

Abstract
With prostate cancer (PCa), circulating tumor cells (CTCs) and disseminated tumor cells (DTCs) portend a poor clinical prognosis. Their unknown biology precludes rational therapeutic design. We demonstrate that CTC and DTC cell lines, established from mice bearing human PCa orthotopic implants, exhibit increased cellular invasion in vitro, increased metastasis in mice, and express increased epithelial to mesenchymal transition biomarkers. Further, they are selectively resistant to growth inhibition by mitoxantrone-like agents. These findings demonstrate that CTC formation is accompanied by phenotypic progression without obligate reversion. Their increased metastatic potential, selective therapeutic resistance, and differential expression of potential therapeutic targets provide a rational basis to test further interventions.
Keywords: Prostate Cancer, Circulating Tumor Cells, EMT, MMP-2, Metastasis, Drug Resistance
1. INTRODUCTION
Prostate cancer (PCa) is a major public health concern. It is the most commonly diagnosed cancer and the second leading cause of cancer death in men in the United States [35]. Localized PCa has a high survival rate and can be managed with localized therapy. However, once PCa has metastasized to distant sites, it is incurable and will inexorably lead to death. In order to move between distant sites throughout the body, cancer cells must move through the circulation. Circulating tumor cells (CTCs) in the blood are the subject of intense investigation because they denote a population at higher risk for poor outcome. However, currently therapy is not altered based upon CTC status. A lack of understanding of the biology of CTCs has served as a barrier to developing rational therapy tailored to these high risk patients.
The presence of CTCs has been linked to poor clinical outcome in several cancer types, including PCa. In patients with metastatic PCa, the presence of CTCs and/or of high numbers of CTCs denote a population with decreased overall survival as measured by either the CellSearch™ system [7, 8, 13, 31, 36] or by RT-PCR based methodology [6]. Further, in patients with metastatic PCa, a decline in CTCs after treatment with cytotoxic chemotherapy portends a more favorable prognosis than those who do not experience a decline [8]. In addition to overall survival, CTCs also predict responsiveness to hormonal therapy in patients with hormone-sensitive PCa [14], and a decline in CTCs is associated with responsiveness to therapy [34]. Finally, CTCs are increasingly recognized as having similar prognostic value in other cancer types, including colorectal [15, 25], breast [25, 42], lung [17], bladder [30], and pancreatic [16]. Despite our rapidly evolving ability to identify CTCs, and accumulating evidence that their presence predicts poor clinical outcome, these findings fail to provide guidance on differential therapeutic strategies for patients with CTCs.
Changes in the epithelial to mesenchymal transition (EMT) state of CTCs have been proposed as a mechanistic driver of CTC formation. Further, the loss of epithelial cell markers which accompany EMT act to offset the efficacy of technology which relies upon them for CTC detection. In multiple studies, breast cancer patients with high CTC counts have correlated with higher levels of EMT proteins, such as twist and phosphoinositide 3-kinase (Pi3K), as well as markers of stem cells [1, 20]. However, the prognostic value of this correlation is still unclear. While some studies have been unable to correlate the presence of CTCs and EMT markers to bone marrow disseminated tumor cells (DTCs) in breast cancer patients [20], others have shown that EMT markers on CTCs change with metastatic progression. In one study, the percentage of twist and vimentin-positive CTCs increased in patients with metastatic versus early stage breast cancer [19], and an additional study demonstrated that patients with metastatic breast and PCa more frequently have CTCs with high levels of vimentin and n-cadherin, with low levels of e-cadherin [2]. This has also been confirmed in hepatocellular carcinomas, where expression of vimentin and twist are increased, while those of e-cadherin decreased, as compared to the primary tumor [24]. Multiple studies have shown the ratio of epithelial to mesenchymal markers on CTCs can be used to determine the likeliness of patient to respond to therapy [1, 40].
In addition to EMT, CTCs constitute cells that are traversing the metastatic cascade. Therefore, it is important to additionally consider factors that drive transformation to a metastatic phenotype. Our group has identified several pathways that, through signaling pathway cross-talk, together serve to regulate cellular motility and transformation to a metastatic phenotype in human PCa (reviewed in [33]). We have demonstrated that heat shock protein 27 (HSP27) is a central downstream mediator of transformation to a metastatic phenotype [38]. HSP27 regulates the expression of matrix metalloproteinase 2 (MMP-2). MMP-2 degrades collagen IV, which is a major component of the prostate basement membrane. Both HSP27 and MMP-2 have been shown to be up regulated in aggressive PCa, and to predict the future development of metastasis in humans [9–12, 28, 29, 37]. Our group has shown that it is possible to inhibit the pathway that activates HSP27 with a small molecule therapeutic [18], and that this will decrease human PCa metastasis in mice bearing orthotopic tumors. Further, we have also shown in a prospective phase II randomized controlled trial in men with localized PCa that therapeutically inhibiting HSP27 activation will decrease MMP-2 expression in human prostate tissue [39]. Therefore if CTCs differentially regulate HSP27 and/or MMP-2, they offer attractive targets to which therapeutic inroads are currently being made.
In this study, we used an orthotopic murine model of human PCa metastasis previously described by us [21, 22, 32] to generate CTC cell lines from blood and disseminated tumor cell (DTC) lines from the bone marrow. Importantly, this allowed us to compare the biological and functional characteristics of prospectively generated CTCs and DTCs, from two different compartments, to the parental cells from which they were derived. CTCs and DTCs exhibited increased invasion, as well as increased metastasis formation, as compared to parental cells. An examination of parental, CTC, and DTC cell lines, and resultant tumors in mice, revealed that CTCs and DTCs exhibited markers of an invasive EMT phenotype, as indicated by increases in Twist1, vimentin, HSP27, and MMP-2. We also evaluated CTC and DTC therapeutic responsiveness to a panel of cancer-relevant therapeutic agents. CTCs and DTCs exhibited selective resistance to growth inhibition by mitoxantrone-like agents. Together, these findings provide new insights into the biology of CTCs and DTCs. Their propensity for increased cell invasion and metastasis provide a mechanistic explanation for their clinical association with both metastatic disease as well as poor clinical outcome. Their selective resistance to specific types of therapeutic agents provides a rationale for implementing studies testing relative effectiveness based upon unique CTC and DTC sensitivity. Further, as HSP27 and MMP-2 can be targeted through several approaches now being developed in human trials, this similarly provides rationale to support CTC-specific interventions. Finally, the current study definitively demonstrates that EMT is a feature of CTC and DTC formation, and that it does not undergo obligate reversion once cells are removed from their circulatory environment.
2. MATERIALS AND METHODS
2.1 Mouse Orthotopic Implantation Model
Orthotopic implantation of human PCa cells was performed as previously described by us [21, 22, 32] . Briefly, 2.5×105 cells were injected into the ventral lobe of the prostate of 4–6 week old inbred Balb/c athymic mice (Charles River Laboratories). Mice were fed ad labium with soy-free chow. All mice were treated, housed and managed under an IACUC approved protocol. Four to five weeks after implantation, blood was obtained via terminal cardiac puncture, bone marrow harvested from femurs, and primary tumor was weighed. In addition, lungs were formalin-fixed, paraffin embedded, serial sectioned, stained with H&E and for green fluorescent protein (GFP) by immunohistochemistry. The number of metastases to lungs was counted under light microscopy.
For the initial generation of CTC and DTC cell lines, mice were implanted with PC3-M cells that were stably transfected with green fluorescent protein (GFP) and constitutively active MAP2K4 as previously described by us[21, 32, 39].
2.2 Isolation and Culture of CTCs
CTCs were isolated from animals as previously described by us [32]. Briefly, at necropsy blood was removed from the animal via terminal cardiac puncture and placed into sterile saline. Samples were centrifuged for 5 min at 3000 rpm, plasma was removed and ACK Lysis Buffer (154.95 mM Ammonium Chloride, 9.99 mM Potassium Bicarbonate, 0.0995 mM EDTA, Gibco) added for 5 minutes. Samples were centrifuged for 5 min at 3000 rpm, and the supernatant removed. The resultant pellet was resuspended in 1ml cell culture media (RPMI media containing 5% FBS, 1% Penicillin-Streptomycin, 1% HEPES Buffer, and 1% L-glutathione, all purchased from Gibco) and added to a T75 flask containing 9ml cell culture media. After 24 hours, media was changed to cell culture media plus 500μg/ml G418 (EMD Millipore). Once emergent colonies were outgrown (8–10 days), cells were sterile cell sorted for GFP-positive status using flow cytometry. Similar procedures were performed for the bone marrow samples. Bone marrow was ejected from the femurs using a syringe and sterile saline. Cells were cultured in cell culture media for 8–10 days, changed to media with G418 after the first 24 hours, and then sterile cell sorted for GFP-positive status using flow cytometry. A schematic of the isolation procedure, with resultant cell lines names is shown in Figure 1.
Figure 1. Schematic of CTC and DTC isolation.
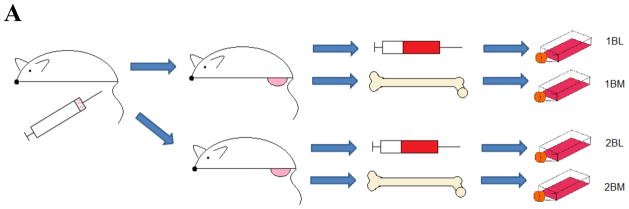
Parental control, P-CON, cells were orthotopically implanted into the prostate. When primary tumors had grown to an advanced size, blood and bone marrow were harvested and cultured. This yielded four cell lines,1BL, 1BM, 2BL, and 2BM, used in future experiments.
2.3 MTT Assay
MTT assays were performed as previously described by us [26]. In brief, for the initial cell growth assay, cell concentrations of 1, 2.5, 5, 10, and 20×103 cells/well in 200μl cell culture media were added to a 96 well plate and allowed to grow for 4 days. Four hours before the end of the experiment, 20μl of 5mg/ml MTT was added to each well. Cells were resuspended in 200μl DMSO and analyzed on a microplate reader at OD550. For the drug treatment assays, 1x104 cells/well in 200μl cell culture media were plated in a 96 well plate, and drug was added 24 hrs after plating.
2.4 Cell Invasion Assay
Cell invasion assays were performed as previously described by us [4]. Briefly, 5×104 cells were plated on the top of a BD Biocoat Growth Factor Reduced Matrigel™ chambers with 8μM pores (BD Biosciences) and allowed to invade for 24 hours toward NIH 3T3 conditioned media in the lower chamber. Wells were plated in quadruplicate, with three wells measured for invasion and one as a loading control for cells in the upper chamber. After removing cells on the top wells for the three invasion wells, invading cells on the bottom membrane were fixed with 100% methanol for 5 min, and then stained with 0.5% crystal violet in 20% methanol for 10 min. Wells were washed with water and dried overnight. Cells were then imaged at 10x using light microscopy and the number of invading cells counted. Experiments were performed in triplicate and a p-value of 0.05 or less as measured by a two-sided t-test were determined to be significant.
2.5 Cell Migration Assay
Cell migration was performed as cellular invasion, but using a BD Falcon uncoated 8μM pore membranes. Chambers were loaded with 2.5×104 cells per well into the upper chamber, and allowed to migrate for 8 hours. Cells were stained, counted, and statistics performed as above.
2.6 Ten Day Colony Formation Assay
Twenty-four hours after plating 103 cells per well, in a six-well plate, 2μM, 200nM, 20nM, 2nM, 0.2nM drug, or DMSO for controls, were added to each well. Colonies were allowed to grow for 10 days. Cells were then fixed with 1 ml of 100% methanol for 5 minutes, and then stained with 0.5% crystal violet in 20% methanol for 5 minutes. Plates were gently washed with dH2O and left to dry overnight. Colonies in each well were counted and taken as a percentage of the control well. Experiments were performed in triplicate.
2.7 Western Blot
Western blots were performed as previously described by us [4]. Antibodies used in this study were HSP27 (catalog #2402), vimentin (#5741), E-cadherin (#3195), all from Cell Signaling, and Desmoplakin (ab71690) from Abcam.
2.8 Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT/PCR)
qRT/PCR was performed as previously described by us [39]. The primers used in these studies were Hs99999905_m1 (GADPH), Hs00234579_m1 (MMP-9), Hs00234422_m1 (MMP-2), and Hs016475818_s1 (Twist1), all purchased from Applied Biosystems. Experiments were performed in at least triplicate and a p-value of 0.05 or less as measured by a two-sided t-test was determined to be significant. Gene expression was normalized to that of GAPDH, and relative gene expression was calculated by the 2−Δ ΔCt method [27].
3. RESULTS
Circulating tumor cells (CTCs) and disseminated tumor cells (DTCs) were isolated from a cohort of 9 mice receiving human PCa orthotopic implants six weeks prior. All mice had primary tumors, and the mean±SEM tumor size was 2.12±0.28 gms. All mice had distant metastasis to the lungs, and the mean±SEM number of metastatic cells was 75.63±22.72 per mouse lung section. A terminal cardiac puncture yielded a mean±SEM 515.6±46.04μl blood, to support blood CTC isolation. Bone marrow was extracted from both femurs of all mice, to support bone marrow DTC isolation.
Two of nine mice (22.2%) yielded CTCs and DTCs. Of the two mice that developed CTCs, each yielded CTCs from their blood (BL) as well as DTCs from their bone marrow (BM). The resultant four CTC and DTC cell lines were labeled 1BL and 1BM, from mouse 1, and 2BL and 2BM from mouse 2 (Figure 1). In all future experiments CTCs were then compared to the parental control cell line that was originally implanted, labeled P-CON.
We first determined if CTCs and DTCs exhibited differential growth characteristics. There were no differences in cell growth amongst any of the cell lines evaluated (Figure 2A). By seeding different numbers of cells, this same study showed that growth conditions were not limited by overcrowding, and alterations in cell density did not differentially affect growth patterns between cell lines. Additionally, no changes in cellular size or morphology were observed in these cells lines. We next examined cell invasion. Increased cell invasion is considered a quintessential characteristic of the metastatic phenotype, and is necessary in order for cells to move from a primary organ to a distant metastatic site. The invasion of all CTCs and DTCs was 50% higher than that of P-CON cells, was statistically significant for three of four CTC and DTC lines, with the fourth exhibiting a strong statistical trend (Figure 2B). We went on to measure cell migration, and demonstrated that there was no difference amongst any of the cells examined (Figure 2D). These findings show that CTCs and DTCs have increased invasive capacity, compared to parental cells, but that this is not due to increased cell migration.
Figure 2. CTCs and DTCs Exhibit Increased Cell Invasion In Vitro.

Cellular phenotypes of CTCs and DTCs were compared to parental control, P-CON, cells. A) Cellular growth as measured by a five day MTT assay. Data are the mean±SEM from 3 independent experiments, each run in replicates of N=5. B) Cellular invasion as measured by a Matrigel™ coated Boyden chamber assay. Data are the mean±SEM from 3 independent experiments, each run in replicates of N=3, and are expressed as a percentage of P-CON cell line. * denotes p value ≤0.05. C) Cellular migration as measured by an uncoated Boyden chamber assay. Data are the mean±SEM from 3 independent experiments, each run in replicates of N=3, and are expressed as a percentage of P-CON cell line.
3.1 CTCs Promote Metastasis Without Altering Tumor Growth
We next assessed whether the increased invasive capacity of CTCs and DTCs would translate into increased metastatic potential. This was tested by orthotopically implanting cells into mice and measuring the resultant formation of distant metastasis after five weeks. Eight mice were implanted with P-CON cells, and cohorts of six mice were each implanted with IBL, 2BL, 1BM or 2BM cells. When considering data, mice were grouped into P-CON, BL-CTC (1BL plus 2BL) and BM-DTC (1BM plus 2BM). After removal of mice due to surgical mortality, post-operative complications (e.g., infighting-related wounds followed by infectious complications) and no tumor take, this gave 6 P-CON, 7 BL-CTC and 9 BM-DTC mice available for analysis at five weeks post-surgery.
As can be seen in Figure 3A, there was no difference in the primary tumor size between the groups. In contrast, however, there was a significant increase in metastasis formation in CTC bearing mice, Figure 3B. Fisher’s exact test p-values were 0.0210 and 0.0278 for comparison of BL-CTC and BM-DTC mice to P-CON mice, respectively. A representative photomicrograph of lung tissue from a CTC- or DTC-implanted mouse depicts metastases in clusters of various sizes, Figure 3C. These findings demonstrate that CTCs and DTCs have an increased ability to form metastasis, without changes in growth kinetics in vivo. Further, by demonstrating no difference between blood-derived CTCs and bone marrow-derived DTCs in both of these measurements, these results indicate that differences in the original microenvironments of the CTCs and DTCs do not translate into altered behavior in vivo. For both measures of cell motility and cell growth, findings in vivo mirror those observed in vitro.
Figure 3. CTCs and DTCs Exhibit Increased Metastatic Potential.

P-CON, BL-CTC, or BM-DTC cells were implanted into the prostate gland, and five weeks later primary tumor weight (A) and metastasis to the lungs (B) were measured. Individual values for each mouse are denoted by individual symbols, with the central line denoting mean value, and whiskers (only for weight) denoting ±SE. Values in brackets denote Fisher’s Exact Test p-values, as compared to P-CON. C) A representative 10x image of a CTC lung with metastatic cells highlighted with an arrow. This image was obtained from a mouse with approximately 1300 total metastatic cells.
In addition to measuring tumor size and metastasis after orthotopic implantation, we also assayed for the presence of CTCs and DTCs in animals (Table 1). In the P-CON group, one animal out of six (17%) developed CTCs and DTCs, which were observed in both the blood and the bone marrow. In the BL-CTC group, two animals out of seven (29%) developed blood CTCs, without the presence of bone marrow DTCs. In the BM-DTC group, one animal out of nine (11%) developed bone marrow DTCs, without the presence of blood CTCs. While it appears that implantation of blood-derived CTCs versus bone marrow-derived DTCs only led to tumor cells being isolated from blood versus bone marrow, respectively, it is very important to consider that the overall number of events is very small. There is no statistically significant difference between these findings. We urge extreme caution in inferring true biological differences.
Table 1.
CTCs detected in mice implanted with blood or bone marrow derived CTCs, or with parental cells
| CTCs detected in mice |
|||
|---|---|---|---|
| Cell type implanted* | Blood derived | Bone marrow derived | Total |
| P-CON | 1/6 (17%) | 1/6 (17%) | 1/6 (17%) |
| BL-CTC | 2/7 (29%) | 0 | 2/7 (29%) |
| BM-CTC | 0 | 1/9 (11%) | 1/9 (11%) |
P-CON-parental, BL-CTC-blood derived, and BM-CTC-bone marrow derived cell line
3.2 CTCs Increase EMT Signaling
We demonstrated above that CTCs and DTCs exhibit increased cell invasion in vitro and increased metastatic potential in vivo, both of which have direct clinical relevance. Therefore, we conducted additional studies at the molecular level to further characterize them. Since CTCs exhibit an increased propensity for metastasis, and DTCs have arrived at a secondary metastatic site, we considered findings by others related to EMT. EMT has been observed in conjunction with transformation to a metastatic phenotype in several cancers, including PCa. Further, emerging evidence indicates that CTCs exhibit markers of mesenchymal transformation, in PCa as well as in other cancer types. As many detection methods of CTCs are dependent on epithelial markers, changes in these proteins may alter the ability to detect CTCs in human patients. Finally, we also considered our prior findings demonstrating the importance of HSP27 and MMP-2 in mediating transformation to a metastatic phenotype and metastasis formation in human PCa [18, 38].
We first measured the mRNA levels of MMP-2 and MMP-9 (Figures 4A and 4B). There was a significant increase in MMP-2 expression in all four CTC and DTC lines measured, compared to P-CON cells, with increases ranging from 3.7- to 6.9-fold. Conversely, MMP-9 expression did not increase (Figure 4B). These findings demonstrate that there is selective upregulation of MMP subtypes in CTCs and DTCs. Further, upregulation of MMP-2 is consistent with our prior findings demonstrating the importance of MMP-2 in regulating human PCa cell invasion and metastasis [18]. In this regard, it is important to consider that CTCs and DTCs have increased cellular invasion in vitro, but no change in cellular migration (Figure 2). Cell invasion is a composite function of cell migration coupled to protease digestion. Increased MMP-2 transcript expression, coupled to our prior reports that changes in MMP-2 transcript expression directly reflect changes in MMP-2 protease activity [38], shows that CTCs and DTCs have an increased capacity to degrade the extracellular matrix, and corroborate that migration is unaffected.
Figure 4. CTCs and DTCs Express Molecular Markers of the EMT and the Metastatic Phenotype.

In the indicated cells, the expression of MMP-2 (A), MMP-9 (B) and Twist1 (C) were measured by qRT/PCR. Samples were normalized to GAPDH and then taken as a percentage of the P-CON cell line. Data are the mean±SEM from 3–5 separate experiments, each in duplicate. * denotes p value ≤0.05 compared to P-CON. The expression of vimentin (D) and HSP27 (E) were measured by Western blot. Experiment was performed 4 times, and a representative blot is shown. Numbers above bands represent the mean relative intensity, normalized to P-CON, as quantified by ImageJ, of 3–4 separate experiments.
With EMT in several cancer types, including PCa, vimentin, Twist1 and HSP27 increase, while E-cadherin and desmoplakin decrease [23]. We therefore examined these in CTCs and DTCs. Twist1 expression is conventionally measured by qRT/PCR, and was increased in all four CTC and DTC lines compared to P-CON, with the increase being statistically significant in three, and a statistical trend in the fourth (p-value 0.08) (Figure 4C). The expression of vimentin and HSP27 were measured by Western blot, and were both increased in all CTC and DTC lines (Figures 4D and 4E). In contrast, the expression of E-cadherin and of desmoplakin measured by Western blot, were not altered in CTCs or DTCs compared to P-CON cells (data not shown). Together, these findings demonstrate that CTCs and DTCs exhibit increased expression of MMP-2 and of several protein markers indicative of EMT.
3.3 CTCs Confer Selective Drug Resistance
Several lines of evidence suggest it would be optimal to identify therapy that directly acts upon CTCs and DTCs themselves. Above, we show that CTCs and DTCs have increased invasive and metastatic potential, which may explain the link between CTCs and poor clinical outcome. We also show these cells are undergoing EMT, which accumulating evidence indicates may impart therapeutic resistance. Taken together, these considerations suggest that primary tumors may have different response profiles than CTCs and DTCs. Therefore, CTCs and DTCs may be resistant to therapy to which the established tumor is responding to. Under such clinical circumstances, therapy-mediated decreases in primary tumor would decrease CTCs and DTCs by virtue of the fact that fewer CTCs are generated from a shrinking primary tumor. However, CTCs which have already formed, and DTCs that have arrived at a secondary site, would have increased resistance to therapy, and may still have the potential to metastasize while otherwise evading clinical detection.
In order to determine if CTCs and DTCs exhibited a differential response to therapy, we evaluated their responsiveness to vinblastine, paclitaxel, doxorubicin and mitoxantrone. These constitute commonly used cytotoxic agents for the treatment of metastatic cancer, and members of these classes of agents are extensively used in the treatment of metastatic prostate and breast cancer. Vinblastine inhibits tubulin polymerization and paclitaxel inhibits depolymerization. Both effects inhibit microtubule function, thereby inhibiting cell division. These agents are well characterized in cell lines and represent a primary class of chemotherapeutic agents as a front line therapy in patients. While mitoxantrone is an anthracenedione and doxorubicin is an anthracycline, they are considered to be in a similar class of agents that induce cytotoxic effects upon cells through a combination of topoisomerase 2 inhibition, free radical generation and DNA intercalation.
We first measured the ability of these drugs to inhibit growth, as measured in a ten day colony formation assay. We observed no difference in the responsiveness between any of the cell lines after treatment with either of the microtubule inhibitors vinblastine or paclitaxel (Figures 5A and 5B). However, when compared to P-CON cells, CTCs and DTCs showed relative resistance to inhibition of cell growth by mitoxantrone (Figure 5C and Table 2). In order to achieve 50% inhibition of cell growth as compared to control, two-to-four times the amount of mitoxantrone was needed. In the case of doxorubicin, there was a more modest increase in IC50 value (1.2 to 1.7 fold increase), but it was increased in all four cell lines (Figure 5D and Table 2). These findings demonstrate that CTCs and DTCs do not possess the same therapeutic response profile as their parental cells. Further, they indicate selective resistance to the mitoxantrone-like class of agents.
Figure 5. CTCs and DTCs Exhibit Differential Responsiveness to Chemotherapeutic Agents.

Cells were treated with different concentrations of vinblastine (A), paclitaxel (B), mitoxantrone (C) or doxorubicin (D), while control cells were treated with vehicle, and resultant growth in a 10 day colony formation assay determined. Data are the mean±SEM from 3–4 separate experiments, each in duplicate, and are expressed as the percentage of untreated controls.
Table 2.
Inhibition of CTCs in a ten day growth assay
| IC50 [95%CI]
|
||
|---|---|---|
| Cell Line | Mitoxantrone, nM | Doxorubicin, nM |
| P-CON | 5.90 [3.47–10.01] | 45.19 [31.57–64.65] |
| 1BL | 12.23 [8.00–18.70] | 54.38 [37.30–79.27] |
| 1BM | 25.37 [15.16–42.46] | 74.25 [49.98–110.35] |
| 2BL | 20.96 [14.24–30.86] | 68.79 [47.46–99.72] |
| 2BM | 16.03 [11.06–23.24] | 74.95 [47.86–117.37] |
To further examine this selective resistance to chemotherapeutic agents observed under ten day growth conditions, we next evaluated doxorubicin and mitoxantrone under three day growth assay conditions (Figure 6). At mitoxantrone concentrations </= 6.25μM, similar cytotoxicity profiles were observed between CTC, DTC, and P-CON cell lines. However, at the higher concentrations of 12.5 and 25μM, the survival of CTCs and DTCs was significantly and substantially higher than that of P-CON cells (Figure 6A). Similar results were observed with doxorubicin (Figure 6B). Here too, with increasing concentration the survival of CTCs and DTCs was higher than that of P-CON cells, with initial differences becoming apparent at 6.25 μM, and becoming statistically significant for all CTC and DTC lines with escalating concentrations. It is worth noting that as concentration increased, the effect on CTC and DTC viability became increasingly flat, and that even at the very high concentration of 100 μM, 10–15% of CTCs and DTCs were still viable. These findings demonstrate that CTCs and DTCs have selective resistance to chemotherapeutic agents, that resistance is maintained even in the face of extremely high concentrations of drug, and that it protects a significant percentage of cells.
Figure 6. CTCs and DTCs are Resistant to Mitoxantrone and Doxorubicin.

Cells were treated with mitoxantrone (A) or Doxorubicin (B), while control cells were treated with vehicle, and cell growth in a 3 day assay measured. Data are the mean±SEM of 4 separate experiments, each run in replicates of N=4, and are expressed that the percentage of untreated controls. * denotes p value ≤0.05, compared to control.
4. DISCUSSION
By comparing CTCs and DTCs to the parental cells from which they were derived, this has allowed us to identify several novel biological attributes of CTCs and DTCs, and to definitively confirm others. Specifically, we show for the first time that PCa CTCs have increased invasive and metastatic potential compared to those of the primary tumor. This new finding is supported by prior work by others which demonstrate that primary breast CTCs from patients will form metastasis in mice [3, 41]. Interestingly, when a similar approach was used with CTCs from patients with PCa, there was no evidence of tumor take in host mice [5].
The phenotypic changes shown in the current study are accompanied by molecular changes, which include increased expression of HSP27, MMP-2, Twist1, and vimentin. Together, they relate to established pathways that regulate cell motility and metastasis in human PCa, as well as changes that relate to EMT. With respect to EMT-related molecular changes, our current findings provide definitive evidence that corroborates prior and extensive associative findings by others. We found that vimentin, Twist1 and HSP27 all increased in CTCs and DTCs, consistent with a typical EMT molecular phenotype. However, we did not find that E-cadherin and desmoplakin are decreased. This may relate to the fact P-CON cells were of PC3-M cell origin, and PC3-M cells are considered to have undergone EMT, at least to some extent. With respect to cell motility-related molecular changes, our findings in the current study are validated by the importance of HSP27 and MMP-2 pathways previously reported by us.
Prior reports have provided some evidence in support of EMT to MET plasticity in cells traversing the metastatic cascade, including some arising from the study of CTCs. In this regard, our findings demonstrate the novel findings that reversion of EMT is not obligate upon removal of cells from circulation. Furthermore, we found that CTCs and DTCs which have not undergone MET reversion in fact have a greater propensity to form metastasis. Therefore, our findings provide support for the notion that EMT-to-MET plasticity is not necessary in order for cells to traverse the metastatic cascade.
Additionally, we demonstrate for the first time that CTCs and DTCs exhibit selective resistance to therapeutic agents and to the mitoxantrone-like agents in particular. In this regard, for many years mitoxantrone was the first line cytotoxic agent recommended for use in the clinic for patients with metastatic hormone refractory PCa. Although it led to clinical responses, it did not prolong survival. In contrast, the taxanes prolonged survival, are widely used clinically, and have also been shown to decrease CTCs. Our findings support the notion that CTCs and DTCs offer a reservoir of cells resistant to mitoxantrone, and thereby provide a potential explanation for the limitation of mitoxantrone in the clinic. Further, future studies aimed at identifying the mechanism of selective resistance by CTCs and DTCs are highly worthy of pursuit. Of more immediate translational potential, our identification of increased HSP27 and MMP-2 in CTCs and DTCs provides targets that can be manipulated by therapy now undergoing evaluation in the clinic. We have previously demonstrated that the natural product, genistein, will inhibit mitogen-activated protein kinase kinase 4 (MEK4/MKK4/MAP2K4) kinase activity, which is an upstream activator of HSP27 and MMP-2. In preclinical murine orthotopic models, genistein inhibits human PCa metastasis. While in prospective phase II trials, genistein decreases MMP-2 expression in prostate tissue in men with PCa. Together with findings from the current study, this supports the notion that a strategy employing genistein along with taxane-based systemic therapy provides a rational strategy that can be tested in the clinic. Similarly, ongoing phase II studies are testing antisense strategies directed at suppressing HSP27 in patients with PCa. Dependent upon the outcome of these studies, this too provides an avenue for a similar strategy in targeting both the primary tumor and CTCs.
It is important to consider that in the current study, despite animals having advanced metastatic disease, a large tumor size, a moribund status, and a loss of an average of 11% body weight, CTCs and DTCs still occurred at low frequency. These findings demonstrate that CTCs and DTCs in the current system are rare events, thus emulating the situation in humans. It is worth noting that we observed little difference in the characteristics of CTCs isolated from either the blood or DTCs isolated from the bone marrow. In the orthotopic mouse model, CTCs derived from blood only formed blood CTCs, and bone marrow DTCs only formed bone marrow DTCs, but the sample size was very low, and thus we caution against any inferences. However, it is possible that with serial repeats of implantation, cells with true organ-specific trafficking may emerge.
In summary, we demonstrate that CTCs and DTCs exhibit increased invasion and metastatic potential. We definitively show that their EMT phenotype differs from that of the parental cells from which they arose. Further, we find that their removal from circulation does not induce obligate phenotypic reversion. We also demonstrate that CTCs and DTCs exhibit selective resistance to therapeutic agents, and use this new information to propose rationally driven therapeutic strategies that can be tested in the future.
Circulating tumor cells (CTCs) and disseminated tumor cells (DTCs) were isolated from a mouse model of human prostate cancer.
CTCs and DTCs increase cellular invasion in vitro and metastatic formation in vivo, without changes in cellular growth.
CTCs and DTCs specifically upregulate epithelial to mesenchymal transition (EMT) proteins.
CTCs and DTCs have resistance to growth inhibition by specific classes of chemotherapeutic agents.
Acknowledgments
FUNDING SOURCES
This work was supported by grants from the National Institutes of Health (NIH) to R.C.B., CA122985 and Prostate SPORE CA90386, and to J.M.P, NIH T32 AG000260 “Drug Discovery Training in Age-Related Disorders”, and by the Walter S. And Lucienne Driskill Graduate Program in Life Sciences at Northwestern University.
Footnotes
Conflict of Interest Statement
None
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11:R46. doi: 10.1186/bcr2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, Scharpff M, Marme F, Sinn HP, Pantel K, Weichert W, Trumpp A. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31:539–544. doi: 10.1038/nbt.2576. [DOI] [PubMed] [Google Scholar]
- 4.Breen MJ, Moran DM, Liu W, Huang X, Vary CP, Bergan RC. Endoglin-mediated suppression of prostate cancer invasion is regulated by activin and bone morphogenetic protein type II receptors. PLoS One. 2013;8:e72407. doi: 10.1371/journal.pone.0072407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carvalho FL, Simons BW, Antonarakis ES, Rasheed Z, Douglas N, Villegas D, Matsui W, Berman DM. Tumorigenic potential of circulating prostate tumor cells. Oncotarget. 2013;4:413–421. doi: 10.18632/oncotarget.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danila DC, Anand A, Schultz N, Heller G, Wan M, Sung CC, Dai C, Khanin R, Fleisher M, Lilja H, Scher HI. Analytic and Clinical Validation of a Prostate Cancer-Enhanced Messenger RNA Detection Assay in Whole Blood as a Prognostic Biomarker for Survival. Eur Urol. 2013 doi: 10.1016/j.eururo.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danila DC, Fleisher M, Scher HI. Circulating tumor cells as biomarkers in prostate cancer. Clin Cancer Res. 2011;17:3903–3912. doi: 10.1158/1078-0432.CCR-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302–6309. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 9.De Cicco C, Ravasi L, Zorzino L, Sandri MT, Botteri E, Verweij F, Granchi D, de Cobelli O, Paganelli G. Circulating levels of VCAM and MMP-2 may help identify patients with more aggressive prostate cancer. Curr Cancer Drug Targets. 2008;8:199–206. doi: 10.2174/156800908784293613. [DOI] [PubMed] [Google Scholar]
- 10.dos Reis ST, Villanova FE, Andrade PM, Pontes J, Jr, de Sousa-Canavez JM, Sanudo A, Antunes AA, Dall'oglio MF, Srougi M, Moreira Leite KR. Matrix metalloproteinase-2 polymorphism is associated with prognosis in prostate cancer. Urol Oncol. 2010;28:624–627. doi: 10.1016/j.urolonc.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Foster CS, Dodson AR, Ambroisine L, Fisher G, Moller H, Clark J, Attard G, De-Bono J, Scardino P, Reuter VE, Cooper CS, Berney DM, Cuzick J. Hsp-27 expression at diagnosis predicts poor clinical outcome in prostate cancer independent of ETS-gene rearrangement. Br J Cancer. 2009;101:1137–1144. doi: 10.1038/sj.bjc.6605227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glaessgen A, Jonmarker S, Lindberg A, Nilsson B, Lewensohn R, Ekman P, Valdman A, Egevad L. Heat shock proteins 27, 60 and 70 as prognostic markers of prostate cancer. APMIS. 2008;116:888–895. doi: 10.1111/j.1600-0463.2008.01051.x. [DOI] [PubMed] [Google Scholar]
- 13.Goodman OB, Jr, Fink LM, Symanowski JT, Wong B, Grobaski B, Pomerantz D, Ma Y, Ward DC, Vogelzang NJ. Circulating tumor cells in patients with castration-resistant prostate cancer baseline values and correlation with prognostic factors. Cancer Epidemiol Biomarkers Prev. 2009;18:1904–1913. doi: 10.1158/1055-9965.EPI-08-1173. [DOI] [PubMed] [Google Scholar]
- 14.Goodman OB, Jr, Symanowski JT, Loudyi A, Fink LM, Ward DC, Vogelzang NJ. Circulating tumor cells as a predictive biomarker in patients with hormone-sensitive prostate cancer. Clin Genitourin Cancer. 2011;9:31–38. doi: 10.1016/j.clgc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Groot Koerkamp B, Rahbari NN, Buchler MW, Koch M, Weitz J. Circulating tumor cells and prognosis of patients with resectable colorectal liver metastases or widespread metastatic colorectal cancer: a meta-analysis. Ann Surg Oncol. 2013;20:2156–2165. doi: 10.1245/s10434-013-2907-8. [DOI] [PubMed] [Google Scholar]
- 16.Han L, Chen W, Zhao Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: a meta-analysis. Tumour Biol. 2014;35:2473–2480. doi: 10.1007/s13277-013-1327-5. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Wang K, Xu J, Zhang T. Prognostic significance of circulating tumor cells in non-small-cell lung cancer patients: a meta-analysis. PLoS One. 2013;8:e78070. doi: 10.1371/journal.pone.0078070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, Chen S, Xu L, Liu Y, Deb DK, Platanias LC, Bergan RC. Genistein inhibits p38 map kinase activation, matrix metalloproteinase type 2, and cell invasion in human prostate epithelial cells. Cancer Res. 2005;65:3470–3478. doi: 10.1158/0008-5472.CAN-04-2807. [DOI] [PubMed] [Google Scholar]
- 19.Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011;13:R59. doi: 10.1186/bcr2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012;14:R15. doi: 10.1186/bcr3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakshman M, Huang X, Ananthanarayanan V, Jovanovic B, Liu Y, Craft CS, Romero D, Vary CP, Bergan RC. Endoglin suppresses human prostate cancer metastasis. Clin Exp Metastasis. 2011;28:39–53. doi: 10.1007/s10585-010-9356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lakshman M, Xu L, Ananthanarayanan V, Cooper J, Takimoto CH, Helenowski I, Pelling JC, Bergan RC. Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Res. 2008;68:2024–2032. doi: 10.1158/0008-5472.CAN-07-1246. [DOI] [PubMed] [Google Scholar]
- 23.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YM, Xu SC, Li J, Han KQ, Pi HF, Zheng L, Zuo GH, Huang XB, Li HY, Zhao HZ, Yu ZP, Zhou Z, Liang P. Epithelial-mesenchymal transition markers expressed in circulating tumor cells in hepatocellular carcinoma patients with different stages of disease. Cell Death Dis. 2013;4:e831. doi: 10.1038/cddis.2013.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ligthart ST, Coumans FA, Bidard FC, Simkens LH, Punt CJ, de Groot MR, Attard G, de Bono JS, Pierga JY, Terstappen LW. Circulating Tumor Cells Count and Morphological Features in Breast, Colorectal and Prostate Cancer. PLoS One. 2013;8:e67148. doi: 10.1371/journal.pone.0067148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Jovanovic B, Pins M, Lee C, Bergan RC. Over expression of endoglin in human prostate cancer suppresses cell detachment, migration and invasion. Oncogene. 2002;21:8272–8281. doi: 10.1038/sj.onc.1206117. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Miyake H, Muramaki M, Kurahashi T, Takenaka A, Fujisawa M. Expression of potential molecular markers in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy. Urologic oncology. 2010;28:145–151. doi: 10.1016/j.urolonc.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Miyake H, Muramaki M, Kurahashi T, Yamanaka K, Hara I, Fujisawa M. Enhanced expression of heat shock protein 27 following neoadjuvant hormonal therapy is associated with poor clinical outcome in patients undergoing radical prostatectomy for prostate cancer. Anticancer Res. 2006;26:1583–1587. [PubMed] [Google Scholar]
- 30.Msaouel P, Koutsilieris M. Diagnostic value of circulating tumor cell detection in bladder and urothelial cancer: systematic review and meta-analysis. BMC Cancer. 2011;11:336. doi: 10.1186/1471-2407-11-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okegawa T, Hayashi K, Hara H, Nutahara K, Higashihara E. Immunomagnetic quantification of circulating tumor cells in patients with urothelial cancer. Int J Urol. 2010;17:254–258. doi: 10.1111/j.1442-2042.2010.02454.x. [DOI] [PubMed] [Google Scholar]
- 32.Pavese J, Ogden IM, Bergan RC. An orthotopic murine model of human prostate cancer metastasis. J Vis Exp. 2013:e50873. doi: 10.3791/50873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavese JM, Farmer RL, Bergan RC. Inhibition of cancer cell invasion and metastasis by genistein. Cancer Metastasis Rev. 2010;29:465–482. doi: 10.1007/s10555-010-9238-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resel Folkersma L, San Jose Manso L, Galante Romo I, Moreno Sierra J, Olivier Gomez C. Prognostic significance of circulating tumor cell count in patients with metastatic hormone-sensitive prostate cancer. Urology. 2012;80:1328–1332. doi: 10.1016/j.urology.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 36.Strijbos MH, Gratama JW, Schmitz PI, Rao C, Onstenk W, Doyle GV, Miller MC, de Wit R, Terstappen LW, Sleijfer S. Circulating endothelial cells, circulating tumour cells, tissue factor, endothelin-1 and overall survival in prostate cancer patients treated with docetaxel. Eur J Cancer. 2010;46:2027–2035. doi: 10.1016/j.ejca.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 37.Trudel D, Fradet Y, Meyer F, Harel F, Tetu B. Membrane-type-1 matrix metalloproteinase, matrix metalloproteinase 2, and tissue inhibitor of matrix proteinase 2 in prostate cancer: identification of patients with poor prognosis by immunohistochemistry. Hum Pathol. 2008;39:731–739. doi: 10.1016/j.humpath.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 38.Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25:2987–2998. doi: 10.1038/sj.onc.1209337. [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Ding Y, Catalona WJ, Yang XJ, Anderson WF, Jovanovic B, Wellman K, Killmer J, Huang X, Scheidt KA, Montgomery RB, Bergan RC. MEK4 function, genistein treatment, and invasion of human prostate cancer cells. J Natl Cancer Inst. 2009;101:1141–1155. doi: 10.1093/jnci/djp227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, Goodman JC, Groves MD, Marchetti D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5:180ra148. doi: 10.1126/scitranslmed.3005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Riethdorf S, Wu G, Wang T, Yang K, Peng G, Liu J, Pantel K. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin Cancer Res. 2012;18:5701–5710. doi: 10.1158/1078-0432.CCR-12-1587. [DOI] [PubMed] [Google Scholar]