

Conspectus
The eukaryotic heme oxygenases (HOs) (E.C. 1.14.99.3) convert heme to biliverdin, iron, and carbon monoxide (CO) in three successive oxygenation steps. Pathogenic bacteria require iron for survival and infection. Extracellular heme uptake from the host plays a critical role in iron acquisition and virulence. In the past decade, several HOs required for the release of iron from extracellular heme have been identified in pathogenic bacteria, including Corynebacterium diphtheriae, Neisseriae meningitides, and Pseudomonas aeruginosa. The bacterial enzymes were shown to be structurally and mechanistically similar to those of the canonical eukaryotic HO enzymes. However, the recent discovery of the structurally and mechanistically distinct noncanonical heme oxygenases of Staphylococcus aureus and Mycobacterium tuberculosis has expanded the reaction manifold of heme degradation. The distinct ferredoxin-like structural fold and extreme heme ruffling are proposed to give rise to the alternate heme degradation products in the S. aureus and M. tuberculosis enzymes. In addition, several “heme-degrading factors” with no structural homology to either class of HOs have recently been reported. The identification of these “heme-degrading proteins” has largely been determined on the basis of in vitro heme degradation assays. Many of these proteins were reported to produce biliverdin, although no extensive characterization of the products was performed. Prior to the characterization of the canonical HO enzymes, the nonenzymatic degradation of heme and heme proteins in the presence of a reductant such as ascorbate or hydrazine, a reaction termed “coupled oxidation”, served as a model for biological heme degradation. However, it was recognized that there were important mechanistic differences between the so-called coupled oxidation of heme proteins and enzymatic heme oxygenation. In the coupled oxidation reaction, the final product, verdoheme, can readily be converted to biliverdin under hydrolytic conditions. The differences between heme oxygenation by the canonical and noncanonical HOs and coupled oxidation will be discussed in the context of the stabilization of the reactive FeIII–OOH intermediate and regioselective heme hydroxylation. Thus, in the determination of heme oxygenase activity in vitro, it is important to ensure that the reaction proceeds through successive oxygenation steps. We further suggest that when bacterial heme degradation is being characterized, a systems biology approach combining genetics, mechanistic enzymology, and metabolite profiling should be undertaken.
Introduction
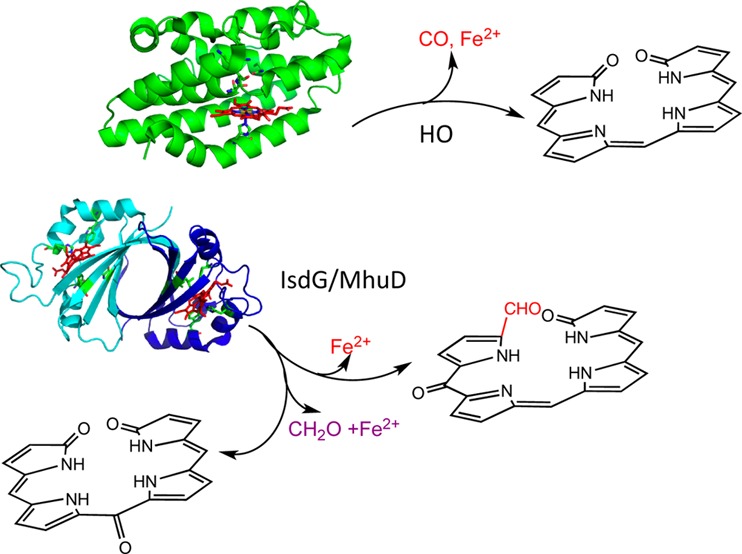
Heme oxidation plays a central role in iron reutilization and cell signaling in mammals, cyanobacteria, and plants.1−3 Bacterial pathogens utilize extracellular heme as an iron source via heme-degrading enzymes.4−6 The ability to utilize heme as an iron source is essential for virulence and pathogenesis. Similar to their eukaryotic counterparts, heme oxygenases (HOs) from pathogens such as Corynebacterium diphtheriae,7Neisseriae meningitides,8 and Pseudomonas aeruginosa(9) catalyze the conversion of heme to biliverdin with the release of iron and CO (Figure 1A). Distinct from the canonical HOs are the noncanonical IsdG/I (iron surface determinant) proteins of Staphylococcus aureus and Bacillus anthracis and MhuD (Mycobacteriumheme utilization) of Mycobacterium tuberculosis.5,10,11 The S. aureus IsdG reaction converts heme to a novel chromophore termed staphylobilin, where the β- or δ-meso-carbon is released as formaldehyde (Figure 1C).12,13 In contrast, the structurally related MhuD cleaves heme to a product called mycobilin that retains the meso-carbon as an aldehyde (Figure 1B).14
Figure 1.
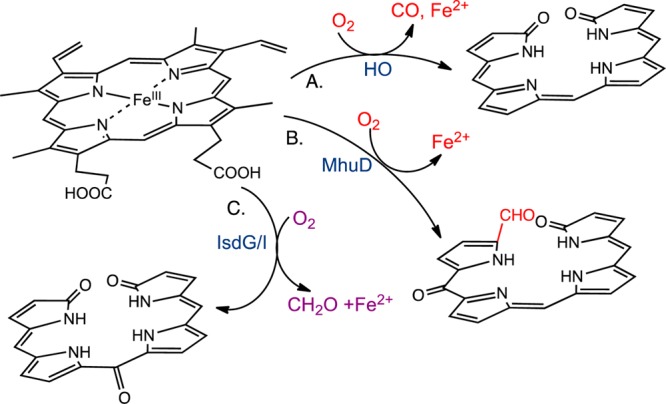
Reaction products of the (A) canonical and (B and C) noncanonical heme oxygenases. The pyrrole substituents have been omitted for simplicity.
It is important to consider the biological significance of the HOs in the context of pathogenesis. The products of the eukaryotic canonical HOs have been reported to have important antioxidant and antiproliferative/anti-inflammatory properties.15−18 Similarly, CO produced as a byproduct of heme utilization may provide an advantage by aiding in the suppression of inflammation. Cystic fibrosis patients with chronic P. aeruginosa infections exhale higher levels of CO.19 The absence of CO production in M. tuberculosis and S. aureus suggests that infection does not favor CO production. In M. tuberculosis, CO can activate the dormancy regulon through the heme-dependent two-component sensor kinases DosS and DosT.20 Thus, avoiding activation of the dormancy genes in an immune-competent state is important for survival of the organism. In contrast, CO-releasing molecules have been shown to produce bactericidal effects in S. aureus.21,22 Induction of colonic HO-1 or exposure to CO in animal models of colitis increases the activity of macrophages and reduces intracellular invasion and bacterial load.23,24 Therefore, in gastroenteric pathogens that cause acute invasive disease, heme utilization may involve a mechanism similar to that of S. aureus. Furthermore, mycobilin, staphylobilin, and/or formaldehyde may have as yet unidentified biological functions in the host–pathogen interaction.
In addition to the previously described enzymes, several heme-binding proteins identified in pathogenic bacteria have been reported to be HOs on the basis of in vitro assays. However, it is not clear whether heme degradation is due to HO activity or the nonenzymatic “coupled oxidation” of heme described almost a century ago.25−27 In the current Account, we discuss the differences between coupled oxidation and heme oxygenation in the context of generating, stabilizing, and directing the FeIII–OOH intermediate toward regioselective heme hydroxylation. In addition, the unique mechanism by which the noncanonical HOs control the intrinsic reactivity of meso-hydroxyheme by suppressing CO production and promoting further oxidation of heme is also addressed.
Coupled Oxidation: A Model for Biological Heme Breakdown
It was shown some 90 years ago that in the presence of O2 a mixture of heme in pyridine/water could be degraded by hydrazine or ascorbate.26,27 The product “green hemin” was formed simultaneously with the release of CO. In such studies it was clear that the product of the reaction was not biliverdin but rather verdohemin, which upon addition of KOH and HCl hydrolyzes to biliverdin. Interestingly, the chemically coupled oxidation of hemin in aqueous pyridine gave an equimolar mixture of all four verdohemin isomers, whereas oxidation of hemoglobin and myoglobin yielded primarily biliverdin IXα.28,29
The first step in the reaction was presumed to be heme hydroxylation at the α-meso position to form the α-oxy derivative (Figure 2).30 Early studies confirmed that FeII–oxymesoporphyrin could be converted stepwise to FeIII–mesobiliverdin by two molecules of O2.31 Chemically synthesized meso-hydroxyheme was similarly converted to biliverdin.32 It was initially thought the reaction of verdoheme to biliverdin proceeded through hydrolysis with insertion of an oxygen atom from water,33 a mechanism at odds with 18O2/16O2-labeling studies identifying the terminal lactam oxygens as being derived from separate O2 molecules.34−36 However, a series of detailed mechanistic studies determined that verdoheme could be oxidatively cleaved to biliverdin (Figure 2).37,38
Figure 2.

Reaction intermediates in the canonical HO-catalyzed degradation of heme to biliverdin.
Analysis of the coupled oxidation of cytochrome b5 variants revealed critical differences between enzymatic and nonenzymatic heme degradation.39 Replacement of the coordinating residue His-63 or His-39 with Val resulted in different end products. The H63V variant was arrested at verdoheme, whereas the final product of the H39V-catalyzed reaction was biliverdin.39 The difference in end products was explained in terms of the relative stability of the FeII–O2 complexes. Coupled oxidation of the H63V variant could be inhibited by the addition of the H2O2-scavenging enzyme catalase.40 This is distinct from the reaction of H2O2 with the FeIII–heme:HO-1 complex, which generates a coordinated FeIII–OOH intermediate that then reacts to form verdoheme.41 In contrast, H39V proceeds to FeIII–biliverdin in the presence of catalase, suggesting that the reaction proceeds through a coordinated FeII–O2 complex. Therefore, although coupled oxidation and heme oxygenation lead to meso hydroxylation, they do so by distinct mechanisms. The fact that heme proteins can undergo nonenzymatic conversion of heme to verdoheme with the release of CO indicates that this step in heme degradation is relatively facile. Therefore, detection of CO as defining enzymatic HO activity should be met with caution.42
What Defines a Heme Oxygenase?
Several structurally unrelated bacterial heme-binding proteins distinct from the canonical and noncanonical HOs have been reported to have HO activity.5,10,42−45 However, this characterization was largely based on a heme toxicity phenotype that could be “complemented” with a gene encoding a canonical HO. The cytoplasmic heme-binding protein ChuS of Escherichia coli O157:H742,46 belongs to a structurally related family that includes Yersinia entercolitica HemS and P. aeruginosa PhuS.46,47 The HO activity of ChuS was based on the detection of CO in the presence of ascorbate. Although the authors ruled out degradation by H2O2 by including catalase in the reaction, they did not characterize the products of the reaction. Given the fact that P. aeruginosa encodes a previously characterized iron-regulated canonical HemO, the Wilks group revisited the role of the cytoplasmic heme-binding protein PhuS. Bacterial genetics and [13C]heme isotopic labeling studies confirmed that heme degradation is driven by the catalytic action of HemO, with PhuS acting as a regulator of extracellular heme flux to HemO.48,49 A recent report suggests that in addition to trafficking heme, PhuS is a heme-degrading enzyme that converts heme to verdoheme.50 The authors further suggested that verdoheme is trafficked to HemO for conversion to biliverdin. However, it should be noted that these studies were performed in vitro and that the stoichiometry of the reaction was not reported. Furthermore, in the ΔhemO strain, PhuS does not compensate for the loss of HemO by converting heme to verdoheme.49
Similarly, several gastroenteric pathogens, including Helicobacter pylori, Vibrio cholera, Campylobacter jejuni, and Plesiomonas shigelloides, have been described as encoding HOs.43,51,52 In P. shigelloides this was based on an inability to efficiently utilize heme in the ΔhugZ (hutZ in V. cholera and chuZ in C. jejuni) strain. Furthermore, while complementation of P. shigelloideshugZ with a canonical HO from Synechosistis PCC6803 alleviated heme toxicity, the HugZ protein showed no detectable HO activity.51 Therefore, it is extremely important when assigning HO function that a systematic approach combining genetics, metabolite analysis, and enzymology is undertaken.
Structural Diversity and the Widening Paradigm of Heme Oxygenation
The canonical HO enzymes from bacteria to mammals have a similar overall α-helical structural fold (Figure 3A).53−55 Heme is held between the proximal and distal helices and anchored in the pocket through interactions of the propionates with surface-exposed Arg and Lys residues. Interestingly, HemO from P. aeruginosa gives rise to a regioselectivity altered from that of all other HOs as a consequence of the in-plane rotation of the heme within the binding site.53 In addition to the conserved proximal His ligand, all canonical HOs retain an ordered hydrogen-bonding network required for proton delivery and activation of the coordinated FeII–O2 (Figure 3B).
Figure 3.
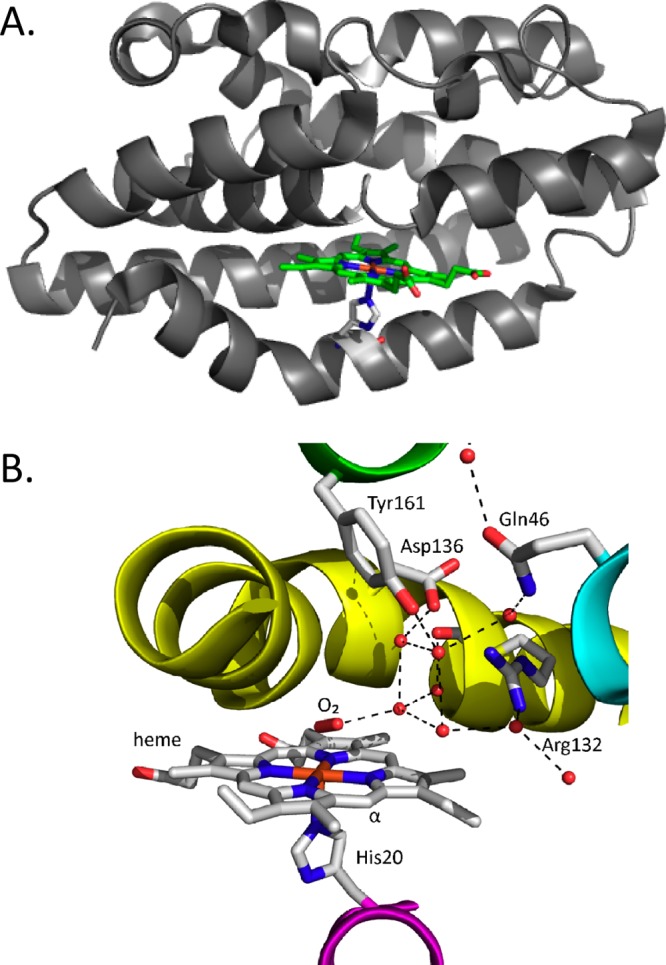
Structure of the bacterial canonical HOs. (A) Overall structural fold of the δ/β-regioselective P. aeruginosa HemO [Protein Data Bank (PDB) code 1SK7]. (B) Active site structure with the C. diphtheriae FeII–O2 heme–HmuO complex (PDB code 1V8X), where the oxygen atoms of the structural waters are represented as red balls and the bound O2 is shown in stick format.
In contrast, IsdG/I and MhuD have an overall fold distinct from that of the classical HOs in which the two monomers adopt ferredoxin-like α/β-sandwich folds that come together to form a β-barrel at the homodimer interface (Figure 4A).11,56 Each monomer binds heme in a hydrophobic cleft on either side of the β-barrel, with the coordinating His residue provided by the surrounding helix/loops. In IsdI, heme is ligated through His-76 and the propionates are anchored through interactions with Arg-21 and Arg-25 (Figure 4B). The ordered hydrogen-bonding network required for activity in the canonical HO enzymes is absent, with Asn-6 being the only residue capable of hydrogen bonding to the ligand. However, the resting-state heme in IsdI undergoes significant distortion from planarity through steric interactions of the heme β-, γ-, and δ-meso-carbon atoms with Trp-66, Phe-22, and Val-79, respectively (Figure 4B).56 Furthermore, mutation of Trp-66 to Tyr leads to less heme ruffling and reduced enzyme activity.57 Therefore, in contrast to the canonical HOs, where the interactions of the coordinated FeIII–OOH with the hydrogen-bonding network modulate the heme reactivity, in the noncanonical IsdG-like proteins this is achieved through sterically induced heme ruffling.
Figure 4.
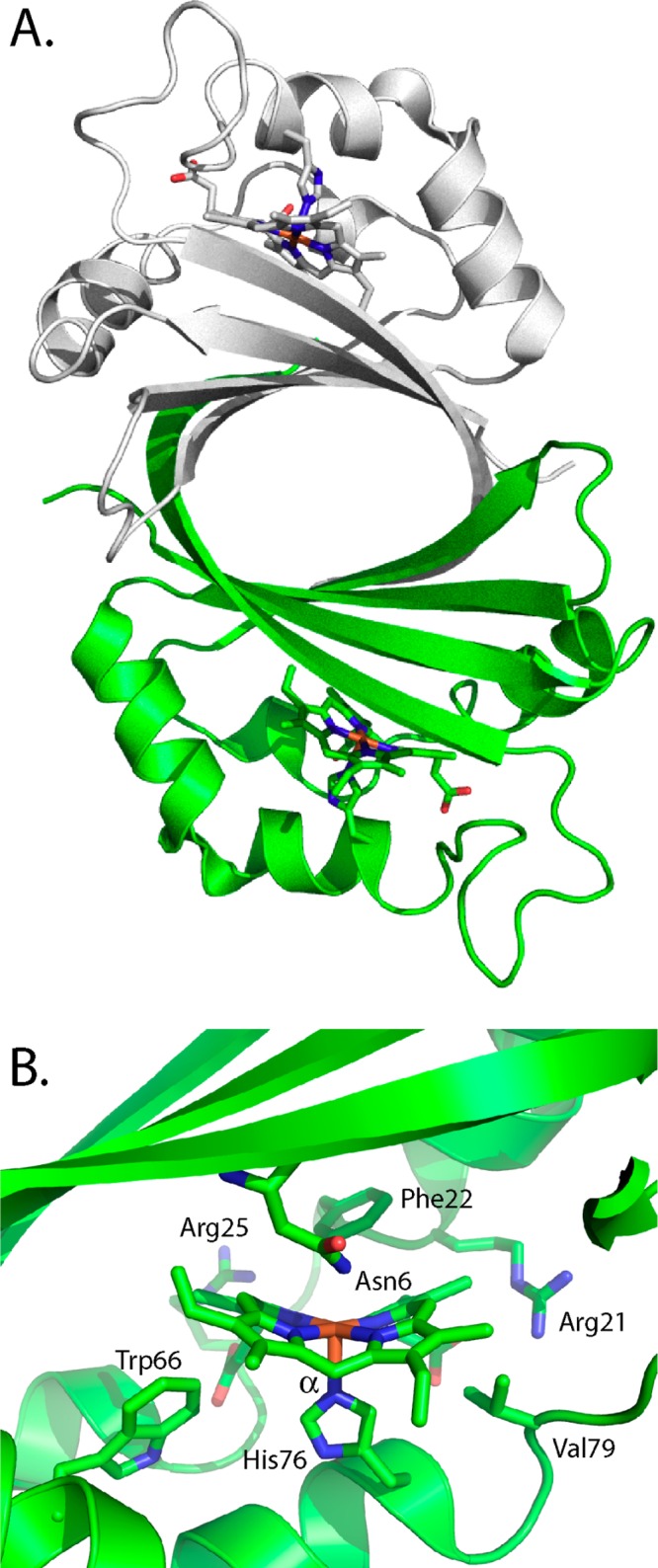
Structure of the IsdI of S. aureus. (A) Overall structural fold of the heme-bound IsdI homodimer. (B) Active site of heme–IsdI. The α-meso-carbon is labeled. PDB code 3LGM.
Oxygen Activation and Heme Hydroxylation: The Initial Step in Heme Degradation
The initial step in the canonical HOs is the reduction of the FeIII–heme:HO complex to the FeII–O2 complex.58−60 Although the absorption spectrum of the HO-1 FeII–O2 complex is similar to that of oxymyoglobin,61 the Raman spectrum shows a unique oxygen-isotope shift suggesting that the bound O2 is highly bent.62 The structure of the bacterial FeII–O2 heme–HmuO complex confirmed that the 110° Fe–O–O bond angle is due to interaction of the O2 ligand with the distal helix (Figure 3B).63 Reduction of the FeII–O2 complex to the activated FeIII–OOH complex leads to the formation of α-meso-hydroxyheme,64 which in the presence of oxygen is rapidly converted to ferric verdoheme.41
In contrast, the noncanonical enzymes degrade heme to bilin products distinct from those for the canonical enzymes in which either the meso-carbon is retained as an aldehyde (as in MhuD) or released as formaldehyde (as in IsdG/I).13,14 It is proposed that the conserved structural fold and resulting heme distortion account for the alternate reaction products.13,14
The Yin and Yang: Steric versus Electronic Contributions to Heme Hydroxylation
Regioselective hydroxylation of heme in the canonical HOs results from a combination of steric constraints placed on the activated oxygen species and electronic perturbation of the heme. On the basis of the fact that H2O2 can substitute for O2 and reducing equivalents, the meso hydroxylation of heme was proposed to proceed by either (a) nucleophilic addition of the terminal oxygen of the ferric peroxo (FeIII–OO–) species to the α-meso-carbon or (b) electrophilic addition of the terminal oxygen of the protonated (FeIII–OOH) species.41 However, ethyl hydroperoxide yielded α-meso-ethoxyheme, ruling out nucleophilic addition.65 Cryogenic electron–nuclear double resonance (ENDOR) and electron paramagnetic resonance (EPR) studies confirmed FeIII–OOH to be the activated intermediate following one-electron reduction of the FeII–O2 complex. Temperature annealing experiments further identified FeIII–OOH as the immediate precursor of α-meso-hydroxyheme (Figure 2).64 The activation of FeII–O2 to FeIII–OOH is a common step in heme-containing monoxygenases and HOs.66 However, the reactions diverge significantly, as in contrast to the self-hydroxylation by heme oxygenases, the P450-type monooxygenases proceed to Compound I, a ferryl (FeV=O) porphyrin π-radical cation.
The stabilization of the O2 ligand in the canonical HOs is a combination of the conserved distal Gly-Gly motif and the hydrogen-bonding network provided through bridging H2O molecules (Figure 3B).53−55,63,67 NMR studies confirmed that the ordered water molecules are required for the donation of a proton to the coordinated FeII–O2.68,69 Disruption of the hydrogen-bonding network upon mutation of Asp-140 in HO-1 resulted in destabilization of the FeIII–OOH intermediate and accelerated decay to the FeV=O species.70,71 This was supported by EPR and ENDOR experiments establishing that Asp-140 forms a hydrogen bond with the ordered water molecule that not only stabilizes the FeII–O–O species but also functions as an efficient conduit to transfer protons required for the formation and activation of FeIII–OOH.72 The positioning of the nearby water was proposed to promote nucleophilic attack by acting as a hydrogen-bond acceptor and/or to control the steric interactions with the α-meso-carbon.73 Interestingly, mutation of the equivalent Asp-136 in HmuO did not lead to complete loss of the water network or activity, highlighting the critical nature of the hydrogen bond to the distal oxygen.74 Collectively, the data support a theory wherein the water promotes nucleophilic attack by acting as a hydrogen-bond acceptor73 or alternatively constrains the bent end-on geometry of FeIII–OOH for interaction with the α-meso-carbon.63,74 Furthermore, while the conserved Asp is absent in the N. meningitides and P. aeruginosa HemOs, the hydrogen-bonding network is conserved.53,55
In contrast to the water-driven oxygen activation of the FeIII–OOH intermediate, Rivera and co-workers proposed that the initial hydroxylation was facilitated by the heme.75,76 Previous studies of model porphyrins have shown a correlation between the sum of the squares of the g values (∑g2) and the electronic structure of the heme in which a ∑g2 value of ∼14 is indicative of the (dxz,dyz)4(dxy)1 state with the unpaired electron residing in the dxy orbital.76 The unique EPR fingerprint of low-spin models of the FeIII–OOH HO intermediate and 13C NMR spectrum of the FeIII–OH HO complex led the authors to propose such an electronic structure for the FeIII–OOH HO intermediate. However, Mössbauer analysis indicated that the FeIII–OOH complex assumed a (dxy)2(dxz,dyz)3 electronic state common to most low-spin ferric species.66 Interestingly, the (dxy)1 electron configuration, which places a large amount of unpaired electron density on the meso-carbons,77 does provide a rationale for heme degradation by the noncanonical enzymes.
In earlier mechanistic studies of the canonical HOs, O–O bond homolysis followed by addition of hydroxyl radical (·OH) to the meso-carbon was ruled out on the basis of the indiscriminate nature of the hydroxyl radical.41 However, recent quantum mechanics/molecular mechanics (QM/MM) studies have suggested that the most favorable pathway is a nonsynchronous concerted mechanism wherein O–O bond homolysis guided by the distal-pocket water cluster leads to attack of ·OH at the meso-carbon.78
In contrast, the ruffled heme in the noncanonical heme–IsdG/I has been proposed to promote hydroxylation through modulation of the heme electronic structure.56,57 An 1H NMR study of the FeIII–OH and FeIII–CN– complexes of IsdI revealed large upfield shifts for the meso-H and smaller downfield shifts for the methyl groups, suggesting that the heme exhibits predominantly the (dxz,dyz)4(dxy)1 electron configuration with the electron density residing on the meso-carbons.79 This is consistent with the inactive IsdI W66Y variant, which shows reduced (dxz,dyz)4(dxy)1 character and less heme ruffling.57 The authors concluded that the heme ruffling in the Isd-like proteins promotes oxidation without the assistance of the hydrogen-bonding network required in the canonical HOs.
Oxidation of meso-Hydroxyheme: A New Paradigm
Beyond the initial hydroxylation step, the mechanism of the noncanonical HOs diverges from that of the canonical HOs. In mechanistic studies of the canonical HOs, the FeIII–meso-hydroxyheme intermediate was shown to decay to FeIII–verdoheme in the presence of oxygen.60,65,80,81 EPR and Raman studies showed that FeIII–meso-hydroxyheme exists as a resonance hybrid of keto (oxophlorin), phenolate, and ferrous neutral radical structures (Figure 5).80,82 Under aerobic conditions, the neutral radical reacts with oxygen to give a ferrous peroxy radical intermediate that interacts with the ferrous iron or via internal electron transfer to give the FeIII–OOH (Figure 5). The FeIII–OOH species decays to an unstable ferryl alkoxy radical that extrudes the meso-carbon as CO. The resulting radical is oxidized to the FeIV=O intermediate. and the carbocation is trapped as verdoheme.
Figure 5.

Proposed mechanism for the conversion of meso-hydroxyheme to verdoheme in the canonical HOs.
Skaar and co-workers invoked a similar mechanism for IsdG in which the conversion of heme to staphylobilin proceeds through either FeIII–dihydroxyheme (Figure 6A) or FeIII–meso-hydroxyverdoheme (Figure 6B).12 The common intermediate in both pathways FeIII-hydroxyverdoheme is converted to 5-oxo-bilirubin (ring cleavage at the β-meso-carbon) or 15-oxo-bilirubin (ring cleavage at the δ-meso-carbon) via a similar mechanism as the canonical HOs. However, neither the stoichiometry of the reaction or verification of the FeIII-dihydroxyverdoheme intermediate has been shown.
Figure 6.

Proposed mechanism for the conversion of heme to staphylobilin via a verdoheme intermediate. Adapted from ref (12).
In contrast, Ikeda-Saito and co-workers have shown that heme oxidation by MhuD does not give rise to CO, precluding verdoheme as an intermediate.14 The MhuD-catalyzed reaction cleaves the porphyrin ring with retention of the α-meso-carbon as an aldehyde and modification of the adjacent meso-carbon to a carbonyl at either the β-meso-carbon (mycobilin-a) or δ-meso-carbon (mycobilin-b).14 On the basis of the structural homology between MhuD and IsdG, the authors proposed that the reaction mechanism for IsdG is similar to that for MhuD, and in subsequent studies they have shown that the conversion of heme to staphylobilin proceeds with the release of the meso-carbon as formaldehyde.13 Furthermore, 18O2-labeling studies confirmed that three O atoms from 18O2 are incorporated into staphylobilin and mycobilin, in contrast to the two O atoms in biliverdin.
Although the detailed mechanism of the noncanonical HO reaction has yet to be elucidated, the unique protein fold and heme electronic structure must play a significant role. In the canonical HOs, the ferrous neutral radical form of FeIII–meso-hydroxyheme reacts directly with O2 to generate verdoheme with the release of CO (Figure 5). This rapid autoxidation of meso-hydroxyheme to verdoheme is suppressed in the IsdG-like proteins. Suppression of the autoxidation step is most likely due to the extreme heme ruffling and altered electronic configuration of the meso-hydroxyheme or equivalent intermediate.79
Despite the similarity between the MhuD and IsdG/I aldehyde intermediates, the enzymes have distinct regioselectivities. The diagonal modification at the β- and δ-meso-carbons in IsdG/I was proposed to occur because the extreme heme ruffling places the terminal oxygen of the putative FeIII–OOH in proximity of both the β- and δ-meso-carbons. In contrast, MhuD cleaves the heme at the α-meso carbon with carbonyl modification of either the adjacent β- or δ-meso-carbon. The regioselectivity is most likely a combination of an in-plane rotation that places the α-meso-carbon at the site of activation and the orientational disorder around the α/γ-axis that places either the β- or δ-meso-carbon in a position for oxidation to the carbonyl.13 This alternate heme orientation is supported in the diheme MhuD structure, where heme I is rotated 90° from that of the heme in IsdI and heme II in MhuD.11
It is unclear at the present time whether the alternate regioselectivity in the MhuD and IsdG proteins plays a role in determining whether the aldehyde is retained or released, respectively. Furthermore, it is not known whether the initial meso-carbon hydroxylation leads to ring opening prior to the carbonyl modification or vice versa. Although the mechanistic details of the noncanonical HO-catalyzed reaction have yet to be revealed, this class of heme-degrading enzymes represents a paradigm shift in oxidative heme cleavage.
Concluding Remarks
For decades the biological degradation of heme was thought to be restricted to oxidative cleavage of heme to biliverdin with the release CO. Although monooxygenases and heme oxygenases share an O2-activation mechanism similar to that of the FeIII–OOH, they diverge through either an oxoferryl species or “self” heme hydroxylation, respectively. In HO, heme hydroxylation is a combination of the decreased tendency to undergo O–O bond cleavage while simultaneously activating the heme. This is achieved by the conformational flexibilty of the heme binding site and the extensive hydrogen-bonding network. Where the IsdG-like proteins lie along this activation pathway is of great interest and will provide further insight into the role of structural distortion in heme reactivity.
Acknowledgments
We thank C. S. Raman, T. Matsui, and T. L. Poulos for helpful conversations. We thank NIH (AI55912 and AI102883 to A.W.) and JSPS (21350087, 2412006, and 24350081 to M.I.-S.) for continued support.
Biographies
Angela Wilks received her Ph.D. in Biochemistry from the University of Leeds in England. She completed her postdoctoral studies at the University of California, San Francisco, on the structure and function of heme oxygenase. She is currently a professor in the Department of Pharmaceutical Sciences at the University of Maryland.
Masao Ikeda-Saito received his Ph.D. in Biophysical Engineering from Osaka University. He was a faculty member at the University of Pennsylvania and Case Western Reserve University prior to his appointment as a professor at Tohoku University.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Frankenberg-Dinkel N. Bacterial heme oxygenases. Antioxid. Redox Signaling 2004, 6, 825–834. [DOI] [PubMed] [Google Scholar]
- Wilks A. Heme oxygenase: Evolution, structure, and mechanism. Antioxid. Redox Signaling 2002, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Wilks A.; Burkhard K. A. Heme and virulence: How bacterial pathogens regulate, transport and utilize heme. Nat. Prod. Rep. 2007, 24, 511–522. [DOI] [PubMed] [Google Scholar]
- Brickman T. J.; Hanawa T.; Anderson M. T.; Suhadolc R. J.; Armstrong S. K. Differential expression of Bordetella pertussis iron transport system genes during infection. Mol. Microbiol. 2008, 70, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar E. P.; Gaspar A. H.; Schneewind O. IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J. Biol. Chem. 2004, 279, 436–443. [DOI] [PubMed] [Google Scholar]
- Morton D. J.; Seale T. W.; Vanwagoner T. M.; Whitby P. W.; Stull T. L. The dppBCDF gene cluster of Haemophilus influenzae: Role in heme utilization. BMC Res. Notes 2009, 2, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks A.; Schmitt M. P. Expression and characterization of a heme oxygenase (HmuO) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle. J. Biol. Chem. 1998, 273, 837–841. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Wilks A.; Stojiljkovic I. Degradation of heme in Gram-negative bacteria: The product of the hemO gene of Neisseriae is a heme oxygenase. J. Bacteriol. 2000, 182, 6783–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratliff M.; Zhu W.; Deshmukh R.; Wilks A.; Stojiljkovic I. Homologues of Neisserial heme oxygenase in Gram-negative bacteria: Degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 6394–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar E. P.; Gaspar A. H.; Schneewind O. Bacillus anthracis IsdG, a heme-degrading monooxygenase. J. Bacteriol. 2006, 188, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chim N.; Iniguez A.; Nguyen T. Q.; Goulding C. W. Unusual diheme conformation of the heme-degrading protein from Mycobacterium tuberculosis. J. Mol. Biol. 2010, 395, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reniere M. L.; Ukpabi G. N.; Harry S. R.; Stec D. F.; Krull R.; Wright D. W.; Bachmann B. O.; Murphy M. E.; Skaar E. P. The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol. Microbiol. 2010, 75, 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Nambu S.; Ono Y.; Goulding C. W.; Tsumoto K.; Ikeda-Saito M. Heme degradation by Staphylococcus aureus IsdG and IsdI liberates formaldehyde rather than carbon monoxide. Biochemistry 2013, 52, 3025–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu S.; Matsui T.; Goulding C. W.; Takahashi S.; Ikeda-Saito M. A New Way to Degrade Heme: The Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J. Biol. Chem. 2013, 288, 10101–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. P.; Ryter S. W.; Choi A. M. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449. [DOI] [PubMed] [Google Scholar]
- Rochette L.; Cottin Y.; Zeller M.; Vergely C. Carbon monoxide: Mechanisms of action and potential clinical implications. Pharmacol. Ther. 2013, 137, 133–152. [DOI] [PubMed] [Google Scholar]
- Snyder S. H.; Baranano D. E. Heme oxygenase: A font of multiple messengers. Neuropsychopharmacology 2001, 25, 294–298. [DOI] [PubMed] [Google Scholar]
- Wegiel B.; Hanto D. W.; Otterbein L. E. The social network of carbon monoxide in medicine. Trends Mol. Med. 2013, 19, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antuni J. D.; Kharitonov S. A.; Hughes D.; Hodson M. E.; Barnes P. J. Increase in exhaled carbon monoxide during exacerbations of cystic fibrosis. Thorax 2000, 55, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Deshane J. S.; Crossman D. K.; Bolisetty S.; Yan B. S.; Kramnik I.; Agarwal A.; Steyn A. J. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 2008, 283, 18032–18039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares A. F.; Nobre L. S.; Saraiva L. M. A role for reactive oxygen species in the antibacterial properties of carbon monoxide-releasing molecules. FEMS Microbiol. Lett. 2012, 336, 1–10. [DOI] [PubMed] [Google Scholar]
- Nobre L. S.; Seixas J. D.; Romao C. C.; Saraiva L. M. Antimicrobial action of carbon monoxide-releasing compounds. Antimicrob. Agents Chemother. 2007, 51, 4303–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y.; Uchiyama K.; Takagi T.; Yoshikawa T. Therapeutic potential of carbon monoxide (CO) for intestinal inflammation. Curr. Med. Chem. 2012, 19, 70–76. [DOI] [PubMed] [Google Scholar]
- Onyiah J. C.; Sheikh S. Z.; Maharshak N.; Steinbach E. C.; Russo S. M.; Kobayashi T.; Mackey L. C.; Hansen J. J.; Moeser A. J.; Rawls J. F.; Borst L. B.; Otterbein L. E.; Plevy S. E. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 2013, 144, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J. E.Porphyrins and Metalloporphyrins; Elsevier: Amsterdam, 1964. [Google Scholar]
- Warburg O.; Negelein E. Grunes Haemin aus Blast-haemin. Chem. Ber. 1930, 63, 1816–1819. [Google Scholar]
- Lemberg R. Transformation of hemins into bile pigments. Biochem. J. 1935, 29, 1322–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carra P.; Colleran E. Coupled oxidation of myoglobin with ascorbate as a model of haem breakdown in vivo. Biochem. J. 1969, 115, 13P–14P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carra P.; Colleran E. Methine-bridge specificity of the coupled oxidation of myoglobin and haemoglobin with ascorbate. Biochem. J. 1970, 119, 42P–43P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg R. Chemical mechanism of bile pigment formation. Rev. Pure Appl. Chem. 1956, 6, 1–23. [Google Scholar]
- Sano S.; Sugiura Y. Penta-co-ordinate iron (II) oxyporphyrin–2-methylimidazole complex; Model of the heme oxygenase reaction. J. Chem. Soc., Chem. Commun. 1982, 750–752. [Google Scholar]
- Kondo T.; Nicholson D. C.; Jackson A. H.; Kenner G. W. Isotopic studies of the conversion of oxophlorins and their ferrihaems into bile pigments in the rat. Biochem. J. 1971, 121, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A. H.Iron in Biochemistry and Medicine; Academic Press: New York, 1974. [Google Scholar]
- Docherty J. C.; Firneisz G. D.; Schacter B. A. Methene bridge carbon atom elimination in oxidative heme degradation catalyzed by heme oxygenase and NADPH-cytochrome P-450 reductase. Arch. Biochem. Biophys. 1984, 235, 657–664. [DOI] [PubMed] [Google Scholar]
- Docherty J. C.; Schacter B. A.; Firneisz G. D.; Brown S. B. Mechanism of action of heme oxygenase. A study of heme degradation to bile pigment by 18O labeling. J. Biol. Chem. 1984, 259, 13066–13069. [PubMed] [Google Scholar]
- King R. F.; Brown S. B. The mechanism of haem catabolism. A study of haem breakdown in spleen microsomal fraction and in a model system by 18O labelling and metal substitution. Biochem. J. 1978, 174, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S.; Itano H. A. Verdohemochrome IX alpha: Preparation and oxidoreductive cleavage to biliverdin IX alpha. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S.; Sano T.; Morishima I.; Shiro Y.; Maeda Y. On the mechanism of the chemical and enzymic oxygenations of alpha-oxyprotohemin IX to Fe.biliverdin IX alpha. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila L.; Huang H. W.; Damaso C. O.; Lu S.; Moënne-Loccoz P.; Rivera M. Coupled oxidation vs heme oxygenation: Insights from axial ligand mutants of mitochondrial cytochrome b5. J. Am. Chem. Soc. 2003, 125, 4103–4110. [DOI] [PubMed] [Google Scholar]
- Sigman J. A.; Wang X.; Lu Y. Coupled oxidation of heme by myoglobin is mediated by exogenous peroxide. J. Am. Chem. Soc. 2001, 123, 6945–6946. [DOI] [PubMed] [Google Scholar]
- Wilks A.; Ortiz de Montellano P. R. Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J. Biol. Chem. 1993, 268, 22357–22362. [PubMed] [Google Scholar]
- Suits M. D.; Pal G. P.; Nakatsu K.; Matte A.; Cygler M.; Jia Z. Identification of an Escherichia coli O157:H7 heme oxygenase with tandem functional repeats. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 16955–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Guo G.; Mao X.; Zhang W.; Xiao J.; Tong W.; Liu T.; Xiao B.; Liu X.; Feng Y.; Zou Q. Functional identification of HugZ, a heme oxygenase from Helicobacter pylori. BMC Microbiol. 2008, 8, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic I.; Hantke K. Transport of haemin across the cytoplasmic membrane through a haemin-specific periplasmic binding-protein-dependent transport system in Yersinia enterocolitica. Mol. Microbiol. 1994, 13, 719–732. [DOI] [PubMed] [Google Scholar]
- Suits M. D.; Lang J.; Pal G. P.; Couture M.; Jia Z. Structure and heme binding properties of Escherichia coli O157:H7 ChuX. Protein Sci. 2009, 18, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suits M. D.; Jaffer N.; Jia Z. Structure of the Escherichia coli O157:H7 heme oxygenase ChuS in complex with heme and enzymatic inactivation by mutation of the heme coordinating residue His-193. J. Biol. Chem. 2006, 281, 36776–36782. [DOI] [PubMed] [Google Scholar]
- Schneider S.; Sharp K. H.; Barker P. D.; Paoli M. An induced fit conformational change underlies the binding mechanism of the heme transport proteobacteria-protein HemS. J. Biol. Chem. 2006, 281, 32606–32610. [DOI] [PubMed] [Google Scholar]
- Barker K. D.; Barkovits K.; Wilks A. Metabolic flux of extracellular heme uptake in Pseudomonas aeruginosa is driven by the iron-regulated heme oxygenase (HemO). J. Biol. Chem. 2012, 287, 18342–18350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill M. J.; Wilks A. The P. aeruginosa heme binding protein PhuS is a heme oxygenase titratable regulator of heme uptake. ACS Chem. Biol. 2013, 8, 1794–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J.; Schep D.; McLaughlin B.; Kaufmann M.; Jia Z. Structural Analysis and Identification of PhuS as a Heme-Degrading Enzyme from Pseudomonas aeruginosa. J. Mol. Biol. 2014, 426, 1936–1946. [DOI] [PubMed] [Google Scholar]
- Oldham A. L.; Wood T. A.; Henderson D. P. Plesiomonas shigelloides hugZ encodes an iron-regulated heme binding protein required for heme iron utilization. Can. J. Microbiol. 2008, 54, 97–102. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Zhang J.; Guo G.; Mao X.; Tong W.; Zhang Y.; Wang D. C.; Hu Y.; Zou Q. Crystal structure of Campylobacter jejuni ChuZ: A split-barrel family heme oxygenase with a novel heme-binding mode. Biochem. Biophys. Res. Commun. 2011, 415, 82–87. [DOI] [PubMed] [Google Scholar]
- Friedman J.; Lad L.; Li H.; Wilks A.; Poulos T. L. Structural basis for novel δ-regioselective heme oxygenation in the opportunistic pathogen Pseudomonas aeruginosa. Biochemistry 2004, 43, 5239–5245. [DOI] [PubMed] [Google Scholar]
- Hirotsu S.; Chu G. C.; Unno M.; Lee D. S.; Yoshida T.; Park S. Y.; Shiro Y.; Ikeda-Saito M. The crystal structures of the ferric and ferrous forms of the heme complex of HmuO, a heme oxygenase of Corynebacterium diphtheriae. J. Biol. Chem. 2004, 279, 11937–11947. [DOI] [PubMed] [Google Scholar]
- Schuller D. J.; Zhu W.; Stojiljkovic I.; Wilks A.; Poulos T. L. Crystal structure of heme oxygenase from the Gram-negative pathogen Neisseria meningitidis and a comparison with mammalian heme oxygenase-1. Biochemistry 2001, 40, 11552–11558. [DOI] [PubMed] [Google Scholar]
- Lee W. C.; Reniere M. L.; Skaar E. P.; Murphy M. E. Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J. Biol. Chem. 2008, 283, 30957–30963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukpabi G.; Takayama S. J.; Mauk A. G.; Murphy M. E. Inactivation of the heme degrading enzyme IsdI by an active site substitution that diminishes heme ruffling. J. Biol. Chem. 2012, 287, 34179–34188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Unno M.; Ikeda-Saito M. Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc. Chem. Res. 2010, 43, 240–247. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R.; Wilks A. Heme Oxygenase Structure and Mechanism. Adv. Inorg. Chem. 2000, 51, 359–402. [Google Scholar]
- Yoshida T.; Noguchi M.; Kikuchi G. Oxygenated form of heme. Heme oxygenase complex and requirement for second electron to initiate heme degradation from the oxygenated complex. J. Biol. Chem. 1980, 255, 4418–4420. [PubMed] [Google Scholar]
- Takahashi S.; Ishikawa K.; Takeuchi E.; Ikeda-Saito M.; Yoshida T.; Rousseau D. L. Oxygen-bound heme−heme oxygenase complex: Evidence for a highly bent structure of the coordinated oxygen. J. Am. Chem. Soc. 1995, 117, 6002–6006. [Google Scholar]
- Unno M.; Matsui T.; Chu G. C.; Couture M.; Yoshida T.; Rousseau D. L.; Olson J. S.; Ikeda-Saito M. Crystal structure of the dioxygen-bound heme oxygenase from Corynebacterium diphtheriae: Implications for heme oxygenase function. J. Biol. Chem. 2004, 279, 21055–21061. [DOI] [PubMed] [Google Scholar]
- Davydov R. M.; Yoshida T.; Ikeda-Saito M.; Hoffman B. M. Hydroperoxy-Heme Oxygenase generated by cryoreduction catalyzes the formation of α-meso-hydroxyheme as detected by EPR and ENDOR. J. Am. Chem. Soc. 1999, 121, 10656–10657. [Google Scholar]
- Wilks A.; Torpey J.; Ortiz de Montellano P. R. Heme oxygenase (HO-1). Evidence for electrophilic oxygen addition to the porphyrin ring in the formation of α-meso-hydroxyheme. J. Biol. Chem. 1994, 269, 29553–29556. [PubMed] [Google Scholar]
- Garcia-Serres R.; Davydov R. M.; Matsui T.; Ikeda-Saito M.; Hoffman B. M.; Huynh B. H. Distinct reaction pathways followed upon reduction of oxy-heme oxygenase and oxy-myoglobin as characterized by Mössbauer spectroscopy. J. Am. Chem. Soc. 2007, 129, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller D. J.; Wilks A.; Ortiz de Montellano P. R.; Poulos T. L. Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 1999, 6, 860–867. [DOI] [PubMed] [Google Scholar]
- Li Y.; Syvitski R. T.; Auclair K.; Ortiz de Montellano P.; La Mar G. N. Solution 1H, 15N NMR spectroscopic characterization of substrate-bound, cyanide-inhibited human heme oxygenase: Water occupation of the distal cavity. J. Am. Chem. Soc. 2003, 125, 13392–13403. [DOI] [PubMed] [Google Scholar]
- Syvitski R. T.; Li Y.; Auclair K.; Ortiz de Montellano P. R.; La Mar G. N. 1H NMR detection of immobilized water molecules within a strong distal hydrogen-bonding network of substrate-bound human heme oxygenase-1. J. Am. Chem. Soc. 2002, 124, 14296–14297. [DOI] [PubMed] [Google Scholar]
- Fujii H.; Zhang X.; Tomita T.; Ikeda-Saito M.; Yoshida T. A role for highly conserved carboxylate, aspartate-140, in oxygen activation and heme degradation by heme oxygenase-1. J. Am. Chem. Soc. 2001, 123, 6475–6484. [DOI] [PubMed] [Google Scholar]
- Lightning L. K.; Huang H.; Moënne-Loccoz P.; Loehr T. M.; Schuller D. J.; Poulos T. L.; Ortiz de Montellano P. R. Disruption of an active site hydrogen bond converts human heme oxygenase-1 into a peroxidase. J. Biol. Chem. 2001, 276, 10612–10619. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Kofman V.; Fujii H.; Yoshida T.; Ikeda-Saito M.; Hoffman B. M. Catalytic mechanism of heme oxygenase through EPR and ENDOR of cryoreduced oxy-heme oxygenase and its Asp 140 mutants. J. Am. Chem. Soc. 2002, 124, 1798–1808. [DOI] [PubMed] [Google Scholar]
- Lad L.; Wang J.; Li H.; Friedman J.; Bhaskar B.; Ortiz de Montellano P. R.; Poulos T. L. Crystal structures of the ferric, ferrous, and ferrous-NO forms of the Asp140Ala mutant of human heme oxygenase-1: Catalytic implications. J. Mol. Biol. 2003, 330, 527–538. [DOI] [PubMed] [Google Scholar]
- Matsui T.; Furukawa M.; Unno M.; Tomita T.; Ikeda-Saito M. Roles of distal Asp in heme oxygenase from Corynebacterium diphteriae, HmuO: A water-driven oxygen activation mechanism. J. Biol. Chem. 2004, 280, 2981–2989. [DOI] [PubMed] [Google Scholar]
- Caignan G. A.; Deshmukh R.; Zeng Y.; Wilks A.; Bunce R. A.; Rivera M. The hydroxide complex of Pseudomonas aeruginosa heme oxygenase as a model of the low-spin iron(III) hydroperoxide intermediate in heme catabolism: 13C NMR spectroscopic studies suggest the active participation of the heme in macrocycle hydroxylation. J. Am. Chem. Soc. 2003, 125, 11842–11852. [DOI] [PubMed] [Google Scholar]
- Rivera M.; Caignan G. A.; Astashkin A. V.; Raitsimring A. M.; Shokhireva T.; Walker F. A. Models of the low-spin iron(III) hydroperoxide intermediate of heme oxygenase: Magnetic resonance evidence for thermodynamic stabilization of the dxy electronic state at ambient temperatures. J. Am. Chem. Soc. 2002, 124, 6077–6089. [DOI] [PubMed] [Google Scholar]
- Walker F. Magnetic spectroscopic (EPR, ESEEM, Mössbauer, MCD, NMR) studies of low-spin ferriheme centers and their corresponding heme proteins. Coord. Chem. Rev. 1999, 185–186, 471–534. [Google Scholar]
- Chen H.; Moreau Y.; Derat E.; Shaik S. Quantum mechanical/molecular mechanical study of mechanisms of heme degradation by the enzyme heme oxygenase: The strategic function of the water cluster. J. Am. Chem. Soc. 2008, 130, 1953–1965. [DOI] [PubMed] [Google Scholar]
- Takayama S. J.; Ukpabi G.; Murphy M. E.; Mauk A. G. Electronic properties of the highly ruffled heme bound to the heme degrading enzyme IsdI. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 13071–13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Moënne-Loccoz P.; Loehr T. M.; Ortiz de Montellano P. R. Heme oxygenase-1, intermediates in verdoheme formation and the requirement for reduction equivalents. J. Biol. Chem. 1997, 272, 6909–6917. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ortiz de Montellano P. R. Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J. Biol. Chem. 2000, 275, 5297–5307. [DOI] [PubMed] [Google Scholar]
- Matera K. M.; Takahashi S.; Fujii H.; Zhou H.; Ishikawa K.; Yoshimura T.; Rousseau D. L.; Yoshida T.; Ikeda-Saito M. Oxygen and one reducing equivalent are both required for the conversion of α-hydroxyhemin to verdoheme in heme oxygenase. J. Biol. Chem. 1996, 271, 6618–6624. [DOI] [PubMed] [Google Scholar]