

Abstract
A number of surface proteins specific to pathogenic strains of Leptospira have been identified. The Lig protein family has shown promise as a marker in typing leptospiral isolates for pathogenesis and as an antigen in vaccines. We used NMR spectroscopy to solve the solution structure of the twelfth immunoglobulin-like (Ig-like) repeat domain from LigB (LigB-12). The fold is similar to that of other bacterial Ig-like domains and comprised mainly of β-strands that form a β-sandwich based on a Greek-key folding arrangement. Based on sequence analysis and conservation of structurally important residues, homology models for the other LigB Ig-like domains were generated. The set of LigB models illustrates the electrostatic differences between the domains as well as the possible interactions between neighboring domains. Understanding the structure of the extracellular portion of LigB and related proteins is important for developing diagnostic methods and new therapeutics directed toward leptospirosis.
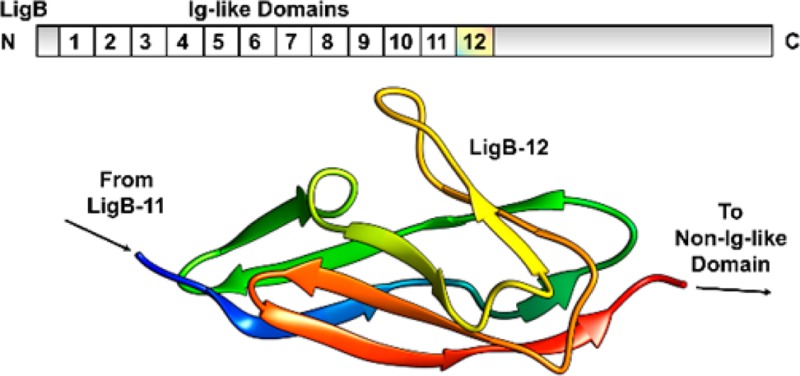
Leptospira spp. are pathogenic spirochetes that can cause multiorgan failure in both humans and animals.1 The associated neglected tropical disease, leptospirosis, is reemerging in the United States but is especially prevalent in developing nations.2,3 While the molecular details of leptospiral infection are poorly understood, the initial steps of host attachment are being uncovered.1,4 Host-interacting surface proteins from virulent bacteria have been implicated in pathogenesis;5 however, these same proteins offer an opportunity for the development of vaccine antigens, serological markers, and attachment blockers. In the outer membrane of Leptospira spp., over 12 proteins have been shown to express at detectable levels,6 yet articles describing high-resolution structures of only two leptospiral surface proteins, LipL32 and Lp49, have been published.7−10 The occurrence of Leptospira immunoglobulin-like genes (Lig) is limited to the pathogenic subset of Leptospira species and is being developed for use as a marker for leptospirosis.11 Lig proteins are promiscuous adhesins and bind to a wide variety of extracellular proteins.12 The Lig protein family is composed of the outer surface proteins LigA, LigB (Figure 1A), and LigC, which contain 13, 12, and 13 immunoglobulin-like (Ig-like) domains, respectively.13−15 The N-terminal 630 amino acids of LigA and LigB (LigCon), covering the first 61/2 Ig-like domains, are highly conserved between the two Lig proteins, but the remaining C-terminal domains are variable (Figure 1A).14,15 In addition, a non-Ig-like region is located at the C-terminus of LigB and LigC. The modular Ig-like domain repeats allow binding to a large number of host proteins since each Ig-like domain may exhibit different specificities. A host-interacting region of LigB is located within the LigBCen2 construct (amino acids 1014–1165 of LigB), which contains part of the 11th and the entire 12th Ig-like domain (LigBCen2R) as well as a disordered region from the non-Ig-like C-terminus (LigBCen2NR).16,17 LigBCen2 binds to fibronectin (Fn), elastin, laminin, and fibrinogen (Fg).18−20 The ability of LigBCen2 to impart variable host binding interactions suggests that LigB plays an important role in Leptospira infections.
Figure 1.

Ig-like domain, LigB-12, interacts with Fg. (A) A schematic showing the location of domains within the LigB protein. (B) Binding of LigB constructs to Fg. Increasing concentrations of His-tagged LigBCen2R (positive control), LigB-12, or LigB-4 (negative control) were added to microtiter plate wells coated with 1 μM of Fg. Bound proteins were detected by ELISA.
Bacterial Ig-like (Big) domain-containing surface proteins have been identified in a number of pathogenic species and provide an evolutionarily tunable protein-binding functionality that is convenient for mimicking natural host interactions.21,22 Although the sequence identity among Big domains is quite low, the structures of intimin from Escherichia coli and invasin from Yersinia pseudotuberculosis are similar and representative of the stably folded β-sheets that are characteristic of many Big domains.23−26 The Big domain β-sandwich is typically stabilized by a conserved hydrophobic core and a Greek key topology.27 Here, we report the NMR solution structure for LigB-12 and highlight features of the Ig-like domain structure from the Lig protein family. LigB-12 is the 12th and most C-terminal Ig-like domain from LigB and the only full Ig-like domain in LigBCen2. In addition, we have identified LigB-12 as the physiologically relevant region involved in LigBCen2 binding to Fg. The LigB-12 structure is compared with the Ig-like domains from other pathogen surface proteins.24,26 The LigB Ig-like domains contribute the scaffold for the Lig family of surface proteins and should aid in the understanding of Leptospira–host interactions as well as in the development of ways to treat and diagnose leptospirosis.
Experimental Procedures
Cloning, Expression, and Purification
Histidine-tagged LigBCen2R (amino acids 1014–1123), LigB-4 (amino acids 309–403), and LigB-12 (amino acids 1029–1123) were cloned from L. interrogans serovar Pomona (L. Pomona) as described previously.16,19 Histidine-tagged, sumo-fused LigB-12 was constructed by inserting the PCR-amplified LigB-12 fragment into the vector pET28-His-Sumo28 between the BamHI and HindIII sites (primers 5′-CGCGGATCCGCAGCAACCCTTTCT-3′ and 5′-CCCAAGCTTCTACGTGTCCGTTTTGTTTAC-3′). LigB-11 (amino acids 939–1033) was also generated in the same way as LigB-12 by using the forward primer 5′-CGCGGATCCGCTGCCACGTTAGAT-3′ and the reverse primer 5′-CCCAAGCTTCTAAAGGGTTGCTGCGCT-3′. Amplified PCR product was digested by BamHI and HindIII and then ligated into pET28-His-Sumo vector. Similarly, two-domain construct LigB-11,12 (amino acids 939–1123) was amplified and constructed with the same forward primer used for LigB-11 and the same reverse primer used for LigB-12. LigB-12 mutations (F1053C and P1040C/F1053C) were generated using the Quik Change protocol (Stratagene). The correct sequences for all constructs were confirmed at the Cornell DNA Sequencing Facility. His-tagged LigBCen2R, LigB-4, and LigB-12 protein constructs were purified using the protocols previously described.16,19 The protocols were modified slightly for the His-sumo protein constructs, LigB-11, LigB-12, and LigB11,12. After isopropyl β-d-thiogalactopyranoside (IPTG)-induced protein expression in E. coli, the bacterial cells were harvested and lysed by high-pressure cell disruption systems (Constant Systems Ltd.) at 20000 psi. After removal of cell debris by centrifugation, the soluble fraction was incubated with phosphate buffered saline (PBS; 137 mM sodium chloride, 10 mM sodium phosphate, 2.7 mM potassium chloride, 1.8 mM potassium phosphate) equilibrated Ni-NTA resin for 2 h. The resin was washed with aliquots of PBS buffer of increasing imidazole concentrations up to 30 mM, and His-sumo-tagged LigB protein was then eluted with 200 mM imidazole–PBS buffer (pH 7.4). The sumo specific protease, His-tagged Ulp-1, was applied to remove the N-terminal His-sumo tag from the LigB protein construct and digestion was allowed to proceed overnight at 4 °C with dialysis against PBS buffer (pH 7.4). Mixtures of His-tagged Ulp-1 and untagged LigB protein were separated by passing the sample over an additional Ni-NTA column. The untagged LigB protein was washed off of the column and concentrated. For NMR experiments, His-sumo-tagged LigB proteins were expressed in E. coli that were grown in isotopically enriched, vitamin-supplemented minimal media.29 Stable isotopes were purchased from Cambridge Isotopes (Cambridge, MA). Prior to NMR sample loading, the labeled proteins were exchanged into PBS, pH 7.0, 1 mM sodium azide, and 10% D2O by successive concentration and dilution and finally concentrated to 0.5 mM.
ELISA Binding Assays
The ability of LigB proteins to bind to human plasma fibrinogen (Fg) (obtained from Sigma-Aldrich) was assessed using an ELISA assay as previously described.20 Various concentrations (0, 0.31, 0.63, 1.25, 2.5, 5, 10, and 20 μM) of histidine-tagged LigBCen2R (positive control), LigB-4 (negative control), and LigB-12 were added to microtiter wells coated with 1 μM Fg or BSA (negative control, data not shown) in PBS buffer. Following 1 h incubation at 37 °C, the wells were washed with PBS buffer containing 0.05% Tween 20 (PBS-T) to remove unbound LigB proteins. To detect the interaction of each LigB truncate with Fg, mouse anti-His tag antibody (1:500) and horseradish peroxidase (HRP) conjugated goat anti-mouse IgG antibody were used as primary and secondary antibody (Eugene). Finally, 100 μL of HRP substrates was applied to develop the color and then the plates were read at 630 nm with an ELISA plate reader (Biotek EL-312, Winooski, VT). To determine the end-point dissociation constant (KD), the binding curves were fit with the following equation using KaleidaGraph software (Abelbeck software, Reading, PA):
Measurement of Protein Thiols (Free Cysteine)
An Ellman’s reagent assay30 was used to assess the presence of free cysteine versus intramolecular disulfide bonds in LigB-12 cysteine mutants. LigB-12 wild type or F1053C or P1040C/F1053C mutant (20 μM) was treated with 1 mM of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) in Tris buffer (pH 7.4) containing 4 M urea for 5 min. The absorption intensity of reaction product, 2-nitro-5-thiobenzoic acid (TNB2–), was measured at 412 nm, which indicated the relative amount of free cysteine in each protein sample. In a complementary assay, the formation of intermolecular disulfide bonds was used to infer the presence of free cysteine in LigB-12 wild-type and mutants. Each set of protein samples was preincubated with 100 μM oxidized glutathione (GSSG) at room temperature for 1 h. Subsequently, the protein samples were heated at 37, 60, and 100 °C for 10 min and then were separated on the nonreducing SDS-PAGE gel. β-mercaptoethanol (5%) was also added to wild type and mutant proteins to serve as a reducing condition counterpart. Proteins involved in intermolecular disulfides migrated with twice the molecular weight as expected for a monomer.
NMR Spectroscopy and Resonance Assignments
NMR spectra were acquired on a 500 MHz Varian spectrometer at 286 K. Spectral assignments were obtained from HNCOCA, HNCACO, HNCA, HNCO, CBCANH, HNCBCACO, CCONH, HCCONH, and TOCSY-NHSQC experiments performed on 15N-only and 15N, 13C-labeled protein. Distance constraints were generated using NOESY–15N-HSQC, NOESY–13C-HSQC, and aromatic NOESY–13C-HSQC. Additional secondary structural constraints were taken from stable hydrogen bonded amides determined from 1H,15N-HSQC spectra of hydrogen–deuterium (H-D) exchanged protein. For H-D exchange, an initial control spectrum was taken in 90% H2O/10% D2O. The sample was lyophilized and resuspended in 100% D2O. Immediately after exchange into D2O, the sample was transferred to an NMR tube and was measured with a series of 12 1H,15N HSQC experiments. In the proton dimension, 2048 points (real plus imaginary) were recorded with 256 points in the nitrogen dimension (real plus imaginary) for 16 scans per increment. Each experiments lasted ∼82 min. For residual dipolar coupling (RDC) measurements, 15N-labeled protein was aligned in 5% stretched acrylamide gels.31 Spectra were processed using NMRPipe32 and analyzed using SPARKY (T. D. Goddard and D. G. Kneller, UCSF). Residue assignments were generated manually and confirmed with the PINE server v1.0.33 The structure of LigB-12 was determined using NIH-Xplor34 with distance constraints, dipolar coupling constraints, hydrogen bond constraints, Promega-determined proline conformations,35 and dihedral angle restraints generated using TALOS-N.36 Structure validation was performed using PROCHECK-NMR,37 MolProbity,38 PSVS,39 and SuperPose.40 Assigned chemical shifts and coordinates were deposited in the PDB and BMRB databases under the PDB accession code 2MOG and the BMRB accession number: 19942.
Sequence Analysis and Modeling
The Ig-like domains from LigA, LigB, and LigC from L. Pomona were aligned with the LigB-12 construct sequence (Figure S1, Supporting Information), and residue conservation was determined and illustrated using the HMMER software suit.41 Homology models of LigB-1 through LigB-11 and LigB-11,12 were generated using Modeller42 with the LigB-12 structure as a template. The pI for each Ig-like domain was computed using the ExPASy server,43 while electrostatic surfaces were calculated using Delphi.44
Results
LigBCen2R and LigB-12 Constructs
Initial attempts to gain structural insight into the Lig protein family focused on the previously characterized LigBCen2R construct, which is composed of the last 15 residues of the 11th Ig-like domain and the entire 12th Ig-like domain (Figure 1A). NMR experiments performed on 15N-only and 15N, 13C-labeled LigBCen2R were used to identify preliminary 15N–1H backbone assignments (results not shown). The regions from partial domains were not well structured and could not be assigned. The start of the LigBCen2R protein corresponding to the C-terminal end of the 11th Ig-like domain has homology to the final two predicted β strands in LigB’s 12th Ig-like domain. When the solvent was exchanged to deuterium oxide, the remaining 15N–1H backbone peaks corresponded to unexchanged hydrogen-bonded secondary structural elements and could be assigned to residues from the 12th Ig-like domain. The partial 11th Ig-like domain at the N-terminus of LigBCen2R probably does not contribute to hydrogen bonded secondary structural elements and may require additional β-strands to complete proper folding of its β-sheet.
The construct, LigB-12, residues S1029–T1123 from LigB, was designed to remove the nonstructured regions and isolate the core Ig-like domain. The alignment of each unique Ig-like domain from LigA (six common with LigB and seven LigA-only), LigB (six unique), and LigC (six unique) suggests that the secondary structure of LigB-12 is shared by the family of 31 domains (Figure S1, Supporting Information). The sequence is well conserved for the predicted β-sheets, which comprise the major elements of Ig-like domain folds. The ability of LigB-12 to retain the binding affinity displayed by LigBCen2R for Fg was investigated using ELISA (Figure 1B). The KD of LigBCen2R and LigB-12 were similar (LigBCen2R, KD = 117 ± 11 μM; LigB-12, KD = 125 ± 20 μM), suggesting that LigB-12 is the Fg-interacting Ig-like domain from the LigB protein. The negative control LigB-4 (residues T309–L403) does not bind to Fg.
NMR Solution Structure of LigB-12
The 15N,1H-HSQC NMR spectrum of LigB-12 (sequence; Figure 2A) was well dispersed suggesting a folded Ig-like domain protein (Figure 2B). NMR spectra of LigB-12 was found to be more stable at 286 than 298 K over long acquisition times (1 week); therefore all data used for structure determination was obtained at 286 K. Although chemical shift resonances were assigned for 95.1% of the backbone, the following residues could not be assigned in the 15N,1H-HSQC NMR spectrum: S1029, A1030, P1040*, T1047, S1050, Q1078, P1101*, and T1103. Chemical-shift derived dihedral angles, NOEs, RDCs, and H-D exchange data from NMR spectra were used to determine the solution structure of the LigB-12 construct. P1040 was determined to favor a cis conformation by Promega.35 Of the 100 calculated structures using NIH-Xplor,34 the 20 lowest energy structures were selected to represent the LigB-12 ensemble (Table 1). The PROCHECK-NMR37 Ramachandran statistics (from PDB submission; includes all nonterminal, non-glycine, and non-proline residues) provide an indication of the overall structural quality with most favored regions at 86.7%, additionally allowed regions at 11.3%, generously allowed regions at 1.7%, and disallowed regions at 0.3%.
Figure 2.

Structure of LigB-12 determined using NMR spectroscopy (PDB ID 2MOG). (A) The primary sequence and secondary structure of LigB-12 is shown. Half circles indicate the core tryptophan (green), residues aligned with the proposed LigC-2 disulfide (cyan) (Figure 4C), and additional residues that are important to the hydrophobic core (yellow) (Figure 3A). (B) Residue assignments are labeled on the 15N,1H-HSQC NMR spectrum of LigB-12 obtained on a Varian 500 MHz NMR spectrometer at 13 °C. (C) Overlay of the 20 lowest energy structures of LigB-12 calculated from the NMR constraints. (D) A schematic of the secondary structure of LigB-12 indicating the position of residues P1040, F1053, and W1073. (E) The lowest energy NMR solution structure of LigB-12.
Table 1. Structural Statistics for the NMR Structure of LigB-12 (PDB ID 2MOG) (20 of 100).
| NMR Distance and Dihedral Constraints | |
| Distance Constraints | |
| total NOE | 1504 |
| intraresidue | 489 |
| inter-residue | |
| sequential (|i – j| = 1) | 437 |
| medium-range (|i – j| ≤ 5) | 137 |
| long-range (|i – j| > 5) | 441 |
| hydrogen bonds | 39 |
| Total Dihedral Angle Restraints | |
| ϕ | 85 |
| ψ | 81 |
| χ1 | 36 |
| total RDCs | 50 |
| Percent Resonance Assignments | |
| backbone, with CA (%) | 96.0 |
| side chain, without CA (%) | 83.8 |
| Structure Statistics | |
| Constraint Violations (mean and SD) | |
| distance constraints | |
| number >0.2 Å | 1.2 ± 1.1 |
| rms deviation (Å) | 0.015 ± 0.002 |
| dihedral angle constraints | |
| number >5° | 1.9 ± 0.9 |
| rms deviation (deg) | 0.857 ± 0.110 |
| RDCs | |
| number >5 (Qa) | 0.0 ± 0.0 |
| rms deviation (Qa) | 0.174 ± 0.052 |
| Deviations from Idealized Geometry | |
| bond lengths (Å) | 0.002 ± 0.000 |
| bond angles (deg) | 0.440 ± 0.010 |
| impropers (deg) | 0.380 ± 0.025 |
| Average Pairwise rms Deviation from Average Model (Å) Residues L1033–V1118 | |
| backbone | 0.397 |
| heavy | 0.723 |
| Ramachandran Statistics | |
| PROCHECK-NMR (includes all nonterminal, non-glycine, and non-proline residues) | |
| most favored regions (%) | 86.7 |
| additionally allowed regions (%) | 11.3 |
| generously allowed regions (%) | 1.7 |
| disallowed regions (%) | 0.3 |
The LigB-12 structure displays the characteristic fold found in other Ig-like domains (Figures 2C,E). The fold is composed of β-strands A–G, which form a β-sandwich based on a Greek key folding arrangement (Figure 2D). Two layers of β-sheets close around a hydrophobic side-chain core. The more extensive layer is divided into sheets 1a (β-strands A, B, and C) and 1b (β-strands B′, D, and E) each containing a portion of β-strand B separated by a break and a slight twist but otherwise composed of distinct β-strands. Sheet 1b participates in forming the β-sandwich with sheet 2 (β-strands C′, F, and G), while sheet 1a extends beyond the complementary surface with sheet 2 leaving both surfaces solvent-exposed. The β-strands A, C, and G are also not continuous with each participating in two sheets. The break in β-strand C is almost a full helical turn allowing for the strand to interact directly with both layers of the β-sandwich. A single helical turn between β-strands C and D is also present. Ramachandran statistics using MolProbity38 (includes glycine and proline) identify P1040 (A–A′ loop), N1042 (A–A′ loop), T1047 (A′–B′ loop), and A1097 (E–F loop) as major torsion angle outliers (disallowed) in 25–100% of models. By limiting the Ramachandran analysis to residues L1033–S1039, S1050–I1096, and N1100–V1118, we improved the overall quality of the structure to 96.3% in favored regions (vs 90.3% for residues L1033–V1118), 3.7% in allowed regions (vs 6.3% for residues L1033–V1118), and 0.0% in disallowed regions (vs 3.4% for residues L1033–V1118). Since the per residue rms deviation for the A–A′ loop to the A′–B′ loop and the E–F loop are higher than the overall rms deviation for residues L1033–V1118, these loops can be described as having a high degree of structural variability relative to the rest of the LigB-12 domain ensemble. P1040 within the A–A′ loop favors a cis conformation but may be present in trans form leading to increased structural variability of the region extending to the A′–B′ loop and possibly affecting the neighboring E–F loop. In addition, the possibility of a calcium ion binding site was investigated on LigB-12 in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer at pH 7.0. No chemical shift changes were observed after a titration of either CaCl2 up to 5 mM or EGTA up to 5 mM suggesting that calcium ions do not induce an overall conformational change in LigB-12 and are not an integral component of the fold.
Based on the identified LigB-12 domain, two additional constructs for LigB-11, the full homologous Ig-like domain neighbor preceding LigB-12, and for LigB-11,12, the double Ig-like domain that includes both LigB-11 and LigB-12, were generated. The 1H,15N-HSQC spectra of LigB-11 (green) and LigB-12 (red) were overlaid onto the LigB-11,12 (black) spectrum (Figure S2, Supporting Information). The high degree of spectral similarity between the isolated domains and the two domain construct suggests that the individual LigB-12 and LigB-11 constructs are representative of its Ig-like domain structure in the context of the multiple linked domains found in the full length protein.
Hydrophobic Core
The residues located between the two layers of the LigB-12 β-sandwich form a hydrophobic core for LigB-12 (Figure 3A). The sequences from the β-strands that line the folded core are generally composed of alternating buried hydrophobic and surface-exposed hydrophilic residues. Aromatic residues from each of the layers, F1053 (sheet 1) and W1073 (sheet 2), interact and contribute to a hydrophobic center (Figures 2A,D and 3A). In addition, the side chain NH of W1073 does not efficiently exchange with solvent as observed by NMR-based deuterium exchange experiments (Figures 3B,C). In the structure, the backbone carbonyl oxygen of T1068 points toward the W1073 side chain nitrogen and is in proximity to be its hydrogen-bonding partner (Figure 3A). The unique hydrogen bond provides an anchor for the twist in β-strand C that rotates almost a full helical turn at the break in sheet 1.
Figure 3.

Hydrophobic core of LigB-12. (A) Structure of LigB-12 showing the residues in the hydrophobic core. (B) A section of the LigB-12 15N,1H NMR spectrum (red) is shown overlaid with a spectrum of the same protein after replacing the solvent with D2O for 12 h (blue). (C) H–D exchange data for the five backbone amide protons shown in panel B. Note the slow exchange of the side chain of W1073.
Lig Protein Ig-like Domain Conservation
An alignment of the 12 LigB repeats reveals a high level of conservation for residues facing the hydrophobic core (Figure S1, Supporting Information). The degree of conservation for the 12 LigB domains was mapped onto the LigB-12 structure in Figure 4A. The residues surrounding the core tryptophan, W1073 (green surface), are well conserved. The high level of sequence consensus decreases as a function of distance from the central tryptophan residue (Figure 4B). The Ig-like domains from Lig proteins are likely derived from a common evolutionary Ig-like domain through gene replication,45 and residue conservation across the domain set can provide clues to a residue’s importance. Conserved residues are clustered in the core of the Lig protein Ig-like fold suggesting that all 12 repeats have homologous structures and common folding pathways that are stabilized by a conserved hydrophobic core.
Figure 4.
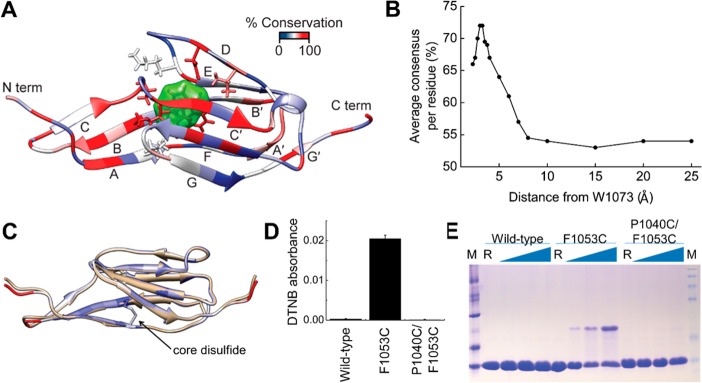
Conservation of Lig Ig-like domains. (A) Residue conservation mapped onto the LigB-12 structure. The core tryptophan, W1073, is shown with a green surface, while atoms for the neighboring residues are also shown in stick representation. (B) The average residue consensus for aligned LigB domains plotted as a function of distance to the core tryptophan (W1073 in LigB-12). (C) The disulfide seen in LigC-2 was transplanted to LigB-12 by making the P1040C/F1053C mutation of LigB-12. Shown is the model of the mutant protein (white) overlaid on the LigB-12 structure (beige). (D) Absorption of the reaction product (TNB2–) at 412 nm after DTNB modification of a free cysteine for wild-type LigB-12 and the F1053C and P1040C/F1053C mutations (n = 3). (E) A nonreducing SDS-PAGE gel was used to detect the formation of interdomain disulfides at increasing temperatures. A significant increase in disulfide-trapped dimers was seen for the single cysteine but not the double cysteine mutant.
LigC-2 Disulfide
Among all of the 31 unique Lig protein Ig-like domains only three cysteines are present with two occurring in LigC-2. The two LigC-2 cysteines are positioned on neighboring β-strands, A and B, and are close enough to form a disulfide bond as suggested by a model of LigB-12 containing cysteine mutations (P1040C, F1053C) at the corresponding sites (Figure 4C). When the mutations are modeled as a disulfide, the LigB-12 structure is left unaltered as illustrated by backbone RMSDs mapped onto the model. The potential for the internal disulfide to exist in LigC-2 and to be transplanted to LigB-12, a more divergent Lig Ig-like domain,45 was tested by generating the P1040C/F1053C mutant of LigB-12.
LigB-12, the F1053C mutant, and the P1040C/F1053C mutant were reacted with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) to investigate the presence of free cysteines. Under denaturing conditions (4 M urea), both the wild-type and P1040C/F1053C mutant exhibited little reactivity while the F1053C mutant reacted with DTNB (Figure 4D). The difference between the F1053C mutant and other constructs is statistically significant (F-test p-value = 0.0), while the difference between wild-type and P1040C/F1053C mutant is not statistically significant (F-test p-value = 0.199). In addition, when the three LigB-12 constructs were incubated at various temperatures under oxidizing conditions, disulfide cross-linked dimers were observable on a nonreducing Coomassie blue-stained gel as the incubation temperature increased for the F1053C mutant (Figure 4E). A decrease in the F1053C mutant monomer band (10 kDa) corresponded to the increase in the dimer band (20 kDa) suggesting that a free cysteine (F1053C) was available for intermolecular cross-linking. Essentially no dimer was observed for the wild-type and P1040C/F1053C mutant. The inability to detect free cysteines for the P1040C/F1053C mutant in either assay suggests that an internal disulfide forms as predicted by the LigB-12 structure-based model.
LigB-12 Specific Divergence
The degree of conservation for the set of LigB Ig-like domains from Figure 4 was further analyzed for residue specific deviation of LigB-12 (Figure 5A). A patch of residues facing the non-Ig-like terminal domain and near to the C-terminus of LigB-12 showed nonconserved changes at positions that are conserved for the other 11 Ig-like domain sequences. Specifically, a conserved stretch of three residues, A–K–G, is located in a loop between β-strands A and B for most of the LigB Ig-like domains; however in LigB-12, the corresponding AB loop sequence was unrelated, N1046–T1047–T1048. While the nonconserved surface provides a potential LigB-12 interaction site with the terminal non-Ig-like domain, the complementary conserved surface in other LigB domains likely interacts with the immediate Ig-like domain neighbor. The conserved lysine (in 15 of 19 LigA and LigB Ig-like domains) projects toward the neighboring C-terminal Ig-like domain. A model of LigB-12 and the preceding (11th) Ig-like domain was generated using Modeller42 with the LigB-12 structure as a template (Figure 5B). The conserved lysine from the 11th Ig-like domain, K957, is readily positioned toward a conserved aspartic acid, D1061, on the loop between β-strands B and C of the 12th Ig-like domain. The conservation of this lysine–aspartic acid pair suggests a possible salt bridge. Electrostatic interactions between similar positions are also present in between the three Big (bacterial Ig-like) domains found in the invasin (Yersinia pseudotuberculosis) structure24 (Figure 5C).
Figure 5.
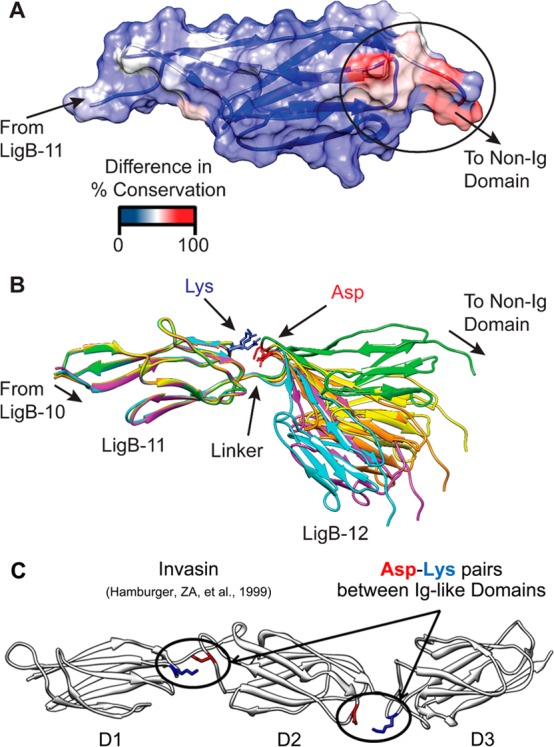
Divergence of Lig Ig-like domains. (A) A surface showing and structure colored to show the divergence between LigB-12 and the consensus sequence for the LigB domains. The deviation occurs at a patch near the C-terminus that would be near the terminal non-Ig-like domain. (B) A set of five models of LigB-11,12 is shown illustrating the linker and potential interdomain interactions. The models depict a range of relative orientations that would allow for the formation of a domain–domain salt bridge. (C) The crystal structure of invasin (1CWV) is depicted highlighting possible salt bridges between neighboring Big domains.
Ig-like Domain Surface Differences
The structural homology of the Lig protein Ig-like domains allows us to generate models of the protein set. A comparison of the modeled structures highlights potentially important differences between the Lig domains. Differences on the surface of the Lig domains are greater than within the hydrophobic core and may reveal regions that interact with host proteins. The calculated pI values for the Lig protein domains are likely to be either acidic (pI = 4–5) or basic (pI = 8–10). Models of the LigB domains were aligned, and the electrostatic surfaces (calculated using Delphi44) are displayed in Figure 6A. The first four repeats are calculated to have an acidic pI and are characterized by patches of negative charge; whereas the next five repeats are much more basic, with corresponding positively charged patches. The distribution of charged domains is similar for both LigA and LigB, that is, a stretch of negatively charged repeats near the membrane followed by a stretch of positively charged domains at the midsection of the Ig-like domain stretch, and finally a stretch of negatively charged domains. In contrast, all of the domains of LigC are negatively charged. The arrangement of LigA, LigB, and LigC is summarized in Figure 6B,C. The difference in surface charges could represent points of electrostatic interaction with host proteins.
Figure 6.
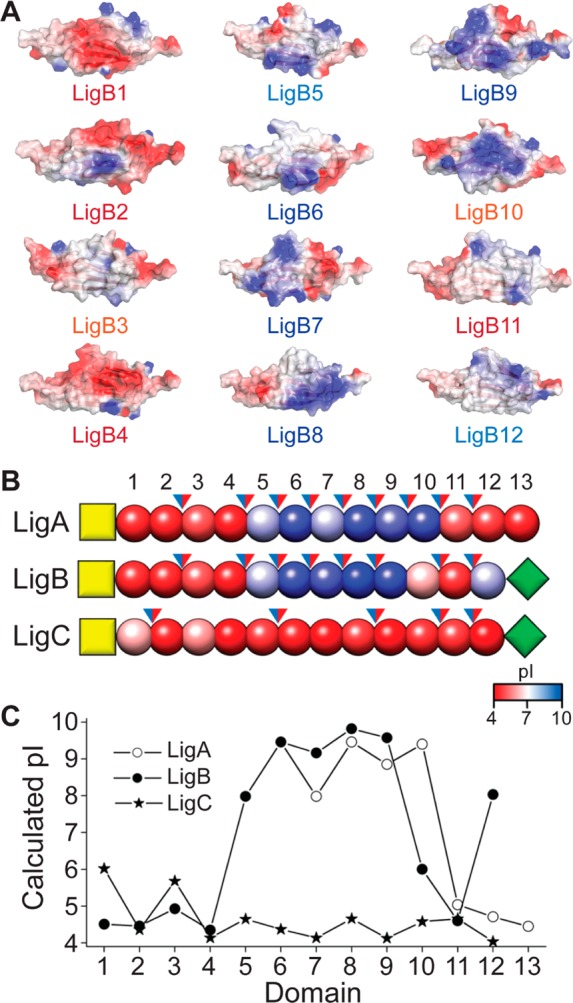
Electrostatics of Lig Ig-like domains. (A) Electrostatic surfaces of modeled repeats of LigB, showing differences in surface charges computed with Delphi (2kT/e (blue), 0kT/e (white), and −2kT/e (red)). (B) A schematic diagram of the domains of LigA, LigB, and LigC (N-terminal anchor, yellow squares; Ig-like domains, red/blue beads; C-terminal domains, green diamonds). The Ig-like domains are colored by calculated pI values, and blue/red arrows indicate positive/negative charges for a potential domain–domain salt bridge. (C) A plot of the calculated pI for each domain of LigA, LigB and LigC.
Bacterial Ig-like Domain Fold Comparison
Insertions and deletions within the set of homologous Lig domains are positioned at the D–E and G–F loops and also the A–A′ and C–C′ transitions. The largest differences between Lig protein Ig-like domains can be seen in the variation in the loop between the D and E β-strands. Interestingly, these positions are located toward the middle of the domain near the interface between sheets 1a and 2. Changes in the length of these loops can affect the extent to which sheet 1a is exposed. Comparing the Lig protein Ig-like domain structure to other bacterial Ig-like (Big) domain structures illustrates the importance of the D–E and the C–C′ β-strands to the shape of the folded protein (Figure 7). Big_1 domains from intimin (Escherichia coli) (1F00)26 and invasin (Yersinia pseudotuberculosis) (1CWV)24 have long D–E β-strands with only three hydrogen bonds connecting the C β-strand with the B β-strand as a subtle hairpin within the loop before the strand analogous to the C′ β-strand initiates a twist to sheet 2. The resulting structure is almost a full β-sandwich with little of sheet 1 uncovered. Big_3 domain structures from a putative bacillolysin (Bacillus cereus) (2KPN)46 and a putative endo-β-N-acetylglucosaminidase (Streptococcus pneumonia) (2L7Y)47 lack the equivalent D–E β-strands and the Greek key structure. The Big_3 domains are therefore narrower along the short axis. While both Bacillus cereus and S. pneumonia Big_3 domains have a turn connecting the C β-strand in sheet 1 to the C′ β-strand in sheet 2, the Bacillus Big_3 domain has a full sheet 2 with a β-sandwich along the entire domain and the Streptococcus Big_3 domain has a short sheet2 with only two-thirds of the domain’s sheet 1 participating in a β-sandwich. The LigB-12 structure is classified as a Big_2 domain and is somewhat intermediate between the Streptococcus Big_3 domain and the Big_1 domains. The comparison across Big domains may yield a better understanding of Ig-like domains for design and bioengineering technologies.
Figure 7.

Comparison of structures of various Big domains (invasin, 1CWV; intimin, 1F00; BC Big_3, 2KPN; SP Big_3, 2L7Y), highlighting folding patterns and secondary structures. A set of structures illustrates the relationship between the position of the C and C′ loop and the length of sheet 2. The Greek key structure of invasin, intimin, and LigB-12 is colored yellow. Note the lack of the Greek key structure due to the absence of D and E β-strands in Big_3 domains. The length of the D–E loop is highly variable among Lig domains. An alignment of sequences for the illustrated structures can be found in the Supporting Information (Figure S3).
Discussion
Surface proteins from pathogenic bacteria offer novel opportunities for combating dynamic and evolving disease threats. Lig proteins function as adhesins and are directly involved in the binding of numerous host cell matrix proteins. The individual Ig-like domains from LigA and LigB possess various affinities for different host proteins. Here, we show that the terminal Ig-like domain from LigB, LigB-12, binds to Fg. Protein constructs containing the LigB-12 domain also provide host protein interactions with Fn and tropoelastin.16−20 Fg, Fn, and tropoelastin are all accessible to invading pathogens and provide important contacts for initiating an infection.
The structure of LigB-12 provides an important view of a surface-expressed Ig-like domain from leptospirosis-causing pathogens as well as insight into the overall architecture of the Lig protein family. Within Lig proteins, the oblong Ig-like domains are organized as multiple repeats of up to 13 homologous domains with only a few amino acids between neighboring domains. Because the domains are tethered together near the antipodal points of its longest diameter, the multiple domains have the potential to form long extensions. Crystal structures of multiple Ig-like domains from various proteins, including fibronectin and invasin, adopt such a linear confirmation.24,48 For similar tandem Ig domains, dynamic conformations have also been shown to exist in solution.49 An outstretched extracellular Lig protein structure could explain the preference for the more C-terminal variable Ig-like domains to interact with host proteins.18,50 Associations between host proteins and the conserved Ig-like domains that are positioned closer to the N-terminal membrane-bound domain may confer special anchoring or cell invasion properties.19
While the main fold of Lig protein Ig-like domains is conserved, the LigB-12 structure can be used to compare differences between the domains that may play a role in pathogenesis. Residue conservation is an indication of importance to stability and function. In contrast, the least conserved residues, those that lie on the surface of the Ig-like domains, are most likely to make adhesive contacts with the host proteins and are also most likely to be evolutionarily tuned to tightly bind targets. The eukaryotic extracellular matrix (ECM) forms a biopolymer hydrogel with a propensity to bind both positive and negative charges and also to allow neutral charges to move more freely.51 The majority of Lig protein Ig-like domains are charged (either acidic or basic) providing a platform for ECM immobilization (Figures 6B,C). Binding assays between LigB domains and ECM proteins reveal that LigB-4 and LigB-8 through LigB-12 have the highest affinity for host proteins.17−20,50,52 Surface electrostatics could influence specific domain interactions with ECM binding partners. For example, tropoelastin, a protein containing a high density of lysine residues, has been shown to bind to constructs containing LigB-4, LigB-8, LigB-9, and LigB-12. The relative binding affinity for the domains, LigB-4 > LigB-12 > LigB-8 > LigB-9.19 correlates well with the theoretical pI of the domains. For tropoelastin, the binding to the more acidic LigB-4 could be influenced by attractive lysine interactions while the binding to the more basic LigB-8 and LigB-9, could be influenced by repulsive lysine interactions. In a second example, a multidomain segment of Fn with an overall negative charge interacts preferentially with basic LigB domains LigB-8, LigB-9, and LigB-12.17
In an attempt to identify potential protein interacting sites on LigB-12, a comparative analysis of LigB-12 against the consensus for LigB Ig-like domains was used to locate the residues that are generally conserved for the set of Ig-like domains but only diverge for LigB-12. A patch of surface residues that are specifically divergent for LigB-12 faces the non-Ig-like terminal domain of LigB (Figure 5A). While the difference in LigB-12 could be important for binding of a host protein, the position of the residues along the terminal surface are probably related to adaptive interactions with the nonrepeat terminal domain. Currently, the structure of the nonrepeat domain is not known, but it may interact directly with LigB-12.16 Conversely, the conserved surfaces in the other LigB Ig-like domains face the neighboring Ig-like domains, suggesting that a domain–domain interaction may be maintained through selective pressure. A conserved lysine is flanked by two small flexible residues (A–K–G). The positive charge is positioned in a loop to extend toward the neighboring Ig-like domain. A homology model of LigB-11 in tandem with the LigB-12 structure reveals a potential electrostatic interaction between the conserved lysine (K957) on LigB-11 and a conserved aspartic acid (D1061) on LigB-12 (Figure 5B). Along with the domain–domain linker, the potential salt bridge offers a second point of contact between the Ig-like domain neighbors. A two-point interaction between domains would function to limit relative rotation of neighboring domains but would be unlikely to limit a hinge motion. A domain–domain interaction network could allow for modulatory control of rigidity in the tandem Ig-like domains through environmental cues or host interactions. Interestingly, three Ig-like domains in the X-ray crystal structure of invasin have two domain–domain interfaces, each of which has a salt bridge that is similar to the ones suggested between the Ig-like domains of LigB (Figure 5C).24 In tandem Ig domains from titin, a network of nonhydrophobic interactions has been demonstrated to impose rigidity on the relative domain orientation in solution.53 The dynamic aspects of tandem Ig and Ig-like neighboring domain interactions may be important for functional regulation of multirepeat containing proteins.
The β-sandwich anchored by a central Greek-key fold is thought to provide Ig and Ig-like domains with a simple and common folding pathway. Big domains represent a large Ig-like domain Pfam54 fold family with many subgroups (e.g., Big_1, Big_2, and Big_3) and are often found in bacterial cell surface proteins.22 The comparison of Big domain folds and sequences shows that the Greek-key is one of the least conserved features with two of the four Greek-key β-strands being absent from two representative Big_3 domains (Figure 7). The two dispensable β-strands border the loop with the largest degree of insertion and deletion variability among Lig domains and are potential targets for engineering the size and shape of the Big domains.
An important feature of all Big domain folds is the hydrophobic core. The residues comprising the hydrophobic core of LigB-12 were recently identified using tryptophan fluorescence spectroscopy for wild-type and aromatic residue mutants.55 The fluorescence of W1073 was shown to be consistent with a hydrophobic environment. Additionally, an alanine mutation of a conserved phenylalanine residue, F1053A, decreases the hydrophobic environment surrounding the tryptophan as well as the stability of the fold. In agreement with fluorescence observations, the majority of the F1053 and W1073 aromatic rings are positioned within 7.5 Å of each other and buried within the hydrophobic core of the LigB-12 NMR structure. Tryptophan side chain hydrogen bonding, as observed for the W1073 side chain nitrogen, could also have an influence on the emission wavelength.56 For the Big domains with structures solved, the tryptophan–phenylalanine core is unique but is also highly conserved among Lig protein domains. Within homologous proteins, conserved residues often play a role in fold stability or function. The tryptophan fluorescence studies55 have been extended to support a hydrophobic tryptophan environment at the core of several Lig protein Ig-like domains with similar results.
Core disulfides are commonly found in some Ig domain families where the disulfides connect the main β-sheets of the β-sandwich.22 The disulfide from LigC-2 is positioned within a single β-sheet and should provide stability near the fold’s N-terminus. Investigations of an Ig-like domain from the muscle protein titin suggest that force-induced unfolding is initiated when hydrogen bonds between β-strands A and B (near the N-terminus) break, followed by those of β-strands A′ and G′ (near the C-terminus), and finally the main domain strands unfold.57 Stabilizing sheet 1 should be important in limiting the type of mechanical unfolding that regulates the functional elasticity of some multi-Ig-like domain proteins. The ability to transplant the disulfide to the LigB-12 domain provides additional evidence of structural homology among Lig domains. Thermal denaturation of LigB-12, the F1053C mutant, and the P1040C/F1053C mutant observed by tryptophan fluorescence shows that the F1053C mutant is less stable than wild-type; however, some stability is recovered in the P1040C/F1053C mutant (C.-L. Hsieh et al., unpublished results). The inability of the P1040C/F1053C mutant to improve wild-type stability is likely related to the high degree of conservation and hydrophobic core proximity of these two residues. Future work directed at stabilizing Big domains for use in biotechnologies could incorporate disulfides at similar positions between β-strands A and B but closer to the N-terminus and further from the hydrophobic core.
Ig-like domains are one of the most common extracellular fold types, and tethering repeats of Ig-like domains produces a simple architecture with the potential for multiple tunable surfaces and rigidity control, features that are highly advantageous for an adhesion molecule. The structure of LigB-12 has, as expected, an Ig-like fold, but the details of the structure and comparison with other Big proteins provides insight into the function of the domains and their interaction with host proteins.
Acknowledgments
We thank Y. Mao (Cornell) for providing the pET28-His-Sumo vector.
Glossary
Abbreviations
- Big
bacterial Ig-like
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- Fg
fibrinogen
- Fn
fibronectin
- Ig
immunoglobulin
- Lig
Leptospira immunoglobulin-like genes
- LigBCen2
amino acids 1014–1165 of LigB
- LigCon
N-terminal 630 amino acids of LigA and LigB
- RDC
residual dipolar coupling
Supporting Information Available
Full sequence alignment of Ig-like domains from LigA, LigB, and LigC, an overlay of the 15N,1H-HSQC NMR spectra for LigB-11, LigB-12, and LigB-11,12, and a sequence alignment for structures depicted in Figure 7. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ C.P.P. and C.-L.H. contributed equally to this work.
The authors declare no competing financial interest.
This work was supported by grants from the Biotechnology Research and Development Corp., the Cornell Center of Advanced Technology program, and the Zweig Foundation to Y.F.C.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Palaniappan R. U. M.; Ramanujam S.; Chang Y.-F. (2007) Leptospirosis: Pathogenesis, immunity, and diagnosis. Curr. Opin. Infect. Dis. 20, 284–292. [DOI] [PubMed] [Google Scholar]
- Vinetz J. M. (2001) Leptospirosis. Curr. Opin. Infect. Dis. 14, 527–538. [DOI] [PubMed] [Google Scholar]
- Bharti A. R.; Nally J. E.; Ricaldi J. N.; Matthias M. A.; Diaz M. M.; Lovett M. A.; Levett P. N.; Gilman R. H.; Willig M. R.; Gotuzzo E.; Vinetz J. M. (2003) Leptospirosis: A zoonotic disease of global importance. Lancet Infect. Dis. 3, 757–771. [DOI] [PubMed] [Google Scholar]
- Meites E.; Jay M. T.; Deresinski S.; Shieh W. J.; Zaki S. R.; Tompkins L.; Smith D. S. (2004) Reemerging leptospirosis, California. Emerging Infect. Dis. 10, 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund B.; Korhonen T. K. (1993) Bacterial proteins binding to the mammalian extracellular-matix. Mol. Microbiol. 9, 687–694. [DOI] [PubMed] [Google Scholar]
- Ko A. I.; Goarant C.; Picardeau M. (2009) Leptospira: The dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat. Rev. Microbiol. 7, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk P.; Guzzo C. R.; Ramos H. R.; Ho P. L.; Farah C. S. (2009) Structure and Calcium-Binding Activity of LipL32, the Major Surface Antigen of Pathogenic Leptospira sp. J. Mol. Biol. 390, 722–736. [DOI] [PubMed] [Google Scholar]
- Tung J.-Y.; Yang C.-W.; Chou S.-W.; Lin C.-C.; Sun Y.-J. (2010) Calcium Binds to LipL32, a Lipoprotein from Pathogenic Leptospira, and Modulates Fibronectin Binding. J. Biol. Chem. 285, 3245–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian J. P.; Beddoe T.; McAlister A. D.; Wilce M. C. J.; Zaker-Tabrizi L.; Troy S.; Byres E.; Hoke D. E.; Cullen P. A.; Lo M.; Murray G. L.; Adler B.; Rossjohn J. (2009) Crystal Structure of LipL32, the Most Abundant Surface Protein of Pathogenic Leptospira spp. J. Mol. Biol. 387, 1229–1238. [DOI] [PubMed] [Google Scholar]
- Giuseppe P. O.; Neves F. O.; Nascimento A.; Guimaraes B. G. (2008) The leptospiral antigen Lp49 is a two-domain protein with putative protein binding function. J. Struct. Biol. 163, 53–60. [DOI] [PubMed] [Google Scholar]
- Croda J.; Ramos J. G. R.; Matsunaga J.; Queiroz A.; Homma A.; Riley L. W.; Haake D. A.; Reis M. G.; Ko A. I. (2007) Leptospira immunoglobulin-like proteins as a serodiagnostic marker for acute leptospirosis. J. Clin. Microbiol. 45, 1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira M. L.; Fernandes L. G.; Domingos R. F.; Oliveira R.; Siqueira G. H.; Souza N. M.; Teixeira A. R. F.; Atzingen M. V.; Nascimento A. (2014) Leptospiral extracellular matrix adhesins as mediators of pathogen-host interactions. FEMS Microbiol. Lett. 352, 129–139. [DOI] [PubMed] [Google Scholar]
- Matsunaga J.; Barocchi M. A.; Croda J.; Young T. A.; Sanchez Y.; Siqueira I.; Bolin C. A.; Reis M. G.; Riley L. W.; Haake D. A.; Ko A. I. (2003) Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily. Mol. Microbiol. 49, 929–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan R. U. M.; Chang Y. F.; Jusuf S. S. D.; Artiushin S.; Timoney J. F.; McDonough S. P.; Barr S. C.; Divers T. J.; Simpson K. W.; McDonough P. L.; Mohammed H. O. (2002) Cloning and molecular characterization of an immunogenic LigA protein of Leptospira interrogans. Infect. Immun. 70, 5924–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan R. U. M.; Chang Y. F.; Hassan F.; McDonough S. P.; Pough M.; Barr S. C.; Simpson K. W.; Mohammed H. O.; Shin S.; McDonough P.; Zuerner R. L.; Ou J. X.; Roe B. (2004) Expression of leptospiral immunoglobulin-like protein by Leptospira interrogans and evaluation of its diagnostic potential in a kinetic ELISA. J. Med. Microbiol. 53, 975–984. [DOI] [PubMed] [Google Scholar]
- Lin Y. P.; Greenwood A.; Nicholson L. K.; Sharma Y.; McDonough S. P.; Chang Y. F. (2009) Fibronectin binds to and induces conformational change in a disordered region of Leptospira immunoglobulin-like protein B. J. Biol. Chem. 284, 23547–23557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-P.; McDonough S. P.; Sharma Y.; Chang Y.-F. (2010) The Terminal Immunoglobulin-Like Repeats of LigA and LigB of Leptospira Enhance Their Binding to Gelatin Binding Domain of Fibronectin and Host Cells. PLoS One 5, e11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-P.; Chang Y.-F. (2007) A domain of the Leptospira LigB contributes to high affinity binding of fibronectin. Biochem. Biophys. Res. Commun. 362, 443–448. [DOI] [PubMed] [Google Scholar]
- Lin Y.-P.; Lee D.-W.; McDonough S. P.; Nicholson L. K.; Sharma Y.; Chang Y.-F. (2009) Repeated Domains of Leptospira Immunoglobulin-like Proteins Interact with Elastin and Tropoelastin. J. Biol. Chem. 284, 19380–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-P.; McDonough S. P.; Sharma Y.; Chang Y.-F. (2011) Leptospira immunoglobulin-like protein B (LigB) binding to the C-terminal fibrinogen alpha C domain inhibits fibrin clot formation, platelet adhesion and aggregation. Mol. Microbiol. 79, 1063–1076. [DOI] [PubMed] [Google Scholar]
- Stebbins C. E.; Galan J. E. (2001) Structural mimicry in bacterial virulence. Nature 412, 701–705. [DOI] [PubMed] [Google Scholar]
- Bodelon G.; Palomino C.; Fernandez L. A. (2013) Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev. 37, 204–250. [DOI] [PubMed] [Google Scholar]
- Batchelor M.; Prasannan S.; Daniell S.; Reece S.; Connerton I.; Bloomberg G.; Dougan G.; Frankel G.; Matthews S. (2000) Structural basis for recognition of the translocated intimin receptor (Tir) by intimin from enteropathogenic Escherichia coli. EMBO J. 19, 2452–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger Z. A.; Brown M. S.; Isberg R. R.; Bjorkman P. J. (1999) Crystal structure of invasin: A bacterial integrin-binding protein. Science 286, 291–295. [DOI] [PubMed] [Google Scholar]
- Kelly G.; Prasannan S.; Daniell S.; Fleming K.; Frankel G.; Dougan G.; Connerton I.; Matthews S. (1999) Structure of the cell-adhesion fragment of intimin from enteropathogenic Escherichia coli. Nat. Struct. Biol. 6, 313–318. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Frey E. A.; Pfuetzner R. A.; Creagh A. L.; Knoechel D. G.; Haynes C. A.; Finlay B. B.; Strynadka N. C. J. (2000) Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature 405, 1073–1077. [DOI] [PubMed] [Google Scholar]
- Lappalainen I.; Hurley M. G.; Clarke J. (2008) Plasticity within the obligatory folding nucleus of an immunoglobulin-like domain. J. Mol. Biol. 375, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manford A.; Xia T.; Saxena A. K.; Stefan C.; Hu F. H.; Emr S. D.; Mao Y. X. (2010) Crystal structure of the yeast Sac1: Implications for its phosphoinositide phosphatase function. EMBO J. 29, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F., and Sambrook J. (1982) Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY. [Google Scholar]
- Habeeb A. F. (1972) Reaction of protein sulfhydryl groups with Ellman’s reagent. Methods Enzymol. 25, 457–464. [DOI] [PubMed] [Google Scholar]
- Ishii Y.; Markus M. A.; Tycko R. (2001) Controlling residual dipolar couplings in high-resolution NMR of proteins by strain induced alignment in a gel. J. Biomol. NMR 21, 141–151. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Bahrami A.; Assadi A. H.; Markley J. L.; Eghbalnia H. R. (2009) Probabilistic Interaction Network of Evidence Algorithm and its Application to Complete Labeling of Peak Lists from Protein NMR Spectroscopy. PLoS Comput. Biol. 5, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Bax A. (2010) Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 46, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Rullmann J. A. C.; MacArthur M. W.; Kaptein R.; Thornton J. M. (1996) AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A.; Tejero R.; Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins: Struct., Funct., Bioinf. 66, 778–795. [DOI] [PubMed] [Google Scholar]
- Maiti R.; Van Domselaar G. H.; Zhang H.; Wishart D. S. (2004) SuperPose: A simple server for sophisticated structural superposition. Nucleic Acids Res. 32, W590–W594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; Clements J.; Eddy S. R. (2011) HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig B.; Nicholls A. (1995) Classical electrostatics in biology and chemistry. Science 268, 1144–1149. [DOI] [PubMed] [Google Scholar]
- McBride A. J. A.; Cerqueira G. M.; Suchard M. A.; Moreira A. N.; Zuerner R. L.; Reis M. G.; Haake D. A.; Ko A. I.; Dellagostin O. A. (2009) Genetic diversity of the Leptospiral immunoglobulin-like (Lig) genes in pathogenic Leptospira spp. Infect. Genet. Evol. 9, 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramini J. M., Wang D., Ciccosanti C. T., Janjua H., Rost B., Acton T. B., Xiao R., Swapna G. V. T., Everett J. K., Montelione G. T., and Northeast Structural Genomics Consortium . Solution NMR structure of a Bacterial Ig-like (Big_3) domain from Bacillus cereus: Northeast Structural Genomics Consortium target BcR147A. Manuscript in preparation, 2014.
- Wang T.; Zhang J.; Zhang X.; Xu C.; Tu X. (2013) Solution structure of the Big domain from Streptococcus pneumoniae reveals a novel Ca2+-binding module. Sci. Rep. 3, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy D. J.; Aukhil I.; Erickson H. P. (1996) 2.0 angstrom crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84, 155–164. [DOI] [PubMed] [Google Scholar]
- Jeffries C. M.; Lu Y. L.; Hynson R. M. G.; Taylor J. E.; Ballesteros M.; Kwan A. H.; Trewhella J. (2011) Human Cardiac Myosin Binding Protein C: Structural Flexibility within an Extended Modular Architecture. J. Mol. Biol. 414, 735–748. [DOI] [PubMed] [Google Scholar]
- Choy H. A.; Kelley M. M.; Chen T. L.; Moller A. K.; Matsunaga J.; Haake D. A. (2007) Physiological osmotic induction of Leptospira interrogans adhesion: LigA and LigB bind extracellular matrix proteins and fibrinogen. Infect. Immun. 75, 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieleg O.; Baumgartel R. M.; Bausch A. R. (2009) Selective Filtering of Particles by the Extracellular Matrix: An Electrostatic Bandpass. Biophys. J. 97, 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy H. A.; Kelley M. M.; Croda J.; Matsunaga J.; Babbitt J. T.; Ko A. I.; Picardeau M.; Haake D. A. (2011) The Multifunctional LigB Adhesin Binds Homeostatic Proteins with Potential Roles in Cutaneous Infection by Pathogenic Leptospira interrogans. PLoS One 6, e16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher R. M.; Svergun D. I.; Muhle-Goll C.; Mayans O. (2010) The Structure of the FnIII Tandem A77-A78 Points to a Periodically Conserved Architecture in the Myosin-Binding Region of Titin. J. Mol. Biol. 401, 843–853. [DOI] [PubMed] [Google Scholar]
- Finn R. D.; Mistry J.; Tate J.; Coggill P.; Heger A.; Pollington J. E.; Gavin O. L.; Gunasekaran P.; Ceric G.; Forslund K.; Holm L.; Sonnhammer E. L. L.; Eddy S. R.; Bateman A. (2010) The Pfam protein families database. Nucleic Acids Res. 38, D211–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman R.; Ptak C. P.; Hsieh C.-L.; Oswald R. E.; Chang Y.-F.; Sharma Y. (2013) The Perturbation of Tryptophan Fluorescence by Phenylalanine to Alanine Mutations Identifies the Hydrophobic Core in a Subset of Bacterial Ig-like Domains. Biochemistry 52, 4589–4591. [DOI] [PubMed] [Google Scholar]
- Reshetnyak Y. K.; Koshevnik Y.; Burstein E. A. (2001) Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 81, 1735–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Schulten K. (2000) The key event in force-induced unfolding of titin’s immunoglobulin domains. Biophys. J. 79, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.