asteropsins have different lengths, and possess a number of different residues, but their tertiary structures are highly comparable. Significant structural deviations are avoided by the length of sequence variances, especially within the coil regions of loop 3 (Fig. 5B). To draw an analogy, the six disulfide-crosslinked Cys residues and the seven highly conserved residues function as ‘nails’ that determine the spatial disposition of the main scaffold, whereas other residues act rather like ‘elastic bands’ around these loci. The identification of the conserved residues that primarily determine the structures of asteropsins is important in the contexts of structure–activity relationships (SARs) and peptide engineering with respect to the developments of bioactive peptides that conserve tertiary structure.
4.1. Structure–activity relationships (SARs) of ASPA and ASPC with respect to neuronal Ca2+ influx
During our investigations of the effects of ASPA and ASPC on neuronal Ca2+ influx, it was found that ASPA significantly enhanced veratridine-induced Ca2+ influx in murine cerebrocortical neuron cells at an EC50 value of 14 nM [20]. However, despite a similar sequence pattern and a highly comparable tertiary structure (Fig. 5), ASPC was found to have no effect on neuronal Ca2+ influx (data not shown). Thus, the different effects of ASPA and ASPC on neuronal Ca2+ influx are probably due to their sequence gap.
Because of the relatively large sizes of knottins, most interactions between knottins and target proteins are attributable to surface exposed residues. Thus, the distribution of active residues in relatively confined regions should be important to determine binding affinities. This is supported by the reported crystal structures of knottin–protein complexes [37–40], and by SAR studies of ω-conotoxin MVIIA [41,42], hainantoxin IV [43], and kalata B1 [44], which showed that all possess a clustered target protein binding region (Fig. 8A and B). Thus, we compared the surface profiles of ASPA and ASPC. In ASPA, Glu6, Glu8, Ser9, Asp31, and Thr33 were distributed on the hemispherical face comprising a clustered hydrophilic polar patch, and a surrounding hydrophobic region composed of Phe5, Phe14, Tyr15, Leu21, Ile26, and Tyr35-Leu37 (Fig. 8C). These clustered potent active residues could increase binding affinity between ASPA and Na+ channels. On the contrary, ASPC displays quite a different surface profile. In ASPC, hydrophobic Tyr29 and Ile31 replace acidic Asp31 and polar Thr33 of ASPA, respectively, and prevent the peptide from forming a hydrophilic region. In addition, ASPC is deficit in the residues (such as Phe5, Tyr15, and Leu21 of ASPA) required to form a hydrophobic region in the same location as ASPA (Fig. 8D). These surface property differences possibly explain the inability of ASPC to induce neuronal Ca2+ influx.
Fig. 8.

Sequences and surface profiles of ASPA, ASPC, and other selected knottins. (A) Sequences of ASPA, ASPC, ω-conotoxin MVIIA (CNTX_MVIIA), hainantoxin IV (HNTX_IV), and kalata B1. Residues: red = acidic, blue = basic, yellow = Cys, purple = Pro. X = pyroglutamic acid. The conserved residues involved in structural maintenance are indicated by boxes with solid lines. Sequence alignment was performed using ClustalX 2.0. (B) Surface profiles of CNTX_MVIIA [41], HNTX_IV [43], and kalata B1 [44]. Target protein binding regions are labeled and colored blue. (C) The surface profile of ASPA. Acidic Glu6, Glu8, and Asp31 and polar uncharged Ser9 and Thr33 compose a clustered hydrophilic polar patch; and Phe5, Phe14, Tyr15, Leu21, Ile26, and Tyr35-Leu37 compose a surrounding hydrophobic region. (D) Surface profile of ASPC. Hydrophobic Tyr29 and Ile31 replace acidic Asp31 and polar uncharged Thr33 of ASPA, respectively. ASPC is deficient in residues that form a hydrophobic region in ASPA. Surface exposed hydrophilic (charged and uncharged) and hydrophobic residues are labeled, and colored red and green, respectively.
4.2. Potential of oral administration
Oral delivery remains the most convenient and compliant method of administering drugs; however, designing orally bioavailable peptide delivery systems has been a persistent challenge to pharmaceutical scientists because of their susceptibility to gastrointestinal enzymatic degradation. Constrained macrocyclics like the cyclotides have extraordinary chemical, thermal, proteolytic stabilities [3,4], and cell-penetrating abilities [45,46], which make them attractive scaffolds for peptide-based oral drugs.
On the contrary, linear knottins did not get the spotlight for orally bioavailable peptide scaffolds because their gastrointestinal stabilities are much lower [13–15]. The presence of N-terminal pGlu and the absence of basic residues make asteropsins inherently stable to aminopeptidase and luminally secreted trypsin, which indicated their potential for an oral peptide drug carrier. In our investigation of gastrointestinal enzymatic stability towards chymotrypsin, elastase, pepsin, and trypsin, asteropsins A–D exhibited almost complete resistance against each enzyme within a time span of 4 h (Fig. 6). To be utilized for oral drug delivery, a carrier should not affect the gastrointestinal enzymatic activities, for protecting their normal biological functions. In chymotrypsin or trypsin inhibitory assays, both ASPA and ASPC showed no effects up to the concentration of 100 μg/mL, indicating their gastrointestinal enzymatic resistance was not caused by enzyme inhibition. Indeed, the asteropsins are the first linear knottin family members found to resist proteolysis by these four major gastrointestinal enzymes.
Furthermore, asteropsins A–D were remarkably stable in human plasma (Fig. 7), which assures their longer half-life in the circulatory system. ASPA and ASPC also exhibited exceptional proteolytic and thermal stability against proteinase K (from Tritirachium album; 50 °C, up to 48 h) and thermolysin (from Bacillus thermoproteolyticus Rokko; 65 °C, up to 12 h), indicative of their ultrastable structural properties. Asteropsins A–D showed no cytotoxicity against five human solid tumor cell lines (up to 30 μg/mL, A-549, SK-OV-3, SK-MEL-2, XF-498, and HCT-15) (Table S2); and ASPA and ASPC elicited no toxic effects or behavior changes in mice [i.p. 40 μg/g] or chicks [i.c.v. 60 μg/g] suggesting that they are acceptable as a potential inert drug carrier. Cystine knot peptides are known to show good permeation through rat intestinal mucosa [13,14]. However, further studies on the cell-penetrating ability of asteropsins are necessary because the drug efficacy is also modulated by their ability to cross the cell membranes.
The cyclotide kalata B1 is a popular choice for drug design and peptide engineering in the context of oral administration [3,4]. However, the recombinant production and chemical synthesis of kalata B1, as well as other cyclotides, require two post-synthetic processing steps involving the formation of three disulfide bonds and head-to-tail cyclization. Although some effective biochemical approaches have been developed for one-pot folding and backbone-cyclization of cyclotides [5], the synthesis of asteropsins will be inherently more efficient (higher yield) due to less reaction steps.
Summarizing, we report three unusual knottins, asteropsins B–D, from the marine sponge Asteropus sp., which with the previously reported ASPA, compose a new knottin subfamily that feature N-terminal blocking, a highly acidic nature, and two cis-prolines. The three-dimensional structures of ASPB and ASPC were determined using solution NMR techniques, whereas the tertiary structure of ASPD was determined by homology modeling. Despite moderate sequence homologies, asteropsins A–D share a highly conserved structural framework due to the presence of a rigid disulfide core and seven conserved residues at specific locations. These asteropsins are the first linear knottin family members found to be stable in gastrointestinal tract as well as human plasma. These findings suggest that these asteropsins could be utilized as novel scaffolds for orally delivered peptides. Furthermore, the identification of residues responsible for structural maintenance provides essential information for SAR studies and for the peptide engineering of asteropsins.
Supplementary Material
Acknowledgments
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (No. 2012043039). This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program C06 RR-14503-01 from the NIH National Center for Research Resources. The authors would like to thank Drs. Frank Mari (Florida Atlantic University), Dong-Soon Im (Pusan National University, Korea), Seung-Yeol Nah (Konkuk University), and Jae Il Kim (GIST, Korea) for help and assistance during the course of this study.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbagen.2013.11.001.
References
- 1.Pichereau C, Allary C. Therapeutic peptides under the spotlight. Eur BioPharm Rev. 2005 Winter;:88–91. [Google Scholar]
- 2.Kolmar H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr Opin Pharmacol. 2009;9:608–614. doi: 10.1016/j.coph.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Getz JA, Rice JJ, Daugherty PS. Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem Biol. 2011;6:837–844. doi: 10.1021/cb200039s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong CT, Rowlands DK, Wong CH, Lo TW, Nguyen GK, Li HY, Tam JP. Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew Chem Int Ed Engl. 2012;51:5620–5624. doi: 10.1002/anie.201200984. [DOI] [PubMed] [Google Scholar]
- 5.Aboye TL, Camarero JA. Biological synthesis of circular polypeptides. J Biol Chem. 2012;287:27026–27032. doi: 10.1074/jbc.R111.305508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- 7.Bogin O. Venom peptides and their mimetics as potential drugs. Modulator. 2005;19:14–20. [Google Scholar]
- 8.Chan LY, Gunasekera S, Henriques ST, Worth NF, Le SJ, Clark RJ, Campbell JH, Craik DJ, Daly NL. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood. 2011;118:6709–6717. doi: 10.1182/blood-2011-06-359141. [DOI] [PubMed] [Google Scholar]
- 9.Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, Neamati N, Camarero JA. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J Med Chem. 2012;55:10729–10734. doi: 10.1021/jm301468k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, Neamati N, Shekhtman A, Camarero JA. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J Am Chem Soc. 2013;135:11623–11633. doi: 10.1021/ja405108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia AE, Camarero JA. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr Mol Pharmacol. 2010;3:153–163. doi: 10.2174/1874467211003030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jagadish K, Camarero JA. Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Biopolymers. 2010;94:611–616. doi: 10.1002/bip.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werle M, Schmitz T, Huang HL, Wentzel A, Kolmar H, Bernkop-Schnürch A. The potential of cystine-knot microproteins as novel pharmacophoric scaffolds in oral peptide drug delivery. J Drug Target. 2006;14:137–146. doi: 10.1080/10611860600648254. [DOI] [PubMed] [Google Scholar]
- 14.Werle M, Kafedjiiski K, Kolmar H, Bernkop-Schnürch A. Evaluation and improvement of the properties of the novel cystine-knot microprotein McoEeTI for oral administration. Int J Pharm. 2007;332:72–79. doi: 10.1016/j.ijpharm.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 15.Werle M, Kolmar H, Albrecht R, Bernkop-Schnürch A. Characterisation of the barrier caused by luminally secreted gastro-intestinal proteolytic enzymes for two novel cystine-knot microproteins. Amino Acids. 2008;35:195–200. doi: 10.1007/s00726-007-0569-1. [DOI] [PubMed] [Google Scholar]
- 16.Williams JA, Day M, Heavner JE. Ziconotide: an update and review. Expert Opin Pharmacother. 2008;9:1575–1583. doi: 10.1517/14656566.9.9.1575. [DOI] [PubMed] [Google Scholar]
- 17.Zheng K, Lubman DM, Rossi DT, Nordblom GD, Barksdale CM. Elucidation of peptide metabolism by on-line immunoaffinity liquid chromatography mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:261–269. doi: 10.1002/(SICI)1097-0231(20000229)14:4<261::AID-RCM868>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed Engl. 2010;49:6545–6548. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- 19.Takada K, Hamada T, Hirota H, Nakao Y, Matsunaga S, van Soest RWM, Fusetani N. Asteropine A, a sialidase-inhibiting conotoxin-like peptide from the marine sponge Asteropus simplex. Chem Biol. 2006;13:569–574. doi: 10.1016/j.chembiol.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Bowling JJ, Fronczek FR, Hong J, Jabba SV, Murray TF, Ha NC, Hamann MT, Jung JH. Asteropsin A: an unusual cystine-crosslinked peptide from porifera enhances neuronal Ca2+ influx. Biochim Biophys Acta. 2013;1830:2591–2599. doi: 10.1016/j.bbagen.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goddard TD, Kneller DG. SPARKY. Vol. 3. University of California; San Francisco: 2007. [Google Scholar]
- 22.Wüthrich K. NMR of Proteins and Nucleic Acids. J. Wiley; New York: 1986. [Google Scholar]
- 23.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 24.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 25.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 26.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.The PyMOL Molecular Graphics System, version 1.3. Schrödinger, LLC; 2010. [Google Scholar]
- 28.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 30.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernkop-Schnürch A. The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J Control Release. 1998;52:1–16. doi: 10.1016/s0168-3659(97)00204-6. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Hung TD, Li J, Sim CJ, Hong J, Kim DK, Jung JH. Pyroglutamyl dipeptides and tetrahydro-beta-carboline alkaloids from a marine sponge Asteropus sp. Biochem Syst Ecol. 2010;38:1049–1051. [Google Scholar]
- 33.Lewis PN, Momany FA, Scheraga HA. Chain reversals in proteins. Biochim Biophys Acta. 1973;303:211–229. doi: 10.1016/0005-2795(73)90350-4. [DOI] [PubMed] [Google Scholar]
- 34.Wilmot CM, Thornton JM. Beta-turns and their distortions: a proposed new nomenclature. Protein Eng. 1990;3:479–493. doi: 10.1093/protein/3.6.479. [DOI] [PubMed] [Google Scholar]
- 35.Klaus W, Broger C, Gerber P, Senn H. Determination of the disulphide bonding pattern in proteins by local and global analysis of nuclear magnetic resonance data. Application to flavoridin. J Mol Biol. 1993;232:897–906. doi: 10.1006/jmbi.1993.1438. [DOI] [PubMed] [Google Scholar]
- 36.Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J Biomol NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 37.Ay J, Hilpert K, Krauss N, Schneider-Mergener J, Hohne W. Structure of a hybrid squash inhibitor in complex with porcine pancreatic elastase at 1.8 Å resolution. Acta Crystallogr D Biol Crystallogr. 2003;59:247–254. doi: 10.1107/s0907444902020887. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Y, Huang Q, Qian M, Jia Y, Tang Y. Crystal structure of the complex formed between bovine beta-trypsin and MCTI-A, a trypsin inhibitor of squash family, at 1.8-Å resolution. J Protein Chem. 1999;18:505–509. doi: 10.1023/a:1020690931043. [DOI] [PubMed] [Google Scholar]
- 39.Rees DC, Lipscomb WN. Refined crystal structure of the potato inhibitor complex of carboxypeptidase A at 2.5 Å resolution. J Mol Biol. 1982;160:475–498. doi: 10.1016/0022-2836(82)90309-6. [DOI] [PubMed] [Google Scholar]
- 40.Kratzner R, Debreczeni JE, Pape T, Schneider TR, Wentzel A, Kolmar H, Sheldrick GM, Uson I. Structure of Ecballium elaterium trypsin inhibitor II (EETI-II): a rigid molecular scaffold. Acta Crystallogr D Biol Crystallogr. 2005;61:1255–1262. doi: 10.1107/S0907444905021207. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen KJ, Adams D, Thomas L, Bond T, Alewood PF, Craik DJ, Lewis RJ. Structure–activity relationships of omega-conotoxins MVIIA, MVIIC and 14 loop splice hybrids at N and P/Q-type calcium channels. J Mol Biol. 1999;289:1405–1421. doi: 10.1006/jmbi.1999.2817. [DOI] [PubMed] [Google Scholar]
- 42.Nadasdi L, Yamashiro D, Chung D, Tarczy-Hornoch K, Adriaenssens P, Ramachandran J. Structure–activity analysis of a Conus peptide blocker of N-type neuronal calcium channels. Biochemistry. 1995;34:8076–8081. doi: 10.1021/bi00025a013. [DOI] [PubMed] [Google Scholar]
- 43.Li D, Xiao Y, Xu X, Xiong X, Lu S, Liu Z, Zhu Q, Wang M, Gu X, Liang S. Structure–activity relationships of hainantoxin-IV and structure determination of active and inactive sodium channel blockers. J Biol Chem. 2004;279:37734–37740. doi: 10.1074/jbc.M405765200. [DOI] [PubMed] [Google Scholar]
- 44.Ireland DC, Clark RJ, Daly NL, Craik DJ. Isolation, sequencing, and structure–activity relationships of cyclotides. J Nat Prod. 2010;73:1610–1622. doi: 10.1021/np1000413. [DOI] [PubMed] [Google Scholar]
- 45.Cascales L, Henriques ST, Kerr MC, Huang YH, Sweet MJ, Daly NL, Craik DJ. Identification and characterization of a new family of cell-penetrating peptides: cyclic cell-penetrating peptides. J Biol Chem. 2011;286:36932–36943. doi: 10.1074/jbc.M111.264424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Contreras J, Elnagar AY, Hamm-Alvarez SF, Camarero JA. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J Control Release. 2011;155:134–143. doi: 10.1016/j.jconrel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.