

Abstract
Thymidylate, a vital DNA precursor, is synthesized by thymidylate synthases (TSs). A second class of TSs, encoded by the thyX gene, is found in bacteria and a few other microbes and is especially widespread in anaerobes. TS encoded by thyX requires a flavin adenine dinucleotide prosthetic group for activity. In the oxidative half-reaction, the reduced flavin is oxidized by 2′-deoxyuridine 5′-monophosphate (dUMP) and (6R)-N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2THF), synthesizing 2′-deoxythymidine 5′-monophosphate (dTMP). dTMP synthesis is a complex process, requiring the enzyme to promote carbon transfer, probably by increasing the nucleophilicity of dUMP and the electrophilicity of CH2THF, and reduction of the transferred carbon. The mechanism of the oxidative half-reaction was investigated by transient kinetics. Two intermediates were detected, the first by a change in the flavin absorbance spectrum in stopped-flow experiments and the second by the transient disappearance of deoxynucleotide in acid quenching experiments. The effects of substrate analogues and the behavior of mutated enzymes on these reactions lead to the conclusion that activation of dUMP does not occur through a Michael-like addition, the mechanism for the activation analogous with that of the flavin-independent TS. Rather, we propose that the nucleophilicity of dUMP is enhanced by electrostatic polarization upon binding to the active site. This conclusion rationalizes many of our observations, for instance, the markedly slower reactions when two arginine residues that hydrogen bond with the uracil moiety of dUMP were mutated to alanine. The activation of dUMP by polarization is consistent with the majority of the published data on ThyX and provides a testable mechanistic hypothesis.
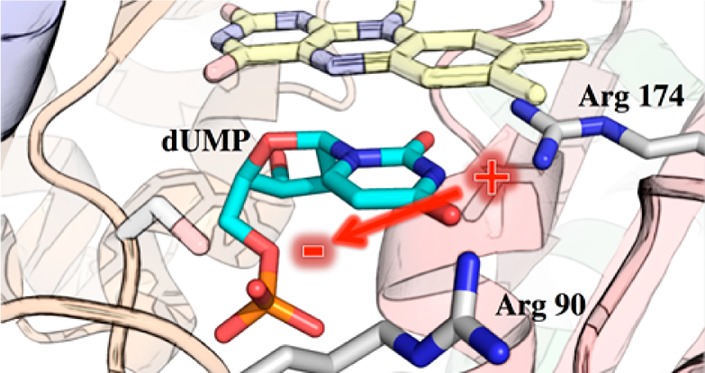
One of the building blocks of DNA, 2′-deoxythymidine 5′-monophosphate (dTMP), is synthesized from 2′-deoxyuridine 5′-monophosphate (dUMP) and (6R)-N5,N10-5,10-methylene-5,6,7,8-tetrahydrofolate (CH2THF) by thymidylate synthase (TS). Flavin-dependent TSs (ThyX), encoded by thyX genes, have been identified in microbes, including many pathogens associated with diseases such as tuberculosis, anthrax, pneumonia, diarrhea, and syphilis.1−5 ThyX catalyzes the reaction shown in Scheme 1. Crystal structures of oxidized ThyX have been determined6−10 from Thermotoga maritima, Paramecium bursaria chlorella virus-1, Mycobacterium tuberculosis, Corynebacterium glutamicum, and Helicobacter pylori. ThyX is a tetramer with each of the four active sites being formed by the juncture of three of the four subunits. dUMP binds in a pocket protected from the solvent by a protein loop (residues 87–91, T. maritima numbering), and the uracil ring of dUMP stacks on the si-face of the isoalloxazine of FAD with its C6 position next to N5 of the flavin (Figure 1).6 Structures of the T. maritima enzyme with bound CH2THF and folinic acid have been determined, showing that dUMP and folates bind to sites separated by the isoalloxazine, an arrangement that does not allow carbon transfer. Thus, catalytically relevant structures with deoxynucleotide and folate have not yet been determined.11
Scheme 1. Overall Reaction Catalyzed by ThyX.
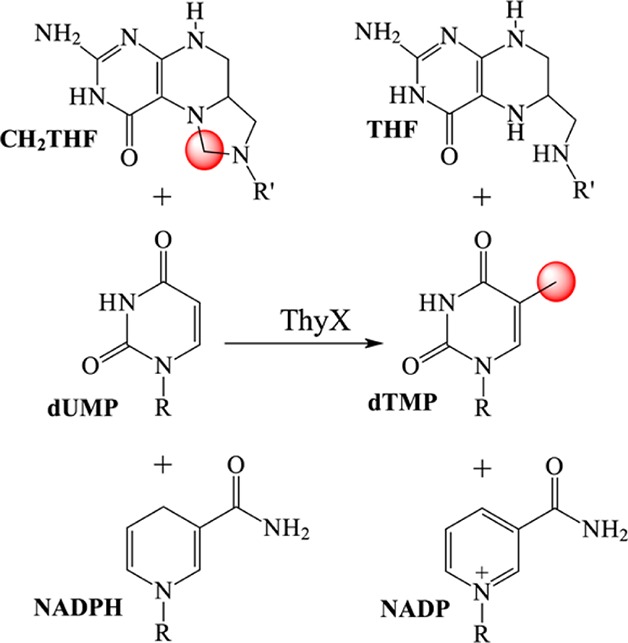
Figure 1.
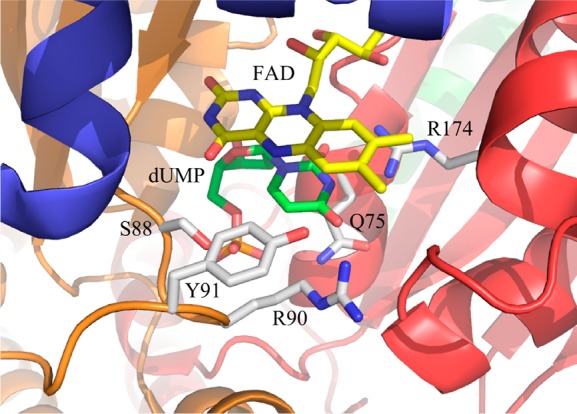
Active site of ThyX from T. maritima. The crystal structure (Protein Data Bank entry 1o26) shows dUMP (green carbons) bound to the active site, stacking directly below the isoalloxazine of the flavin (yellow, of course). The different subunits that make up the active site are colored differently. The residues mutated in this work are also shown (white carbons); others have been omitted for the sake of clarity.
Synthesizing dTMP from dUMP and CH2THF is complex. The well-studied TSs, which do not use a prosthetic group, accomplish the synthesis through a sequence of several steps:12 activation of C5 of dUMP as a nucleophile so that it can react with CH2THF, activation of the methylene carbon of CH2THF to its electrophilic iminium form, transfer of methylene from CH2THF to the nucleotide, and reduction by THF of the transferred carbon to form the methyl of dTMP (summarized in Figure S1 of the Supporting Information). ThyX accomplishes the same synthesis using the same substrates but has inserted a flavin prosthetic group into the reaction sequence in a way that is not yet fully understood. The analogy of the classic TS has guided investigations into the mechanism of ThyX. It is especially interesting to learn whether ThyX accomplishes particular chemical tasks in the same way as the classic TS and to determine whether similar intermediates are formed.
A key catalytic step in the classic TS is the activation of dUMP by the Michael-like addition of a cysteine residue so that the pyrimidine becomes nucleophilic.12 No similar cysteine residue is found in ThyX. Several mechanisms have been proposed for the activation of dUMP, generally invoking an alternate nucleophile (e.g., see ref (27)). We propose here that dUMP is not activated by covalent catalysis but instead is activated by the polarization of the uracil moiety upon binding and that carbon transfer occurs before oxidation of the flavin. This proposal is based on transient kinetic studies (both stopped-flow and chemical quenching) using the natural ThyX–substrate system and from the changes caused by using mutated enzymes, substrate analogues, or an artificial flavin. This proposal is consistent with a number of published studies13−15 and is supported by the recently reported trapping of an intermediate.16
Experimental Procedures
Materials
dUMP, dTMP, 5F-dUMP, sodium dithionite, EDTA, formaldehyde, and l-cysteine were purchased from Sigma Chemical Co. dUMPS was from Axxora, LLC. CH2THF was kindly provided by R. Moser from Merk-Eprova AG. Protocatechuate dioxygenase, purified from Pseudomonas cepacia DB01, was a gift from D. P. Ballou (University of Michigan, Ann Arbor, MI).17 Prepurified dry argon from Matheson Coleman and Bell was passed through a column of OxiClear (LabClear) to remove traces of oxygen.
WT ThyX from T. maritima (TM 0449) and five different variants containing substitutions S88A, R90A, R174A, Q75A, and Y91A were expressed with an N-terminal His tag in Escherichia coli BL21 grown in terrific broth, induced by adding l-(+)-arabinose,18 and purified with TALON metal affinity resin (Clontech). ThyX was purified further by being incubated at 65 °C for 15 min, followed by removal of the precipitate by centrifugation for 10 min at 23400 g.
Preparation of Enzyme for Reactions
Enzyme was prepared for oxidative half-reactions by being exchanged into 0.1 M Tris-HCl (pH 8.0) with 1 mM EDTA using Bio-Rad PD10 desalting columns. Enzyme solutions were made anaerobic19 in glass tonometers fitted with a cuvette (for rapid reaction experiments) or in anaerobic cuvettes with side arms (for slow scanning experiments). A syringe containing ∼6 mM dithionite was attached, and the enzyme was titrated to complete reduction.
Substrates
For rapid reaction experiments, 300 μM deoxynucleotide (dUMP, the phosphothioate analogue dUMPS or the fluorinated analogue 5F-dUMP) was added to the reduced enzyme before the stopped-flow instrument was loaded and reactions were initiated by the rapid mixing of CH2THF (400 μM). For slow reactions, the two substrates were added sequentially from the side arms of an anaerobic cuvette. Formaldehyde (15 mM) was added to solutions of CH2THF; no reaction was observed spectrophotometrically between free reduced enzyme and CH2O.
Spectrophotometry
Slow reactions were studied in anaerobic cuvettes and monitored in a Shimadzu UV-2501PC scanning spectrophotometer by repeated scanning from 700 to 300 nm. Rapid reactions were monitored spectrophotometrically in a Hi-Tech Scientific SF-61 DX2 double-mixing instrument (TGK Scientific) using either a monochromator and photomultiplier tube to collect single-wavelength absorbance traces or a photodiode-array detector (1.5 ms integration time) to collect absorbance spectra from 350 to 700 nm. The stopped-flow instrument was made anaerobic by flushing the system with an oxygen-scrubbing solution containing ∼0.1 unit mL–1 protocatechuate dioxygenase17 and 1 mM protocatechuate (3,4-dihydroxybenzoate) in 0.1 M KPi buffer (pH 7.0) and soaked overnight. The apparatus was thoroughly rinsed with anaerobic buffer prior to being used. Reaction traces were fit to sums of exponentials using Kinetic Studio (Hi-Tech Scientific), KaleidaGraph (Synergy Software), or Program A (R. Chang, C.-Y. Chiu, J. Dinverno, and D. Ballou, University of Michigan). Spectra collected by the photodiode array were analyzed by singular-value decomposition to calculate the spectrum of a reaction intermediate using KinTek Explorer (KinTek Corp.).
Acid Quenching
For chemical quenching, the optical cell and detectors of the double-mixing instrument were replaced with a prototype of a qPod attachment (TGK Scientific). The oxygen-scrubbing solution was included with the enzyme in acid-quench experiments. Reactions were quenched with 1 M HCl after different aging times (0.045–199.96 s). The quenched reaction mixtures (80 μL) were collected by flushing the cell with 500 μL of anaerobic buffer. The pH was adjusted to 7.0 by adding 6 μL of 2.5 M NaOH. The solutions were spun in a microcentrifuge, and the supernatants were filtered with Millex-GV filter units for analysis of soluble components. Pellets (denatured enzyme) were stored at −80 °C for later analysis by digestion with trypsin and were subjected to matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis by the University of Michigan Biomedical Mass Spectroscopy Facility. To analyze soluble components of the quenched reaction mixtures, 25 μL of each filtrate was injected into a Waters high-performance liquid chromatography (HPLC) system [Waters C-18 Symmetry column, 3.9 mm × 150 mm, 5 μm pore size; 200 mM triethylammonium bicarbonate (pH 7.0)]. The chromatograms showed many peaks, corresponding to dTMP, dUMP, FAD, CH2THF, and breakdown products (e.g., THF). In this system, dUMP and dTMP eluted at 7.34 and 15.0 min, respectively. The assignments of the dUMP and dTMP peaks were confirmed by co-injecting standards. Concentrations of dUMP and dTMP were determined from standard curves of peak area as a function of nucleotide concentration.
dTMP Production by Mutant Enzymes
Oxidative half-reactions of mutant enzymes (∼25 μM) were performed in anaerobic cuvettes using 500 μM dUMP and 500 μM CH2THF. After oxidation, the enzyme was removed using a 10000 molecular weight cutoff microcentrifugal spin filter (Millipore), and 10 μL of the filtrate was injected onto a Shimadzu HPLC system fitted with a 3.9 mm × 150 mm, 5 μm Symmetry C18 column (Waters) and run with isocratic elution [1 mL min–1 with 200 mM ammonium phosphate (pH 5.0)]; dTMP had a retention time of 5.3 min.20
Results
Spectrophotometric Detection of Intermediates
In the oxidative half-reaction of ThyX, the reduced flavin is oxidized only when both dUMP and CH2THF are available to the enzyme.13 The reaction was studied at 25 °C by mixing the reduced WT ThyX·dUMP complex (14 μM, final concentration) with CH2THF (400 μM) in 0.1 M Tris-HCl (pH 8.0) and 1 mM EDTA. A diode-array spectrometer was used to collect spectra from 350 to 700 nm (Figure 2A). These data were analyzed by singular-value decomposition and fitting to a two-step reaction (Figure 2B). The initial spectrum calculated in this analysis was different from that of the WT ThyX·dUMP complex, indicating CH2THF binding in the dead-time of the instrument (1.6 ms) to form the reduced WT ThyX·dUMP·CH2THF complex. After CH2THF binding, a small spectral change—a decrease in absorbance centered at 420 nm—followed, indicating the formation of an intermediate (I1 in Figure 2B). The spectrum of I1 is clearly that of a flavin hydroquinone,21 indicating that non-redox reaction(s) changed the flavin environment and, therefore, its spectrum. The spectra of reduced isoalloxazines vary considerably among flavoenzymes and are sensitive to their environments, but not in a way that is readily interpretable. After its formation, I1 reacted causing a large increase in the absorbance at wavelengths greater than ∼340 nm as the enzyme-bound flavin became oxidized (Figure 2B). Rate constants for these reactions were determined at 420 nm by collecting single-wavelength traces using a monochromator and photomultiplier tube (Figure 3A). The first reaction phase had a rate constant of 28.4 ± 0.8 s–1, and the second phase, corresponding to flavin oxidation, had a rate constant of 0.20 ± 0.01 s–1 (Table 1). These reactions were dependent on pH. In 100 mM glycine (pH 10.0), observed rate constants of 1.2 and 0.025 s–1 were obtained for the decrease and increase in A420, respectively. In 100 mM potassium phosphate (pH 6.0), these observed rate constants were 60 and 0.77 s–1, respectively.
Figure 2.

Spectrum of the intermediate detected in the oxidative half-reaction. An anaerobic solution of reduced WT ThyX (14 μM active sites, after mixing) and dUMP (300 μM) in 0.1 M Tris-HCl (pH 8.0) and 1 mM EDTA was mixed with 400 μM CH2THF and 15 mM formaldehyde using a stopped-flow spectrophotometer in diode-array mode. (A) Stereoview of spectra as a function of time. Spectra were recorded with an integration time of 1.5 ms at various intervals out to 10 s. Note the logarithmic time scale. (B) Deconvoluted intermediate spectra calculated by singular-value decomposition using the raw data in panel A and a two-step mechanism, giving rate constants of 28.4 and 0.2 s–1.
Figure 3.
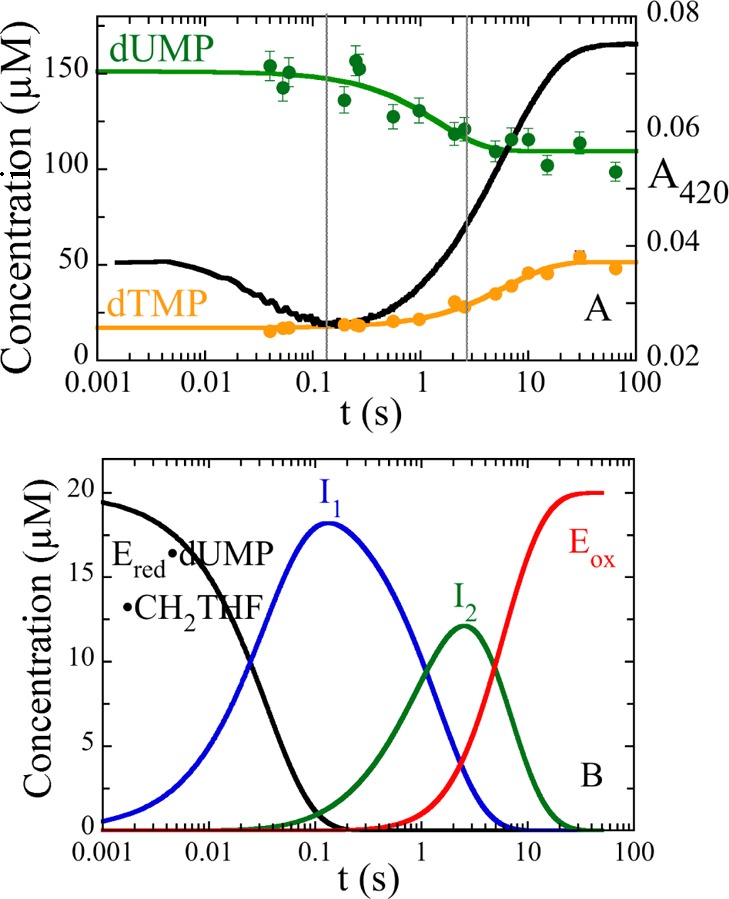
Chemical quenching. (A) An anaerobic solution of reduced WT ThyX (50 μM) and dUMP (300 μM) was mixed with 400 μM CH2THF and 15 mM formaldehyde in 0.1 M Tris-HCl (pH 8.0) and 1 mM EDTA. The reaction was quenched with 1 M HCl at different times, and the concentrations of dUMP (green) and dTMP (orange) were calculated from the area under the peaks of HPLC chromatograms. An absorbance trace at 420 nm obtained in stopped-flow experiments (black) is shown for comparison. The vertical lines at 0.13 and 2.6 s indicate the times of maximal accumulation of intermediates I1, detected by the flavin spectral change, and I2, detected by the consumption of dUMP. (B) Calculated concentrations of species during the oxidative half-reaction. The rate constants from stopped-flow experiments, 28.4 and 0.2 s–1, and the rate constant from the consumption of dUMP, 0.7 s–1, observed by quenching, were used to simulate consecutive reactions. The simulation used an enzyme concentration of 20 μM and shows that intermediates accumulate maximally at 0.13 and 2.6 s.
Table 1. Comparison of the Oxidative Half-Reaction Rate Constants of Variantsa.
| enzyme | k1 (s–1) | k2 (s–1) | k3 (s–1)b |
|---|---|---|---|
| WT | 28.4 ± 0.8 | 0.20 ± 0.01 | – |
| Q75A | 2.2 ± 0.3 | 0.074 ± 0.002 | 0.003 ± 0.0005 |
| R90A | 0.33 ± 0.01 | 0.026 ± 0.01 | – |
| R174A | 6.8 × 10–5 ± 1.6 × 10–7 | – | – |
| Y91A | 0.53 ± 0.04 | 0.007 ± 0.0001 | 0.001 ± 0.0002 |
| S88A | 0.82 ± 0.05 | 0.06 ± 0.002 | 0.019 ± 0.0005 |
An anaerobic solution of reduced enzyme (14 μM active sites) and dUMP (300 μM) was mixed with 400 μM CH2THF using a stopped-flow spectrophotometer. The reactions were monitored at 420 nm, and the traces were fit to a sum of exponentials. Reactions were conducted in 0.1 M Tris-HCl (pH 8.0) with 1 mM EDTA and 15 mM CH2O at 25 °C.
Some reaction traces required a third exponential to fit a small increase in absorbance at the end of the reaction. We speculate that this small phase was caused by the reassociation and subsequent oxidation of a small amount of free FAD that dissociated during the preparation.
Acid Quenching
The reaction of the reduced WT ThyX·dUMP complex with 400 μM CH2THF in 0.1 M Tris-HCl (pH 8.0) and 1 mM EDTA was quenched with 1 M HCl at different times. Deoxynucleotides were quantified by HPLC. dUMP consumption takes place after the small initial decrease in absorbance at 420 nm observed in stopped-flow experiments (Figure 3A), and dTMP formation occurs with a concomitant increase in absorbance at 420 nm, corresponding to FAD oxidation. dUMP was consumed with a single exponential giving an observed rate constant of 0.7 s–1. There was a single-exponential rise of dTMP with an observed rate constant of 0.15 s–1. The rate constant for the loss of dUMP is larger than that for the formation of dTMP; if dUMP were converted directly to dTMP, these rate constants would be equal. Therefore, conservation of mass requires the existence of a deoxynucleotide intermediate. This intermediate was not detected during HPLC analysis. With rate constants of 0.7 s–1 for formation and 0.15 s–1 for decay, it would form maximally at 2.6 s (Figure 3B). The precipitated protein from quenched samples was analyzed by MALDI-TOF in an attempt to detect a protein adduct to deoxynucleotide or folate, but no change in the molecular weight of the protein was observed. Scheme 2 shows a minimal kinetic scheme describing the stopped-flow and quenching experiments.
Scheme 2. Reactions Detected by Transient Kinetics.
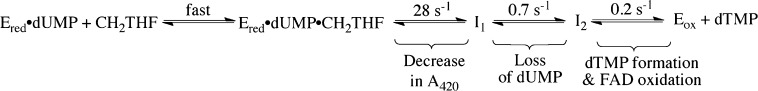
Oxidative Half-Reaction with 5-dUMPS
The phosphate moiety of dUMP is in an unusual conformation for a nucleotide, curling close to the uracil moiety, suggesting a possible role in the mechanism. Its importance during the oxidative half-reactions was examined using the phosphothioate analogue of dUMP (5-dUMPS). The sulfur atom of the analogue is more nucleophilic than the oxygen it replaces and also disperses the negative charge compared to the concentrated charge on the smaller, more electronegative oxygen. Therefore, substituting oxygen with sulfur should drastically speed step(s) in which phosphate acts as a nucleophile but slow steps requiring a localized charge. The reaction of the reduced WT ThyX·5-dUMPS complex with a saturating concentration of CH2THF (400 μM) was monitored at 420 nm using a stopped-flow spectrophotometer (Figure 4). As with the reaction of the WT ThyX·dUMP complex, there was an initial decrease in absorbance followed by a large increase, with an observed rate constant for the first phase of 3 s–1 and an observed rate constant for the second phase of 0.08 s–1, 9- and 2.5-fold slower than with dUMP, respectively. The production of the phosphothioate analogue of dTMP was confirmed by HPLC as a new peak in the chromatogram. These data suggest that the reaction is favored by the more concentrated negative charge of oxygen in dUMP rather than by the more nucleophilic sulfur of dUMPS.
Figure 4.
Reaction of the reduced WT ThyX·5-dUMPS complex with CH2THF. Anaerobic solutions of reduced WT ThyX (14 μM active sites) and dUMP [300 μM (black)] or with 5-dUMPS [300 μM (blue)] were mixed with 400 μM CH2THF and 15 mM formaldehyde using a stopped-flow spectrophotometer. The reaction mixtures were in 0.1 M Tris-HCl (pH 8.0) with 1 mM EDTA and 15 mM CH2O at 25 °C.
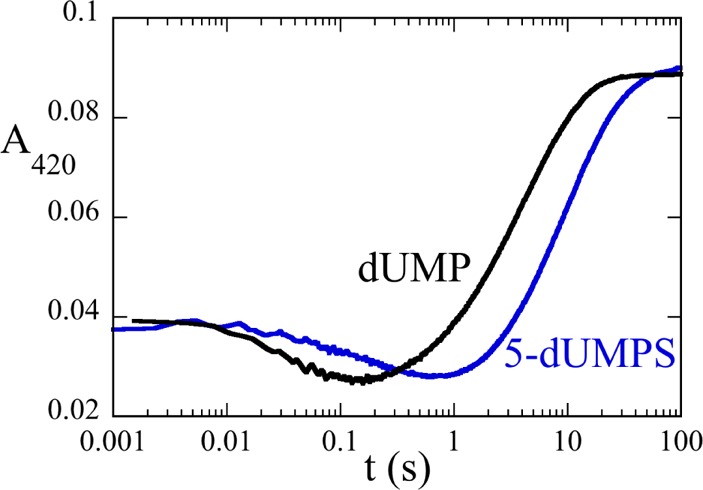
Formation of the WT ThyX·5F-dUMP·CH2THF Complex
The analogue 5F-dUMP is useful for probing the relative timing of flavin oxidation and carbon transfer.13 While the fluorouracil moiety of 5F-dUMP can be reduced, it cannot be dehalogenated because fluorine would need to leave as F+, a species too unstable for biochemical conditions. In classic TS, the inability to dehalogenate 5F-dUMP prevents C–C bond scission between the 5-methylene adduct of 5F-dUMP and THF.12 The effect of fluorine substitution was tested in T. maritima ThyX. Reduced WT ThyX was incubated with 5F-dUMP and CH2THF and monitored over time using a scanning spectrophotometer. Spectral changes associated with the binding of both 5F-dUMP and CH2THF were observed immediately (Figure 5), but flavin oxidation was not observed even after many hours, showing that the fluorine substituent blocks the oxidative half-reaction at a step prior to or at redox chemistry.
Figure 5.
Formation of the reduced WT ThyX·5F-dUMP·CH2THF complex. An anaerobic solution of WT ThyX [87 μM active sites (green)] was reduced with 1 equiv of dithionite (blue). An anaerobic solution of 5F-dUMP (300 μM) was added to form the reduced WT ThyX·5F-dUMP complex (red). The anaerobic addition of 400 μM CH2THF and 15 mM formaldehyde formed the reduced WT ThyX·5F-dUMP·CH2THF complex (black). The flavin remained reduced indefinitely. These spectra were recorded using a scanning spectrophotometer.
Reactions of Variant Enzymes
The roles of active site residues making contact with dUMP (Figure 1) were investigated by replacing them with alanine and determining the effects on the oxidative half-reaction in stopped-flow experiments (Table 1). All variant enzymes synthesized dTMP, as detected by HPLC. The reactions of all mutant enzymes, except R174A, showed an initial decrease in absorbance at 420 nm followed by a large increase (Figure 6). R174 hydrogen bonds to dUMP at N3 and at the C4 carbonyl oxygen. Substituting R174 with alanine had a huge effect on the oxidative half-reaction. Only one phase, a large increase in absorbance, was observed. Its rate constant was 6.8 × 10–5 s–1, ∼400000-fold slower than that of the oxidation phase of the WT reaction. The loss of the initial phase and the drastic decrease in the rate constant for flavin oxidation suggest that a reaction before flavin oxidation has become severely rate-limiting. R90 also hydrogen bonds to the C4 oxygen of dUMP. The oxidative half-reaction of the R90A enzyme has a greater effect on the first reaction phase (86-fold) than on the second phase (flavin oxidation) (7.6-fold). Residues Q75 and S88 are within hydrogen bonding distance of the phosphate oxygen of dUMP, 2.9 and 2.6 Å, respectively. Substitutions of these residues gave rate constants that were lower by 13- and 35-fold on the first phase and by 2.7- and 3.3-fold on the second phase, respectively, compared to the WT reaction. Y91 is 4.6 Å from N5 of the flavin. The Y91A mutation also had a greater effect on the first phase (53-fold slower) than on the second phase (28-fold slower).
Figure 6.
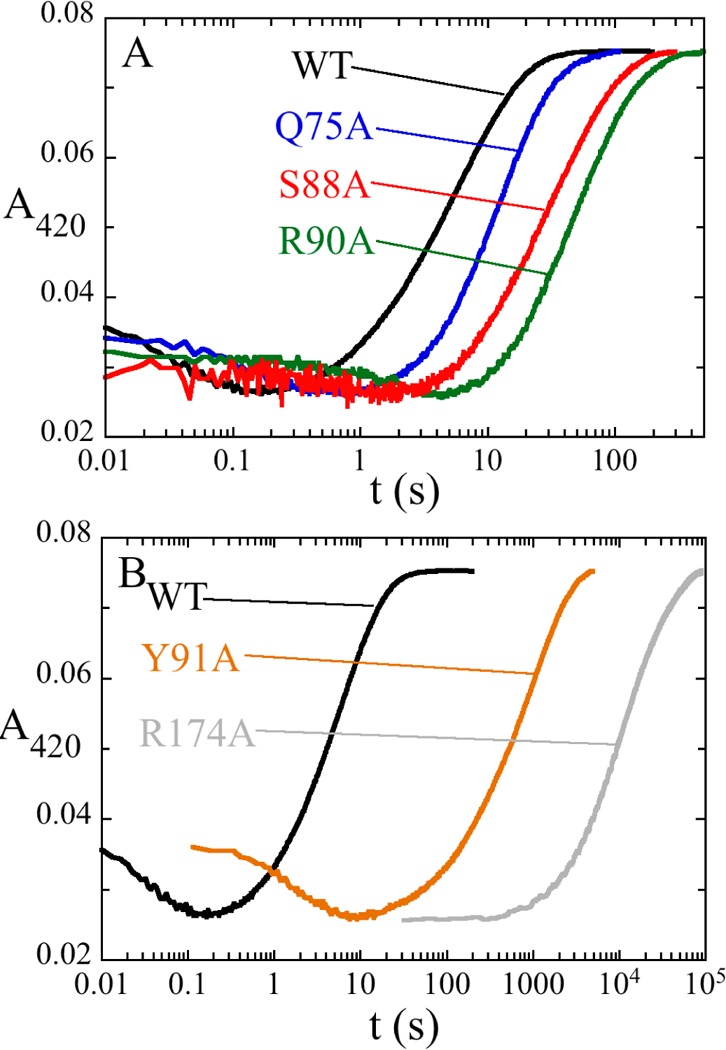
Oxidative half-reactions of variant enzymes. The reduced variant enzyme·dUMP complexes were mixed with saturating concentrations CH2THF using a stopped-flow spectrophotometer. The reactions were monitored by their absorbance at 420 nm. The reaction mixtures were in 0.1 M Tris-HCl (pH 8.0) with 1 mM EDTA and 15 mM CH2O at 25 °C. Traces, labeled by ThyX variant, are displayed in two panels for the sake of clarity.
Discussion
Two intermediates were detected in the oxidation of reduced ThyX by dUMP and CH2THF; however, the complexity of the chemistry dictates that any mechanism proposed for the transfer and reduction of the methylene must include additional species to account for all chemical steps. One important reaction in the sequence is the nucleophilic attack of the pyrimidine on the methylene of CH2THF. How ThyX activates dUMP as a nucleophile for this reaction is an important question. The classic TSs12,22 use an active site cysteine to activate dUMP through a Michael-like addition at C6 to increase the nucleophilicity of C5 (Figure S1 of the Supporting Information). Activation by Michael-like addition is also thought to occur in other enzymes that methylate pyrimidines, and each uses an active site cysteine.23−26 There has been an extensive search for an activating nucleophile in ThyX. There is no cysteine in the active site. The only conceivable nucleophilic residue close to the uracil moiety of dUMP is a conserved active site serine.27 Substituting S88 in T. maritima ThyX with an alanine to eliminate the putative nucleophile did not have a strong effect on the oxidative half-reaction, with both phases slowed only 10-fold, confirming the observation that S88A was still active14,24 and showing that S88 is not a critical nucleophile (Figure 6A).
Nucleophiles other than protein residues have been considered and also rejected. Conceivably, water could act as a nucleophile, but the available data do not support this. Activation of a water molecule would require its deprotonation, but the active site has no bases or metals that would deprotonate water.6−9 The closest potential base is Y91, but it points away from the reaction site rather than toward it; in addition, C5 of dUMP is not accessible to water. Also, the rate constants of all the phases of the oxidative half-reaction increase with a decrease in pH, behavior that is the opposite of that predicted if water were a nucleophile. The possibility that the phosphate of dUMP might act as a nucleophile was considered because the structure of ThyX shows that dUMP is in an unusual curled conformation, with phosphate oxygens 3.5 and 4 Å from C6 of the uracil moiety (Figure 1). However, the phosphothioate analogue of dUMP, with a more nucleophilic sulfur atom available to attack the uracil moiety, reacted slower, not faster, than dUMP, which is not expected if the phosphate oxygen of dUMP is the nucleophile. N5 of reduced flavin has also been considered as a possible nucleophile, based on analogies with some other flavoenzymes.28,29 However, the enzyme substituted with reduced 5-deaza-FAD, which does not have a lone electron pair at position 5 of the flavin for nucleophilic attack, was still oxidized by dUMP and CH2THF,14 leading to the rejection of reduced flavin as a nucleophile. (Also, the reaction of the 5-deaza-FAD-substitued enzyme is strong evidence against mechanisms invoking flavin radicals because 5-deaza-FAD radicals are extremely unfavorable.30)
The transfer of a hydride from the reduced flavin to dUMP has been considered as the activation mechanism of dUMP.14 Nuclear magnetic resonance (NMR) analysis clearly shows that a hydrogen derived from the solvent labels C6 and C7 of dTMP, suggesting that it washes-in via exchange at N5 of the reduced flavin.14,31 However, several experiments show that the transfer of the hydride to dUMP cannot take place until after other events occur, likely including the transfer of the methylene from CH2THF to dUMP. Activation of dUMP by hydride transfer would mean that flavin oxidation is the first step of the oxidative half-reaction sequence. This is not observed; the addition of dUMP alone does not oxidize the reduced flavin, while dUMP and CH2THF together oxidize the flavin only after at least two other reactions form I1 and I2 (Figure 3). Importantly, dUMP is consumed by forming I2, but before flavin oxidation. The reduced enzyme is also not oxidized by 5F-dUMP and CH2THF, although fluorine substitution would not prevent the reduction of the pyrimidine. However, fluorine substitution would prevent the breakdown of a nucleotide–folate adduct because C5 could not be deprotonated to expel the leaving-group (THF) in the elimination reaction, which would block redox chemistry if it occurs in a subsequent step. This is consistent with previously published data obtained for Campylobacter jejuni ThyX that show 5F-dUMP and CH2THF do not oxidize the reduced enzyme.13 All of these results strongly suggest that dTMP synthesis cannot be initiated by reductive enolization of dUMP.
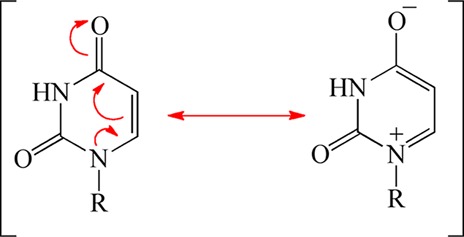
The extensive search for a nucleophile that activates dUMP has been futile. We propose that there actually is no activating nucleophile; instead, bound dUMP is activated by electrostatic polarization. Crystal structures show that the negative charge from the phosphate of dUMP is close to N1 and C6 of dUMP, and the positive charges from R90 and R174 are close to the carbonyl oxygen of C4 (Figure 1). This electrostatic environment would favor the polarized resonance description of dUMP in Scheme 3, which has a electron density on C5 higher than the usual nonpolarized resonance description. The higher electron density on C5 accelerates attack on the iminium carbon of CH2THF. It is noteworthy that in aqueous solution uracil can react with formaldehyde without nucleophilic catalysis.32−34 The conclusion that there is no enzymatic nucleophile in this mechanism is corroborated by our inability to detect a protein adduct by MALDI-TOF and by the absence of radiolabeled protein in the sodium dodecyl sulfate–polyacrylamide gel electrophoresis results of quenched reactions using radioactive dUMP.16
Scheme 3. Polarization of dUMP.
This concept leads to the chemical mechanism for the oxidative half-reaction shown in Scheme 4. Steps whose arrows are colored red (1–3) are suggested to occur in the first phase observed in stopped-flow experiments, while those colored blue (4–6) occur in the second phase as the flavin becomes oxidized. Candidates for I1 and I2 are indicated in maize and blue boxes, respectively. The first phase, the small flavin spectral change, could comprise polarization of dUMP upon binding, activation of CH2THF by ring opening (step 1), nucleophilic attack of dUMP on CH2THF (step 2), and deprotonation of C5 (step 3). None of these reactions involve the flavin directly but could change its immediate environment leading to changes in the spectrum of the flavin hydroquinone, as observed. I1 represents an enzyme form (or forms) after the spectral change to the flavin hydroquinone but from which dUMP can still be recovered in an acid quench. These criteria are met by the species in the maize box in Scheme 4. Thus, the spectral change might be caused by the formation of the iminium from CH2THF (Step 1) or by the formation of the dUMP–CH2THF adduct that, prior to deprotonation of C5 of uracil, could be cleaved by acid quench, but after deprotonation of the nucleotide–folate adduct (step 3), a stable adduct is expected; an analogous methylene-bridged adduct between uracil and THF has been synthesized.35 Thus, the deprotonated adduct formed in step 3 is rejected as a candidate for I1.
Scheme 4. Proposed Mechanism of the Oxidative Half-Reaction.
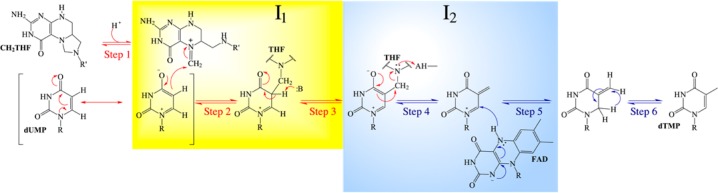
Flavin oxidation occurs in the second phase observed in stopped-flow experiments, which is the step that forms dTMP from I2. Our analytical methods did not affirmatively detect the putative intermediate I2, but its presence was inferred by the loss of total identified deoxynucleotide (dUMP and dTMP). Scheme 4, on the basis of our data, suggests that I2 could be either the methylene-bridged dUMP–THF adduct or the cleaved adduct (THF and the exocyclic enone deoxynucleotide). Importantly, Mishanina and colleagues16 trapped 5-hydroxymethyl-dUMP, detected by radioactivity and mass spectroscopy, at times when I2 should be maximal. 5-Hydroxymethyl-dUMP likely forms upon acid quench from the exocyclic enone deoxynucleotide16 before hydride transfer occurs (step 5), suggesting that I2 is the cleaved adduct. The elimination of THF from the methylene-bridged complex between THF and dUMP immediately precedes flavin oxidation (step 4) and would be catalyzed by the protonation of N5 of THF. Preliminary stopped-flow experiments show that flavin oxidation is faster at lower pH, consistent with the acid-catalyzed step 4 determining the rate of flavin oxidation. A hydride is then transferred from the flavin to the nucleotide in step 5, accounting for the incorporation of solvent-derived hydrogen at C6 of the nucleotide observed previously14,31 and forming the penultimate nucleotide intermediate, an exocyclic enone tautomer of dTMP. Lastly, a 1,3-hydride shift has been previously proposed as the final step (step 6) based on NMR analysis of label incorporation.14 This reaction is unlikely to perturb the absorbance spectrum of the oxidized flavin by much and so may not be detected in stopped-flow experiments. Furthermore, if tautomerization to dTMP is fast compared to the time required for HPLC analysis of acid-quenched samples, only dTMP would be detected.
In Scheme 4, it is proposed that the pyrimidine moiety is deprotonated (step 3) and then THF is eliminated (step 4), followed by hydride transfer (step 5). Other sequences might be imagined and are not firmly excluded by the available data but seem less likely than the reaction sequence in Scheme 4. For instance, steps 3 and 4 might be combined into a single concerted step; however, that would eliminate the explanation for acid catalysis of flavin oxidation. Alternatively, concerted expulsion of THF and hydride transfer, combining steps 4 and 5 of Scheme 4, would exclude the enone deoxynucleotide as a distinct intermediate, but this would eliminate from the mechanism the species most likely to lead to 5-hydroxymethyl-dUMP identified in quenching experiments.16
Many of the kinetic perturbations to the first reaction phase caused by mutagenesis or substrate analogues can be rationalized by the mechanism in Scheme 4. The replacement of R90 or R174 with alanine diminishes polarization of dUMP so that the more nucleophilic resonance form (Scheme 3) is not stabilized. Thus, the first phase for these variants is slower than the WT phase. Likewise, the correctly positioned phosphate helps to efficiently polarize dUMP by stabilizing a positive charge at N1. Substitution of either Q75 or S88 slows the first phase, presumably because the lost hydrogen bonds from the phosphate to the protein disrupt the optimal positioning, decreasing the level of polarization. The nucleotide analogue 5-dUMPS, which has a more dispersed negative charge, should also be less effective in polarizing the uracil moiety, consistent with the slower first reaction phase observed with this analogue (Figure 4).
Conclusion
The search for a nucleophile to initiate the ThyX reaction by a Michael-like addition to dUMP has been unsuccessful because, we believe, there is no nucleophile. We propose a chemical mechanism in Scheme 4 for the synthesis of dTMP that does not invoke nucleophilic activation. It is consistent with the data presented here and most of the thyX literature. This scheme is useful as a working hypothesis, and further investigation could show that some of the reactions might in fact be more complex. This novel scheme adds to the rich repertoire of mechanisms that have evolved to methylate pyrimidine rings.
Acknowledgments
We thank Meran M. Ebadi-Tehrani for purifying ThyX, Rudolf Moser from Merk-Eprova AG for the kind gift of (6R)-5,10-CH2THF, and Professor David P. Ballou and Dr. Michael Tarasev for the use of an HPLC system. We are especially grateful to Ted King of TGK Scientific for the qPod quenching module and for guidance in its use, and we thank Professor Kenneth A. Johnson for the use of an early version of KinTek Explorer. We also thank Eric Koehn, Tatiana Mishanina, and Professor Amnon Kohen (University of Iowa, Iowa City, IA) for critical readings of the paper and endless stimulating scientific debates.
Glossary
Abbreviations
- TS
thymidylate synthase
- 5F-dUMP
5-fluoro-2′-deoxyuridine 5′-monophosphate
- dUMP
2′-deoxyuridine 5′-monophosphate
- 5-dUMPS
2′-deoxyuridine 5′-monophosphothioate
- dTMP
2′-deoxythymidine 5′-monophosphate
- THF
(6S)-5,6,7,8-tetrahydrofolate
- CH2THF
(6R)-N5,N10-methylene-5,6,7,8-tetrahydrofolate
- FolA
dihydrofolate reductase
- Tris-HCl
tris(hydroxymethyl)aminomethane hydrochloride
- 5-deaza-FAD
5-carba-5-deaza-flavin adenine dinucleotide.
Supporting Information Available
A reaction scheme showing the mechanism of the classic thymidylate synthase. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ J.A.C.: Department of Chemistry, University of Nebraska—Omaha, Omaha, NE 68182-0109.
Author Present Address
∥ M.O.-M.: Genencor, Palo Alto, CA 94304.
Author Present Address
⊥ S.W.H.: Wayne State University School of Medicine, Detroit, MI 48201.
Author Contributions
J.A.C. and M.O.-M. contributed equally to this work.
The authors declare no competing financial interest.
This research was supported by National Science Foundation Grant CHE 1213620 to B.A.P. and National Institutes of Health Training Grant GM08270 to J.A.C.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Myllykallio H.; Lipowski G.; Leduc D.; Filee J.; Forterre P.; Liebl U. (2002) An Alternative Flavin-Dependent Mechanism for Thymidylate Synthesis. Science 297, 105–107. [DOI] [PubMed] [Google Scholar]
- Kuhn P.; Lesley S. A.; Mathews I. I.; Canaves J. M.; Brinen L. S.; Dai X.; Deacon A. M.; Elsliger M. A.; Eshaghi S.; Floyd R.; Godzik A.; Grittini C.; Grzechnik S. K.; Guda C.; Hodgson K. O.; Jaroszewski L.; Karlak C.; Klock H. E.; Koesema E.; Kovarik J. M.; Kreusch A. T.; McMullan D.; Robb A.; Rodrigues K.; Selby T. L.; Spraggon G.; Stevens R. C.; Taylor S. S.; Bedem H. v. d.; Valasquez J.; Vincent J.; Wang X.; West B.; Wolf G.; Wooley J.; Wilson I. A. (2002) Crystal Structure of Thy1, a Thymidylate Synthase Complementing Protein from Thermotoga maritima at 2.25 Å Resolution. Proteins 49, 142–145. [DOI] [PubMed] [Google Scholar]
- Giladi M.; Bitan-Banin G.; Mevarech M.; Ortenberg R. (2002) Genetic Evidence for a Novel Thymidylate Synthase in the Halophilic Archeon Halobacterium salinarum and in Campylobacter jejuni. FEMS Microbiol. Lett. 216, 105–109. [DOI] [PubMed] [Google Scholar]
- Myllykallio H.; Leduc D.; Filee J.; Liebl U. (2003) Life Without Dihydrofolate Reductase FolA. Trends Microbiol. 11, 220–223. [DOI] [PubMed] [Google Scholar]
- Leduc D.; Escartin F.; Nijhout H. F.; Reed M. C.; Liebl U.; Skouloubris S.; Myllykallio H. (2007) Flavin-dependent thymidylate synthase ThyX activity: Implications for the folate cycle in bacteria. J. Bacteriol. 189, 8537–8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews I. I.; Deacon A. M.; Canaves J. M.; McMullan D.; Lesley S. A.; Agarwalla S.; Kuhn P. (2003) Functional Analysis of Substrate and Cofactor Complex Structures of a Thymidylate Synthase-Complementing Protein. Structure 11, 677–690. [DOI] [PubMed] [Google Scholar]
- Graziani S.; Bernauer J.; Skouloubris S.; Graille M.; Zhou C. Z.; Marchand C.; Decottignies P.; van Tilbeurgh H.; Myllykallio H.; Liebl U. (2006) Catalytic mechanism and structure of viral flavin-dependent thymidylate synthase ThyX. J. Biol. Chem. 281, 24048–24057. [DOI] [PubMed] [Google Scholar]
- Sampathkumar P.; Turley S.; Ulmer J. E.; Rhie H. G.; Sibley C. H.; Hol W. G. J. (2005) Structure of Mycobacterium tuberculosis flavin-dependent thymidylate synthase (MtbThyX) at 2.0 Å resolution. J. Mol. Biol. 352, 1091–1104. [DOI] [PubMed] [Google Scholar]
- Wang W. C., Liu J. S., and Chang C. M. (2009) Protein Data Bank entry 3FNN, DOI: 10.2210/pdb3fnn/pdb.
- Wang K.; Wang Q.; Chen J.; Chen L.; Jiang H.; Shen X. (2011) Crystal structure and enzymatic characterization of thymidylate synthase X (ThyX) from Helicobacter pylori strain SS1. Protein Sci. 20, 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn E. M.; Perissinotti L. L.; Moghram S.; Prabhakar A.; Lesley S. A.; Mathews I. I.; Kohen A. (2012) Folate binding site of flavin-dependent thymidylate synthase. Proc. Natl. Acad. Sci. U.S.A. 109, 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras C. W.; Santi D. V. (1995) The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 64, 721–762. [DOI] [PubMed] [Google Scholar]
- Gattis S. G.; Palfey B. A. (2005) Direct Observation of the Participation of Flavin in Product Formation by thyX-Encoded Thymidylate Synthase. J. Am. Chem. Soc. 127, 832–833. [DOI] [PubMed] [Google Scholar]
- Koehn E. M.; Fleischmann T.; Conrad J. A.; Palfey B. A.; Lesley S. A.; Mathews I. I.; Kohen A. (2009) A Novel Chemical Mechanism of Thymidylate Biosynthesis in Human Pathogens Containing the thyX Gene. Nature 458, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyshev A.; Fleischmann T.; Koehn E. M.; Lesley S. A.; Kohen A. (2007) The relationships between oxidase and synthase activities of flavin dependent thymidylate synthase (FDTS). Chem. Commun. 2861–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishanina T.; Koehn E. M.; Conrad J. A.; Palfey B. A.; Lesley S. A.; Kohen A. (2012) Flavin-Dependent Thymidylate Synthase: Trapping of a Reaction Intermediate. J. Am. Chem. Soc. 134, 4442–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil P. V.; Ballou D. P. (2000) The use of protocatechuate dioxygenase for maintaining anaerobic conditions in biochemical experiments. Anal. Biochem. 286, 187–192. [DOI] [PubMed] [Google Scholar]
- Lesley S. A.; Kuhn P.; Godzik A.; Deacon A. M.; Mathews I.; Kreusch A.; Spraggon G.; Klock H. E.; McMullan D.; Shin T.; Vincent J.; Robb A.; Brinen L. S.; Miller M. D.; McPhillips T. M.; Miller M. A.; Scheibe D.; Canaves J. M.; Guda C.; Jaroszewski L.; Selby T. L.; Elsliger M. A.; Wooley J.; Taylor S. S.; Hodgson K. O.; Wilson I. A.; Schultz P. G.; Stevens R. C. (2002) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U.S.A. 99, 11664–11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfey B. A. (2003) Time Resolved Spectral Analysis. In Kinetic Analysis of Macromolecules: A Practical Approach (Johnson K. A., Ed.) Chapter 9, pp 203–228, Oxford University Press, New York. [Google Scholar]
- Martinez-Valdez H.; Kothari R. M.; Hershey H. V.; Taylor M. W. (1982) Rapid and Reliable Method for the Analysis of Nucleotide Pools by Reversed-Phase High-Performance Liquid Chromatography. J. Chromatogr. 247, 307–314. [DOI] [PubMed] [Google Scholar]
- Ghisla S.; Massey V.; Lhoste J. M.; Mayhew S. G. (1974) Fluorescence and optical characteristics of reduced flavines and flavoproteins. Biochemistry 13, 589–597. [DOI] [PubMed] [Google Scholar]
- Atreya C. E.; Anderson K. S. (2004) Kinetic characterization of bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) from Cryptosporidium hominis: A paradigm shift for ts activity and channeling behavior. J. Biol. Chem. 279, 18314–18322. [DOI] [PubMed] [Google Scholar]
- Nishimasu H.; Ishitani R.; Yamashita K.; Iwashita C.; Hirata A.; Hori H.; Nureki O. (2009) Atomic structure of a folate/FAD-dependent tRNA T54 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 106, 8180–8105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Santi D. V. (2000) m5C RNA and m5C DNA methyl transferases use different cysteine residues as catalysts. Proc. Natl. Acad. Sci. U.S.A. 97, 8263–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi D. V. (2000) Mechanistic studies of RNA modifying enzymes. RNA pseudouridine synthase and m5Cytosine methyl transferase. Nucleic Acids Symp. Ser. 2000, 147–148. [DOI] [PubMed] [Google Scholar]
- Kealey J. T.; Gu X.; Santi D. V. (1994) Enzymatic mechanism of tRNA (m5U54)methyltransferase. Biochimie 76, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Leduc D.; Graziani S.; Lipowski G.; Marchand C.; Le Maréchal P.; Liebl U.; Myllykallio H. (2004) Functional evidence for active site location of tetrameric thymidylate synthase X at the interphase [sic] of three monomers. Proc. Natl. Acad. Sci. U.S.A. 101, 7252–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltero-Higgin M.; Carlson E. E.; Gruber T. D.; Kiessling L. L. (2004) A unique catalytic mechanism for UDP-galactopyranose mutase. Nat. Struct. Mol. Biol. 11, 539–543. [DOI] [PubMed] [Google Scholar]
- Hamdane D.; Bruch E.; Un S.; Field M.; Fontecave M. (2013) Activation of a Unique Flavin-Dependent tRNA-Methylating Agent. Biochemistry 52, 8949–8956. [DOI] [PubMed] [Google Scholar]
- Blankenhorn G. (1976) Nicotinamide-dependent one-electron and two-electron (flavin) oxidoreduction: Thermodynamics, kinetics, and mechanism. Eur. J. Biochem. 67, 67–80. [DOI] [PubMed] [Google Scholar]
- Agrawal N.; Lesley S. A.; Kuhn P.; Kohen A. (2004) Mechanistic Studies of a Flavin-Dependent Thymidylate Synthase. Biochemistry 43, 10295–10301. [DOI] [PubMed] [Google Scholar]
- Dworkin J. P. (1997) Attempted prebiotic synthesis of pseudouridine. Origins Life Evol. Biospheres 27, 345–355. [DOI] [PubMed] [Google Scholar]
- Choughuley A. S. U.; Subbaraman A. S.; Kazi Z. A.; Chada M. S. (1977) A Possible Prebiotic Synthesis of Thymine: Uracil-Formaldehyde-Formic Acid Reaction. BioSystems 9, 73–80. [DOI] [PubMed] [Google Scholar]
- Alegria A. H. (1967) Hydroxymethylation of Pyrimidine Mononucleotides with Formaldehyde. Biochim. Biophys. Acta 149, 317–324. [DOI] [PubMed] [Google Scholar]
- Gupta V. S.; Huennekens F. M. (1967) Thyminyl Derivatives of Tetrahydrofolate. Biochemistry 6, 2168–2177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.