

Abstract
The Ca2+ sensor for rapid synaptic vesicle exocytosis, synaptotagmin I (syt), is largely composed of two Ca2+-sensing C2-domains, C2A and C2B. We have investigated the apparent synergy between the tandem C2 domains by altering the length and rigidity of the linker that connects them. The behavior of the linker mutants revealed a correlation between the ability of the C2-domains to penetrate membranes in response to Ca2+ and to drive evoked neurotransmitter release in cultured mouse neurons, uncovering a step in excitation-secretion coupling. Atomic force microscopy experiments indicate that the synergy between these C2-domains involves intra-molecular interactions between them. Thus, syt function is profoundly affected by changes in the physical nature of the linker that connects its tandem C2-domains. Moreover, the linker mutations uncoupled syt-mediated regulation of evoked and spontaneous release, revealing that syt also acts as a fusion clamp prior to the Ca2+ trigger.
INTRODUCTION
Excitation-secretion coupling is the process by which electrical signals are converted into the release of neurotransmitters from neurons. Excitation opens voltage-gated Ca2+ channels and the subsequent influx of Ca2+ ions triggers the rapid fusion of neurotransmitter-filled synaptic vesicles (SV) with the presynaptic plasma membrane, releasing transmitters into the synaptic cleft, where they bind and activate post-synaptic receptors. In many synapses, there are two forms of evoked release: a rapid synchronous component, that mediates fast, point-to-point communication within the nervous system, and a slower asynchronous component1,2, that underlies aspects of circuit function, including persistent reverberation3. In addition to evoked release, neurotransmitters are also secreted in the absence of electrical stimulation. These spontaneous release events give rise to ‘miniature’ (mini) postsynaptic currents that regulate numerous aspects of synaptic function, including post-synaptic protein synthesis and the maturation and stability of synaptic contacts4,5.
All three modes of SV fusion are mediated by a conserved set of membrane proteins called soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs)6. Vesicular (v-) SNAREs interact with target membrane (t-) SNAREs, forming four-helix bundles that pull the bilayers together to directly catalyze bilayer fusion7,8. For rapid secretion, fusion is thought to be triggered by the binding of Ca2+ to the synaptic vesicle protein, synaptotagmin I (syt)9. Syt is anchored to the vesicle membrane via a single transmembrane domain and binds multiple Ca2+ ions via tandem C2-domains, C2A and C2B, which comprise most of the cytoplasmic domain of the protein. These tandem C2-domains are connected via a short flexible linker10,11.
Syt has been proposed to accelerate SNARE-mediated fusion via the rapid Ca2+-triggered partial penetration of its C2-domains into the target membrane12–14, resulting in localized bending of the bilayer15,16. Ca2+•syt might also trigger release by driving the assembly of SNAREs into SNARE complexes17–19. However, neither putative mechanism has been demonstrated in nerve terminals. Recent studies indicate that asynchronous release requires another Ca2+ binding protein, Doc2, which is also largely composed of tandem C2-domains that interact with anionic phospholipids and SNARE proteins, and thus might operate in a way analogous to syt during evoked release19,20.
Syt and Doc2 have also both been proposed to function as Ca2+ sensors for spontaneous release (minis)20,21, but the question of whether glutamatergic minis are regulated by Ca2+ is currently the subject of debate22,23. Interestingly, loss of syt results in an increase in mini frequency (see Ref.9,24 and references therein). Moreover, in reconstituted membrane fusion reactions the cytoplasmic domain of syt efficiently inhibits SNARE-catalyzed membrane fusion in the absence of Ca2+, so syt was also proposed to function as a clamp that inhibits SV exocytosis prior to the Ca2+ trigger14. Alternatively, the observed increase in mini frequency in KO neurons might be a compensatory response that is secondary to the loss of evoked transmission.
There are seventeen distinct isoforms of syt, each encoded by distinct genes25,26. While the overall homology among isoforms is limited (as low as 17%), the length, but not the sequence, of the linker segments that connect C2A and C2B, is conserved. Specifically, linkers are always 8 to 11 residues long (except in syt 14 and syt 16, which have longer linker segments, but do not appear to bind Ca2+)27. This conservation of length suggests that the linker might be a key determinant of syt function. Moreover, the tandem arrangement is not simply a repeat of a C2 domain; in syt, C2A and C2B share only 40% similarity and bind to Ca2+ and effectors with distinct affinities13,28–31.
Interestingly, a number of biochemical studies indicate that C2A and C2B functionally interact, endowing the protein with properties that were not anticipated from the independent action of each C2-domain. For example, the C2A domain of syt has been shown to strongly influence the membrane penetration activity of the adjacent C2B domain, even when C2A is unable to bind Ca2+ or membranes29,32. How this occurs is unclear, as structural studies assaying physical interactions between C2A and C2B have led to conflicting conclusions.10,11,33–35
The current study is focused on the function of the linker segment that connects the tandem C2-domains of syt, and on the mechanism of the apparent synergy between these domains. We systematically altered the length and rigidity of the linker, and analyzed each mutant via in vitro biochemical and biophysical assays, and by expression of these mutants in neurons lacking the wild type (WT) protein. We show that the tandem C2-domains of syt physically interact with one another, and that these intra-molecular interactions strongly affect the ability of the protein to penetrate membranes. Strikingly, the graded changes in membrane penetration activity, observed for the linker mutants, were well correlated with the ability of each mutant to mediate excitation-secretion coupling in neurons. These findings reveal that partial insertion of the Ca2+-binding loops of syt into lipid bilayers corresponds with a step in excitation-secretion coupling. Finally, these linker mutations also dissociated the roles of syt in evoked and spontaneous release, revealing that syt also functions as a ‘fusion clamp’ prior to the arrival of the Ca2+ trigger.
RESULTS
Analysis of the linker that connects the C2-domains of syt
To investigate the function of the linker that connects the C2A and C2B domains of syt (residues 264 to 272, SAEKEEQEK) during SV exocytosis, we designed a series of mutant forms of the protein to make the linker shorter, longer, more flexible, or less flexible (Fig. 1a). We first shortened the linker by deleting five residues, EKEEQ, in the center of this segment, which we term 0.5xWT. Next, we extended the linker by doubling or tripling the native sequence, termed 2xWT and 3xWT, respectively. Finally, we changed the rigidity of the linker by replacing it with highly flexible, or highly rigid, segments. For the former, we used poly-glycine segments of either nine or eighteen residues (1xGly, 2xGly), and for the latter we used segments of nine or eighteen proline residues (1xPro, 2xPro), since polyproline forms rigid rod structures36. Each of these constructs was expressed, using lentivirus, in primary hippocampal neurons derived from syt KO mice. Neurons from WT mice from the same litters were used as controls. Co-immunostaining with a synaptic vesicle marker confirmed that each of the linker mutants was correctly targeted to synapses (Fig. 1b and c).
Figure 1.

Syt linker mutants are targeted to synapses. (a) The linker between the C2A and C2B domains of syt (residues 264–272, SAEKEEQEK) was modified as indicated. TMD stands for Transmembrane Domain. (b) Syt KO hippocampal neurons were infected with lentivirus expressing each mutant, and immunostained for syt (red) and synapsin (green). WT littermates, that harbor native endogenous syt, were used as controls (WT). (c) Colocalization of WT and linker mutant forms of syt with synapsin; in all cases the mutant forms of syt were well targeted to synaptic sites. For each condition, two distinct random regions were imaged from each of three coverslips that were derived from three independent litters of mice, yielding an N of six images for each construct. The total number of puncta was 1,180–1,320 per condition. The mean values ± SEM are indicated. Statistical significance was determined using a bootstrap approach (see Online Methods); no significant differences were observed (Supplementary Statistics).
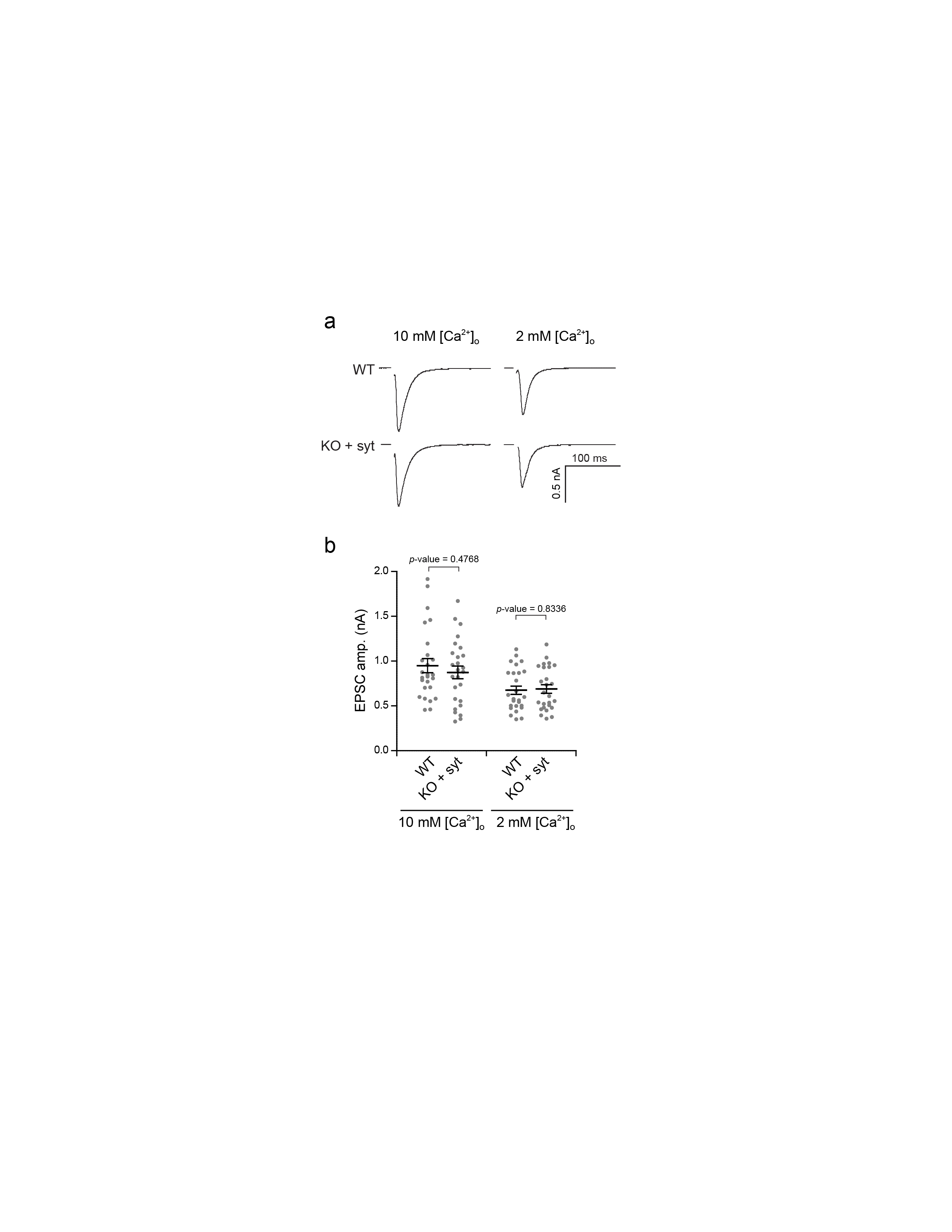
We then carried out whole-cell voltage clamp recordings to examine the impact of the linker mutations on evoked SV release. Briefly, we stimulated one neuron with a bipolar glass electrode placed against the soma and recorded excitatory post-synaptic currents (eEPSCs) from a second synaptically connected neuron; for all electrophysiology measurements, we used 10 mM [Ca2+]o to maximize synaptic responses, unless otherwise indicated. Each construct was able to rescue some degree of fast release, with the exception of the 2xPro mutant, which completely failed to rescue the syt KO phenotype (Fig. 2a–d). Among the constructs that did rescue, the degree of rescue varied in terms of the fast release amplitude (Fig. 2b) and total charge transfer (Fig. 2c). 1xGly, 2xGly and 1xPro fully rescued fast release, while 0.5xWT, 2xWT and 3xWT only partially rescued the fast component of release. Interestingly, the 2xWT linker mutant restored more fast release than did 0.5xWT or 3xWT. Together, these data reveal that the native linker is optimized for release: making the native linker longer, or shorter, diminishes excitation-secretion coupling. We repeated these experiments using physiological [Ca2+]o, 2 mM, and observed the same trends for each mutant (Supplementary Fig. 1a–c). We note that WT neurons were used as controls; this is valid since their synaptic responses were identical to syt KO neurons rescued with a syt cDNA (Supplementary Fig. 2).
Figure 2.

The linker mutants exhibit marked differences in their abilities to support evoked transmission. (a) Representative eEPSC traces recorded from WT, syt KO and KO neurons that expressed each linker mutant. (b–d) Scatter plots of eEPSC amplitude (b), total charge (c), and latency to peak (d; the time between the start of stimulation and the peak current). (e) Normalized cumulative total charge transfer, averaged for each group. (f, g) Fraction of total release (upper panels), and release time constants (lower panels) for the fast (f) and slow release components (g), which were calculated by fitting the normalized cumulative charge traces in panel e with a double exponential function. For each condition, we recorded from 33–41 cells from a total of six coverslips, where two coverslips were obtained from each of three independent litters of mice. The mean values ± SEM are indicated. Results from bootstrap statistical analysis are listed in Supplementary Statistics. Asterisks indicate differences compared with WT; * p < 0.05, ** p < 0.01, *** p < 0.001.
We emphasize that while the highly flexible 2xGly linker mutant fully rescued fast release, the rigid 2xPro linker mutant, failed to rescue. These results suggest that holding C2A and C2B apart, via a proline rod, impairs the function of the protein, implying that the tandem C2-domains might act via physical intra-molecular interactions, a point we return to below.
The inefficient rescue by some of the linker mutants could result from reductions in the size of the readily releasable pool (RRP), as previously described for syt KO neurons24. To address this, hypertonic sucrose (500 mM) was used to drive release of the RRP24; all of the linker mutants successfully restored the loss of the RRP characteristic of syt KO neurons (Supplementary Fig. 3a and b). Thus, while some of the linker mutants diminish the function of syt as a Ca2+ sensor for release, they do not disrupt upstream steps in the action of syt in the formation, or stabilization, of the RRP37.
We analyzed the total charge transfer using a double exponential function (Fig. 2e)24 and plotted the fractions of the fast (upper panel in Fig. 2f) and slow release components (upper panel in Fig. 2g), along with the time constants for each (lower panels of Fig. 2f and Fig. 2g, respectively). Again, transmission in neurons expressing the 2xPro linker mutant was virtually identical to that observed in the KOs. In contrast, the kinetics of the fast component of release, and the relative amounts of fast and slow transmission, were similar among 0.5xWT, 2xWT, 3xWT, 1xGly, 2xGly, 1xPro linker mutants, and WT neurons, indicating that these mutants synchronized fast release while inhibiting asynchronous release (Fig. 2f and g). In all cases, the time constants for the slow phase of release were the same (Fig. 2g, lower panel).
From the experiments described in this section, we conclude that: 1) the composition of the nine-residue linker between C2A and C2B is not important for syt to trigger fast release; however, 2) the length and rigidity of the linker, and thus the distance between C2A and C2B, plays a critical role in rapid SV exocytosis.
Syt linker mutants alter short-term plasticity
We further addressed the function of the syt linker by examining the paired pulse ratio (PPR) of neurons expressing each of the mutants, detailed above, by dividing the second eEPSC by the first eEPSC (Fig. 3a and b). Consistent with the full rescue of SV release mediated by the 1xGly, 2xGly and 1xPro mutants, the PPR from these three linker mutants were reduced to almost the same level as observed for WT neurons; 1xGly and 2xGly were only slightly different from WT syt (Fig. 3b). As expected, the 2xPro mutant, which failed to rescue, displayed a PPR that was similar to the nulls (Fig. 3b). The 0.5xWT and 3xWT mutants gave rise to only low levels of rescue and also displayed PPRs that were similar to the nulls. Finally, the 2xWT mutant yielded intermediate rescue and an intermediate PPR. Together, these data are consistent with lower release probabilities for the loss-of-function linker mutants, as compared with WT neurons (given their similar RRPs). The same trends were observed when recordings were carried out in physiological, 2 mM, [Ca2+]o (Supplementary Fig. 1d and e).
Figure 3.

Syt linker mutants alter short-term plasticity. (a) Representative eEPSCs recorded in paired-pulse experiments with 50 ms inter-stimulus intervals. (b) Scatter plots of paired-pulse ratios (PPR), calculated by dividing the amplitude of the second response (Amp2) by the first response (Amp1); the amplitude right before the second stimulation served as the new baseline for Amp 2. (c) Example eEPSCs in response to a stimulus train of 50 pulses at 20 Hz. (d) The peak amplitude of each eEPSC in the train was normalized to the first eEPSC and plotted versus time to show SV release potentiation and depression. eEPSCs from the KO and 2xPro linker mutant are not shown because there was no fast release in these two groups. For each condition, we recorded from 33–41 cells from a total of six coverslips, where two coverslips were obtained from each of three independent litters of mice. The mean values ± SEM are indicated. Statistical significance was determined using a bootstrap approach (Supplementary Statistics). Asterisks indicate differences compared with WT; * p < 0.05, *** p < 0.001.
We then measured synaptic depression during high frequency stimulation (HFS; 50 APs at 20 Hz) (Fig. 3c and d), and observed that the 1xGly, 2xGly and 1xPro mutants exhibited fast depression that was analogous to WT neurons, whereas the 0.5xWT, 2xWT and 3xWT linker mutants displayed potentiation, as compared with WT neurons. The 2xWT mutant exhibited less potentiation, as compared with 0.5xWT and 3xWT (Fig. 3d), which is consistent with the smaller PPR in 2xWT versus the other two linker mutants. In summary, the length and rigidity of the linker between C2A and C2B are critical determinants of the function of syt during short term plasticity.
Membrane penetration activity of syt triggers exocytosis
As detailed above, electrophysiological recordings demonstrated that the linker region between C2A and C2B plays a crucial role in the ability of syt to function as a Ca2+ sensor for rapid exocytosis. But what is the molecular basis for these observations? Both C2A and C2B have been implicated in regulating release through interactions with two sets of effectors: membranes that harbor anionic phospholipids38, and t-SNAREs17,39,40. We therefore determined the impact of the linker mutations on these interactions.
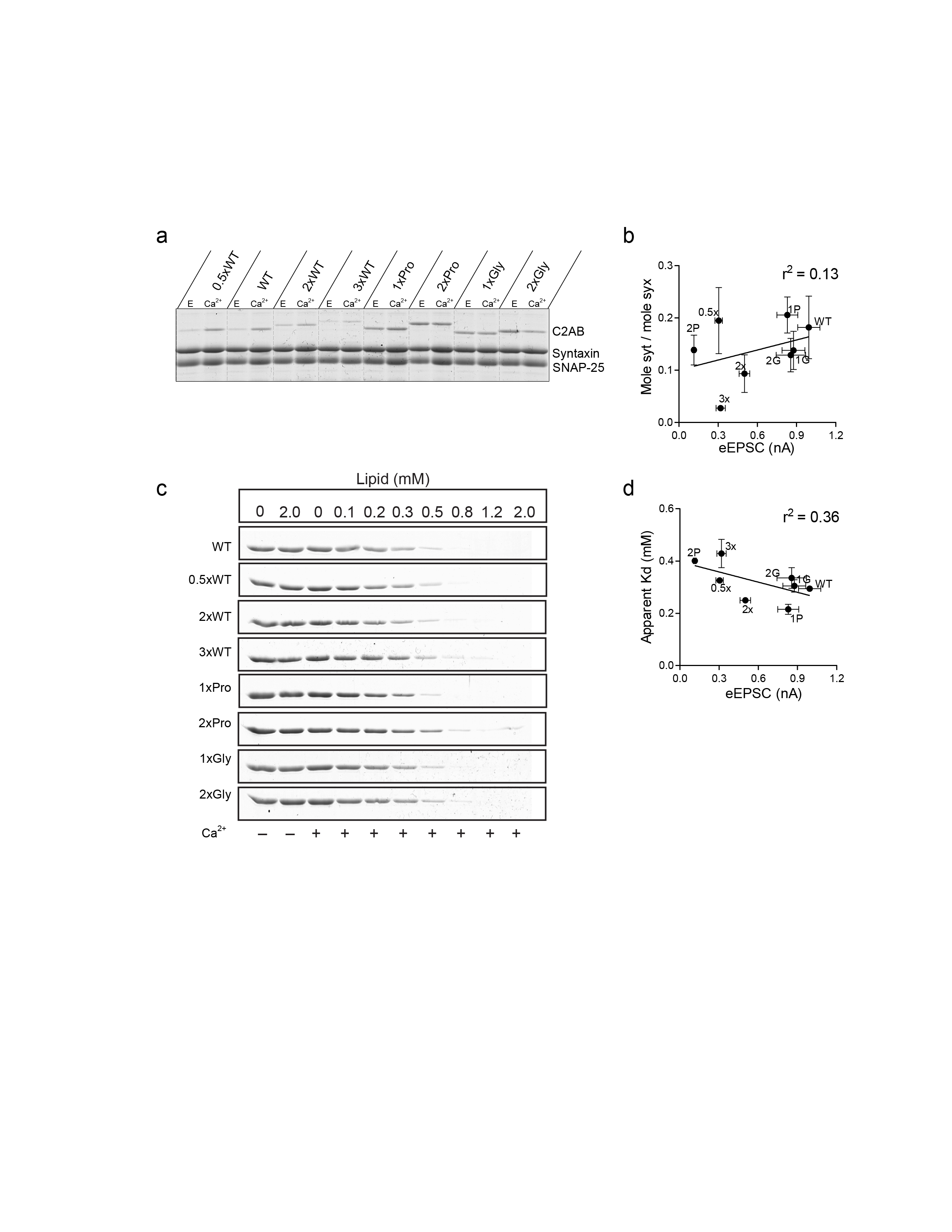
We first analyzed the binding of the linker mutants to t-SNARE- (Supplementary Fig. 4a) or PS-bearing liposomes (Supplementary Fig. 4c) as described previously using the cytoplasmic domain of the protein, C2AB16,41. The extent of binding was plotted versus the eEPSC amplitudes from Fig. 2b. Surprisingly, there was no correlation between the extent of t-SNARE binding activity and magnitude of the synaptic currents (r2 = 0.13, Supplementary Fig. 4b), and only a weak correlation with PS binding activity was apparent (r2 = 0.36, Supplementary Fig. 4d).
In the course of these experiments, we confirmed that the 2xWT and 3xWT linker mutants exhibit impaired t-SNARE binding activity, as reported previously42, but inclusion of the other five linker mutants in our analysis, again, revealed an overall lack of correlation between mutant syt activity in neurons and the ability of these mutants to bind to SNAREs. At present, a consensus regarding the physiological significance of syt-SNARE interactions has not been reached, with data that both support17,40 and that argue against43 this interaction as being important for secretion, so this remains an open question. Interestingly, while all of the linker mutants were able to bind to liposomes in a Ca2+-dependent manner, the 2xPro, 1xGly, and 2xGly bound to t-SNAREs in a constitutive, Ca2+-independent manner.
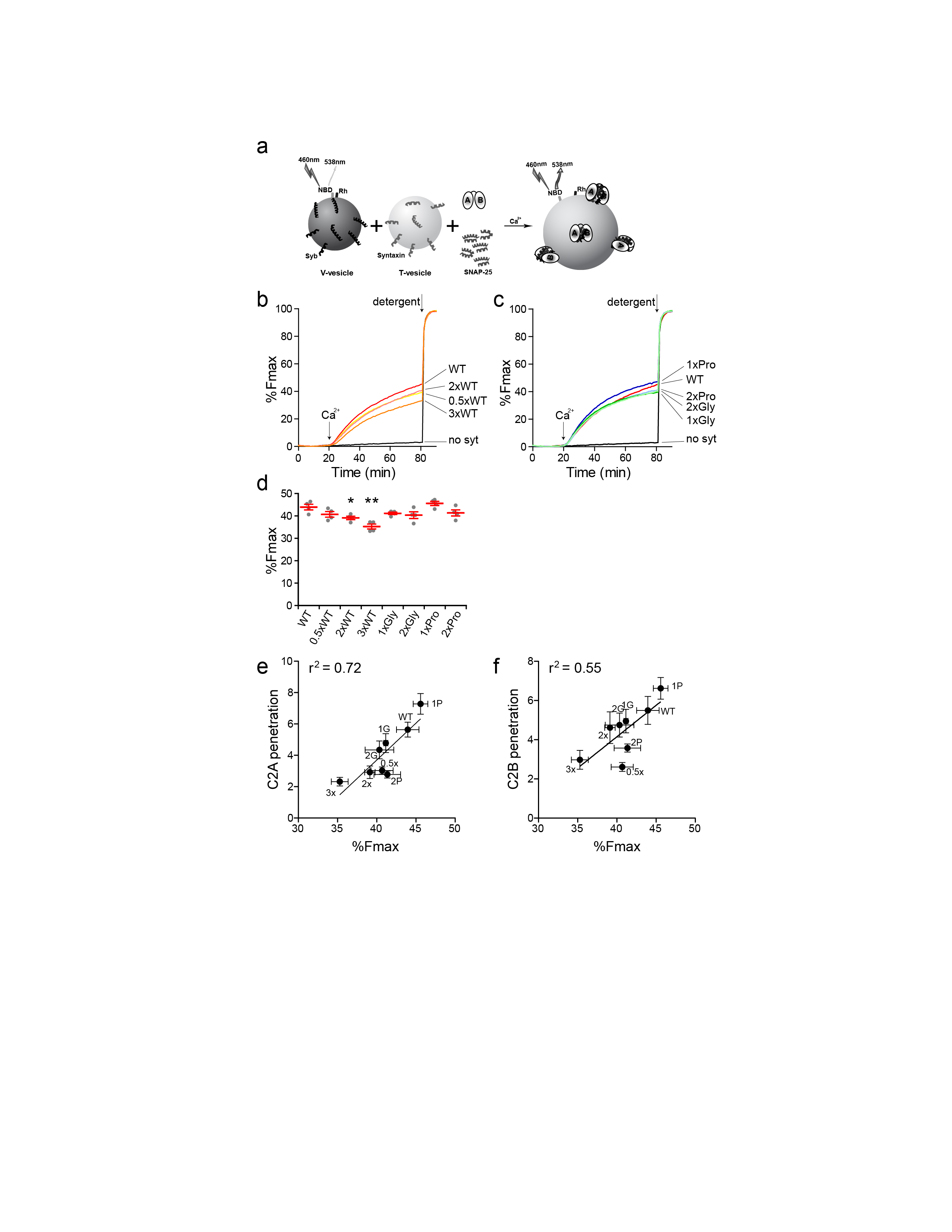
Upon binding to membranes, the distal tips of the Ca2+ binding loops of syt penetrate into the bilayer, and this has been proposed to drive lipid rearrangements to accelerate the rate of fusion, potentially by juxtaposing the membranes destined to fuse, or by bending the target (i.e. plasma) membrane at sites of fusion15,16,44. We therefore conducted in vitro membrane penetration assays using an environmentally sensitive fluorescent probe, NBD, which earlier studies have shown reports penetration as an increase, and a blue shift, in emission spectra18. A cysteine mutation was first introduced in the distal tip of Ca2+ binding loop 3 of C2A (F234C) or C2B (I367C), and labeled with NBD. Upon addition of Ca2+, in the presence of PS-bearing liposomes, the NBD emission from C2A (Fig. 4a and b) and C2B (Fig. 4d and e) increased and was shifted to shorter wavelengths. However, the strength of the fluorescence intensity (FI) increase differed among the linker mutants (Fig. 4c and f). We plotted the relative degree of membrane penetration activity against the eEPSC amplitudes from Fig. 2b (Fig. 4g and h). A key finding was that a clear correlation between the ability of either C2-domain to penetrate membranes and to drive fast release emerged (Fig. 4g, r2 = 0.64 for C2A; Fig. 4h, r2 = 0.65 for C2B). We further investigated this correlation by repeating the membrane penetration assays at lower [Ca2+], 100 μM, and plotting these data against the synaptic currents recorded at physiological, 2 mM, [Ca2+]o (Supplementary Fig. 5a–d). These experiments further confirmed the relationship between the membrane penetration activity of mutant forms of syt with their ability to drive synaptic transmission (Supplementary Fig. 5c, r2 = 0.65 for C2A; Supplementary Fig. 5d, r2 = 0.77 for C2B).
Figure 4.

Correlation between the membrane penetration activity of syt linker mutants with their abilities to trigger rapid SV release. (a–f) Linker mutations affect the membrane penetration activity of syt. The tip of Ca2+ and membrane-binding loop 3, in the C2A or C2B domain of the cytoplasmic domain of syt, was labeled with NBD; these constructs are denoted as C2A*(3)-C2B or C2A-C2B*(3), respectively. The fluorescence of NBD exhibited an increase in intensity and a blue shift in its emission spectra upon Ca2+-triggered penetration of loop 3 into lipid bilayers harboring 15% PS. (a, b) Representative normalized NBD fluorescence spectra of C2A*(3)-C2B obtained in 0.2 mM EGTA or 1 mM Ca2+. (c) Scatter plot of peak values of the normalized NBD fluorescence spectra of C2A*(3)-C2B in 1 mM Ca2+. (d, e) Representative normalized NBD fluorescence spectra of C2A-C2B*(3) obtained in 0.2 mM EGTA or 1 mM Ca2+. (f) Scatter plot of peak values of the normalized NBD fluorescence spectra of C2A-C2B*(3) in 1 mM Ca2+. N = 5 independent experiments. (g, h) A clear correlation was observed between the membrane penetration activity of C2A (g), and C2B (h), and eEPSC amplitude (Fig. 2b). The mean values ± SEM are indicated. Statistical significance was determined using a bootstrap approach. Asterisks indicate differences compared with WT; * p < 0.05, ** p < 0.01. All statistical analysis and results, including the equations and p-values for the linear regressions, are provided in Supplementary Statistics.
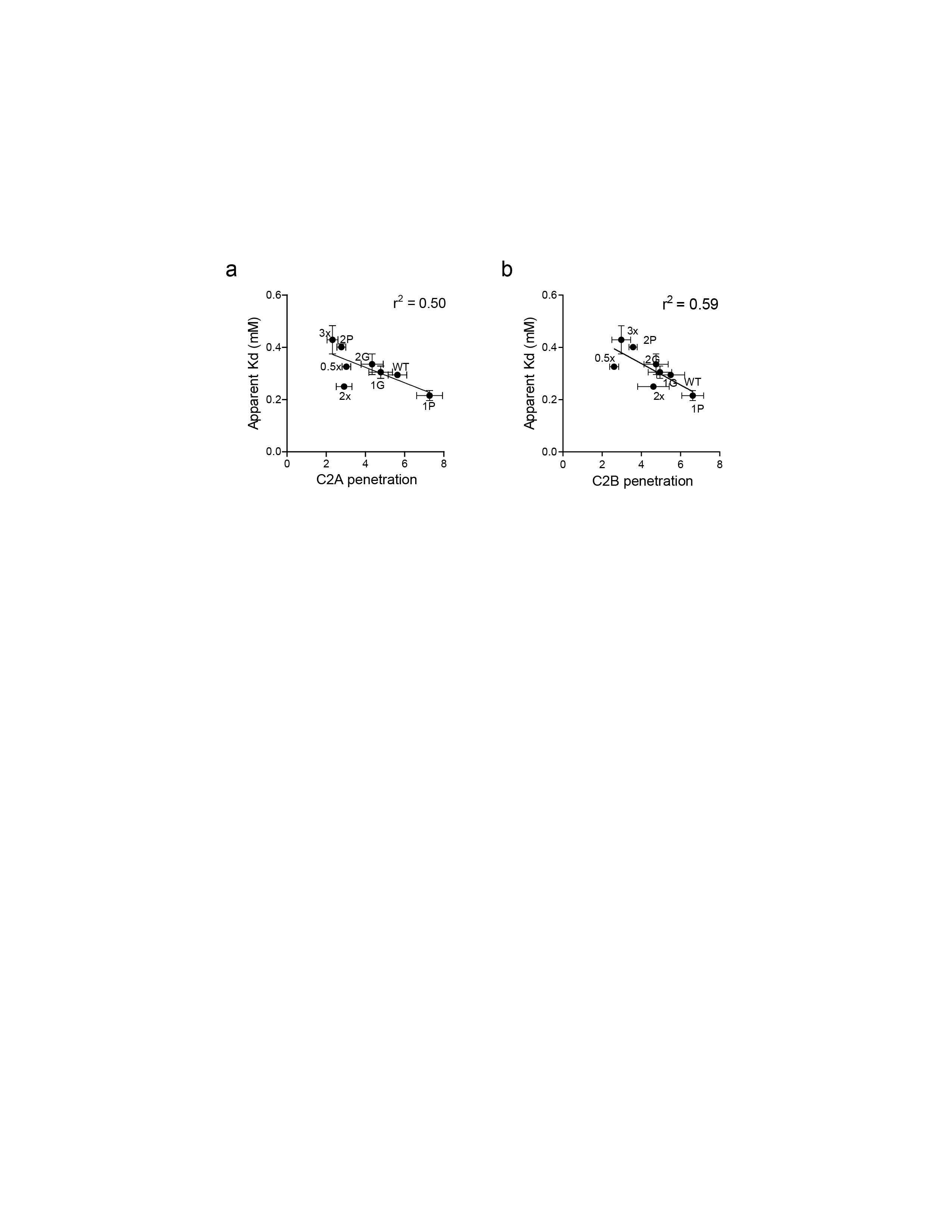
We note that the apparent membrane binding affinities of the syt variants were correlated, to some degree, with membrane penetration activity (r2 = 0.50 for C2A, r2 = 0.59 for C2B; Supplementary Fig. 6), but there were exceptions (e.g. the 2xWT linker), suggesting that penetration can be partially dissociated from binding to membranes.
We extended this work to analyze the linker mutants via in vitro fusion assays utilizing reconstituted SNARE proteins (Supplementary Fig. 7a–d) and then plotting the extent of fusion versus the extent of membrane penetration (Supplementary Fig. 7e and f). As in the physiology experiments, the abilities of the linker mutants to stimulate Ca2+-triggered fusion in the in vitro system correlated with their abilities to penetrate membranes (r2 = 0.72 for C2A and r2 = 0.55 for C2B; Supplementary Fig. 7e and f).
Interaction between the tandem C-domains domains of syt
To address the unresolved issue as to whether C2A and C2B physically interact with each other10,11,33–35, and to explore the molecular basis for the observation that the 2xPro linker mutant failed to rescue evoked release, we conducted atomic force microscopy (AFM) experiments using the cytoplasmic domain of syt harboring various linker segments (WT, 3xWT, 1xPro, and 2xPro). In the AFM images (Fig. 5a). WT C2AB often adopted a horseshoe shaped structure, which likely represents the two interacting C2 domains; schematic representations of these horseshoe structures are provided in Supplementary Fig. 8a. Frequency distributions of the end-to-end distances of the four constructs, measured as illustrated in Supplementary Fig. 8b, are shown in Fig. 5b. The 3xWT linker mutant was also globular and had a distance distribution that was similar to the WT protein (Fig. 5b). This latter observation strongly indicates that C2A and C2B physically interact; if inter-domain interactions did not occur, the 3xWT linker mutant would be expected to exhibit a broader distance distribution. However, these experiments are near the resolution for AFM, so we examined additional linkers to further determine whether changes in structure can be observed with this method. The 1xPro and 3xWT linker mutants were similar to the WT protein, in terms of having an overall globular shape with a nearly identical distance distribution (Fig. 5a and b). However, the 2xPro linker was clearly different, having a dumbbell shape and a significant increase in the average longest dimension of the protein (Fig. 5a and b). Each lobe of the dumbbell likely corresponds to a C2-domain, and the rigid nature of the 2xPro linker serves to hold the tandem C2-domains apart. Gaussian fits of the four distance distributions are overlaid in Fig. 5c to highlight the fact that the 2xPro mutant is distinct from the other constructs. It should be noted that the lengths determined by AFM are larger than the actual size of the protein45, so we emphasize the relative differences between each mutant rather than the absolute dimensions.
Figure 5.

A long, rigid proline rod holds C2A and C2B apart. (a) Representative AFM images of WT and linker mutant forms (3xWT, 1xPro and 3xPro) of the cytoplasmic domain of syt on mica. Scale bars are 20 nm. (b) Frequency distributions of the maximum lengths of the syt constructs, as determined from the AFM images. Errors are SD; 132–140 particles were measured. (c) Gaussian fits of the four distance distributions, overlaid to illustrate the increase in length of the 2xPro linker mutant. (d) Paired-distance distribution function plots from SAXS measurements of each construct shown in panel a, where r is the paired-distance, which is the distance between random pairs of atoms, and p(r) is the relative probability density for each r value. The shape of a protein determines the shape of the p(r) trace. For example, a molecule with a perfect spherical shape yields a bell-shaped p(r) trace with a single peak; a molecule with dumbbell shape gives rise to two peaks; and a molecule with a rod-like shape results in a distribution that is skewed to the right. Thus, 3xWT and 2xPro have more rod-like structures as compared with WT and 1xPro.
To gain further insight into the structure of the linker mutants, we carried out small angle X-ray scattering (SAXS) experiments. Due to the tendency of some of the syt constructs to self-associate, differences in the radius of gyration could not be determined, but information was garnered from the paired distance distribution plots shown in Fig. 5d. The 3xWT clearly differed from the WT protein, with a distribution indicative of a more rod-like overall structure. The paired distance distribution plot for the 1xPro clearly indicates the presence of two distinct domains (which were obscured in the AFM images); the C2-domains of this construct are even more clearly delineated than in the WT protein. Finally, the distribution of the 2xPro indicates a more rod-like overall structure, akin to the 3xWT. These findings are consistent with the idea that alterations in the linker affect the relative orientation of the tandem C2-domains of syt, and allow, for example, the 2xPro and 3xWT mutants to adopt a more rod-like structure.
Finally, we carried out FRET experiments to address the question of whether Ca2+ and membranes influence the interaction between C2A and C2B. These experiments revealed that the intramolecular interaction between C2A and C2B is constitutive and is not influenced by either Ca2+ or membranes (Supplementary Fig. 9).
The finding that C2A and C2B physically interact provides a physical explanation for earlier findings showing that mutations in one C2-domain appear to alter the function of the adjacent C2-domain, as detailed in the Discussion. Disruption of this inter-domain interaction, via the 2xPro rod, completely disrupts the function of syt.
Uncoupling syt function: evoked versus spontaneous release
In addition to functioning as a Ca2+ sensor for fast release46, syt has been proposed to function as a fusion clamp that arrests SNARE-catalyzed fusion prior to the arrival of the Ca2+ trigger14,24. However, the increase in mini frequency observed for syt KO neurons could be a compensatory response that occurs due to the loss of evoked transmission. The linker mutants characterized in the current study make it possible to address this question in an unambiguous manner, as detailed below.
We monitored excitatory minis (mEPSCs) under Ca2+-free conditions as described previously14,24 in syt KO neurons expressing each linker mutant (Fig. 6a). The amplitude (Fig. 6b) and quantal release size of mEPSCs (Fig. 6c) were unaffected by alterations in the linker, as compared with the WT and KO groups. However, differences in mEPSC frequency were observed. While the 0.5xWT, 2xWT, 3xWT, 1xGly, and 2xGly linker mutant exhibited putative fusion clamp activity, as revealed by the suppression of mini frequency observed in the KO neurons, the two rigid linker mutants, 1xPro and 2xPro, surprisingly, were devoid of this apparent clamping activity (Fig. 6d). A key observation is that the 1xPro linker mutant yields normal evoked release, yet this mutant fails to suppress mini frequency. This finding strongly indicates that syt does in fact serve as a fusion clamp under resting conditions, and that the observed increase in mini frequency observed in syt KO neurons is not a compensatory response due to loss of evoked transmission. Moreover, the 0.5xWT, 2xWT, and 3xWT linker mutants displayed impaired evoked release, but were still able to serve as clamps to reduce mini frequency. Thus, the loss of evoked release observed for these latter mutants did not result in compensatory increases in mini frequency. To address this issue systematically, we plotted the ability of the linker mutants to clamp fusion (mini frequency) in the absence of Ca2+ against their ability to stimulate exocytosis in response to 10 mM Ca2+ (Supplementary Fig. 10a). Again, no correlation was observed (r2 = 0.17), further demonstrating that distinct structural features of syt are involved in clamping versus evoked fusion activity; yet both functions involve physical interactions between the tandem C2-domains of syt.
Figure 6.

Syt linker mutants uncouple the function of syt during evoked versus spontaneous release. (a) Example mEPSC trace recorded from WT neurons and syt KO neurons expressing each linker mutant. (b, c) Scatter plots of the mEPSC amplitude (b) and quantal charge (c). (d) The rigid linker mutants, 1xPro and 2xPro, were unable to clamp spontaneous release. However, the 1xPro linker mutant gave rise to normal evoked release (Fig. 2b), indicating that the increase in mini frequency is not secondary to a loss of evoked release. For each condition we recorded from 9–12 neurons from a total of three coverslips; each coverslip was derived from an independent litter of mice. Two to three hundred events were recorded from each neuron. The mean values ± SEM are indicated. Statistical significance was determined using a bootstrap approach. No differences among groups were found for either mEPSC amplitudes or charge; however, significant differences among groups were found for mEPSC freq. (Supplementary Statistics). Asterisks indicate differences compared with WT; ** p < 0.01, *** p < 0.001.
We explored the idea that Ca2+-independent interactions with t-SNAREs underlie the clamping activity of syt, by plotting Ca2+-independent binding activity versus mini frequency. Interestingly, all of the linker mutants that harbored variations of the native linker sequence (0.5x WT, 2x WT and 3x WT) were tightly clustered in a correlation plot of mini frequency versus Ca2+-independent t-SNARE binding activity (Supplementary Fig. 10b). There were small but significant differences in Ca2+-independent t-SNARE binding activity42, but mini frequencies were virtually identical among this group. A weak correlation, among all the linker mutants, was observed for this plot, but this was not statistically significant (r2 = 0.40). So, while syt can clamp fusion, the underlying mechanism remains an open question.
Together with the evoked responses, our findings regarding the impact of the linker mutants on mini frequency indicate that syt has a dual function in the regulation of synaptic transmission.
DISCUSSION
In the current study we have carried out a structure-function analysis of the linker that connects the tandem C2-domains of syt. From these experiments we draw three major conclusions: 1) Ca2+-triggered penetration of the C2-domains of syt into lipid bilayers constitutes an essential step in excitation-secretion coupling; 2) C2A and C2B exhibit intra-molecular interactions, providing a physical basis for the observed functional interactions between them, and, finally 3) by uncoupling the roles of syt in evoked versus spontaneous release, we show that syt also functions as a fusion clamp.
In an earlier study, it was suggested that the membrane penetration activity of syt was important for its function at the Drosophila neuromuscular junction47. While this previous work clearly showed that mutations in the Ca2+ and effector binding loops of Drosophila syt were correlated with a loss of transmission, direct membrane penetration assays were not carried out, and thus it remained unknown as to whether penetration per se was correlated with the extent of SV release. In the current study, we utilized hippocampal neurons from syt KO mice to study the ability of syt linker mutants to mediate synaptic transmission in a central synapse, and in parallel we conducted membrane penetration assays for each of these mutant proteins. By analyzing a panel of seven distinct linker mutations that exhibit graded alterations in membrane penetration activity, we report for the first time a clear correlation between membrane insertion activity of the tandem C2-domains and the ability of syt to drive efficient, rapid SV exocytosis. These findings support a model in which syt acts, at least in part, by ‘dipping’ into the target membrane to mediate close apposition of the bilayers and/or to drive local bending, thereby accelerating the rate of fusion16.
The experiments reported here further document a functional interaction between the tandem C2-domains of syt. Previously, the physical basis for this synergy was unclear. In some structural studies it appeared that C2A and C2B do not physically interact with each other33, while other structural studies concluded that an intramolecular interaction does occur11. These reports were based on NMR and X-ray crystallography approaches, which introduce certain caveats: the size of C2AB limits analysis by NMR, and the crystallography approach provides a view of a limited number of states of the protein. We have therefore addressed this problem using two novel approaches, AFM and SAXS. Our results indicate that C2A and C2B do in fact physically interact with one another, thus providing a potential mechanism to explain how one C2-domain can affect the properties of the adjacent domain. These data are consistent with the structure of C2AB reported by Fuson et al. (2007)11; future work, using this crystal structure as a guide, in conjunction with mutagenesis, will identify the residues that mediate contacts between the tandem C2-domains.
While the tandem C2-domains of syt must cooperate in order to drive rapid evoked release, syt has also been implicated in the regulation of spontaneous vesicle fusion events. Minis have recently attracted considerable attention as they are thought to play important roles in synaptic physiology4,5. Relatively little is known concerning the means by which mini frequency is regulated, but complexin and syt have been implicated as fusion clamps that serve to suppress minis under resting conditions9,48. While it is well documented that loss of syt results in a significant increase in mini frequency (see Ref.9,24 and references therein), it remained possible that the increase was a compensatory response that was secondary to a loss of evoked transmission. In the current study, we observed that the 1xPro mutant fully rescued evoked transmission yet failed to suppress the elevated mini frequency in syt KO neurons. This result provides two novel insights into syt function: syt does indeed serve as a fusion clamp that normally suppresses minis, and within the structure of syt, distinct molecular determinants regulate evoked versus spontaneous release. Indeed, analysis of all the linker mutants revealed a complete lack of correlation between the ability of these constructs to clamp minis and to drive evoked release.
A crucial point is that the 2xPro is the only mutation that clearly held C2A and C2B apart based on the AFM (Fig. 5), and this mutation completely disrupted the ability of syt to regulate both evoked and spontaneous SV release. In this light, we note that in the 3xWT linker mutant, C2A and C2B still appear to interact, since this alteration in the linker does not affect the overall length of the protein. The 3x linker can fully rescue the clamping function of syt, but only partially rescues the function of syt during evoked transmission. The reasons for this loss of activity are not entirely clear, but might involve alterations in the disposition of C2A with respect to C2B. In this light we reiterate that only WT syt, and not the 1xPro, 2xPro, or 3xWT linker mutant, exhibited a horseshoe shaped structure that is likely due to a specific intramolecular interactions between C2A and C2B11 (Supplementary Fig. 8a). Indeed, the SAXS measurements indicate that changes in the linker do in fact alter the overall structure of syt, and thus likely affect the relative orientation of the tandem C2-domains. It is tempting to speculate that the loss-of-function exhibited by these mutants in the electrophysiological experiments results from the inability of C2A and C2B to engage in the specific intramolecular contacts that would normally result in the horseshoe shape.
Thirty to forty percent of all C2-domain proteins harbor more than one copy of this conserved protein motif49. In all isoforms of syt that act as Ca2+ sensors27, the length of the connecting linker is conserved25. However, the length of the linker varies among syt isoforms that do not sense Ca2+, and this length varies enormously among different families of proteins. For example, in Doc2, which is also a Ca2+ sensor that regulates exocytosis19,20, the linker length is roughly twice the length of the syt, but in the ferlin family there are linker segments that are ~500–600 residues long50. It remains to be determined whether C2-domains connected by such long linkers engage in intramolecular interactions within the ternary structure of their parent proteins.
As shown in the current study, the C2-domains of syt engage in intramolecular interactions that endow the protein with novel biochemical and functional properties that would not be anticipated from studies of the isolated C2-domains. Thus, during evolution, duplications of C2-domains not only provide the possibility of diverging these motifs to yield a greater range of independent functions, but also result in tandem sets of C2-domains that have evolved to work together to impart an even richer repertoire of functional properties, including the regulation of both evoked and spontaneous release. Future studies are needed to explore the generality of our finding that C2-domains can functionally interact within a single parent molecule. In the case of syt, the tandem C2-domains have evolved to work in a synergistic and interdependent manner to regulate SV exocytosis.
ONLINE METHODS
Cell Culture
Primary hippocampal cultures from heterozygous syt KO mouse matings were prepared as described previously24 in accordance with the guidelines of the National Institutes of Health, as approved by the Animal Care and Use Committee of the University of Wisconsin-Madison. In brief, heterozygous syt KO mice were bred, and hippocampal neurons from newborn pups (postnatal day 0) were isolated. Neurons were plated at 25,000 – 50,000 cells/cm2 on poly-lysine coated coverslips (Carolina Biologicals), and cultured in Neurobasal media supplemented with 2% B-27 and 2 mM Glutamax (Gibco/ Invitrogen). Tails from pups were used for genotyping, and electrophysiological recordings were carried out using neurons from KO and WT littermates.
Molecular Biology
cDNA encoding rat syt51 was provided by T.C. Südhof (Palo Alto, CA ); the D374 mutation was corrected by replacement with a glycine residue52. Various linker mutations were generated via PCR using the overlapping primer method53. Full-length linker mutants were sub-cloned into pLox Syn-DsRed-Syn-GFP using BamHI and Not I, thus replacing DsRed, to generate lentiviral particles. The cytoplasmic domain (C2AB) of WT syt and each linker mutant were subcloned into pGEX-2T or -4T-1 vectors. For membrane penetration assays using fluorescence, the lone native cysteine (Cys277) in C2AB was substituted with an alanine. Then, a single cysteine was introduced at Phe234 of C2A, or Ile367 of C2B. For FRET experiments, the lone native tryptophan (Trp259) in the C2A domain of C2AB was substituted with a leucine residue; again the lone native cysteine (Cys277) was substituted with an alanine, then, a single cysteine was introduced at Phe234 of C2A. Point mutations were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene).
WT and linker mutant forms of C2AB were expressed as glutathione S-transferase (GST) fusion proteins, purified using glutathione–Sepharose beads (GE healthcare), and the GST tag was removed by thrombin cleavage, as described13. For SAXS measurements, the protein samples were further purified by size-exclusion chromatography on a Superdex 200 (GE healthcare) column using FPLC.
cDNA for full-length syntaxin-1A and full-length SNAP-25B were provided by R.H. Scheller (Genentech) and M.C. Wilson (Univ. New Mexico), respectively. Each t-SNARE cDNA was subcloned into a pTrcHis vector (Invitrogen), resulting in an N-terminal His6 tag, and proteins were purified as described17.
Infection of Hippocampal Neurons
Lentiviral particles were generated by co-transfecting pLox Syn-DsRed-Syn-GFP with two other packaging vectors encoding VSV-G, and gag and pol, into HEK293T cells. The supernatant was collected after 48–72 h, purified by filtration through a 0.45 μm filter and centrifuged at 70,000 g for 2 h to concentrate virus. Viral particles were resuspended in phosphate-buffered saline (PBS), and were used to infect syt KO neurons at DIV 5.
Electrophysiology
Whole-cell patch-clamp recordings were made from dissociated hippocampal cultures. All recordings were made 12–17 days after neurons were plated on coverslips. The pipette solution consisted of 130 mM K-gluconate, 1 mM EGTA, 5 mM Na-phosphocreatine, 2 mM Mg-ATP, 0.3 mM Na-GTP, and 10 mM HEPES, pH 7.3 (290 mOsm). Evoked transmission was measured as described previously24. Briefly, 5 mM QX-314 (lidocaine N-ethyl bromide) was added to the pipette solution, and neurons were continuously perfused with extracellular solution consisting of 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.3 (300 mOsm), 50 μM DL-2-amino-5-phosphonopentanoate (DL-AP5), 0.1 mM picrotoxin, and 10 (Fig. 2 and 3) or 2 (Supplementary Fig. 1) mM Ca2+. A bipolar electrode, made from theta tubing (Warner instruments, model: TG150-4) and filled with extracellular solution, was placed against the soma and used to trigger action potentials via a 1 ms −20V pulse, and postsynaptic responses were measured via a whole-cell patch electrode in a connected neuron. mEPSC recordings in Ca2+-free conditions (Fig. 6) were carried out as described previously24. Briefly, 10 mM EGTA replaced Ca2+; 1 μM tetrodotoxin (TTX), 25 μM 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid acetoxymethyl ester (BAPTA-AM), and 30 μM cyclopiazonic acid (CPA) were added to the bath solution. Cultures were incubated in bath solution for 30 min before recording to allow BAPTA-AM and CPA to exert their effects.
The size of the readily releasable pool (RRP) was measured using 500 mM sucrose as described previously24, in the presence of 2 mM [Ca2+]o. A Pico Spritzer (Parker Hannifin Corporation, U.S.A) system was used to puff the hypertonic sucrose solution over the entire area viewed under a 40x objective lens, which includes virtually all presynaptic boutons contacting the patched cells. The sucrose was applied for 8 s so that we could acquire the steady-state response, indicating depletion of the RRP; signals were integrated during the fast response phase to calculate the RRP size.
To make stock solutions, picrotoxin (50 mM) was dissolved in ethanol, DL-AP5 (20 mM) was dissolved in 0.1 N NaOH, and BAPTA-AM (30 mM) and CPA (100 mM) were dissolved in DMSO. All drugs were from Sigma-Aldrich. Neurons were voltage clamped at −70 mV with an EPC-10/2 amplifier (HEKA). Only cells with series resistances of <15 MΩ, with 70–80% of this resistance compensated, were analyzed. Currents were acquired using PATCHMASTER software (HEKA), filtered at 2.9 kHz, and digitized at 10 kHz. Data were analyzed using MiniAnalysis software (Synaptosoft), Clampfit (Molecular Devices), and Igor (Wavemetrics). All experiments were performed at room temperature.
Immunostaining
Neuronal cultures were immunostained with a mouse monoclonal antibody directed against syt (Synaptic Systems, 105 011, 1:400) to determine the localization of the linker mutants. Nerve terminals were identified using a polyclonal rabbit anti-synapsin antibody (Millipore, AB1543P, 1:1000). At 12–14 DIV, neurons were fixed with 4% paraformaldehyde in PBS, permeabilized and blocked in 10% goat serum plus 0.1% Triton X-100, and immunostained with primary antibodies for 2 h. Samples were washed with PBS three times, and then incubated with either Cy3-tagged anti-mouse or Alexa 647-tagged anti-rabbit (Jackson ImmunoResearch Laboratories) secondary antibodies for 1 h at room temperature. Coverslips were then mounted in Fluoromount (Southern Biotechnology Associates) and images were acquired on an Olympus FV1000 upright confocal microscope with a 60x 1.10 numerical aperture water-immersion lens. To quantify colocalization, images were acquired under identical laser and gain settings and imported into Metamorph (Improvision). Channels were thresholded separately to include all recognizable puncta. The threshold was determined separately for each channel to include all boutons with intensities that were at least 3-fold above noise and had diameters between 3 and 7 pixels (0.7 ~ 1.6 μm), as described previously54,55. The degree of co-localization was calculated by using the “Measure Colocalization” function in MetaMorph. The percentage of colocalization was determined as the percentage of the total thresholded area of one channel that overlapped with the thresholded area of the other channel. Using this approach, markers that fail to overlap yield co-localization values < 0.254,55.
Liposomes
Protein-free SUVs (small unilamellar vesicles) were prepared as follows: lipids (15% PS + 30% PE + 55% PC or 25% PS + 30% PE + 45% PC, as indicated) were mixed and dried under nitrogen and lyophilized for 1 h. The dried lipid film was resuspended in HEPES buffer (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl) and extruded 20 times through 100 nm polycarbonate filters. SNARE-bearing SUVs were prepared as described previously17 using 15% PS + 30% PE + 55% PC for syntaxin vesicles (SYXr) and 15% PS + 27% PE + 55% PC + 1.5% NBD-PE + 1.5% rhodamine-PE for v-SNARE vesicles (Vr). All lipids were synthetic and were obtained from Avanti Polar Lipids.
Labeling of the cytoplasmic domain of syt with fluorescent dyes
For labeling with NBD for membrane penetration experiments, the appropriate purified cysteine mutants were adjusted to ~10 μM in Tris buffer (50 mM Tris, pH 7.4, 150 mM potassium chloride). N-(7-nitro-2,1,3-benzoxadiazol-4-yl) (NBD)(Molecular Probes) was then added drop-wise to each protein solution to reach a final concentration of ~150 μM, followed by incubation at room temperature for 2 h with rotation. Free dye was removed by extensive dialysis in Tris buffer (50 mM Tris, pH 7.4, 150 mM potassium chloride). The NBD concentration was determined by absorbance using a molar extinction coefficient of 25,000 M−1cm−1 at 480 nm. The protein concentration was determined by SDS-PAGE and staining with Coomassie blue, using BSA as a standard. The labeling stoichiometry ranged from 0.7–0.9 mole of dye per mole of protein.
Labeling with AEDANS for FRET experiments was carried out in the same manner with the following modifications: 5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (IAEDANS)(Molecular Probes) was used at ~150 μM, labeling was carried out for 3 h, and free dye was removed by using Zeba Spin Desalting Columns (Thermo, 7K MWCO). The AEDANS concentration was determined by absorbance using a molar extinction coefficient of 6,000 M−1cm−1 at 337 nm. The labeling stoichiometry was ~ 1 mole of dye per mole of protein.
Fluorescence measurements
Membrane penetration and FRET assays were performed at room temperature using a PTI QM-1 fluorometer and FELIX software. For membrane penetration assays, NBD-labeled protein was mixed with SUVs (113 μM total lipid) in Tris buffer (50 mM Tris, pH 7.4, 150 mM potassium chloride), in the presence of either EGTA (0.2 mM final concentration) or Ca2+ (100 μM or 1 mM final concentration). NBD was excited at 470 nm, and the emission spectra were collected from 490 to 630 nm (4-nm slits), and were corrected for blank and instrument response. For FRET measurements, AEDANS-labeled syt C2AB was either mixed with EGTA (100 μM final concentration) or Ca2+ (100 μM or 1 mM final concentration) in Tris buffer (50 mM Tris, pH 7.4, 150 mM potassium chloride), either in the presence or absence of liposomes (113 μM total lipid). Native tryptophans in the C2B domain were excited at 295 nm, and the emission spectra were collected from 320 to 570 nm (4-nm slits), and corrected for blank and instrument response.
Co-sedimentation assays
WT or linker mutant C2AB domains (4 μM) were mixed with liposomes (at the indicated concentrations) in HEPES buffered (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl), in the presence of 1 mM Ca2+ or 0.2 mM EGTA, and then centrifuged at 170,000g at 4 °C for 45 min in a Beckman Optima MAX-E (Beckman Coulter) tabletop ultracentrifuge. The supernatant from each sample was collected and analyzed by SDS-PAGE and Coomassie blue staining. Bands were quantified by densitometry to reveal the amount of free protein, and these data were used to calculate the amount of bound protein which was then plotted versus [lipid]; apparent Kd values were determined by fitting the data with ‘one-site specific binding curves’ using Prism 6 (Graphpad Software).
Co-flotation assays
As described previously39, 100 μl reactions were prepared containing 25 μM WT or linker mutant forms of C2AB, 45 μl PS-free t-SNARE vesicles, 0.2 mM EGTA, and reconstitution buffer (25 mM HEPES-KOH, pH 7.4, 100 mM KCl, 10% glycerol, 1 mM dithiothreitol) in the presence or absence of 1 mM free Ca2+. Components were incubated at room temperature for 45 min with shaking, loaded onto an Accudenz gradient, centrifuged, and collected at the 0–30% Accudenz interface. Vesicles were then subjected to SDS-PAGE and stained with Coomassie blue, and bands were quantified by densitometry.
In vitro fusion assays
Fusion assays using v-SNARE vesicles (Vr) and syntaxin vesicles (SYXr) were carried out as described17 with one modification: both vesicle populations were scaled-down to 10% of the previously used concentrations (0.5 μl of Vr, 4.5 μl of SYXr); 3 μM soluble SNAP-25 was added, reconstitution buffer (25 mM HEPES-KOH, pH 7.4, 100 mM KCl, 10% glycerol, 1 mM dithiothreitol) was used, and the total volume was 75 μl. This configuration bypasses the ability of syt to stimulate fusion by merely aggregating v- and t-SNARE vesicles when the t-SNAREs, syntaxin and SNAP-25, are reconstituted as pre-formed heterodimers18. Rather, syt must direct the folding of SNAP-25 onto syntaxin, resulting in the assembly of functional trans SNARE complexes17, thus driving fusion. Optimal folding activity requires Ca2+•syt•PS interactions17, so this assay system involves both syt-membrane interactions as well as syt-mediated assembly of SNARE complexes. Samples were incubated at 37°C for 20 min in the presence of 0.2 mM EGTA, followed by addition of Ca2+ (1 mM final concentration). Fusion was monitored, as dequenching of NBD fluorescence, for an additional 1 h. For normalization, after each run 0.5% of the detergent β-D-maltoside was added, resulting in ‘infinite’ dilution of the NBD-rhodamine FRET pair to produce the maximum fluorescence signal.
AFM imaging
Purified C2AB was diluted to 100 ng/ml in HEPES buffered saline and deposited onto freshly-cleaved mica (5-mm diameter disks) fixed by epoxy (Aron Alpha type 102, Agar Scientific, UK) to 15-mm steel SPM specimen disks (Agar Scientific, UK). After a 3-min adsorption period, the sample was rinsed with 1 ml of Biotechnology Performance Certified (BPC) water (Sigma) to remove unabsorbed protein, and dried under a stream of nitrogen gas. Samples were then imaged. AFM imaging was carried out at room temperature (20°C) using a Digital Instruments Multimode atomic force microscope equipped with a E-scanner and a Nanoscope IIIa Controller, with an in-line electronics extender module (Bruker AXS GmbH, Karlsruhe, Germany). All images were collected using tapping mode in air, using oxide-sharpened silicon nitride probes (OTESPA; Bruker AFM Probes, Camarillo, CA). The cantilevers (typically exhibiting a spring constant of 42 N/m) were tuned to 10–20% below the resonance frequency, generally found between 290 and 320 kHz. The drive amplitude was set to generate a root-mean-square amplitude of 2.0–2.5 V. The microscope was engaged with a 0-nm scan area to allow for tuning. The set-point was adjusted to the highest setting that allowed imaging with little noise, to minimize the force applied to the sample. Images were captured at a scan rate of 2 Hz, and with 512 scan lines per area. Data analysis was performed using commercially available software (NanoScope III software, Digital Instruments).
SAXS
All SAXS data were generated at The National Magnetic Resonance Facility at Madison (NMRFAM). For each run, scattering data were collected for 4 h using 50 μM protein in Tris buffer (50 mM Tris, pH 7.4, 150 mM sodium chloride, 0.2 mM EGTA). Signals were corrected for background scattering and the data were analyzed using ATSAS 2.5.0 (European Molecular Biology Laboratory).
Statistical analysis
Statistical significance was determined using a bootstrap approach as detailed in ref.56. Briefly, we generated 10,000 resampled (with replacement) distributions from the each data set (after first transforming the original data, as detailed in Supplementary Statistics), and computed an Fi or ti statistic for each comparison (i ∈ [1,10000]). The Fobserved, or tobserved, statistics computed from the original data were compared to this distribution of test statistics to compute a p-value, as follows:
Details of the bootstrap approach utilized throughout this study, and the complete results of all statistical analysis, are listed in a Supplementary file as an Excel spreadsheet (Supplementary Statistics); all raw data are also included in a Supplementary file (Raw Data). No statistical methods were used to predetermine sample sizes; however, our sample sizes conform to those generally used in similar studies24,57. All data are presented as the mean ± SEM unless otherwise indicated. In all the electrophysiological experiments, n refers to the number of individual neurons from which the recordings were made. Correlations were determined via linear regression.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank D. Ruhl for help with bootstrap analysis, and we thank S. Butcher and J. Burke for help with the SAXS measurements. HT was supported by a Newton International Fellowship. This study was supported by a grant from the National Institutes of Health (MH 61876). ERC is an Investigator of the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
H.L. and R.X. performed and analyzed the electrophysiological experiments; H.B., performed and analyzed the biochemical experiments; H.T., J.M.E., performed and analyzed the AFM measurements; E.R.C., designed experiments, analyzed the data; H.L., H.B., and E.R.C. co-wrote the paper.
References cited
- 1.Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–94. doi: 10.1016/s0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- 2.Iremonger KJ, Bains JS. Integration of asynchronously released quanta prolongs the postsynaptic spike window. J Neurosci. 2007;27:6684–91. doi: 10.1523/JNEUROSCI.0934-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau PM, Bi GQ. Synaptic mechanisms of persistent reverberatory activity in neuronal networks. Proc Natl Acad Sci U S A. 2005;102:10333–8. doi: 10.1073/pnas.0500717102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kavalali ET, et al. Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology (Bethesda) 2011;26:45–53. doi: 10.1152/physiol.00040.2010. [DOI] [PubMed] [Google Scholar]
- 5.Sutton MA, et al. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–99. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 6.Sollner T, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–24. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 7.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–53. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 8.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 9.Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–41. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- 10.Sutton RB, Ernst JA, Brunger AT. Crystal structure of the cytosolic C2A-C2B domains of synaptotagmin III. Implications for Ca(+2)-independent snare complex interaction. J Cell Biol. 1999;147:589–98. doi: 10.1083/jcb.147.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuson KL, Montes M, Robert JJ, Sutton RB. Structure of human synaptotagmin 1 C2AB in the absence of Ca2+ reveals a novel domain association. Biochemistry. 2007;46:13041–8. doi: 10.1021/bi701651k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–4001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 13.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 14.Chicka MC, Hui EF, Liu HS, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca(2+) Nat Struct Mol Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 16.Hui EF, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-Mediated Bending of the Target Membrane Is a Critical Step in Ca2+-Regulated Fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–30. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- 18.Hui E, et al. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol. 2011;18:813–21. doi: 10.1038/nsmb.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao J, Gaffaney JD, Kwon SE, Chapman ER. Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell. 2011;147:666–77. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groffen AJ, et al. Doc2b Is a High-Affinity Ca2+ Sensor for Spontaneous Neurotransmitter Release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Pang ZPP, Shin OH, Sudhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nature Neuroscience. 2009;12:759–U111. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kochubey O, Schneggenburger R. Synaptotagmin increases the dynamic range of synapses by driving Ca(2)+-evoked release and by clamping a near-linear remaining Ca(2)+ sensor. Neuron. 2011;69:736–48. doi: 10.1016/j.neuron.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Vyleta NP, Smith SM. Spontaneous Glutamate Release Is Independent of Calcium Influx and Tonically Activated by the Calcium-Sensing Receptor. Journal of Neuroscience. 2011;31:4593–4606. doi: 10.1523/JNEUROSCI.6398-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci. 2009;29:7395–403. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craxton M. Synaptotagmin gene content of the sequenced genomes. BMC Genomics. 2004;5:43. doi: 10.1186/1471-2164-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon OJ, Gainer H, Wray S, Chin H. Identification of a novel protein containing two C2 domains selectively expressed in the rat brain and kidney. FEBS Lett. 1996;378:135–9. doi: 10.1016/0014-5793(95)01430-6. [DOI] [PubMed] [Google Scholar]
- 27.Bhalla A, Chicka MC, Chapman ER. Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. J Biol Chem. 2008;283:21799–807. doi: 10.1074/jbc.M709628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008;283:31763–75. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hui E, Bai J, Chapman ER. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J. 2006;91:1767–77. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez I, et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–69. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- 31.Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–30. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca(2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 2002;99:1665–70. doi: 10.1073/pnas.032541099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arac D, et al. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–17. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 34.Choi UB, et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–24. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Cafiso DS. Conformation and membrane position of the region linking the two C2 domains in synaptotagmin 1 by site-directed spin labeling. Biochemistry. 2008;47:12380–8. doi: 10.1021/bi801470m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doose S, Neuweiler H, Barsch H, Sauer M. Probing polyproline structure and dynamics by photoinduced electron transfer provides evidence for deviations from a regular polyproline type II helix. Proc Natl Acad Sci U S A. 2007;104:17400–5. doi: 10.1073/pnas.0705605104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voets T, et al. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A. 2001;98:11680–5. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–64. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–8. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XD, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TFJ. Ca2+-dependent synaptotagmin binding to SNAP–25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/s0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 41.Tucker WC, Chapman ER. Role of synaptotagmin in Ca2+-triggered exocytosis. Biochem J. 2002;366:1–13. doi: 10.1042/BJ20020776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron. 2004;41:929–42. doi: 10.1016/s0896-6273(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z, Hui E, Chapman ER, Jackson MB. Regulation of exocytosis and fusion pores by synaptotagmin-effector interactions. Mol Biol Cell. 2010;21:2821–31. doi: 10.1091/mbc.E10-04-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozlov MM, Chernomordik LV. The protein coat in membrane fusion: lessons from fission. Traffic. 2002;3:256–67. doi: 10.1034/j.1600-0854.2002.030403.x. [DOI] [PubMed] [Google Scholar]
- 45.Sun S, et al. Receptor binding enables botulinum neurotoxin B to sense low pH for translocation channel assembly. Cell Host Microbe. 2011;10:237–47. doi: 10.1016/j.chom.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–22. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- 47.Paddock BE, et al. Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J Neurosci. 2011;31:2248–57. doi: 10.1523/JNEUROSCI.3153-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue MS, et al. Tilting the Balance between Facilitatory and Inhibitory Functions of Mammalian and Drosophila Complexins Orchestrates Synaptic Vesicle Exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–22. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–13. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Perin MS, Fried VA, Mignery GA, Jahn R, Sudhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–3. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 52.Desai RC, et al. The C2B domain of synaptotagmin is a Ca(2+)-sensing module essential for exocytosis. J Cell Biol. 2000;150:1125–36. doi: 10.1083/jcb.150.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-Directed Mutagenesis by Overlap Extension Using the Polymerase Chain-Reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 54.Dean C, et al. Axonal and dendritic synaptotagmin isoforms revealed by a pHluorin-syt functional screen. Mol Biol Cell. 2012;23:1715–27. doi: 10.1091/mbc.E11-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwon SE, Chapman ER. Glycosylation is dispensable for sorting of synaptotagmin 1 but is critical for targeting of SV2 and synaptophysin to recycling synaptic vesicles. J Biol Chem. 2012;287:35658–68. doi: 10.1074/jbc.M112.398883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Efron B, Tibshirani R. An introduction to the bootstrap. Chapman & Hall; New York: 1993. p. xvi.p. 436. [Google Scholar]
- 57.Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–81. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.