

Background: Gα subunit mutants cause different human diseases.
Results: We characterized the Albright's Hereditary Osteodystrophy mutant Gαs-R265H and the equivalent Gαo-R243H cancer mutant to find that they have different biochemical properties.
Conclusion: The same mutation in two homologous Gα-subunits causes either gain-of-function or loss-of-function in two unrelated diseases.
Significance: Not all disease-associated mutations in conserved residues have similar consequences among homologous G proteins.
Keywords: Cancer, Enzyme Kinetics, G Protein-coupled receptor (GPCR), Heterotrimeric G Protein, Oncogene, Albright's Hereditary Osteodystrophy, Pseudohypoparathyroidism Ia
Abstract
There is an increasing number of disease-associated Gα mutations identified from genome-wide sequencing campaigns or targeted efforts. Albright's Hereditary Osteodystrophy (AHO) was the first inherited disease associated with loss-of-function mutations in a G protein (Gαs) and other studies revealed gain-of-function Gα mutations in cancer. Here we attempted to solve the apparent quandary posed by the fact that the same mutation in two different G proteins appeared associated with both AHO and cancer. We first confirmed the presence of an inherited Gαs-R265H mutation from a previously described clinical case report of AHO. This mutation is structurally analogous to Gαo-R243H, an oncogenic mutant with increased activity in vitro and in cells due to rapid nucleotide exchange. We found that, contrary to Gαo-R243H, Gαs-R265H activity is compromised due to greatly impaired nucleotide binding in vitro and in cells. We obtained equivalent results when comparing another AHO mutation in Gαs (D173N) with a counterpart cancer mutation in Gαo (D151N). Gαo-R243H binds nucleotides efficiently under steady-state conditions but releases GDP much faster than the WT protein, suggesting diminished affinity for the nucleotide. These results indicate that the same disease-linked mutation in two different G proteins affects a common biochemical feature (nucleotide affinity) but to a different grade depending on the G protein (mild decrease for Gαo and severe for Gαs). We conclude that Gαs-R265H has dramatically impaired nucleotide affinity leading to the loss-of-function in AHO whereas Gαo-R243H has a mild decrease in nucleotide affinity that causes rapid nucleotide turnover and subsequent hyperactivity in cancer.
Introduction
Heterotrimeric G proteins are molecular switches that alternate between “on” (GTP-bound) and “off” (GDP-bound) states to control signal transduction in eukaryotes (1–3). Signal relay by heterotrimeric G proteins is determined by the lifetime of the GTP-bound form, which is in turn controlled by a delicate equilibrium between the rates of nucleotide exchange and hydrolysis. In resting G proteins, the α-subunit is loaded with GDP and tightly bound to the Gβγ obligatory dimer. G protein-coupled receptors (GPCRs)3 as well as some other non-receptor proteins (4–6) work as guanine nucleotide exchange factors (GEFs) that accelerate the rate of GDP release. In cells, GTP is loaded spontaneously upon GDP release due to the physiological ∼10:1 GTP:GDP concentration ratio, which ensures the directionality of the reaction toward G protein activation. Both GTP-bound Gα and uncomplexed Gβγ subunits regulate the activity of a wide array of effectors. G protein signaling is terminated by the intrinsic GTPase activity of the Gα subunits, which can be further enhanced by GTPase-activating proteins (GAPs). Upon conversion of GTP to GDP + Pi, Gα reassociates with Gβγ to complete a cycle. Given the pivotal role of heterotrimeric G proteins in physiology it is not surprising that dysregulation of the biochemical reactions involved in this process gives rise to human disease. In fact, multiple germline and somatically acquired mutations in Gα subunits that alter their biochemical properties have been described in different human diseases (7, 8).
Albright's Hereditary Osteodystrophy (AHO) was the first hereditary disease associated with mutations in G protein α-subunits (9). Heterozygous loss-of-function mutations in Gαs cause AHO, a syndrome characterized by skeletal and developmental abnormalities including brachydactyly, short stature, obesity, and mental deficits, among others (10). More than 100 different mutations have been described in AHO to date, out of which ∼40 are missense changes (11, 12). An interesting feature of AHO is that maternal inheritance of the Gαs loss of function mutations also produces multihormone resistance whereas paternal inheritance does not. This is caused by imprinting of the gene encoding for Gαs (10–12). The multihormone resistance associated with maternal inheritance of Gαs AHO mutations is termed pseudohypoparathyrodism Ia (PHP-Ia), whereas pseudopseudohypoparathyroidism (PPHP) refers to patients with the features of AHO lacking hormonal resistance. PHP-Ia is characterized by resistance to hormones that exert their action via Gαs-coupled GPCRs, and it is most prominent for thyroid-stimulating hormone (TSH) and parathyroid hormone (PTH). The molecular basis for the loss of function associated with several Gαs mutations found in AHO have been characterized and include defective GTP binding, enhanced GTP hydrolysis, impaired GPCR coupling, and decreased protein stability (10, 13–17).
As opposed to loss of function mutations in AHO, gain of function mutations have been described in cancer for multiple Gα subunits. In addition to mutations in Gαs codons 201 (R→C) and 227 (Q→L), which are found in ∼25% of pituitary adenomas (18), mutations in the same codons have been described for other Gα subunits (19). A notorious example of this is the high frequency (up to 80%) of Gαq or Gα11 mutations in uveal melanomas (20–22). The amino acid residues present in these mutated positions are universally conserved across Gα subunits as is the mechanism by which they trigger oncogenic transformation: i.e. R→C and Q→L mutations promote a gain of function by impairing GTPase activity and rendering the G proteins constitutively active in the GTP-bound conformation. In fact, this high degree of sequence and functional conservation has been traditionally exploited as a tool to investigate the function of G proteins. Artificial mutations equivalent to the oncogenic R→C or Q→L are routinely used to investigate the biological functions of virtually every Gα subunit. For example, introduction of equivalent mutations in Gαo, Gα12, Gα13, or Gαz, which are not found in carcinomas, induces a gain of function and promotes oncogenic transformation when artificially introduced into cultured cells (23–28). R→C and Q→L mutants of Gαi1 were also used in the pioneering crystallographic studies that revealed the structural basis of GTP hydrolysis that are common to all Gα subunits (29).
Cancer genome sequencing campaigns are revealing a far more complex landscape of G protein mutations (8) that contrasts with the reduced number of “classical” G protein oncomutants described above. More than a hundred different mutations have been identified and although some of them occur at conserved positions in the Gα protein sequences, it is still unclear how conserved their impact on G protein function is. Here we compared the functional consequences of a Gαo cancer somatic mutation (R243H) with an identical Gαs-inherited mutation (R265H) present in a family with AHO. We provide evidence indicating that the same mutation in two homologous Gα subunits causes either a gain-of-function or loss-of-function phenotype associated with two unrelated human diseases.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise indicated all reagents were of analytical grade and obtained from Sigma-Aldrich or Fisher Scientific. Escherichia coli strain BL21(DE3) codon+ was purchased from Invitrogen. Pfu ultra DNA polymerase was purchased from Agilent. [γ-32P]GTP and [α-32P]GTP were from Perkin-Elmer.
Patients and Sequence Analysis
Two members of a single family (mother and son, patients II.1 and III.1 in Fig. 2A, respectively) presented skeletal abnormalities of AHO, i.e. brachydactyly assessed by x-ray radiography (30). The son (patient III.1) presented characteristics of abnormal endocrine function (elevated TSH with normal thyroxine (T4)) and was diagnosed with PHP-Ia whereas the mother (patient II.1) had normal endocrine markers and was diagnosed with PPHP. Another member of the family (grandmother, patient I.2, deceased) presented external signs of AHO but was not formally diagnosed.4 Samples from patients II.1 and III.1 were provided by Dr. Bastida Eizaguirre (Hospital Santiago Apostol, Vitoria, Spain) with written consent from the patients and approval by the institutional bioethics committee. We amplified exon 10 of the human GNAS gene by PCR using primers annealing the flanking introns using as template genomic DNA extracted from peripheral blood. The amplicon was run in an agarose gel, purified using a GenJet Gel extraction kit (Thermo) and sequenced.
FIGURE 2.

Gαs R265H is a bona fide loss-of-function mutation in AHO. A, left, pedigree of the family affected by the Gαs R265H mutation. Squares, male; circles, female; black symbols, AHO. The question mark indicates a deceased patient who had external signs of AHO but was not formally diagnosed before death. The table below summarizes clinical features of the two patients with confirmed AHO. Right, sequence analysis of a fragment of Gαs exon 10 displaying a heterozygous substitution of A for G in codon 265. B, Gαs R265H protein expression levels in mammalian cells are similar to Gαs WT. HEK293 cells were transfected with equal amounts of plasmids encoding for Gαs WT or Gαs R265H, lysed, electrophoresed, and immunoblotted with the indicated antibodies. Equal Gαs WT and Gαs R265H plasmid transfection is verified by the equal levels of GFP expression, which is driven by the same promoter as Gαs via an IRES. One experiment representative of three is shown. C and D, Gαs R265H protein half-life in mammalian cells is similar to Gαs WT. HEK293 transfected with Gαs WT or Gαs R265H were treated with cycloheximide (20 μg/nl) for the indicated times, lysed, and immunoblotted with Gαs antibodies. A representative blot is shown in C, and the quantification of two independent experiments (mean ± S.D.) fitted to a single exponential decay curve is shown in D. E, Gαs R265H expressed in mammalian cells has impaired nucleotide binding as assessed by trypsin protection assays. HEK293 cells expressing Gαs WT or Gαs R265H were lysed and incubated in the presence of GDP, GTPγS, or GDP plus AlCl3/NaF (AlF4−) before treatment with trypsin as described in “Experimental Procedures.” Samples were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. One experiment representative of three is shown.
Bioinformatics
Cancer somatic mutations in Gα subunits were searched on Jan 1st 2014 in the cBioPortal (31) or COSMIC (32) databases using the query terms: GNAO1, GNAI1, GNAI2, GNAI3, GNAT1, GNAT2, GNAT3, GNAZ, GNAQ, GNA11, GNA14, GNA15, GNA12, GNA13, GNAL, and GNAS. Visualization and display of protein structure images was done using MolsoftICM (San Diego, CA).
Plasmid Constructs
Rat Gαo (isoform 1; Gαo1, hereafter referred to as Gαo) with a short N-terminal hexahistidine (His) tag was cloned into pET28b as described previously (33). His-tagged bovine Gαs with a short N-terminal His tag cloned into pHIS6 was a kind gift from Dr. Artemyev (University of Iowa) (34). To generate constructs for mammalian expression, Gαs was PCR amplified from pHIS6-Gαs and cloned into the EcoRI and XhoI sites of the plasmid pMSCV-IRES-GFP (Addgene 20672). In this construct GFP expression is driven by the same promoter as the G protein by virtue of an internal ribosome entry site (IRES), making the levels of both transcripts proportional. Point mutations were generated using specific primers following the manufacturer's instructions (QuikChange II, Agilent). All constructs were verified by DNA sequencing.
Cell Culture and Immunoblotting
HEK293 cells were grown at 37 °C in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 1% l-glutamine, and 5% CO2. Equal amounts DNA plasmids were transfected using the calcium phosphate method and cells harvested after 48 h. For the experiments to assess the stability of Gαs WT and Gαs R265H proteins, HEK293 cells transfected with the indicated plasmids were incubated with 20 μg/ml cycloheximide for 0, 0.5, 1, 2, 4, or 6 h prior harvesting. Lysates were prepared by resuspending the cells in lysis buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 0.4% Triton X-100, 1 mm DTT) supplemented with a protease inhibitor mixture (Sigma) and cleared at 14,000 × g for 10 min. Samples were boiled after addition of Laemmli buffer, run by SDS-PAGE and immunoblotted with mouse anti-Gαs (1:500, Gαs (12) Santa Cruz Biotechnology), mouse anti-GFP (1:2,000, Invitrogen), and mouse anti-tubulin (1:2,500, Sigma) antibodies.
cAMP Measurements
Changes in intracellular levels of cAMP were measured by the cAMP-Glo Assay kit (Promega) following the manufacturer's instructions. Briefly, HEK293 cells were transfected with Gαs WT, Gαs R265H, or an empty vector and harvested 48 h later. 20-μl aliquots in duplicate of a 700 × 103 cell/ml suspension were incubated in the presence of IBMX (1 mm) for 15 min at 37 °C. Reactions were stopped using the lysis buffer provided with the kit, and subsequent steps carried out following the manufacturer's instructions to determine the increase in cAMP compared with control cells (empty vector-transfected). Aliquots of the cells were processed in parallel to quantify total amount of protein and verify the expression of the transfected constructs. cAMP levels were normalized by the total amount of protein.
Limited Proteolysis Assay with HEK293 Cell Lysates
Approximately half of the HEK293 cells of a 10 cm dish transfected with Gαs WT or Gαs R265H were lysed in 45 μl of buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 0.4% Triton X-100, 1 mm DTT) supplemented with 125 μm GDP, 125 μm GTPγS, or 125 μm GDP plus 125 μm AlCl3 and 10 mm NaF. Samples were vortexed, incubated in ice for 10 min and centrifuged at 14,000 × g for 10 min. 40 μl of the supernatants were incubated at 30 °C for 30 min to allow the loading of nucleotide. Trypsin (12.5 μg/ml) was added to the tubes and incubated at 30 °C for 5 min. Reactions were stopped by addition of Laemmli sample buffer and boiling. Samples were run by SDS-PAGE and immunoblotted with a rabbit anti-Gαs antibody (1:500, Gαs (C-18) Santa Cruz Biotechnology).
Protein Purification
His-Gαo was purified exactly as described previously (33). His-Gαs was purified from BL21(DE3) codon+ bacteria by nickel affinity chromatography followed by ion exchange chromatography as described in Ref. 35 with minor modifications. Briefly, His-Gαs expression was induced by 0.1 mm IPTG at 23 °C overnight when the OD600 reached ∼0.5. Subsequent steps were exactly as in (35) except that a HiTrapQ HP column (GE Healthcare) was used for the ion exchange chromatography and that proteins were buffer exchanged into 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm MgCl2, 10 μm GDP, 5% (v:v) glycerol, and 5 mm β-mercaptoethanol before storage at −80 °C.
Limited Proteolysis Assay with Purified Proteins
This assay was carried out exactly as described previously (36) for His-Gαo and with minor modifications for His-Gαs. Briefly, His-Gαs (0.2 mg/ml) was incubated for 60 min at 30 °C in the presence of GDP (30 μm), GTPγS (30 μm) or GDP·AlF4− (30–100,000 μm GDP, 30 μm AlCl3, 10 mm NaF). After this incubation, trypsin was added to the tubes (final concentration 80 μg/ml), and samples were incubated for 20 min at 30 °C. Reactions were stopped by adding Laemmli sample buffer and boiling. Proteins were separated by SDS-PAGE and stained with Coomassie Blue.
Steady-state GTPase Assay
This assay was performed as described previously (6, 33, 36).
Measurement of GTPγS Binding by Intrinsic Tryptophan Fluorescence
Purified His-Gαo (1 μm) was equilibrated at room temperature in a cuvette with 1 ml of buffer (20 mm Na-HEPES, pH 8, 100 mm NaCl, 1 mm EDTA, 2 mm MgCl2, 1 mm DTT, 0.05% (w:v) C12E10). GTPγS (2 μm) was added to the cuvette after ∼3 min, and the G protein activation rate monitored by measuring the change in the intrinsic fluorescence (excitation at 284 nm, emission at 340 nm) due to structural rearrangement of the Switch II tryptophan residue Trp-212 (37). Data were collected with a Hitachi F-4500 Fluorescence Spectrophotometer, background corrected (buffer fluorescence) and expressed as % of maximal binding (fluorescence plateau at >40 min). Each protein (WT or mutant) was normalized to its own maximal binding to facilitate the interpretation of the results. Maximal binding values were similar for WT and mutants and are indicated in the figure legends of the corresponding experiment. Fluorescence measurements for His-Gαs (0.25 μm) were performed identically except that the buffer composition was 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm MgCl2, 0.5 μm GDP, 5% (v:v) glycerol, and 5 mm β-mercaptoethanol and that the results were expressed as a ratio of the basal fluorescence (F/F0) because mutants did not reach a maximum.
GDP Release
This assay was carried out essentially as described in Ref. 38. Briefly, His-Gαo (50 nm) was incubated with 0.5 μm, ∼50 cpm/fmol [α-32P]GTP (which is rapidly converted to [α-32P]GDP) for 60 min at 30 °C in 20 mm Na-HEPES, pH 8, 100 mm NaCl, 1 mm EDTA, 25 mm MgCl2, 1 mm DTT, 0.05% (w:v) C12E10. Reactions were initiated by addition of excess (100 μm) unlabeled GDP or GTPγS at 30 °C. Duplicate aliquots (50 μl) were removed at different times, rapidly passed through BA-85 nitrocellulose filters (GE Healthcare) and washed with 4 ml of wash buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 25 mm MgCl2). Filters were dried, and [α-32P]GDP remaining bound to Gαo was quantified by liquid scintillation counting.
RESULTS AND DISCUSSION
Identification of Two Analogous Mutations in Cancer and AHO
We recently characterized a novel oncogenic mutation in Gαo (36). We found that the molecular mechanism by which this gain-of-function mutation, namely R243H, triggers oncogenic transformation differs from any previously characterized oncogenic mutations in other Gα subunits: i.e. it renders the G protein hyperactive by accelerating the rate of nucleotide exchange instead of by impairing GTP hydrolysis (36). The residue corresponding to Gαo Arg-243 is conserved in all 16 human Gα subunits. Bioinformatics searches in the cancer genomics databases cBioPortal (31) and COSMIC (32) revealed somatic mutations affecting this residue in 6 different Gα subunits (Fig. 1A). Remarkably, we did not find in these databases any other residue that was mutated in as many different G proteins (not shown). The 6 mutated α-subunits collectively belong to 3 out of the 4 Gα subfamilies (Gi/o, Gq/11, and G12/13) and the missense changes are either identical (R→H) or different (R→C, W, Q, or L) from that originally found in Gαo (R243H). The high conservation of this residue and the fact that it is altered in different G protein families suggests an impact of its mutation on G protein function. It is tempting to speculate that mutation of this conserved arginine in different Gα-subunits promotes a gain-of-function, hyperactive phenotype analogous to that observed for the previously characterized Gαo R243H oncomutant (36). This would be analogous to what occurs with the “classical” R→C/Q→L oncomutants. However, we came across a clinical case report of an AHO patient (30) bearing a Gαs mutation, namely R265H, analogous to Gαo R243H (Fig. 1A). Because AHO is caused by the acquisition of inherited Gαs mutations leading to a loss-of-function, hypoactive phenotype, this suggests that identical, naturally occurring mutations in homologous α-subunits cause different phenotypes associated with two unrelated human diseases. We set out to investigate this quandary by comparing the properties of the disease-associated mutants Gαo R243H and Gαs R265H.
FIGURE 1.

Identification of disease-linked mutations in a conserved arginine of Gα proteins. A, left, list of cancer somatic mutations in the position corresponding to Gαo Arg-243 in different Gα subunits. Mutations were identified by bioinformatics searches as described in “Experimental Procedures.” The Gαs R265H mutation found in AHO is shown below. Right, alignment of sequences flanking the conserved arginine (in red) from all 16 human Gα subunits. B, left, overlay of the P-loop and SwIII/α3 of active Gαo (blue, PDB: 3C7K) and active Gαs (red, PDB: 1AZT) with a ball and stick representation of Arg-243/265 and Glu-43/50. Nucleotide is shown in yellow. Right, alignment of the P-loop sequence from all 16 human Gα subunits.
As a first step to compare these two mutants we investigated the structural features of this conserved residue. Arg-243 is located in the switch III region of Gαo and the most prominent feature associated with it in the structure of active Gαo is its close proximity to Glu-43, suggestive of an electrostatic interaction between the two residues (Fig. 1B). Glu-43 is located in the P-loop, a highly conserved sequence in Gα subunits which is required for nucleotide binding (39). Because of this, we previously postulated (36) that the increased rate of nucleotide exchange observed in Gαo R243H was due to an alteration of the P-loop associated with the disruption of the electrostatic interaction with Glu-43. However, when we overlaid the structure of active Gαs (40) over that of Gαo (41) no significant differences were observed: i.e. the proximity and orientation of the Arg-265 and Glu-50 in Gαs is almost identical to Arg-243 and Glu-43 in Gαo (Fig. 1B). The overall conformation of the P-loop and SwIII are also virtually identical. These observations suggest that the different functional consequences of the R→H mutation on either Gαo or Gαs do not arise from a dramatic structural difference. In fact, Glu-43 is highly conserved across all Gα subunits with the exception of Gαz (which has unique biochemical properties among α-subunits (42) (Fig. 1B) and the electrostatic contact with Arg-243 is observed in all the Gα subunits crystallized to date (not shown).
Validation of Gαs R265H as a Bona Fide Loss of Function Mutation in AHO
Next we decided to verify the previously described Gαs R265H mutation to rule out a possible error in the assignment of the mutation, which would explain the difference in behavior from Gαo R243H. This mutation, which was reported 12 years ago in a non-English written journal (30) and cited only once (43), has not been seen in other AHO patients or confirmed by others. More importantly, the sequencing data result was not shown in the original report and the description of the mutation was only stated (30). We obtained samples of the two living members of the family with diagnosed AHO, i.e. mother and son (Fig. 2A) directly from the physician in charge of the case. It was not possible to obtain samples of a third member of the family (grandmother) with possible signs of AHO because she was deceased. We amplified by PCR and sequenced a region of the genomic DNA spanning GNAS exon 10. We found two overlapping peaks corresponding to A and C at position 794 of the Gαs coding sequence in the sequencing chromatogram for both mother and son. These findings are consistent with a heterozygous 794A>C mutation and confirm the presence of R265H as an inherited mutation in this family.
AHO-linked mutations are frequently associated with destabilization of Gαs and decreased expression in the cellular environment (10). To test if the stability of Gαs was affected by the R265H mutation we compared the levels of Gαs WT and R265H exogenously expressed in HEK293 cells. To control for the amount of plasmid transfected we monitored GFP expression from an internal ribosome entry site (IRES) under the control of the promoter driving Gαs expression in the same plasmid. We found that Gαs R265H expression was comparable to WT upon transfection of equal plasmid amounts (Fig. 2B), suggesting that the stability of the mutant in cells is not compromised. We further validated this by assessing the half-life of Gαs WT or Gαs R265H proteins expressed in HEK293 cells after blocking protein translation with cycloheximide. Gαs WT and Gαs R265H protein levels decreased at similar rates (t1/2 ∼1.5 h) in the course of the next hours after cycloheximide treatment (Fig. 2, C and D). Taken together, our results indicate that protein destabilization is not the underlying cause of the loss of function in AHO associated with the R265H mutation.
To further investigate the underlying cause of the impaired Gαs function associated with the R265H mutation we analyzed its ability to bind nucleotides in cells. For this we used a well-established assay that monitors G protein activation upon nucleotide binding by measuring its susceptibility to limited trypsinolysis (33, 44). Briefly, inactive, GDP-bound Gα is readily digested by trypsin whereas activation upon GTPγS or GDP-AlF4− binding renders it into a conformation in which only a short N-terminal sequence can be cleaved and the rest remains trypsin-resistant. We expressed Gαs WT and Gαs R265H in HEK293 cells and assessed their susceptibility to proteolysis upon loading with different nucleotides. We found that Gαs R265H protection from trypsinolysis upon GTPγS or GDP-AlF4− loading was dramatically decreased compared with Gαs WT (Fig. 2E). As an alternative assessment of Gαs function in cells we measured changes in intracellular cAMP levels. Expression of Gαs WT in HEK293 cells caused an increase of 60 ± 13 fmol of cAMP/μg protein over the levels in control cells (empty vector-transfected), whereas with Gαs R265H it was only 15 ± 7 fmol of cAMP/μg protein (average ± S.E., n = 3, p < 0.05). These results indicate that Gαs R265H activity in cells is impaired and together with the previous results validate Gαs R265H as a bona fide loss-of-function mutation in AHO.
Gαs R265H Has Impaired Activity in Vitro due to Defective GTP Binding
Next we investigated the biochemical properties of purified His-Gαs R265H and compared it side by side with His-Gαo R243H, which had been previously characterized (36). We first measured the steady-state GTPase activity of His-Gαo and His-Gαs mutants and their corresponding wild-types (WT). It is important to note that Gαo and Gαs WT have very similar rates of nucleotide exchange (k = 0.27 and k = 0.34 min−1, respectively) and nucleotide hydrolysis (k = 2.4 and k = 3.5 min−1, respectively) (45, 46). The steady-state GTPase activity of Gα subunits reflects a reaction that proceeds in two major steps: GDP for GTP nucleotide exchange followed by GTP hydrolysis. It is known (45, 46) that for both Gαo and Gαs WT the GTP hydrolysis step is faster than nucleotide exchange and that GDP release is rate-limiting in this process. Thus, the steady-state GTPase activity of Gαo and Gαs WT reflects the rate of GDP release and subsequent nucleotide exchange. Consistent with our previous findings (36), steady-state GTP hydrolysis by His-Gαo R243H was ∼4–5 fold faster than by His-Gαo WT (Fig. 3A), which is in good agreement with its increased rate of nucleotide exchange (36). On the contrary, steady-state GTP hydrolysis by His-Gαs R265H was virtually suppressed compared with His-Gαs WT (Fig. 3A). These results suggest that R265H mutation dramatically impairs GTP binding by Gαs. However, it is possible that the diminished steady-state GTPase activity observed for Gαs R265H arises from a defect in nucleotide hydrolysis rather than a defect in nucleotide binding/exchange. To rule out this possibility we measured directly the rate of nucleotide binding by monitoring the increase of Gα intrinsic tryptophan fluorescence induced upon GTPγS binding (37). The His-Gαo R243H tryptophan fluorescence time course revealed a rate of GTPγS binding 5–6-fold faster than WT (Fig. 3B), which is in excellent agreement with previous observations using a radioligand binding assay (36). On the other hand, the increase of His-Gαs tryptophan fluorescence upon GTPγS binding was dramatically impaired by the R265H mutation (Fig. 3B). These results indicate that the decreased steady-state GTPase activity observed for Gαs R265H (Fig. 3A) is most likely caused by impaired GTP binding rather than impaired GTP hydrolysis. Moreover, this interpretation is fully consistent with the loss-of-function phenotype commonly associated with AHO mutations, whereas GTPase deficiency would be expected to promote a gain-of-function.
FIGURE 3.
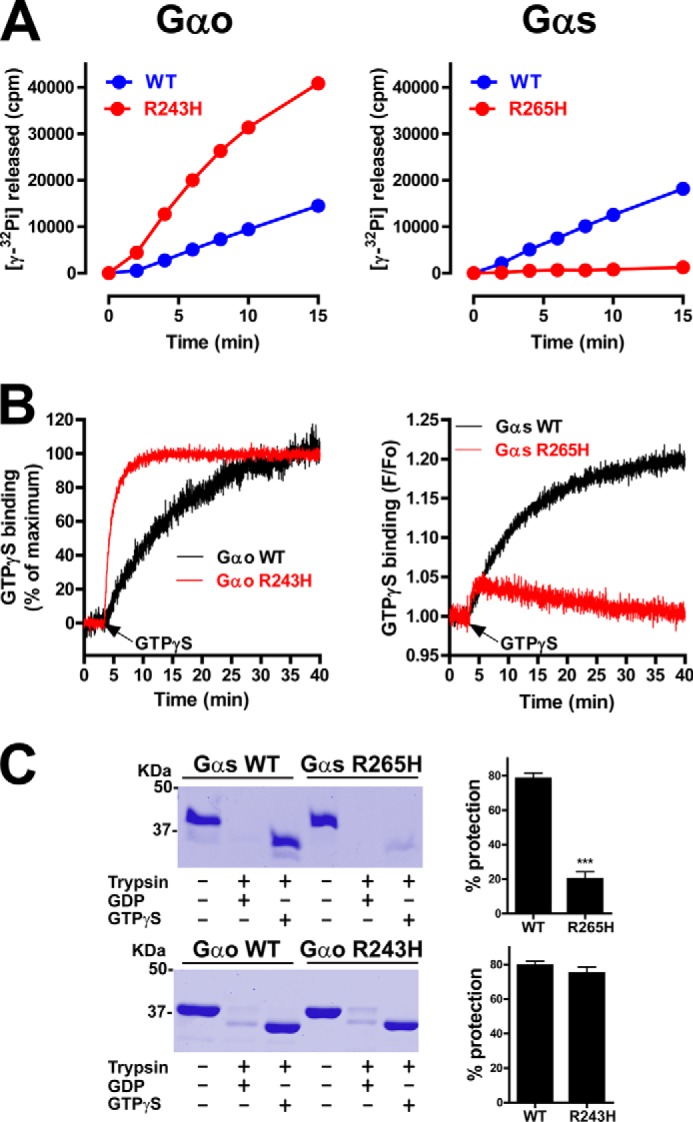
Comparison of the nucleotide binding properties of Gαo R243H and Gαs R265H. A, mutation of Arg-243 to His accelerates the steady-state GTPase activity of His-Gαo whereas mutation of Arg-265 to His in His-Gαs dramatically impairs it. The amount of [γ-32P]GTP (25–50 cpm/fmol) hydrolyzed by Gαo (left) or Gαs (right) WT (blue) or R→H mutant (red) was determined as described in “Experimental Procedures.” Results are shown as mean ± S.D. of one experiment representative of three performed in duplicate. B, mutation of Arg-243 to His accelerates GTPγS binding to His-Gαo whereas mutation of Arg-265 to His in His-Gαs dramatically impairs it. GTPγS binding by Gαo (left) or Gαs (right) WT (black traces) or R→H mutant (red traces) was determined by intrinsic fluorescence measurements as described in “Experimental Procedures.” For Gαo proteins, data are expressed as % of their own maximal binding which was F/Fo = 1.20 and F/Fo = 1.26 for WT and R243H, respectively. One experiment representative of three is shown. C, mutation of R265 to H dramatically impairs His-Gαs protection from trypsin hydrolysis upon GTPγS incubation whereas mutation of Arg-243 to His has no effect on His-Gαo. His-Gαs WT or R265H (top) and His-Gαo WT or R243H (bottom) were incubated in the presence of 30 μm GDP or 30 μm GTPγS before treatment with trypsin as described in “Experimental Procedures.” One representative experiment is shown on the left, and a graph with the mean ± S.E. of three independent experiments is shown on the right.
To further confirm the defect in GTP binding we used limited proteolysis assays as in Fig. 2C but with purified His-Gαs. We found that both His-Gαs WT and His-Gαs R265H were readily digested when loaded with GDP, as expected, but that His-Gαs R265H incubated with GTPγS was only marginally protected from trypsin digestion compared with His-Gαs WT (Fig. 3C). This result is in excellent agreement with that obtained with the same assay on Gαs expressed in cells (Fig. 2C). On the other hand, and consistent with previous observations (36), Gαo WT and R243H were equally protected from trypsin upon incubation with GTPγS. These results not only confirm that Gαs R265H has impaired GTP binding but also that purified His-Gαs R265H recapitulates the biochemical properties of the protein expressed in cells.
Both Gαs R265H and Gαo R243H Bind GDP with Diminished Affinity
To characterize if the impaired GTP binding observed for Gαs R265H is associated with an overall defect in nucleotide binding we performed additional limited proteolysis experiments. Gα subunits are activated by AlF4− in addition than by GTPγS. Because AlF4−-mediated activation requires the presence of GDP in the nucleotide binding pocket (to form the GTP mimetic GDP-AlF4−), we used AlF4− induced protection from trypsinolysis as a surrogate measurement of GDP binding. We found that the R265H mutation dramatically impairs AlF4−-induced protection from trypsinolysis at physiological concentrations (25 μm) of GDP (Fig. 4A). Once again, this is consistent with the result of analogous experiments with Gαs expressed in cells (Fig. 2C). This defect is most likely due to decreased affinity for GDP rather than an intrinsic inability of His-Gαs R265H to adopt an active conformation because it was partially rescued at higher, supraphysiological concentrations of the nucleotide (Fig. 4A). Taken together these results indicate that Gαs R265H has dramatically impaired affinity for both GDP and GTP nucleotides.
FIGURE 4.

GDP binding affinity of Gαs R265H is severely impaired whereas it is mildly reduced for Gαo R243H. A, Gαs R265H is partially protected from trypsin digestion in the presence of AlF4− only at high, supraphysiological concentrations of GDP. His-Gαs WT or R265H were incubated in the presence of 30 μm AlCl3, NaF 10 mm, and the indicated concentrations of GDP before treatment with trypsin as described in “Experimental Procedures.” One representative experiment is shown on the top, and a graph with a graph with the mean ± S.D. of two independent experiments is shown on the bottom. B, mutation of Arg-243 to His accelerates the rate of [α-32P]GDP release from Gαo. [α-32P]GDP-loaded Gαo WT (blue) or Gαo R243H (red) was challenged with excess unlabeled GDP (solid lines) or GTPγS (dashed lines) and [α-32P]GDP remaining bound to Gαo was measured at different time points as described in “Experimental Procedures.” One experiment representative of three is shown. C, schematic diagram summarizing the proposed molecular mechanism underlying the involvement of Gαo R243H and Gαs R265H in disease.
In a previously published work (36), we showed that, contrary to His-Gαs R265H, His-Gαo R243H binds GDP and GTPγS at physiological concentrations as efficiently as His-Gαo WT under steady-state conditions. However, the faster rate of GTPγS binding described for this mutant (36) suggests that it probably binds GDP loosely and releases it at a faster rate than in the WT protein to allow rapid GTP loading. We demonstrated that this is actually the case by directly measuring the rate of GDP-release (Fig. 4B): GDP release in His-Gαo R243H is ∼5-fold faster than in His-Gαo WT. This acceleration of the rate of GDP release is identical when either GTPγS or GDP are used as the displacing competitor (Fig. 4B), further confirming that the faster rate of Gαo R243H activation is due to the unfavorable retention of GDP in its nucleotide binding pocket.
Based on these results we propose a model (Fig. 4C) in which the overall phenotype and disease associated with either Gαs R265H or Gαo R243H arise from a decrease in nucleotide affinity but that it occurs to a different grade depending on the G protein. In Gαo, the R→H mutation promotes a modest decrease in nucleotide affinity that does not preclude nucleotide loading at physiological concentrations. Instead, this mild decrease in affinity favors the rapid exchange of nucleotides and subsequent hyperactivation of the G protein that triggers oncogenesis (36, 43). On the other hand, in Gαs, the R→H mutation translates into a profound decrease of nucleotide affinity that impairs the overall binding of nucleotides even at concentrations several orders of magnitude larger than physiological. As a consequence, Gαs R265H is hypoactive and causes the impaired hormonal responsiveness associated with AHO.
We attempted to dissect the structural basis for the different functional consequences of the R→H mutation in Gαs versus Gαo. We reasoned that this difference could arise from a difference in the amino acid sequence of the two proteins in the region adjacent to the mutation and the nucleotide binding pocket. Among the divergent residues between Gαs and Gαo in this region, only Gαs Arg-258 (which corresponds to His-236 in Gαo) seemed to be able to directly contribute to the nucleotide binding properties: it forms an electrostatic contact with Glu-50 (supplemental Fig. S1A), and its mutation in AHO impairs nucleotide binding (14). We hypothesized that the presence of an Arg in position 258 of Gαs could contribute to the impaired nucleotide binding observed upon R265→H mutation, and that Gαo R243H does not manifest such defect because the corresponding residue is replaced by a histidine (His-236). If this was true, the double Gαo mutant H236R/R243H would mimic the properties of Gαs R265H by displaying impaired nucleotide binding. However, we found that this is not the case because Gαo H236R/R243H bound GTPγS efficiently and with properties analogous to Gαo R243H (supplemental Fig. S1B). This result suggests that other amino acid differences among Gαs and Gαo may account for the observed differences, but this was not investigated further because we could not rationally design additional substitutions predicted to influence nucleotide binding.
Divergent Functional Consequences in Other Disease-related Gα Mutations
To our knowledge the results presented in this work provide the first example of two naturally occurring, identical mutations associated with human disease that render two homologous G proteins with opposite behavior (gain-of-function versus loss-of-function). This contrasts with the “classical” G protein cancer mutants R→C and Q→L, which have highly conserved functional consequences when introduced in virtually any G protein. In addition, the only Gαs mutation associated with AHO (and testotoxicosis) (17) that has been introduced in another Gα subunit (Gαi1, Ref. 47) also seems to have conserved functional consequences. Nevertheless, the behavioral disparity of disease-associated mutants described here may not be an exception. Analysis of the literature (48–50) and the cBioportal/COSMIC databases revealed additional examples of conserved Gα residues that are mutated both in Gαs of AHO patients and in other Gα proteins in cancer (Fig. 5). It is likely that these mutations shared by AHO and cancer represent additional examples of differing mechanisms analogous to that described for the Gαs R265H/Gαo R243H pair.
FIGURE 5.

Identification of mutations in conserved positions of Gα proteins linked to both AHO and cancer. Gα sequence alignment indicating highly conserved residues that are mutated in both cancer and AHO (in red). Mutations were identified by bioinformatics searches as described in “Experimental Procedures.”
To test this idea we investigated the biochemical properties of the Gαs D173N mutant described in AHO, for which analogous mutations have been found in other Gα subunits in cancer (Fig. 5). It is important to note that Gαs D173N has been previously characterized in cells from AHO patients (49), in which it is expressed at normal levels but its function is compromised. Based on this and the fact that Asp-173 is located in the αd-αE loop important for the stabilization of the nucleotide binding pocket we investigated the nucleotide handling properties of the purified protein. We found that Gαs D173N, like Gαs R265H, has greatly impaired GTPγS binding as determined by tryptophan fluorescence measurements (Fig. 6A) and trypsin protection assays (Fig. 6B). It also displayed reduced protection from trypsin upon GDP-AlF4− loading (Fig. 6B), suggesting a defect in GDP binding as well. We compared side by side the properties of the corresponding mutant in Gαo (i.e. D151N) as a surrogate for the cancer mutation found in Gαt2, Gα16, and Gαz. We decided to investigate Gαo instead of the Gα subunits actually mutated in cancer (Gαt2, Gα16, Gαz) because they are extremely challenging to purify in the required quantities. Moreover, their rates of nucleotide binding and hydrolysis are very different from those of Gαs, which would complicate the interpretation of the results. We found that GTPγS binding to Gαo D151N was much faster than to WT (Fig. 6C) and that trypsin protection upon GTPγS or GDP-AlF4− was indistinguishable from that observed in Gαo WT (Fig. 6D). This suggests that the D151N mutation in Gαo leads to a gain-of-function due to accelerated nucleotide exchange. Thus, the results with Gαs D173N/Gαo D151N pair highly resemble the same loss/gain of function dichotomy observed for the Gαs R265H/Gαo R243H pair, thereby supporting the concept that analogous mutation in different Gα subunits can be associated with unrelated diseases.
FIGURE 6.

Comparison of the nucleotide binding properties of Gαs D173N and Gαo D151N. A and C, mutation of Asp-173 to N in His-Gαs dramatically impairs GTPγS binding (A) whereas mutation of Asp-151 to Asn in His-Gαo accelerates it (C). GTPγS binding by Gαs (A) or Gαo (C) WT (black traces) or D→N mutant (red traces) was determined by intrinsic fluorescence measurements as described in “Experimental Procedures.” For Gαo proteins, data are expressed as % of their own maximal binding, which was F/Fo = 1.19 and F/Fo = 1.24 for WT and D151N, respectively. One experiment representative of three is shown. B and D, D173N dramatically impairs Gαs protection from trypsin hydrolysis upon GTPγS or GDP/AlF4− incubation whereas D151N has no effect on Gαo. His-Gαs WT or R265H (B) and His-Gαo WT or R243H (D) were incubated in the presence of 30 μm GDP or 30 μm GTPγS (top) or 30 μm GDP plus AlCl3/NaF (AlF4−) (bottom) before treatment with trypsin as described in “Experimental Procedures.” Gels from representative experiment are shown along with graphs displaying the mean ± S.E. of three independent experiments.
Final Remarks
Overall, the observations reported in this work raise an important issue related to large genome sequencing campaigns like those carried out by The Cancer Genome Atlas. Although the recent advances in DNA sequencing technologies are bound to provide a wealth of information on the molecular basis of disease, the data generated through these efforts must be analyzed with caution to avoid misinterpretation of datasets. Assumptions on the functional outcome of given mutations in systems level analyses must be subjected to rigorous scrutiny even if conserved among homologous proteins,
There are previous examples of identical mutations having significantly different functional consequences depending on the G protein, although all of them are synthetic, man-made mutations. Particularly relevant to the work presented here was the investigation of Gαs R265E in comparison to the same mutation in a Gαt/i chimera (51). The R→E mutation was originally reported to render the Gαt/i chimera with dominant-negative properties, i.e. it is incompetent for GTPγS-mediated signaling activation and irreversibly binds to receptors (52). On the contrary, the same mutation in Gαs renders it a G protein capable of activating effectors upon GTPγS treatment and binds to receptors with unaltered affinity (51). However, a common feature of the R→E mutants among Gαs and Gαt/i as well as with the R→H mutants in the same position described in this work for Gαo and Gαs, is the apparent reduced affinity for nucleotides. Thus, the position corresponding to Arg-265 in Gαs appears to play a critical role in controlling the nucleotide binding properties of multiple, if not all, G proteins, although the precise consequences of mutating it vary from G protein to G protein. It is possible that such differences arise from minor sequence differences found in the SwIII region of different G proteins (51). In summary, this work provides novel information about the importance of conserved G protein mutations in the molecular basis of disease and raises a cautionary note for future research on this topic.
Supplementary Material
Acknowledgments
We thank Dr. Bastida Eizaguirre (Hospital Santiago Apostol, Vitoria, Spain) for providing the AHO patient samples and Dr. Artemyev (U. of Iowa) for providing the pHIS6-Gαs plasmid. We thank Prof. Polgar and Prof. Simmons for giving access to equipment.
This work was supported by the American Cancer Society (RGS-13-362-01-TBE) and the Elsa U. Pardee Foundation (to M. G. M.).

This article contains supplemental Fig. S1.
Dr. Bastida Eizagurre, personal communication.
- GPCR
- G protein-coupled receptor
- GAP
- GTPase-activating protein
- GEF
- guanine nucleotide exchange factor
- AHO
- Albright's Hereditary Osteodystrophy
- PHP-Ia
- pseudohypoparathyroidism Ia
- PPHP
- pseudopseudohypoparathyroidism
- TSH
- thyroid-stimulating hormone
- PTH
- parathyroid hormone
- T4
- thyroxine
- His
- hexahistidine tag
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate.
REFERENCES
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 2. Birnbaumer L. (2007) Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 α subunits plus βγ dimers. Biochim. Biophys. Acta 1768, 772–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Birnbaumer L. (2007) The discovery of signal transduction by G proteins: a personal account and an overview of the initial findings and contributions that led to our present understanding. Biochim. Biophys. Acta 1768, 756–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sato M., Blumer J. B., Simon V., Lanier S. M. (2006) Accessory proteins for G proteins: partners in signaling. Annu. Rev. Pharmacol. Toxicol. 46, 151–187 [DOI] [PubMed] [Google Scholar]
- 5. Tall G. G., Krumins A. M., Gilman A. G. (2003) Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 [DOI] [PubMed] [Google Scholar]
- 6. Garcia-Marcos M., Ghosh P., Farquhar M. G. (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farfel Z., Bourne H. R., Iiri T. (1999) The expanding spectrum of G protein diseases. N. Engl. J. Med. 340, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 8. O'Hayre M., Vázquez-Prado J., Kufareva I., Stawiski E. W., Handel T. M., Seshagiri S., Gutkind J. S. (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patten J. L., Johns D. R., Valle D., Eil C., Gruppuso P. A., Steele G., Smallwood P. M., Levine M. A. (1990) Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright's hereditary osteodystrophy. N. Engl. J. Med. 322, 1412–1419 [DOI] [PubMed] [Google Scholar]
- 10. Weinstein L. S., Yu S., Warner D. R., Liu J. (2001) Endocrine manifestations of stimulatory G protein α-subunit mutations and the role of genomic imprinting. Endocr. Rev. 22, 675–705 [DOI] [PubMed] [Google Scholar]
- 11. Aldred M. A. (2006) Genetics of pseudohypoparathyroidism types Ia and Ic. J. Pediatr. Endocrinol. Metab. 19, 635–640 [DOI] [PubMed] [Google Scholar]
- 12. Bastepe M., Jüppner H. (2005) GNAS locus and pseudohypoparathyroidism. Horm. Res. 63, 65–74 [DOI] [PubMed] [Google Scholar]
- 13. Warner D. R., Romanowski R., Yu S., Weinstein L. S. (1999) Mutagenesis of the conserved residue Glu259 of Gsα demonstrates the importance of interactions between switches 2 and 3 for activation. J. Biol. Chem. 274, 4977–4984 [DOI] [PubMed] [Google Scholar]
- 14. Warner D. R., Weng G., Yu S., Matalon R., Weinstein L. S. (1998) A novel mutation in the switch 3 region of Gsα in a patient with Albright hereditary osteodystrophy impairs GDP binding and receptor activation. J. Biol. Chem. 273, 23976–23983 [DOI] [PubMed] [Google Scholar]
- 15. Warner D. R., Gejman P. V., Collins R. M., Weinstein L. S. (1997) A novel mutation adjacent to the switch III domain of G(Sα) in a patient with pseudohypoparathyroidism. Mol. Endocrinol. 11, 1718–1727 [DOI] [PubMed] [Google Scholar]
- 16. Iiri T., Farfel Z., Bourne H. R. (1997) Conditional activation defect of a human Gsα mutant. Proc. Natl. Acad. Sci. U.S.A. 94, 5656–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iiri T., Herzmark P., Nakamoto J. M., van Dop C., Bourne H. R. (1994) Rapid GDP release from Gsα in patients with gain and loss of endocrine function. Nature 371, 164–168 [DOI] [PubMed] [Google Scholar]
- 18. Landis C. A., Masters S. B., Spada A., Pace A. M., Bourne H. R., Vallar L. (1989) GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340, 692–696 [DOI] [PubMed] [Google Scholar]
- 19. Lyons J., Landis C. A., Harsh G., Vallar L., Grünewald K., Feichtinger H., Duh Q. Y., Clark O. H., Kawasaki E., Bourne H. R. (1990) Two G protein oncogenes in human endocrine tumors. Science 249, 655–659 [DOI] [PubMed] [Google Scholar]
- 20. Lamba S., Felicioni L., Buttitta F., Bleeker F. E., Malatesta S., Corbo V., Scarpa A., Rodolfo M., Knowles M., Frattini M., Marchetti A., Bardelli A. (2009) Mutational profile of GNAQQ209 in human tumors. PLoS One 4, e6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Raamsdonk C. D., Bezrookove V., Green G., Bauer J., Gaugler L., O'Brien J. M., Simpson E. M., Barsh G. S., Bastian B. C. (2009) Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Raamsdonk C. D., Griewank K. G., Crosby M. B., Garrido M. C., Vemula S., Wiesner T., Obenauf A. C., Wackernagel W., Green G., Bouvier N., Sozen M. M., Baimukanova G., Roy R., Heguy A., Dolgalev I., Khanin R., Busam K., Speicher M. R., O'Brien J., Bastian B. C. (2010) Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 363, 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voyno-Yasenetskaya T. A., Pace A. M., Bourne H. R. (1994) Mutant alpha subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene 9, 2559–2565 [PubMed] [Google Scholar]
- 24. Wong Y. H., Chan J. S., Yung L. Y., Bourne H. R. (1995) Mutant α subunit of Gz transforms Swiss 3T3 cells. Oncogene 10, 1927–1933 [PubMed] [Google Scholar]
- 25. Kroll S. D., Chen J., De Vivo M., Carty D. J., Buku A., Premont R. T., Iyengar R. (1992) The Q205LGo-α subunit expressed in NIH-3T3 cells induces transformation. J. Biol. Chem. 267, 23183–23188 [PubMed] [Google Scholar]
- 26. Xu N., Bradley L., Ambdukar I., Gutkind J. S. (1993) A mutant α subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 90, 6741–6745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu N., Voyno-Yasenetskaya T., Gutkind J. S. (1994) Potent transforming activity of the G13 alpha subunit defines a novel family of oncogenes. Biochem. Biophys. Res. Commun. 201, 603–609 [DOI] [PubMed] [Google Scholar]
- 28. Vara Prasad M. V., Shore S. K., Dhanasekaran N. (1994) Activated mutant of Gα 13 induces Egr-1, c-fos, and transformation in NIH 3T3 cells. Oncogene 9, 2425–2429 [PubMed] [Google Scholar]
- 29. Coleman D. E., Berghuis A. M., Lee E., Linder M. E., Gilman A. G., Sprang S. R. (1994) Structures of active conformations of Gi α1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 30. Bastida Eizaguirre M., Iturbe Ortiz De Urbina R., Arto Urzainqui M., Ezquerra Larreina R., Escalada San Martin J. (2001) [Albright hereditary osteodystrophy: identification of a novel mutation in a family]. An. Esp. Pediatr. 54, 598–600 [PubMed] [Google Scholar]
- 31. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forbes S. A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J. W., Futreal P. A., Stratton M. R. (2008) The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr. Protoc. Hum. Genet. Chapter 10, Unit 10 11, John Wiley and Sons, Inc., New Jersey: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garcia-Marcos M., Ghosh P., Ear J., Farquhar M. G. (2010) A structural determinant that renders G α(i) sensitive to activation by GIV/girdin is required to promote cell migration. J. Biol. Chem. 285, 12765–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Natochin M., Artemyev N. O. (1998) A single mutation Asp229 –> Ser confers upon Gsα the ability to interact with regulators of G protein signaling. Biochemistry 37, 13776–13780 [DOI] [PubMed] [Google Scholar]
- 35. Skiba N. P., Bae H., Hamm H. E. (1996) Mapping of effector binding sites of transducin α-subunit using G alpha t/G αi1 chimeras. J. Biol. Chem. 271, 413–424 [DOI] [PubMed] [Google Scholar]
- 36. Garcia-Marcos M., Ghosh P., Farquhar M. G. (2011) Molecular basis of a novel oncogenic mutation in GNAO1. Oncogene 30, 2691–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Higashijima T., Ferguson K. M., Smigel M. D., Gilman A. G. (1987) The effect of GTP and Mg2+ on the GTPase activity and the fluorescent properties of Go. J. Biol. Chem. 262, 757–761 [PubMed] [Google Scholar]
- 38. Ross E. M., Higashijima T. (1994) Regulation of G-protein activation by mastoparans and other cationic peptides. Methods Enzymol. 237, 26–37 [DOI] [PubMed] [Google Scholar]
- 39. Sprang S. R. (1997) G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66, 639–678 [DOI] [PubMed] [Google Scholar]
- 40. Sunahara R. K., Tesmer J. J., Gilman A. G., Sprang S. R. (1997) Crystal structure of the adenylyl cyclase activator Gsα. Science 278, 1943–1947 [DOI] [PubMed] [Google Scholar]
- 41. Slep K. C., Kercher M. A., Wieland T., Chen C. K., Simon M. I., Sigler P. B. (2008) Molecular architecture of Gαo and the structural basis for RGS16-mediated deactivation. Proc. Natl. Acad. Sci. U.S.A. 105, 6243–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Casey P. J., Fong H. K., Simon M. I., Gilman A. G. (1990) Gz, a guanine nucleotide-binding protein with unique biochemical properties. J. Biol. Chem. 265, 2383–2390 [PubMed] [Google Scholar]
- 43. Kan Z., Jaiswal B. S., Stinson J., Janakiraman V., Bhatt D., Stern H. M., Yue P., Haverty P. M., Bourgon R., Zheng J., Moorhead M., Chaudhuri S., Tomsho L. P., Peters B. A., Pujara K., Cordes S., Davis D. P., Carlton V. E., Yuan W., Li L., Wang W., Eigenbrot C., Kaminker J. S., Eberhard D. A., Waring P., Schuster S. C., Modrusan Z., Zhang Z., Stokoe D., de Sauvage F. J., Faham M., Seshagiri S. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 [DOI] [PubMed] [Google Scholar]
- 44. Kleuss C., Raw A. S., Lee E., Sprang S. R., Gilman A. G. (1994) Mechanism of GTP hydrolysis by G-protein α subunits. Proc. Natl. Acad. Sci. U.S.A. 91, 9828–9831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Linder M. E., Ewald D. A., Miller R. J., Gilman A. G. (1990) Purification and characterization of Goα and three types of Giα after expression in Escherichia coli. J. Biol. Chem. 265, 8243–8251 [PubMed] [Google Scholar]
- 46. Graziano M. P., Freissmuth M., Gilman A. G. (1989) Expression of Gsα in Escherichia coli. Purification and properties of two forms of the protein. J. Biol. Chem. 264, 409–418 [PubMed] [Google Scholar]
- 47. Posner B. A., Mixon M. B., Wall M. A., Sprang S. R., Gilman A. G. (1998) The A326S mutant of Giα1 as an approximation of the receptor-bound state. J. Biol. Chem. 273, 21752–21758 [DOI] [PubMed] [Google Scholar]
- 48. Miric A., Vechio J. D., Levine M. A. (1993) Heterogeneous mutations in the gene encoding the α-subunit of the stimulatory G protein of adenylyl cyclase in Albright hereditary osteodystrophy. J. Clin. Endocrinol. Metab. 76, 1560–1568 [DOI] [PubMed] [Google Scholar]
- 49. Freson K., Izzi B., Labarque V., Van Helvoirt M., Thys C., Wittevrongel C., Bex M., Bouillon R., Godefroid N., Proesmans W., de Zegher F., Jaeken J., Van Geet C. (2008) GNAS defects identified by stimulatory G protein α-subunit signalling studies in platelets. J. Clin. Endocrinol. Metab. 93, 4851–4859 [DOI] [PubMed] [Google Scholar]
- 50. Aldred M. A., Trembath R. C. (2000) Activating and inactivating mutations in the human GNAS1 gene. Hum. Mutat. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 51. Zurita A. R., Birnbaumer L. (2008) The same mutation in Gsα and transducin α reveals behavioral differences between these highly homologous G protein α-subunits. Proc. Natl. Acad. Sci. U.S.A. 105, 2363–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pereira R., Cerione R. A. (2005) A switch 3 point mutation in the α subunit of transducin yields a unique dominant-negative inhibitor. J. Biol. Chem. 280, 35696–35703 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.