

Abstract
The solution chemistry and solid-state structures of the CoII, FeII, and NiII complexes of 7,13-bis(carbamoylmethyl)-1,4,10-trioxa-7,13-diazacyclopentadecane (L) are reported as members of a new class of paramagnetic chemical exchange saturation transfer (paraCEST) MRI contrast agents that contain transition metal ions. Crystallographic data show that nitrogen and oxygen donor atoms of the macrocyclic ligand coordinate to the metal ions to generate complexes with distorted pentagonal bipyramidal geometry for [Co(L)]Cl2·2H2O or [Fe(L)](CF3SO3)2. The NiII complex [Ni(L)](CF3SO3)2·H2O features a hexadentate ligand in a distorted octahedral geometry. The proton NMR spectra of all three complexes show highly dispersed and relatively sharp proton resonances. The complexes were further characterized by monitoring their dissociation under biologically relevant conditions including solutions containing phosphate and carbonate, ZnCl2, or acidic conditions. Solutions of the paraCEST agents in 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (pH 7.4) and 100 mM NaCl showed highly shifted and intense CEST peaks at 59, 72, and 92 ppm away from bulk water for [Co(L)]2+, [Ni(L)]2+, and [Fe(L)]2+, respectively at 37 °C on a 11.7 T NMR spectrometer. CEST spectra with corresponding rate constants for proton exchange are reported in 4% agarose gel (w/w), rabbit serum, egg white, or buffered solutions. CEST phantoms of 4 mM complex in buffer, 4% agarose gel (w/w), or rabbit serum on a 4.7 T MRI scanner at 37 °C, are compared. The most substantial change was observed for the reactive [Ni(L)]2+, which showed reduced CEST contrast in rabbit serum and egg white. The complexes with the least highly shifted CEST peaks ([Co(L)]2+ and [Ni(L)]2+) showed a reduction in CEST contrast in 4% agarose gel (w/w) compared to that in buffered solutions, while the CEST effect for [Fe(L)]2+ in 4% agarose gel (w/w) was not substantially different.
Short abstract
The crystal structures, solution chemistry, and CEST properties of FeII, CoII, and NiII complexes of 7,13-bis(carbamoylmethyl)-1,4,10-trioxa-7,13-diazacyclopentadecane ligand (L) in rabbit serum, 4% agarose, egg white, and buffer solutions are compared. The [Ni(L)]2+ complex is the most reactive of all the complexes in serum and egg white, corresponding to the misfit of the NiII ion in the macrocyclic ligand cavity.
Introduction
Divalent first-row transition metal ion complexes have great potential for development as paramagnetic chemical exchange saturation transfer (paraCEST) contrast agents for magnetic resonance imaging (MRI). ParaCEST agents contain protons (−NH, −OH, or bound H2O) that exchange with bulk water protons. Exchange of the protons must be sufficiently slow to produce two independent pools of protons on the NMR time scale, a pool of bulk water protons, and a pool of contrast-agent protons. Selective irradiation with a presaturation pulse at the resonant frequency of the exchangeable protons on the contrast agent gives rise to a decrease in the water proton signal.1−3 Certain transition metal ions including FeII, CoII, or NiII have paramagnetic properties that are generally well-suited for their application as paraCEST agents.4−6 These metal ion complexes may produce relatively narrow and highly shifted proton resonances through interaction with the paramagnetic center. These hyperfine proton shifts are attributed to both contact (through-bond) and pseudocontact (through-space) contributions.4−6 Contact contributions to the paramagnetic proton shift in transition metal ion complexes may be quite substantial. For example, the magnetically inequivalent protons in amide pendent groups bound to paramagnetic FeII, CoII, and NiII centers are shifted far apart (≥54 ppm), signifying substantial through-bond contributions (labeled as NHa and NHb in Scheme 1).7−10 In addition to amides, there is a wide selection of suitable donor groups for transition metal ions that contain exchangeable NH or OH protons including amides, alcohols, pyridines, imidazoles, and pyrazoles.3,11−13 Each of these donor groups have distinct proton exchange rate constants (kex) and proton chemical shifts versus bulk water (Δω) that may be optimized for pH and biological conditions. Despite the initial success of these agents, studies directed toward realizing in vivo applications are lacking.
Scheme 1. Structure of the ParaCEST Agents [M(L)]2+ Where M = FeII or CoII.

CoII, NiII, and FeII amide-appended macrocyclic complexes were recently shown to be paraCEST agents at physiological pH and temperature.7,9,10,14 Amide-appended transition metal paraCEST agents based on various macrocyclic backbones including triaaza-, tetraaza-, and mixed aza–oxa frameworks have different geometries, paramagnetically shifted proton resonances, and numbers of inequivalent amide NH protons. Notably, paraCEST agents containing CoII or NiII complexes of the 7,13-bis(carbamoylmethyl)-1,4,10-trioxa-7,13-diazacyclopentadecane (L) ligand (Scheme 1) exhibited the most intense CEST image and lowest T1 relaxivities in experiments on a 4.7 T MRI scanner, despite the fact that the complexes of L contain a relatively low number of amide NH protons in comparison to other complexes in the study. It was speculated that their low T1 water relaxivity was an important contributing factor to the efficacy of these complexes as paraCEST agents, because T1 relaxation is a competing pathway for CEST contrast.3 Another important consideration is the electronic relaxation time constants of the metal ion, which influences the efficiency of proton relaxation by the paramagnetic center.5,6 These electronic relaxation time constants vary for different coordination environments of the transition metal ion, especially for NiII.4,15−18 The macrocyclic ligand L is of interest because it has seven donor atoms and may potentially form seven-coordinate complexes with first row divalent transition metal ions. Although seven-coordinate complexes of these metal ions are not commonly found in literature, seven-coordinate complexes of first-row transition metals with the related aminobenzyl- appended 1,10-diaza-15-crown-5 has been reported.19,20
Studies of transition metal ion-based paraCEST agents in biological media to date are scarce but have recently been reported for a CoII agent.13 Such studies are important for the identification of interactions that may interfere with CEST contrast. For example, binding of the complexes to macromolecules might modulate the CEST effect.2 Blood serum contains high concentrations of the protein albumin and is also redox buffered by cysteine/cysteine and pH buffered by carbonate.21,22 Any of these components may react with the paraCEST agent. Furthermore, macromolecules in tissue contribute to the magnetization transfer (MT) effect, which gives rise to a broadened peak centered at the bulk water resonance, which spans tens of kilohertz.2,3,23 The MT effect is expected to reduce CEST contrast for complexes that produce CEST peaks within the MT frequency range. Hence it is of interest to develop paraCEST agents with highly shifted exchangeable protons compared to bulk water. Increasing the frequency difference (Δω) between the bulk water resonance and the contrast agent resonance through modification of the ligands or paramagnetic metal ion may serve to minimize MT interference of the CEST effect.2,24
Here, we compare the structures, solution chemistry, CEST properties, and propensity toward dissociation of the [Co(L)]2+, [Fe(L)]2+, and [Ni(L)]2+ complexes. These paraCEST agents are studied in the presence of biologically relevant ions such as phosphate, carbonate, ZnII, or acidic conditions. CEST experiments in different media including 4% agarose gel (w/w), egg white, and rabbit serum are presented to assess the suitability of these complexes for future in vivo studies. This study is, to the best of our knowledge, the first structural comparison of paraCEST agents containing all three of the first-row transition metal ions, FeII, CoII, and NiII.
Experimental Section
General Instrumentation
Evans measurements of magnetic susceptibility, CEST data, and 1H NMR spectra were acquired on a Varian Inova 500 MHz spectrometer. Thermo Finnigan LCQ Advantage Ion Trap LC/MS equipped with a Surveyor HPLC system was used to collect mass spectral data. All pH measurements were obtained by using an Orion 8115BNUWP Ross Ultra Semi Micro pH electrode connected to a 702 SM Titrino pH meter.
Material
Ni(CF3SO3)2 and Fe(CF3SO3)2 were purchased from Strem Chemicals, and CoCl2·6H2O was purchased from Alfa Aesar. Rabbit serum and albumin from porcine serum were purchased from Sigma-Aldrich. Chicken egg white was used, and agarose LF pulse field application PRFG grade was purchased from Amresco.
Synthesis of Complexes
L was prepared using a previously reported procedure.7 Metal salts were complexed to L in equimolar ratio in either ethanol (CoII) or acetonitrile (NiII), stirred at room temp for over 1 h, and isolated as previously reported.7,10 Fe(CF3SO3)2 and L were added under argon to prevent the oxidation of the free FeII to FeIII in an acetonitrile solution. Fe(CF3SO3)2 (0.47 mmol) and ligand (0.47 mmol) were placed in a two-neck round-bottom flask equipped with rubber septa and a magnetic stir bar. A syringe was used for the addition of acetonitrile (5 mL), and the solution was stirred at room temp for over 1 h. The solvent was removed, and the precipitate was dried under vacuum. [Fe(L)]2+ was isolated as a slightly yellowish tan powder. Yield: 63%. Electrospray ionization-mass spectroscopy (ESI-MS): m/z: 194.3 [M/2]+, 387.2 [M–H]+, 537.0 [M–CF3SO3–]+. Solutions of the complexes were standardized versus 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt by using proton NMR spectroscopy.
Preparation of 4% Agarose Gel (w/w)
A slightly modified procedure was used to prepare the 4% agarose gel (w/w) from that reported previously.25 In a 125 mL flask, agarose powder (2.002 g) was added to water (48.048 mL) and stirred at room temperature. The cloudy solution was allowed to boil for 10 min until the solution became clear. The clear solution was weighed, and hot distilled water was added to adjust the solution back to the original mass to compensate for evaporation.
Determination of Magnetic Moment
The effective magnetic moment (μeff) was calculated by using the Evans method (Supporting Information, eqs S1 and S2).26,27 Samples contained 3–5 mM complex and 5% t-butanol by volume in an NMR insert, while the outer NMR tube contained 5% by volume t-butanol in D2O. Evans measurements of magnetic susceptibility were acquired at 298 K (T). In buffered solutions, the calculated magnetic moments for all complexes remained constant over a period of 72 h. Magnetic moments were also measured in the presence or absence of porcine serum albumin or rabbit serum for NiCl2 and for [Ni(L)]2+.
Dissociation of Complexes
Complexes incubated with biologically relevant ions or under acidic conditions were monitored via 1H NMR spectroscopy. For experiments done under acidic conditions, solutions contained 9.8–10 mM complex, 100 mM NaCl, and 3–5 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt as a standard at pD 3.9–4.3. For studies with competing ions, samples contained 10 mM complex, 100 mM NaCl, 0.40 mM Na2HPO4, 25 mM K2CO3, and 1–5 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt as a standard with a pD of 7.5–8.0 or 10 mM complex, 10 mM ZnCl2, 100 mM NaCl, and 5 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt as a standard at pD 6.9–7. All samples were incubated at 37 °C and monitored over a 12 h period.
CEST Experiments
CEST data were acquired with a presaturation pulse power (B1) of 1000 Hz (24 μT) applied for 2 s at 37 °C. Data were acquired in 1 ppm increments and plotted as normalized water signal intensity (Mz/Mo%) against frequency offset (ppm) to produce a CEST spectrum. For CEST in a buffered medium, NMR inserts contained 10 mM complex, 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), and 100 mM NaCl. For experiments in biological media, NMR inserts contained 10 mM complex in either rabbit serum or egg white. The pH values of rabbit serum and egg white samples were adjusted with a dilute solution of HCl. To lock the sample, d6-dimethyl sulfoxide (d6-DMSO) was placed in the outer NMR tube. Agarose samples were prepared by diluting a 0.50 mL sample of 20 mM complex, 40 mM HEPES, and 200 mM NaCl in 0.50 mL of 4% agarose gel (w/w). The solutions were mixed and transferred to NMR tubes placed in a warm water bath (80–90 °C). Samples were allowed to settle in the bath for a few seconds to prevent formation of air bubbles. An NMR insert containing d6-DMSO was placed in the NMR tube to serve as a lock.
Determination of Exchange Rate Constants
The kex values were calculated following a previously reported procedure.28 The magnetization on-resonance (Mz) and off-resonance (Mo) values were acquired at different presaturation pulse powers between 350 and 1000 Hz (8–24 μT) applied for 4 s at 37 °C. The kex value is calculated from the x-intercept (−1/ kex2) from the plot of Mz/(Mo – Mz) against 1/ω12 (ω1 in rad/s). The average kex and standard deviation (≥4 experiments) were calculated using linear regression lines obtained from Microsoft Excel plots, with correlation coefficients of r2 ≥ 0.990. Samples contained 10 mM complex, 20 mM HEPES, and 100 mM NaCl, 10 mM complex in rabbit serum, 10 mM complex in egg white or 10 mM complex, 20 mM HEPES, and 100 mM NaCl in 4% agarose gel (w/w).
ParaCEST Imaging
CEST MR images were acquired at 4.7 T by using a 35 mm transceiver coil (ParaVision 3.0.2, BrukerBiospin, Billerica, MA) as detailed elsewhere.9 Two spoiled gradient-echo images (echo time/repetition time = 2.1/5010 ms, flip angle = 90 deg) were acquired at 37 °C after employing a pulse train composed of five Gauss pulses (12 μT for 1 s each, interpulse delay of 200 μs) applied symmetrically about the bulk water resonance ([Co(L)]2+: ± 59 ppm, [Ni(L)]2+: ± 72 ppm, and [Fe(L)]2+: ± 92 ppm).
| 1 |
Image processing was carried out using in-house software algorithms developed in MATLAB (MathWorks, Natick, MA). Each image was normalized to the mean intensity of the buffer/salt phantom, and the mean signal intensity of each compound was sampled. The percent change in signal, or CEST effect, was calculated with eq 1, where SIon and SIoff represent the mean signal intensity of each sample with the presaturation pulse applied on- and off-resonance of the exchangeable protons, respectively. CEST images were calculated by determining the CEST effect on a pixel-by-pixel basis in MATLAB. To increase the signal-to-noise and decrease spatial variability within each sample, raw data sets were zero-filled to a 512 × 512 matix, a two-dimensional Gaussian windowing function (σ = matrix size)29 was applied to the raw data in the frequency domain prior to Fourier transform into the spatial domain. Noise was removed using a binary mask of the sample tubes, and a “hot iron” color lookup table was applied.
T1/T2 Relaxivity
Using serial dilutions, T1/T2 relaxivity values were determined at 4.7 T and 37 °C, as previously described.9T1 relaxation rates were measured using an inversion–recovery TrueFISP acquisition, while T2 relaxation rates were measured using a multiecho, Carr–Purcell–Meiboom–Gill spin–echo sequence with a fixed TR of 3000 ms and TE times ranging from 20 to 1200 ms. Nonlinear regression analysis in MATLAB was used to calculate the T1 and T2 relaxation rates, and relaxivities were then determined by linear regression fitting of the concentration versus T1/T2 rate in Microsoft Excel.
X-ray Diffraction Data
Single crystals of [Fe(L)](CF3SO3)2, [Co(L)]Cl2·2H2O, and [Ni(L)](CF3SO3)2·H2O were grown over several days by vapor diffusion. Milligram quantities of [Fe(L)](CF3SO3)2 and [Ni(L)](CF3SO3)2 were each dissolved in a vial containing acetonitrile and placed in a larger vial with a solution of hexane. Crystals of [Co(L)]Cl2·2H2O were obtained using methanol as the mother liquor. Suitable crystals were selected and mounted on glass fibers with oil on a Bruker SMART APEX2 CCD diffractometer installed at a rotating anode source (Mo Kα radiation, λ = 0.710 73 Å). The crystals were kept at 90(2) K during data collection using an Oxford Cryosystems nitrogen gas-flow apparatus.
For compounds [Fe(L)](CF3SO3)2, [Co(L)]Cl2·2H2O, and [Ni(L)](CF3SO3)2·H2O, the data were collected by the rotation method with 0.5° frame width (ω scan) and 15, 3, and 30 s exposure times per frame, respectively. Three sets of data (360 frames in each set) were collected for [Fe(L)](CF3SO3)2, and five sets (360 frames in each set) were collected for [Co(L)]Cl2·2H2O, and [Ni(L)](CF3SO3)2·H2O nominally covering complete reciprocal space. The structures were solved with the olex2.solve structure solution program using the Charge Flipping method and refined with the ShelXL refinement package using Least Squares minimization.30,31 The structures were refined by full-matrix least-squares against F2.
Results
FeII, CoII, and NiII complexes of L are highly soluble and air-stable in aqueous solution over a period of several days. However, differences in the reactivity of the CoII and NiII complexes under more stringent conditions such as in solutions containing high concentrations of phosphate, carbonate, or competing cations such as ZnCl2 suggested that there were substantive differences in the coordination spheres of these complexes.7,10 To better understand the coordination chemistry of these complexes, their structures were characterized by using X-ray crystallography.
Crystal Structures
X-ray diffraction data for [Co(L)]Cl2·2H2O and [Fe(L)](CF3SO3)2 (Table 1) indicate that the cobalt(II) complex crystallizes to give a tetragonal unit cell with a space group of P41212, while the iron(II) complex crystallizes to give a monoclinic unit cell with a space group of P21/c. [Co(L)]2+ and [Fe(L)]2+ are both seven-coordinate complexes, with all nitrogen and oxygen donor atoms of the 1,10-diaza-15-crown-5 macrocycle and amide pendent groups bound to the metal ion. Each metal ion binds the carbonyl oxygen of the amide pendent in axial position with the five macrocyclic backbone donors in a planar arrangement to produce a distorted pentagonal bipyramidal geometry (Figures 1 and 2 and Supporting Information, Figure S1). The [Co(L)]2+ cation has an axis of symmetry that gives rise to three types of Co–O bonds and one type of Co–N bond with bond lengths of 2.2288(14), 2.2595(19), 2.800(13), and 2.0631(12) Å for Co1–N1, Co1–O1, Co1–O2, and Co1–O3, respectively. In the [Fe(L)]2+complex, the distances between the FeII and the oxygen donor atoms in the macrocyclic backbone range from 2.198 to 2.295 Å with FeII amine (Fe–N) bond distances of 2.29 Å (Table 2). The amide oxygens bind to both CoII and FeII with shorter bond lengths than those in the macrocycle backbone (2.06–2.09 Å). The bond angle for trans pendent groups O31–Co1–O3 and O5–Fe1–O4 are 176.09(8) and 167.38(4) degrees, respectively.
Table 1. Crystal Data, Collection, and Structure Refinement Parameters for [Fe(L)](CF3SO3)2, [Co(L)]Cl2·2H2O, and [Ni(L)](CF3SO3)2·H2O.
| [Fe(L)](CF3SO3)2 | [Co(L)]Cl2·2H2O | [Ni(L)](CF3O3)2·H2O | |
|---|---|---|---|
| empirical formula | C16H28N4O11F6S2Fe | C14H32N4O7Cl2Co | C16H30N4O12F6S2Ni |
| formula weight | 686.39 | 498.25 | 707.23 |
| crystal system | monoclinic | tetragonal | monoclinic |
| space group | P21/c (No. 14) | P41212 (No. 92) | C2/c (No. 15) |
| crystal size (mm3) | 0.1 × 0.08 × 0.04 | 0.3 × 0.1 × 0.1 | 0.1 × 0.05 × 0.02 |
| temperature (K) | 90 | 90 | 90 |
| a (Å) | 14.7576(6) | 7.3537(4) | 11.6016(6) |
| b (Å) | 9.6299(4) | 7.3537(4) | 23.6949(11) |
| c (Å) | 18.3995(9) | 38.8642(18) | 20.8143(10) |
| α (deg) | 90 | 90 | 90 |
| β (deg) | 92.5065(13) | 90 | 95.4010(14) |
| γ (deg) | 90 | 90 | 90 |
| V (Å3) | 2612.3(2) | 2101.66(19) | 5696.4(5) |
| Z | 4 | 4 | 8 |
| ρcalc (g cm–3) | 1.74 | 1.57 | 1.65 |
| μ (mm–1) | 0.841 | 1.114 | 0.929 |
| F000 | 1408.0 | 1044.0 | 2912.0 |
| R1_obs | 0.032 | 0.028 | 0.037 |
| R1_all | 0.048 | 0.030 | 0.057 |
| wR2_obs | 0.070 | 0.064 | 0.088 |
| wR2_all | 0.077 | 0.064 | 0.097 |
| goodness-of-fit | 1.013 | 1.060 | 1.020 |
Figure 1.
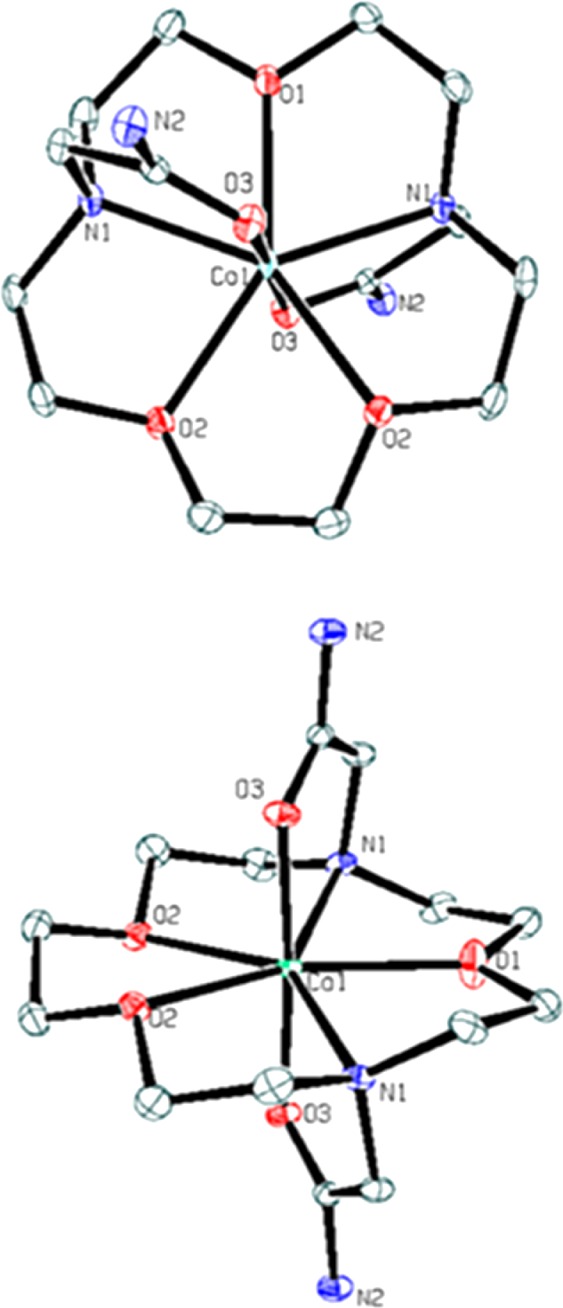
X-ray crystal structure of the complex cation of [Co(L)]Cl2·2H2O. For clarity, the hydrogen atoms, solvent, and counterions were omitted in the structure. Ellipsoids were set at 50%.
Figure 2.
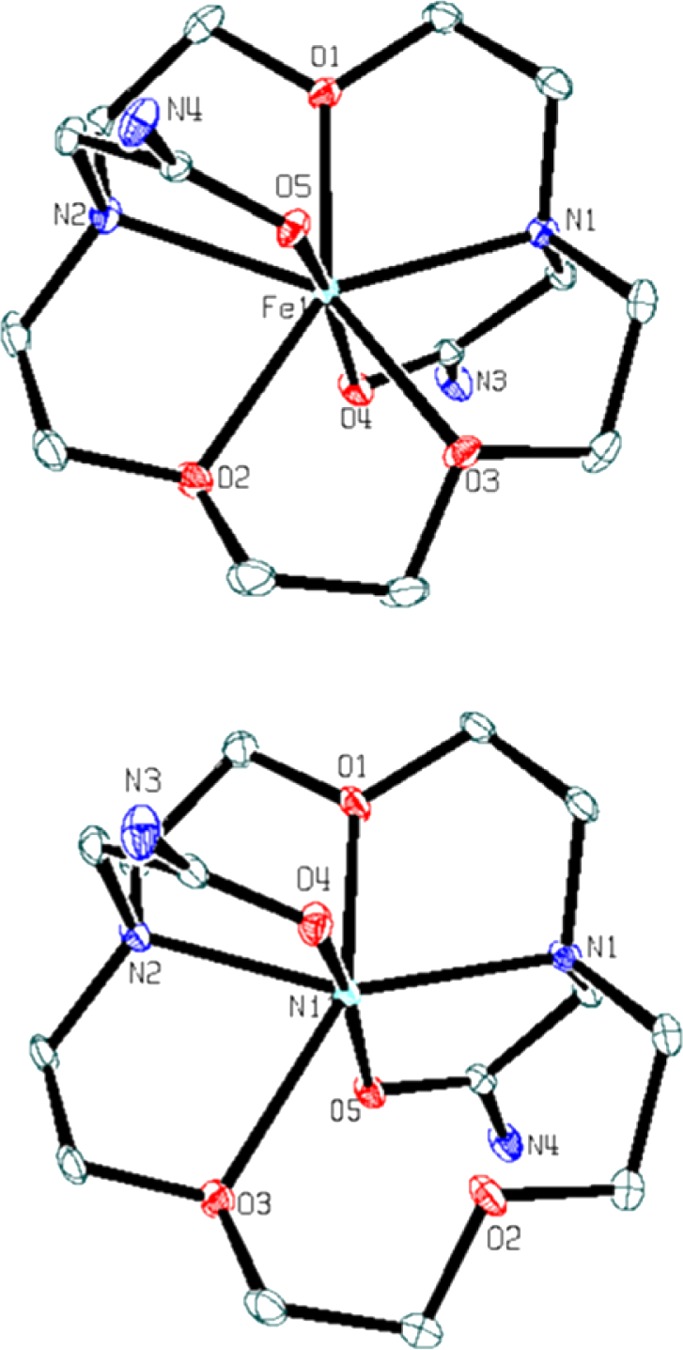
ORTEP plot of the complex cations of [Fe(L)](CF3SO3)2 (top) and [Ni(L)](CF3SO3)2·H2O (bottom). Hydrogen atoms, solvent, and counterions are omitted for clarity. Ellipsoids were set at 50%.
Table 2. Selected Bond Lengths (Å) and Bond Angles (deg) for [Fe(L)](CF3SO3)2, [Co(L)]Cl2·2H2O, and [Ni(L)](CF3SO3)2·H2O.
| [Fe(L)](CF3SO3)2 | [Co(L)]Cl2·2H2O | [Ni(L)](CF3O3)2·H2O | |||
|---|---|---|---|---|---|
| Bond Lengths | |||||
| M1–O1 | 2.1978(9) | M1–O1 | 2.2595(19) | M1–O1 | 2.0421(12) |
| M1–O2 | 2.2952(9) | M1–O2 | 2.2800(13) | M1–O2 | 2.6000(12) |
| M1–O3 | 2.2690(9) | M1–O2a | 2.2799(13) | M1–O3 | 2.4006(12) |
| M1–O4 | 2.0938(9) | M1–O3 | 2.0631(12) | M1–O4 | 2.0042(12) |
| M1–O5 | 2.0866(9) | M1–O3a | 2.0631(12) | M1–O5 | 1.9980(12) |
| M1–N1 | 2.2873(11) | M1–N1 | 2.2288(14) | M1–N1 | 2.1653(14) |
| M1–N2 | 2.2896(11) | M1–N1a | 2.2287(14) | M1–N2 | 2.1381(14) |
| Bond Angles | |||||
| O1–Fe1–O2 | 144.15(4) | O1–Co1–O2 | 144.69(3) | O1–Ni1–O3 | 149.94(5) |
| O1–Fe1–O3 | 145.01(3) | O1–Co1–O2a | 144.70(3) | O1–Ni1–N2 | 76.34(5) |
| O1–Fe1–N1 | 71.18(4) | O2a–Co1–O2 | 70.61(6) | O1–Ni1–N1 | 76.29(5) |
| O1–Fe1–N2 | 72.28(4) | O3–Co1–O1 | 91.96(4) | O4–Ni1–O3 | 88.47(5) |
| O3–Fe1–O2 | 70.78(2) | O3–Co1–O2 | 83.91(5) | O4–Ni1–O1 | 96.69(5) |
| O3–Fe1–N1 | 75.17(4) | O3–Co1–O2a | 92.89(5) | O4–Ni1–N2 | 82.77(5) |
| O3–Fe1–N2 | 140.16(4) | O3–Co1–N1 | 101.36(5) | O4–Ni1–N1 | 102.01(5) |
| O4–Fe1–O1 | 92.48(4) | O3a–Co1–O2 | 92.88(5) | O5–Ni1–O4 | 167.87(5) |
| O4–Fe1–O2 | 84.02(4) | O3a–Co1–O2a | 83.91(5) | O5–Ni1–O3 | 80.53(4) |
| O4–Fe1–O3 | 89.25(4) | O3a–Co1–O3 | 176.09(8) | O5–Ni1–O1 | 95.40(5) |
| O4–Fe1–N1 | 77.29(4) | O3a–Co1–N1 | 79.97(5) | O5–Ni1–N2 | 99.13(5) |
| O4–Fe1–N2 | 106.11(4) | O3a–Co1–N1a | 101.36(5) | O5–Ni1–N1 | 81.87(5) |
| O5–Fe1–O1 | 100.13(4) | N1–Co1–O1 | 70.60(4) | N1–Ni1–O3 | 131.63(5) |
| O5–Fe1–O2 | 85.39(4) | N1–Co1–O2 | 75.88(5) | N2–Ni1–O3 | 74.97(5) |
| O5–Fe1–O3 | 80.77(3) | N1–Co1–O2a | 141.82(5) | N2–Ni1–N1 | 152.58(5) |
| O5–Fe1–O4 | 167.38(4) | N1–Co1–N1a | 141.19(8) | ||
| O5–Fe1–N1 | 107.25(4) | N1a–Co1–O1 | 70.64(4) | ||
| O5–Fe1–N2 | 77.52(4) | N1a–Co1–O2 | 141.82(5) | ||
| N1–Fe1–O2 | 141.14(4) | N1a–Co1–O2a | 75.88(5) | ||
| N1–Fe1–N2 | 143.41(4) | ||||
| N2–Fe1–O2 | 74.51(4) | ||||
+Y,+X,–1–Z.
[Ni(L)](CF3SO3)2·H2O crystallizes in a monoclinic unit cell with a space group C2/c. In contrast to the other two complexes, the [Ni(L)]2+ complex cation has a six-coordinate NiII center. The NiII ion is bound to the N1, N2, O3, and O1 of the macrocycle and O4 and O5 of the amide pendent (Figure 2 and Table 2). The pendent groups are oriented in trans configuration to give a distorted octahedral geometry. [Ni(L)]2+ has the shortest metal-to-oxygen bond lengths of 1.9980(12) and 2.0042(12) Å for the amide pendent groups. The distance between the NiII ion and the O2 is 2.600(12) Å, too long for bond formation.
Crystallographic data, atomic coordinates, equivalent isotropic displacement parameters, anisotropic displacement parameters, bond lengths, bond angles, hydrogen atom coordinates, and isotropic displacement parameters for the complexes are compiled in the Supporting Information (Tables S1–S19) and in Tables 1 and 2.
Solution Chemistry
In D2O, the effective magnetic moments of [Ni(L)]2+, [Co(L)]2+, and [Fe(L)]2+ are 3.4, 4.1, and 5.9 μB at 25 °C as measured by using the Evans method (Supporting Information, eqs S1 and S2).26,27 These values are within the expected range for paramagnetic NiII, CoII, or FeII complexes, respectively.5,6 The paramagnetic complexes produce 12 narrow macrocyclic (CH) proton resonances. The 1H NMR resonances of the complexes range from −60 to 240 ppm with proton resonances at full width half-maximum (FWHM) of 150–400 Hz, 70–350 Hz, and 200–615 Hz for [Ni(L)]2+, [Co(L)]2+, and [Fe(L)]2+, respectively (Figure 3). Variable-temperature 1H NMR spectroscopy experiments on [Fe(L)]2+ (Supporting Information, Figure S2) show relatively little change in the peak widths of the proton resonances in the temperature range of 5 to 50 °C. This suggests dynamic processes do not have a large contribution to proton resonance line broadening over this temperature range.
Figure 3.

1H NMR spectra of (a) [Ni(L)]2+, (b) [Co(L)]2+, and (c) [Fe(L)]2+ in deuterium oxide.
In acetonitrile-d3, [Fe(L)]2+ exhibits 12 macrocyclic (CH) proton resonances and two sets of two inequivalent exchangeable amide (NH) proton resonances at 29 and 103 ppm (Figure 4). The resonances of the amide (NH) protons for [Ni(L)]2+ were observed at 16 and 82 pm in acetonitrile-d3, while the proton resonances of [Co(L)]2+ appeared at −10 and 72 ppm in d6-DMSO.7,10
Figure 4.

1H NMR spectrum of [Fe(L)]2+ in acetonitrile-d3. The peaks at 103 and 29 ppm are the exchangeable amide (NH) protons (*) of L.
Dissociation of the Complexes
Both the diamagnetic and paramagnetic regions of the 1H NMR spectra of the paraCEST agents were monitored to determine dissociation of metal ion (Supporting Information, Figures S3–S9). Under acidic conditions (pD 3.9–4.3), [Ni(L)]2+, [Co(L)]2+, and [Fe(L)]2+ dissociate by 18, 16, and 21%, respectively, over 12 h at 37 °C (Table 3 and Supporting Information, Figures S3 and S4). Samples incubated with equimolar concentrations of ZnCl2 for 12 h also show evidence of metal ion dissociation at 37 °C, pD 6.9–7.0 (Table 3 and Supporting Information, Figures S5–S8). The NiII complex is the most labile in the presence of ZnII and shows 54% dissociation. In contrast, samples containing [Co(L)]2+ or [Ni(L)]2+ incubated in phosphate (0.40 mM) and carbonate (25 mM) showed no detectable dissociation. Notably, changes in the paramagnetic region of the NMR spectrum of [Ni(L)]2+ in the presence of carbonate were observed and were attributed to coordination of carbonate to the intact NiII complex without inducing dissociation.7 In the presence of carbonate and phosphate anions, [Fe(L)]2+ dissociates by approximately 11% over 12 h (Supporting Information, Figure S9).
Table 3. Magnetic Moments and Dissociation of the Complexes in D2O.
| complex | μeffa | % dissociation acidicb | % dissociation anionsc | % dissociation Zn∥d |
|---|---|---|---|---|
| [Ni(L)]2+ | 3.4 | 18 ± 0.3 | 0 | 54 ± 0.6 |
| [Co(L)]2+ | 4.1 | 16 ± 10 | 0 | 13 ± 0.1 |
| [Fe(L)]2+ | 5.9 | 21 ± 0.1 | 11 | I5 ± 0.1 |
The effective magnetic moment in solution at 25 °C.
9.8–10 mM complex, 3–5 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt as standard, 100 mM NaCl in D2O pD 3.9–4.3.
10 mM complex, 100 mM NaCl, 1–3 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt, 0.4 mM Na2HPO4, and 25 mM K2CO3 in D2O pD 7.5–8.
10 mM complex, 100 mM NaCl, 5 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt, and 10 mM ZnCl2 in D2O pD 6.9–7.0. All samples were incubated for 12 h at 37 °C.
CEST Spectra in Biological Media
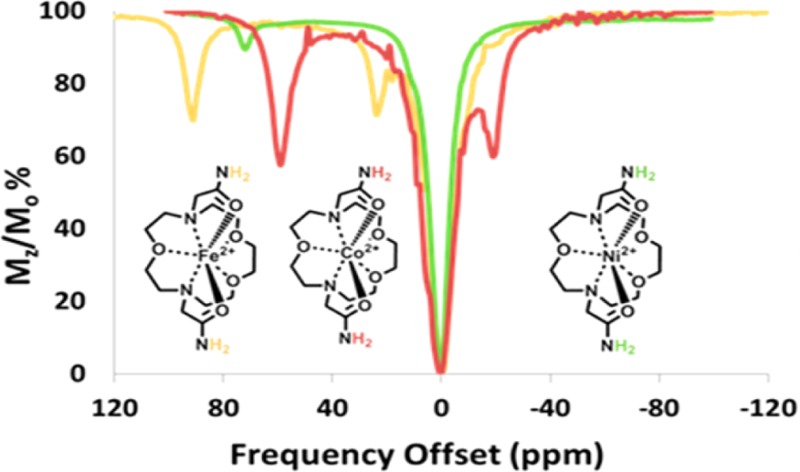
A CEST spectrum is the plot of the normalized water signal (Mz/Mo) against the offset frequency (ppm) of the presaturation pulse. CEST data were acquired in 1 ppm increments with a presaturation pulse (B1 = 24 μT) applied for 2 s at 37 °C. In aqueous media, two CEST peaks are observed for all three complexes, corresponding to two sets of two inequivalent amide protons on the same pendent. These amide protons are labeled NHa and NHb in Scheme 1. [Co(L)]2+and [Ni(L)]2+ CEST peaks are intense, with their most highly shifted peaks at 59 and 72 ppm from bulk water (Figure 5). The most highly shifted [Fe(L)]2+ CEST peak was located at 92 ppm versus bulk water (Figures 5 and 6). This CEST peak roughly corresponds to the exchangeable proton resonance identified at 103 ppm in acetonitrile-d3 versus the trimethylsilyl propanoic acid reference. The magnitude of the CEST effect for [Fe(L)]2+ is only 25 ± 3.0% compared to 38 ± 0.3 or 39 ± 0.2% of the [Co(L)]2+and the [Ni(L)]2+ at pH 7.4, respectively (Figures 5 and 8). The magnitude of the CEST effect for all three complexes increases with pH over the pH range of 6.5 to 7.7, consistent with base-catalyzed proton exchange (Figure 6 and Supporting Information, Figure S10).10
Figure 5.
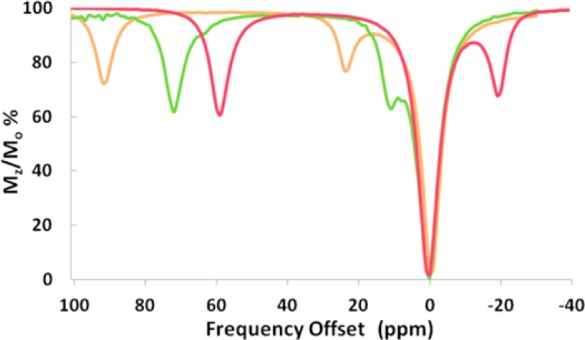
Overlaid CEST spectra of 10 mM complex ([Co(L)]2+(red), [Ni(L)]2+(green), [Fe(L)]2+(light brown)), 100 mM NaCl, 20 mM HEPES, pH 7.4. Radio frequency presaturation pulse applied for 2 s, B1 = 24 μT at 37 °C.
Figure 6.
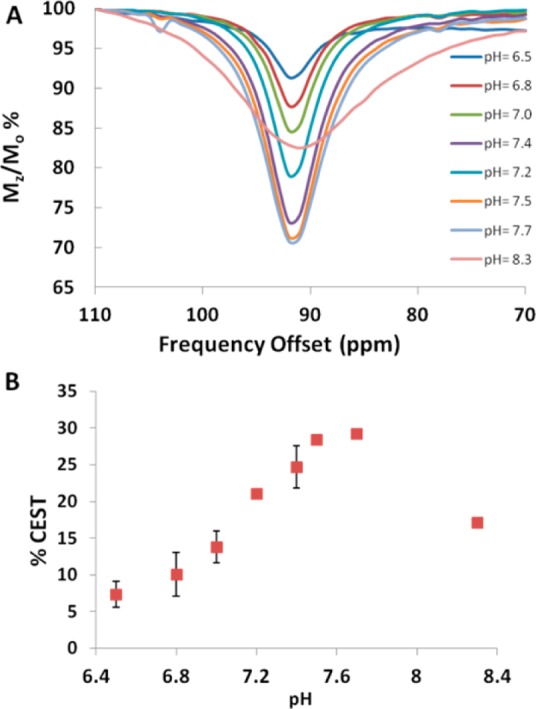
The pH dependence of the magnitude of the CEST peak at 11.7 T of solutions containing (A) 10 mM [Fe(L)]2+, 20 mM buffer pH 6.5–8.3, and 100 mM NaCl. (B) Plot of the CEST effect for 10 mM [Fe(L)]2+, 20 mM buffer pH 6.5–8.3, and 100 mM NaCl. The radio frequency presaturation pulse was applied for 2 s, B1 = 24 μT at 37 °C. Error bars represent standard deviations and are measured for all points.
Figure 8.
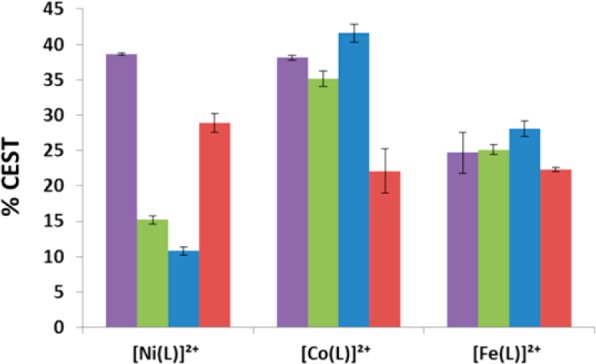
CEST effect of 10 mM complex in 100 M NaCl, 20 mM HEPES (purple), egg white (green), 100 mM NaCl, 20 mM HEPES in 4% agarose gel (w/w) (red), and rabbit serum (blue) with a radio frequency presaturation pulse applied for 2 s, B1 =24 μT at 37 °C, pH 7.3–7.5.
Rate constants for amide proton exchange were determined by using the Omega plot method.28 At 37 °C, the kex of the furthest shifted amide (NH) protons of 10 mM [Ni(L)]2+, [Co(L)]2+, or [Fe(L)]2+ in 20 mM HEPES pH 7.4 and 100 mM NaCl were 240, 240, and 500 s–1, respectively. At the more basic pH value of 8.3, the larger rate constant leads to exchange broadening and a decrease of the CEST signal for the FeII complex (Figure 6). Similar trends were observed for [Ni(L)]2+ (Supporting Information, Figure S10) and [Co(L)]2+.10
The CEST peak intensity and kex values for the complexes were monitored in rabbit serum, egg white, or 4% agarose gel (w/w) and compared to those in buffered aqueous solution (Figures 7 and 8, Table 4, and Supporting Information, Figures S11–13). [Co(L)]2+ and [Fe(L)]2+ in egg white had a CEST effect of 35 ± 1.1 and 25 ± 0.7% at pH 7.3, similar to values in solutions containing only buffer and NaCl. In rabbit serum, the CEST effect increased very slightly for both [Fe(L)]2+ and [Co(L)]2+ at 37 °C (Figures 7 and 8). This corresponds to an increase of the rate constants for exchange of the amide proton of [Co(L)]2+ and [Fe(L)]2+ in egg white to 1600 and 630 s–1, respectively, at 37 °C. [Ni(L)]2+ showed a large decrease in the CEST effect both in egg white (15 ± 0.6%) and in rabbit serum (11 ± 0.6%) despite faster amide (NH) proton exchange rates (Table 4 and Supporting Information, Figure S11).
Figure 7.
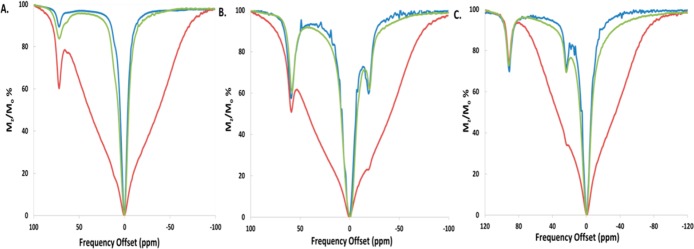
CEST spectra (11.7 T) of solutions containing (A) 10 mM [Ni(L)]2+ in rabbit serum at pH 7.3 (blue), 100 mM NaCl, 20 mM HEPES in 4% agarose gel (w/w) (red), and egg white pH 7.5 (green). (B) 10 mM [Co(L)]2+ in rabbit serum pH 7.5 (blue), 100 mM NaCl, 20 mM HEPES in 4% agarose gel (w/w) (red), and in egg white pH 7.3 (green). (C) 10 mM [Fe(L)]2+ in rabbit serum pH 7.4 (blue), 100 mM NaCl, 20 mM HEPES in 4% agarose gel (w/w) (red), and egg white pH 7.3 (green). Radio frequency presaturation pulse applied for 2 s, B1 = 24 μT at 37 °C.
Table 4. Proton Exchange Rate Constants of ParaCEST Agents in Different Media.
| complex | buffera (s–1) | rabbit seruma (s–1) | egg whitea (s–1) | agarosea (s–1) |
|---|---|---|---|---|
| [Co(L)]2+ | 240 ± 70 | 1600 ± 650 | 860 ± 160 | 850 ± 70 |
| [Ni(L)]2+ | 240 ± 20 | 3200 ± 300 | 1700 ± 400 | 890 ± 190 |
| [Fe(L)]2+ | 500 ± 50 | 630 ± 200 | 760 ± 180 | 520 ± 30 |
Exchange rate constants obtained for solutions containing 10 mM complex in 20 mM HEPES pH 7.3–7.4 and 100 mM NaCl, rabbit serum, egg white or 4% agarose gel (w/w) at 11.7 T. B1 varied between 8 and 24 μT, with an radio frequency presaturation pulse applied for 4 s at 37 °C.
To further probe the identity of the NiII species, the magnetic moment of solutions containing 10 mM [Ni(L)]2+ in rabbit serum or albumin was measured and compared to analogous solutions containing NiII salts by using the Evans method. The magnetic moments of the [Ni(L)]2+ complex in serum (3.1 μB) decreased slightly compared to that of the complex in buffered solution (3.4 μB). Previous measurements on free NiII ion in albumin and in serum were consistent with a diamagnetic NiII complex.32 This comparison suggests that the [Ni(L)]2+ complex interacts with serum proteins to give partial release of NiII ion.
CEST spectra of samples in 4% agarose gel (w/w) exhibited a large MT effect between −80 to +80 ppm (Figure 7). The CEST effect was calculated by taking the difference in percent reduction in water signal (Mz/Mo%) at two frequencies symmetrical about the water resonance. For example, the difference in the Mz/Mo% values at +59 ppm and −59 ppm was used to determine the 22 ± 3.1% CEST effect of [Co(L)]2+ at 11.7 T (Figure 8). Signals that fell within the MT effect such as those of [Co(L)]2+ and [Ni(L)]2+ had a reduced CEST effect. The magnitude of the CEST effect for [Fe(L)]2+ was not affected by the MT effect produced by the 4% agarose gel (w/w). This is attributed to the large Δω of the FeII complex, which places the CEST peak outside of the MT band. The exchange rate constants for the amide protons of the complexes as determined by Omega plots increased slightly in agarose at 37 °C for both the CoII and NiII complexes (Supporting Information, Figure S13).28
Phantom images of the three complexes (4 mM) in 100 mM NaCl, 20 mM HEPES, pH 7.3–7.5 at 37 °C on a 4.7 T MRI scanner produced a CEST contrast of 5.6–11% at a presaturation pulse power of 12 μT (Table 5 and Supporting Information, Figure S14). [Co(L)]2+ and [Ni(L)]2+ produced the largest CEST effects of 9.1 and 11%, respectively, while [Fe(L)]2+ exhibited a lower CEST effect of 5.6% (Supporting Information, Figure S14). In rabbit serum, [Ni(L)]2+ produced no discernible CEST signal, and the T1 and T2 relaxivities increased substantially to 0.526 and 0.784 mM–1 s–1, respectively. Both [Fe(L)]2+ and [Co(L)]2+ exhibited slightly lower CEST contrast in serum (Table 4 and Supporting Information, Figure S15). CEST contrast for [Co(L)]2+ and [Ni(L)]2+ in 4% agarose gel (w/w) exhibited a 50% decrease in their CEST effect compared to buffered samples (Figure 9 and Table 5), whereas [Fe(L)]2+ maintained a similar magnitude of CEST contrast in agarose and buffered solutions (Supporting Information, Figure S15).
Table 5. T1 Relaxivitiy, T2 Relaxivitiy, and CEST Contrast of Complexes in Different Media at 4.7 T.
| buffer | rabbit serum | agarose | ||||
|---|---|---|---|---|---|---|
| complex | Δωa | T1 relaxivityb | T2 relaxivityc | CESTd | CESTe | CESTf |
| (ppm) | (mM·s–1) | (mM·s–1) | (%) | (%) | (%) | |
| [Ni(L)]2+ | 72 | 0.012g,h | 0.092g,h | 11 ± 0.3 | 0 | 4.6 ± 1.6 |
| [Co(L)]2+ | 59 | 0.038g,h | 0.119g,h | 9.1 ± 2.7 | 5.4 ± 0.6 | 6.7 ± 0.5 |
| [Fe(L)]2+ | 92 | 0.097 | 0.203 | 5.6 ± 1.4 | 2.5 ± 1.1 | 5.4 ± 0.2 |
The chemical shift of the furthest downfield shifted amide (NH) exchangeable proton versus the water proton resonance.
T1 relaxivity for 0.25–8 mM paraCEST agent, 100 mM NaCl, 20 mM HEPES, pH 7.3–7.4.
T2 relaxivity for 0.25–8 mM paraCEST agent, 100 mM NaCl, 20 mM HEPES, pH 7.3–7.4.
% CEST of 4 mM paraCEST agent, 100 mM NaCl, 20 mM HEPES pH 7.3–7.4.
% CEST of 4 mM paraCEST agent rabbit serum pH 7.3–7.5.
% CEST of 4 mM paraCEST agent in 20 mM HEPES pH 7.3–7.4, 100 mM NaCl in 4% agarose gel (w/w). CEST images were acquired on a 4.7 T MRI scanner with B1 = 12 μT at 37 °C.
Figure 9.
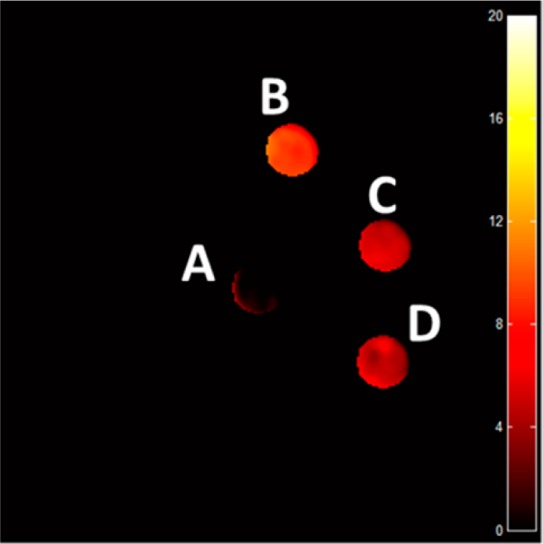
CEST images of phantoms on an MRI 4.7 T scanner with a pulse train composed of five Gauss pulses at 12 μT for 1 s each, interpulse delay of 200 μs applied symmetrically about the bulk water resonance (±59 ppm). Sample A consisted of 20 mM HEPES pH 7.4 and 100 mM NaCl. All other solutions contained 4 mM [Co(L)]2+ in (B) 20 mM HEPES pH 7.4 and 100 mM NaCl, (C) rabbit serum, (D) 20 mM HEPES pH 7.4 and 100 mM NaCl in 4% agarose gel (w/w) pH 7.3–7.4 at 37 °C.
Discussion
Structure of the Complexes
Seven-coordinate complexes of CoII and FeII are formed with the amide-appended macrocyclic ligand L. The complex cations [Co(L)]2+ and [Fe(L)]2+ have the metal ion bound to the five donor atoms of the 1,10-diaza-15-crown-5 macrocyclic backbone and also bound to the carbonyl oxygens of the pendent amides in trans configuration. The geometry that best describes the FeII and CoII complexes is distorted pentagonal bipyramidal. The two nitrogens and the three oxygens of the macrocycle ring form the pentagon base, and the pendent amide groups are in the apical positions. The bond angles between O5–Fe1–O4 (167.38°) and O31–Co1–O3 (176.09°) in these complexes are close to 180°. Similar geometries were observed for analogous complexes containing benzimidazol-2-ylmethyl and 2-aminobenzyl-appended 1,10-diaza-15-crown-5 bound to CoII and MnII.20
NiII coordinates to both amide pendent oxygens, two ring nitrogens, and only two ring oxygens to form a six-coordinate complex best described as having distorted octahedral geometry [Ni(L)]2+. The Ni–O2 distance is too long (2.600 Å) to be considered as a formal bond. In the NiII complex of the benzimidazol-2-ylmethyl-appended 1,10-diaza-15-crown-5 macrocycle, a similar trend was observed with a distance of 3.33 Å between the metal center to one of the oxygens in the ring. Interestingly, the NiII complex of the 2-aminobenzyl-appended analogue had two NiII–oxygen distances in the macrocycle ring that were long (≥2.44 Å).19,20 This shows that seven-coordinate complexes of NiII tend to be highly distorted for L or related macrocyclic ligands.
The 1H NMR spectra of the three complexes studied here are consistent with one predominant conformation in solution. The 12 relatively narrow macrocyclic (CH) proton resonances are consistent with a C2 axis of symmetry that bisects the O1 donor and runs between the equivalent O2 donor groups as viewed in the [Co(L)]2+ cation (Figures 1 and 3). In contrast, the [Ni(L)]2+ complex cation lacks a C2 axis of symmetry in the solid state, but still produces 12 proton resonances in solution. This suggests a dynamic process, perhaps involving the tandem ether oxygen donors, that maintains the higher level of symmetry of the complex in solution.
The NMR spectra of the complexes show proton resonances ranging from −60 to 240 ppm with fairly narrow peaks and FWHM peak widths in the range of 150–615 Hz. The proton resonances for the [Fe(L)]2+ complex were slightly broadened in comparison to the other complexes. The lack of apparent dynamic processes on the NMR spectroscopy time scale suggests that other factors may contribute, such as more efficient proton relaxation enhancement by the paramagnetic FeII center. In comparison, the [Co(L)]2+ complex has relatively narrow proton resonances. CoII complexes other than those that are tetrahedral generally have short electronic relaxation times and sharp proton resonances.6 However, it was unanticipated that the [Ni(L)]2+ complex would produce such narrow proton resonances. Many common geometries including octahedral NiII complexes are known to have broad proton resonances due to their large electronic relaxation time constants.18 The proton NMR spectra of all the other NiII macrocyclic complexes we studied as paraCEST agents showed very broad, overlapping proton resonances, unlike that of [Ni(L)]2+.7
All three complexes produce a similar range of proton resonances including 205 ppm for the NiII complex and approximately 250 ppm for the CoII and FeII complexes (Figure 3). These hyperfine proton shifts are due to a combination of dipolar and contact contributions, which cannot be easily separated without theoretical calculations. However, the magnitude of the hyperfine shifted proton resonances for the three complexes is similar despite the large differences in magnetic moments of the three complexes, which would seem to support differing contact contributions. It is also of interest that two distinct proton peaks for the inequivalent amide (NH) protons can be located for all of the complexes. The inequivalent amide proton peaks of the complexes are separated by 66, 74, and 82 ppm for [Ni(L)]2+, [Fe(L)]2+, and [Co(L)]2+, respectively. This large and nearly constant difference in chemical shift is attributed to a large paramagnetic contact shift contribution. Notably, paramagnetic lanthanide(III) ion complexes with amide pendent groups do not have such large separations in amide proton chemical shifts.3 Contact contributions to protons several bonds removed from the metal ion center are anticipated to be much smaller for LnIII ion complexes compared to those of transition metal ions. The two magnetically inequivalent protons on each complex give rise to two CEST peaks in solution as discussed below.
Dissociation of Complexes
Complexes to be used for in vivo imaging should exhibit minimal dissociation to prevent the accumulation of free metal ion in the body. The complexes studied here are relatively air-stable and inert to oxidation over a period of 24 h in buffered solution at 37 °C. Under more stringent conditions including high concentrations of phosphate and carbonate, which are typically found in blood serum, [Co(L)]2+ and [Ni(L)]2+ remain undissociated over 12 h, but [Fe(L)]2+ does dissociate to a small extent. Notably, under these conditions [Ni(L)]2+ reacts with carbonate, but the complex does not dissociate and still produces a CEST peak.7 However, in the presence of competing ZnII ion, [Ni(L)]2+ shows substantial dissociation. Of all three complexes, [Ni(L)]2+ is also the most reactive in serum and in albumin-rich egg white. The higher reactivity of the NiII complex is consistent with the long bond to one of the ether groups, which suggests that arrangement of donor groups in L is not optimal for coordination of NiII.
CEST Spectra and CEST Images
Each of the complexes produced two CEST peaks from the two inequivalent amide NH protons, one downfield and the other closer to bulk water. [Fe(L)]2+ produced the most highly downfield-shifted set of CEST peaks at 92 and 24 ppm, whereas [Co(L)]2+ produced the most upfield-shifted peaks at 59 and −19 ppm. The [Ni(L)]2+ CEST peaks were observed at 72 and 11 ppm. Further experiments and analysis are in regard to the most highly downfield-shifted CEST peaks of each complex.
The [Co(L)]2+ and [Ni(L)]2+ complexes produced the most intense CEST contrast in comparison to the [Fe(L)]2+ complex on both the 11.7 T NMR and the 4.7 T MRI scanner. There are two factors that are likely to contribute to this trend. First, the NiII and CoII complexes had relatively low T1 relaxivities at 37 °C and physiological pH (7.3–7.4) as shown in Table 5 in comparison to the FeII complex. The bulk water T1 value for the complex should preferably be low in an effective paraCEST agent because it corresponds to a competing pathway for the CEST effect.3 Alternatively, a larger ligand proton relaxation enhancement or dynamic process, which produces more extensive line broadening in the FeII complex, may contribute to the lowered CEST effect.
The three complexes have a similar pH-dependence of the CEST effect. Base-catalyzed proton exchange of the amide protons results in an increase in CEST between pH 6.5 and 7.7. At more basic pH values, the gradual decrease in CEST peak intensity is attributed to faster exchange, leading to exchange broadening for amide protons. Similar trends in the pH dependence of CEST are observed for other amide-appended paraCEST agents for both LnIII and transition metal ions.3,11 It is notable that at pH 7.4, the amide proton exchange rate constant for the FeII complex is 2-fold larger than that of the CoII or NiII complexes, corresponding to the lower Lewis acidity of FeII as an earlier transition metal ion. More strongly Lewis acidic metal ions would be anticipated to increase the N–C amide double bond character, leading to decreased amide proton acidity and lowered base-catalyzed proton exchange rate constants. Further solution characterization of the complexes by measurement of amide pKa values may be warranted to give insight into these differences.
To simulate the MT effect observed in vivo, samples were placed in 4% agarose gel (w/w). In the absence of agarose, the water peak appears symmetrical and spans the range from +5 to −5 ppm. In the presence of agarose, there is a broad peak centered at water that mimics the effect observed for macromolecules and aliphatic protons within the tissue.2,3,23 The MT band in our studies is quite broad, corresponding to the relatively high saturation pulse powers used in our experiments. In our studies, only the [Fe(L)]2+ complex, which has the most highly shifted peak at 92 ppm, was relatively unaffected by agarose. [Co(L)]2+ and, to a lesser extent, [Ni(L)]2+ gave reduced CEST effects for both phantom images on the 4.7 T MRI scanner and by CEST NMR in 4% agarose gel. This result is reminiscent of a recent study with two isomers of the LnIII −DOTAM-based paraCEST agent (DOTAM = 1,4,7,10-tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane) that were shown to produce CEST peaks at −68 ppm and −102 ppm in agar.23 The isomer with the less highly shifted peak at −68 ppm experienced greater interference from the MT effect and exhibited a reduced CEST signal. This highlights the importance of having the CEST peak shifted by ±90 ppm from bulk water to place it outside of the MT band.
For in vivo applications, complexes should tolerate biologically relevant molecules that may act as ligands. Human plasma contains many proteins that might bind to the metal ion complexes, thereby affecting their CEST properties. CEST experiments were conducted in different media that would simulate in vivo interactions. The three complexes were incubated in egg white or rabbit serum, and their CEST spectra and images were recorded. The [Ni(L)]2+ complex showed the largest decrease in the CEST effect in the presence of serum or egg white. Albumin is the major protein in egg white and human plasma. CEST experiments of [Ni(L)]2+ confirmed that the metal complex interacts with albumin (Supporting Information, Figure S16). Also consistent with this interpretation is the larger rate constant for proton exchange in the presence of both serum and egg white as well as the larger T1 and T2 relaxivities. This suggests that the NiII complex that produces the CEST peak is bound to protein. Further experiments that tracked NiII speciation by monitoring magnetic moments were consistent with a partial release of free NiII ion from the complex in solutions containing albumin or in serum. For the CoII and FeII complexes, the NMR experiments at 11.7 T showed little change in intensity of the CEST peak in egg white or in rabbit serum, within experimental error. The larger proton exchange rate constants for the CoII complex in serum led to some exchange broadening of the peak. On the MRI scanner at 4.7 T, both the CoII and FeII complexes show a slightly reduced CEST effect in serum, but values were almost within experimental error.
Conclusions
Our study of the solution and solid-state structures of a series of transition metal ion complexes highlights properties that are important for the design of effective paraCEST MRI contrast agents. Desirable properties of FeII, CoII, and NiII complexes of L include the production of relatively sharp and highly dispersed proton resonances. This demonstrates that the complexes are rigid and not highly fluxional on the NMR time scale, unlike most of the amide-appended transition metal ion paraCEST agents reported to date.11 The T1 relaxivity values for the CoII and NiII complexes are substantially lower than those for our previously reported complexes. Low relaxivity correlates to favorably short electronic relaxation time constants, narrow proton resonances, and potentially more intense CEST contrast. Many transition metal ions, NiII in particular, have electronic relaxation time constants that are highly dependent on geometry. The [Ni(L)]2+ complex is one of the few reported NiII macrocyclic complexes that has sharp proton resonances and correspondingly sharp and intense CEST peaks. Unfortunately, complexes of L are not as inert toward interaction with anions or acid compared to other complexes we have studied.7,10,13
CEST imaging studies in biological media are illustrative of the some of the difficulties that need to be overcome for the complexes to be useful in vivo. CEST imaging experiments in agarose show that MT interferes to some extent with the signal from the NiII and CoII complexes. As anticipated, the complex that has the most highly shifted CEST peak, [Fe(L)]2+, is the least affected by signal interference from MT. CEST spectra and images of the complexes in serum showed unexpected results, including the reactivity of the [Ni(L)]2+ complex in serum and with albumin to give a greatly reduced CEST effect. Future studies will focus on the design of complexes that combine the favorable paraCEST properties of the FeII, CoII, and NiII complexes of L with the requisite properties for successful MRI contrast agents in biological media.
Acknowledgments
J.R.M. and A.O.O. thank the Bruce Holm Catalyst Fund, the NIH (CA-173309) and NSF (CHE-1310374) for support. MR imaging was supported in part by Roswell Park’s NCI Support Grant (P30CA16056) and the Roswell Park Alliance Foundation (J.A.S.).
Supporting Information Available
Methods, NMR spectra, phantom images, additional crystal structures, and CEST spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Merbach A.; Helm L.; Tóth É.. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2 ed.; Wiley: Hoboken, NJ, 2013. [Google Scholar]
- Vinogradov E.; Sherry A. D.; Lenkinski R. E. J. Magn. Reson. 2013, 229, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S.; Kovacs Z.; Green K. N.; Ratnakar S. J.; Sherry A. D. Chem. Rev. 2010, 110, 2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.. NMR of Paramagnetic Molecules in Biological Systems; Benjamin/Cummings: San Francisco, CA, 1986. [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Pierattelli R. ChemBioChem 2005, 6, 1536. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Turano P.; Vila A. J. Chem. Rev. 1993, 93, 2833. [Google Scholar]
- Olatunde A. O.; Dorazio S. J.; Spernyak J. A.; Morrow J. R. J. Am. Chem. Soc. 2012, 134, 18503. [DOI] [PubMed] [Google Scholar]
- Ming L. J.; Lauffer R. B.; Que L. Inorg. Chem. 1990, 29, 3060. [Google Scholar]
- Dorazio S. J.; Tsitovich P. B.; Siters K. E.; Spernyak J. A.; Morrow J. R. J. Am. Chem. Soc. 2011, 133, 14154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorazio S. J.; Olatunde A. O.; Spernyak J. A.; Morrow J. R. Chem. Commun. 2013, 49, 10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorazio S.; Olatunde A.; Tsitovich P.; Morrow J. J. Biol. Inorg. Chem. 2014, 19, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitovich P. B.; Morrow J. R. Inorg. Chim. Acta 2012, 393, 3. [Google Scholar]
- Tsitovich P. B.; Spernyak J. A.; Morrow J. R. Angew. Chem., Int. Ed. 2013, 52, 13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorazio S. J.; Morrow J. R. Inorg. Chem. 2012, 51, 7448. [DOI] [PubMed] [Google Scholar]
- Belle C.; Bougault C.; Averbuch M.-T.; Durif A.; Pierre J.-L.; Latour J.-M.; Le Pape L. J. Am. Chem. Soc. 2001, 123, 8053. [DOI] [PubMed] [Google Scholar]
- La Mar G. N. J. Am. Chem. Soc. 1965, 87, 3567. [Google Scholar]
- Roquette P.; Maronna A.; Reinmuth M.; Kaifer E.; Enders M.; Himmel H.-J. r. Inorg. Chem. 2011, 50, 1942. [DOI] [PubMed] [Google Scholar]
- Comprehensive Coordination Chemistry; Sacconi L., Mani F., Bencini A., Eds.; Pergamon Press: Oxford, U.K., 1987; Vol. 5. [Google Scholar]
- Platas-Iglesias C.; Vaiana L.; Esteban-Gómez D.; Avecilla F.; Real J. A.; de Blas A.; Rodríguez-Blas T. Inorg. Chem. 2005, 44, 9704. [DOI] [PubMed] [Google Scholar]
- Vaiana L.; Regueiro-Figueroa M.; Mato-Iglesias M.; Platas-Iglesias C.; Esteban-Gómez D.; de Blas A.; Rodríguez-Blas T. Inorg. Chem. 2007, 46, 8271. [DOI] [PubMed] [Google Scholar]
- Moriarty-Craige S. E.; Jones D. P. Annu. Rev. Nutr. 2004, 24, 481. [DOI] [PubMed] [Google Scholar]
- Banerjee R. J. Biol. Chem. 2012, 287, 4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T. K.; Milne M.; Elmehriki A. A. H.; Suchý M.; Bartha R.; Hudson R. H. E. Contrast Media Mol. Imaging 2013, 8, 289. [DOI] [PubMed] [Google Scholar]
- Terreno E.; Castelli D. D.; Viale A.; Aime S. Chem. Rev. 2010, 110, 3019. [DOI] [PubMed] [Google Scholar]
- Buck F. M.; Bae W. C.; Diaz E.; Du J.; Statum S.; Han E. T.; Chung C. B. Am. J. Roentgenol. 2011, 196, W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. F. J. Chem. Soc. 1959, 2003. [Google Scholar]
- Schubert E. M. J. Chem. Educ. 1992, 69, 62. [Google Scholar]
- Dixon W. T.; Ren J.; Lubag A. J. M.; Ratnakar J.; Vinogradov E.; Hancu I.; Lenkinski R. E.; Sherry A. D. Magn. Reson. Med. 2010, 63, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R.Machine Vision; McGraw-Hill: New York, NY, 1995. [Google Scholar]
- Sheldrick G. M.SHELXL; University of Gottingen: Göttingen, Germany, 1997. [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. J. Appl. Crystallogr. 2009, 42, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel and its Role in Biology; Sigel H., Sigel A., Eds.; Marcel Dekker: New York, 1988; Vol. 23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.