

Background: A new SNCA mutation, H50Q, has been linked to familial Parkinson disease (PD).
Results: The H50Q mutation does not affect the structure, membrane binding, or subcellular localization of α-Syn but alters its pathogenic properties.
Conclusion: The H50Q mutation increases α-Syn aggregation, secretion, and extracellular toxicity.
Significance: α-Syn mutations contribute to the pathogenesis of PD via multiple mechanisms.
Keywords: α-Synuclein, Amyloid, Membrane, Metal Ion-Protein Interaction, Mitochondria, Protein Aggregation, Mitochondrial Dysfunction, Mutation
Abstract
Over the last two decades, the identification of missense mutations in the α-synuclein (α-Syn) gene SNCA in families with inherited Parkinson disease (PD) has reinforced the central role of α-Syn in PD pathogenesis. Recently, a new missense mutation (H50Q) in α-Syn was described in patients with a familial form of PD and dementia. Here we investigated the effects of this novel mutation on the biophysical properties of α-Syn and the consequences for its cellular function. We found that the H50Q mutation affected neither the structure of free or membrane-bound α-Syn monomer, its interaction with metals, nor its capacity to be phosphorylated in vitro. However, compared with the wild-type (WT) protein, the H50Q mutation accelerated α-Syn fibrillization in vitro. In cell-based models, H50Q mutation did not affect α-Syn subcellular localization or its ability to be phosphorylated by PLK2 and GRK6. Interestingly, H50Q increased α-Syn secretion from SHSY5Y cells into culture medium and induced more mitochondrial fragmentation in hippocampal neurons. Although the transient overexpression of WT or H50Q did not induce toxicity, both species induced significant cell death when added to the culture medium of hippocampal neurons. Strikingly, H50Q exhibited more toxicity, suggesting that the H50Q-related enhancement of α-Syn aggregation and secretion may play a role in the extracellular toxicity of this mutant. Together, our results provide novel insight into the mechanism by which this newly described PD-associated mutation may contribute to the pathogenesis of PD and related disorders.
Introduction
α-Synuclein (α-Syn)4 is a 140-residue natively unfolded protein that is abundantly expressed at the presynaptic terminals (1). Although its physiological functions are not fully known, α-Syn has been implicated in the regulation of synaptic transmission (1) and dopamine biosynthesis (2). Accumulating evidence from neuropathological, animal models, and cell-based studies supports the hypothesis that increased α-Syn expression, misfolding, oligomerization, and fibril formation play central roles in the pathogenesis of neurodegenerative disorders that are collectively referred to as synucleinopathies and include Parkinson disease (PD), PD with dementia, dementia with Lewy bodies, and multiple system atrophy.
In addition to the neuronal degeneration in different brain regions, synucleinopathies are characterized by the presence of intraneuronal inclusions called Lewy bodies and Lewy neurites of which aggregated forms of α-Syn represent the main constituent (3, 4). During the last two decades, three missense mutations linked to familial forms of PD have been identified in the gene encoding α-Syn: A30P (5), E46K (6), and A53T. Subsequent studies in vitro and in vivo revealed that these mutations influence the physiological properties of α-Syn and enhance its oligomerization, fibril formation, and toxicity (7–9).
Recently, two independent groups reported a novel mutation in SNCA encoding a histidine-to-glutamine substitution (H50Q) in patients with a familial form of PD and dementia (10–12). This new PD-linked α-Syn mutation is associated with late onset parkinsonism (60–63 years), and the patients exhibit neuropathological features similar to those observed in carriers of the E46K or A53T mutation, notably the rapidly progressive course of motor symptoms and dementia. Although the mechanism through which the H50Q mutation causes familial PD remains unexplored, the location of the mutated residue (His-50) in the proximity of the short protein loop connecting the two α-helices of the lipid vesicle-bound state suggests that it may affect the conformational properties of α-Syn. His-50 participates in Cu(II) binding through its imidazole group, suggesting that the mutation at this residue may alter α-Syn metal binding in a way that increases the pathogenicity of the protein (13). Together, these observations suggest that the new PD-linked H50Q mutation could significantly affect the structural, aggregation, and physiological properties of α-Syn in ways that may contribute to accelerating neuron loss and the development of PD.
In this study, we set out to determine the effect of the H50Q mutation on the structure, aggregation, fibril formation, and membrane binding of monomeric α-Syn in vitro using nuclear magnetic resonance (NMR), circular dichroism (CD), and a battery of imaging and aggregation assays. To determine how this mutation influences the pathophysiological properties of α-Syn, we examined its effect on α-Syn subcellular localization, secretion, toxicity, and phosphorylation at different residues including Ser-87, Ser-129, and Tyr-125.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant α-Syn
BL21(DE3) cells transformed with a pT7-7 plasmid encoding WT human α-Syn or H50Q mutant were freshly grown on an ampicillin agar plate. Then a single colony was transferred to 50 ml of LB medium with 100 μg/ml ampicillin (AppliChem, Darmstadt, Germany) and incubated overnight at 37 °C with shaking (preculture). The next day, the preculture was used to inoculate 2–4 liters of LB/ampicillin medium. When the absorbance at 600 nm (A600) of the cultures reached 0.7–0.8, protein expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside (AppliChem), and the cells were further incubated at 37 °C for 4 h before harvesting by centrifugation at 5000 rpm in a JLA 8.1000 rotor (Beckman Coulter, Bear, CA) for 20 min at 4 °C. Lysis was performed on ice by resuspending the cell pellet in 40 mm Tris acetate buffer, pH 8.3 containing 1 mm EDTA and 1 mm phenylmethylsulfonyl fluoride (PMSF). Cells were then ultrasonicated (VibraCell VCX130, Sonics, Newtown, CT) with an output power of 12 watts applied in 30-s pulses between 30-s pauses for a total ultrasonication time of 5 min. Cell debris was then pelleted by centrifugation at 20,000 rpm in a JA-20 rotor (Beckman Coulter) for 20 min at 4 °C. The supernatant was then boiled at 100 °C in a water bath for 10 min to precipitate most cellular proteins, which were removed by a second centrifugation step (JA-20 rotor, 20,000 rpm, 4 °C, 20 min). Lysates were finally filtered through 0.45-μm membranes and applied at 1 ml/min on a HiPrep 16/10 Q FF anion-exchange column connected to an ÄKTA FPLC system (GE Healthcare) and equilibrated with 20 mm Tris, pH 8.0. α-Syn was eluted at 3 ml/min by applying increasing concentrations of up to 1.0 m NaCl in 20 mm Tris, pH 8.0 using a linear gradient applied over 10 column volumes. α-Syn eluted around 300 mm NaCl. α-Syn-enriched fractions (as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)/Coomassie Blue analysis) were then pooled and further purified by gel filtration chromatography using a HiLoad 26/60 Superdex 200 column (GE Healthcare) equilibrated with a solution of 50 mm Tris, pH 7.5 and 150 mm NaCl. Proteins were eluted at 2 ml/min; pure fractions were combined and dialyzed against deionized water at 4 °C using a 7-kDa-cutoff dialysis membrane. The protein was subsequently purified by reversed-phase high performance liquid chromatography (HPLC) on a C4 preparative column (Proto 300 C4, 20-mm inner diameter × 250 mm, 10-μm average bead diameter) with a linear gradient of 20–70% B in solvent A over 30 min at a flow rate of 15 ml/min (solvent A was water and 0.1% TFA, and solvent B was acetonitrile and 0.1% TFA). Pure fractions were combined, lyophilized, and stored at −20 °C under inert atmosphere until use. The purity of the proteins was verified by SDS-PAGE, electrospray ionization MS, and reversed-phase ultrahigh performance liquid chromatography.
In Vitro Fibrillization Assay
H50Q and WT α-Syn were incubated at concentrations in the range of 5–45 μm in an initial volume of 800 μl under constant agitation at 1000 rpm for up to 120 h at 37 °C. ThT fluorescence reading was carried out with a ThT concentration of 10 μm and a protein concentration of 1.5 μm in a volume of 70 μl in a pH 8.5 buffer containing 50 mm glycine. A Bucher Analyst AD plate reader was used to measure ThT fluorescence at an excitation wavelength of 450 nm and an emission wavelength of 485 nm. Aliquots taken at different time points were measured in triplicate. ThT kinetic plots were analyzed by nonlinear regression using the following model (14).
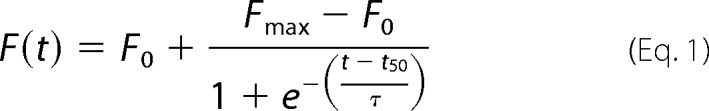 |
where F0 is the initial ThT fluorescence baseline and Fmax is the final ThT fluorescence plateau. Apparent fibril growth rates were calculated as kapp = 1/τ. Fibrillization assays were also monitored by analyzing soluble protein content. To determine the amount of remaining soluble protein, we performed sedimentation assays as follows. 20-μl aliquots withdrawn at different time points were centrifuged at 20,000 × g for 10 min at 4 °C to pellet insoluble aggregates, and then 10 μl of the supernatant was mixed with 10 μl of 2× Laemmli sample buffer (60 mm Tris, pH 6.8, 3.6% (w/v) SDS, 20% (v/v) glycerol, 713 mm 2-mercaptoethanol, 0.004% (w/v) bromphenol blue). 10 μl of the mixture was loaded on 15% polyacrylamide-SDS gels, which were stained with a Coomassie Blue R-250 solution. The relative amounts of soluble protein with respect to the initial conditions were determined by densitometry analysis of the scanned gels with NIH ImageJ (Bethesda, MD). Protein solubility time courses were also fitted with a sigmoidal function.
Preparation of Crude WT and H50Q α-Syn
Recombinant WT or H50Q α-Syn was dissolved in a Tris/NaCl solution (50 mm Tris, pH 7.4, 100 mm NaCl). The protein solution was filtered through a 100-kDa filter to remove small aggregates that might form. The concentration was measured by UV absorption and adjusted with Tris buffer (50 mm Tris, pH 7.4, 100 mm NaCl) to a final concentration of 360 μm. The crude WT or H50Q α-Syn mixture was formed by incubating 70 μl of α-Syn solution (360 μm) under constant agitation (1000 rpm) (Thriller thermoshaker, PEQLAB Ltd., Germany) at 37 °C for 24 h.
Transmission Electron Microscopy
Aliquots taken at various time points were analyzed by transmission electron microscopy. From each sample, 5 μl was spotted on Formvar/carbon-coated 200 mesh copper grids (Electron Microscopy Sciences). The grids were washed twice with 5 μl of ultrapure water, then stained twice with 5 μl of an aqueous 2% (w/v) uranyl formate solution (Electron Microscopy Sciences), and finally vacuum-dried from the edges of the grids. Grids were imaged using a Tecnai Spirit BioTWIN electron microscope operated at 80 kV with a LaB6 source. Digital micrographs were recorded with a 4096 × 4096 FEI Eagle charge-coupled device camera (FEI, Hillsboro, OR).
Gel Filtration Chromatography/Static Light Scattering
Samples (100 μl) were applied at 30 μm onto a Superdex 200 10/300 GL column equilibrated with 50 mm Tris, 150 mm NaCl, and 0.05% (w/v) NaN3, pH 7.5 and eluted at 0.4 ml/min using an Agilent 1200 series HPLC pump (Agilent, Santa Clara, CA). Proteins were detected by monitoring the absorbance at 280 nm (Agilent 1200 VWD). Absolute molecular weights were determined by static light scattering using a Wyatt Dawn Heleos II multiangle light scattering detector (Wyatt Technology Europe GmbH, Dernbach, Germany) connected in series with the UV detector. The protein concentration data used to obtain molecular weights from light scattering data (using the Zimm model) were derived from either refractive index measurements (Wyatt Optilab rEX connected downstream of the light scattering detector) or UV measurements (Agilent 1200 VWD upstream of the light scattering detector). Interdetector delays and band-broadening effects were corrected using the Astra 5.3 analysis software (Wyatt Technology Europe GmbH).
Circular Dichroism Measurements
Protein samples (diluted to 10 μm with water when required) in pH 7.5 phosphate buffer were analyzed either alone or mixed with large unilamellar lipid vesicles using the following protein:lipid mass ratios: 1:0.5, 1:2, 1:5, and 1:10. Samples were analyzed at room temperature using a Jasco J-815 CD spectrometer. An average of 10 spectra was collected in the range of 195–250 nm using a 1.0-mm-optical path length quartz cuvette. Data points were acquired every 0.2 nm in the continuous scanning mode at a speed of 50 nm/min with a digital integration time of 2 s and a bandwidth of 1 nm. Processed spectra were obtained by subtracting the baseline signal due to the buffer and cell contribution from the protein spectra, and a final smoothing was applied (Savitzky-Golay filter; convolution width of 25 data points). For the preparation of large unilamellar vesicles, the negatively charged phospholipid 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-racemic glycerol), denoted 16:0–18:1 phosphatidylglycerol and hereafter referred to as POPG, was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Vesicles were prepared using the extrusion method. Briefly, phospholipids in chloroform were dried using an argon stream to form a thin film on the wall of a glass vial. Any remaining chloroform was removed by placing the vial under vacuum (<0.05 millibar) overnight. The phospholipids were then resolubilized in phosphate buffer to their final concentrations by brief sonication. The solution was then extruded through Avestin LiposoFastTM (Avestin Inc., Ottawa, Ontario, Canada) (membrane pore size, 100 nm) as described previously (15).
NMR Experiments
Isotopically labeled α-Syn H50Q was produced as reported previously (16). The H50Q mutation was introduced into the WT or E20C α-Syn plasmid using a Stratagene site-directed mutagenesis kit (Agilent Technologies, Switzerland). Escherichia coli BL21(DE3) cells expressing the α-Syn H50Q plasmid were first grown in rich medium and then transferred to minimal medium containing either 15N-labeled ammonium chloride or 15N-labeled ammonium chloride and 13C-labeled glucose for production of 15N-labeled or 15N-,13C-labeled protein, respectively, before induction (17). After protein expression, bacteria were lysed by sonication, and the lysate was either directly used for NMR experiments (free-state HNCA) or subjected to further purification via ammonium sulfate precipitation, anion-exchange chromatography, reversed-phase HPLC, and lyophilization (all other NMR experiments). The use of cell lysates for NMR experiments takes advantage of the high NMR signal arising from overexpressed and dynamic synuclein to save time and avoid the possible effects of purification methods.
NMR experiments were carried out on a Varian Inova 600-MHz spectrometer, a Bruker Avance 800-MHz spectrometer, and a Bruker Avance 900-MHz spectrometer, all equipped with cryogenic probes. All experiments were carried out at 10 °C except for SDS-containing samples, which were collected at 40 °C. In all cases, dissolved lyophilized protein was filtered through 100-kDa-cutoff centrifugal filters prior to preparing NMR samples. Protein concentrations were ∼200 μm in the samples for HNCA and paramagnetic relaxation enhancement (PRE) experiments, ∼100 μm in the samples for lipid binding and iron and zinc binding experiments, and ∼70 μm in the samples for copper binding experiments. Backbone amide assignments were transferred from previously published WT assignments (16, 18) for the free and SDS-bound states of the protein using HNCA experiments. The free-state HNCA was collected from fresh lysate of cells overexpressing doubly labeled α-Syn H50Q and had spectral widths of 12, 26, and 26 ppm in the 1H, 15N, and 13C dimensions, respectively. The SDS-bound HNCA was collected on lyophilized purified protein, dissolved in NMR buffer (100 mm NaCl, 10 mm Na2HPO4, pH 6.8), and mixed with SDS to a final concentration of 40 mm. It had spectral widths of 13, 29, and 24 ppm in the 1H, 15N, and 13C dimensions, respectively.
PRE NMR experiments were carried out on α-Syn E20C/H50Q. The lyophilized protein was dissolved in NMR buffer and incubated with 10-fold excess of the paramagnetic spin label S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (Toronto Research Chemicals) to effect conjugation of the spin label. Excess unbound spin label was removed via a buffer exchange column, and the sample was split into paramagnetic (+H2O) and diamagnetic (+2 mm DTT) samples. The intramolecular PRE effect was assayed by comparing cross-peak intensity in matched 1H/15N HSQC spectra of paramagnetic and diamagnetic samples.
Lipid binding was assayed by comparing cross-peak intensity in matched 1H/15N HSQC spectra of lipid-free and lipid-containing (3 mm 15% 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), 25% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 60% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) small unilamellar vesicle (SUV)) samples of α-Syn H50Q. SUVs were prepared by mixing chloroform-dissolved lipids, drying under nitrogen, resuspending in NMR buffer, sonicating, and clarifying by ultracentrifugation. Lyophilized α-Syn H50Q was dissolved in NMR buffer and mixed with SUV stock solutions. Binding to Cu(II) was also assayed by comparing cross-peak intensity in matched 1H/15N HSQC spectra of metal-free and metal-containing (final CuCl2 concentrations, 70, 140, and 280 μm) samples of α-Syn H50Q. Binding to Fe(III) and Zn(II) was assayed by measuring chemical shift differences between matched 1H/15N HSQC spectra of metal-free and metal-containing (final FeCl3 and ZnSO4 concentrations, 1 mm) samples of WT and H50Q α-Syn. Metal ion solutions were prepared in 20 mm PIPES, 20 mm NaCl, pH 6.8 and mixed with lyophilized α-Syn H50Q dissolved in the same buffer following published protocols (18).
Atomic Force Microscopy Imaging
Aggregated α-Syn samples for atomic force microscopy (AFM) studies were prepared by incubating a 45 μm α-Syn solution at 37 °C with shaking at 400 rpm. The starting material was prepared as described above (i.e. including a filtration step through 100-kDa-molecular mass cutoff filters). Analysis by AFM was performed on two differently charged substrates: freshly cleaved negatively charged mica and positively functionalized mica. In the latter case, after the cleaving, the mica substrate was incubated with a 10-μl drop of 0.05% (v/v) (3-aminopropyl)triethoxysilane (APTES; Fluka) in Milli-Q water (Millipore, Switzerland) for 1 min at room temperature, rinsed with Milli-Q water, and then dried by the passage of a gentle flow of gaseous nitrogen. AFM samples were prepared at room temperature by deposition of a 10-μl aliquot in the following manner. For bare mica surfaces, a diluted protein sample (0.5 μm) was deposited on the surface and incubated for 1 min. In the case of APTES-functionalized surfaces, an undiluted sample (45 μm) was deposited and incubated for 10 min. AFM images were acquired on two instruments in ambient conditions: a Nanoscope IIIa (Bruker) operating in tapping mode and equipped with an antimony (type n)-doped silicon tip (Bruker, MPP-12120-10, 5 newtons m−1) with a nominal radius of 8 nm and a Park NX10 operating in true non-contact mode and equipped with a silicon tip (Nanosensor, PPP-NCHR, 40 newtons m−1) with a nominal radius of 7 nm. The first microscope was used to monitor the entire fibrillation process and in particular the early stages, whereas the second was used to consistently compare the morphology of fibrils after 6 days. Image flattening and analysis were performed by SPIP (Image Metrology, Hørsholm, Denmark) software.
In Vitro Phosphorylation Assay
H50Q or WT α-Syn (6 μm) was incubated in a solution of 50 mm Tris pH 7.5, 10 mm DTT, 1 mm ATP, and 1 mm MgCl2 in the presence of 0.42 μg (1 μl) of Polo-like kinase 3 (PLK3) (Invitrogen) in a total volume of 50 μl. For the phosphorylation by casein kinase 1 (CK1), 6 μm protein was incubated in 1× CK1 buffer supplemented with 1 mm ATP and 1000 units of CK1 (1 μl) in a final volume of 50 μl. Phosphorylation assays using the SYK kinase were conducted as described previously (19). The extent of phosphorylation was monitored by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy and Western blotting using anti-Ser(P)-87 (20), -Ser(P)-129 (pSyn 64, Wako), -Tyr(P)-125 (BD Transduction Laboratories), and -Tyr(P)-133 (BD Transduction Laboratories) antibodies.
Mammalian Cell Culture
HEK cells were maintained in GlutaMAX-pyruvate-DMEM (Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen), 100 μg/ml streptomycin, and 100 units/ml penicillin (Invitrogen).
M17 cells were maintained in a nutrient mixture composed of 50% GlutaMAX-pyruvate-DMEM and 50% F-12 (Invitrogen) supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin. HEK cells were transiently transfected using the standard calcium phosphate transfection protocol (21). HeLa and M17 cells were transfected with Effectene (Qiagen, Switzerland) following the manufacturer's instructions.
α-Syn Phosphorylation in Mammalian Cell Lines
HEK cells were transiently co-transfected with 1 μg of α-Syn and 0.5 μg of PLK2 or GRK6 using the standard calcium phosphate transfection protocol. Cells were harvested 24 h post-transfection and lysed in 20 mm Tris base, pH 7.4 containing 150 mm NaCl, 1 mm EDTA, 0.25% Nonidet P-40, 0.25% Triton X-100, and 1 mm PMSF (Sigma) and 1:200-diluted protease inhibitor mixture (Sigma). Insoluble particles were pelleted by centrifugation at 14,000 × g for 20 min at 4 °C. Cleared lysates were collected, and the total amount of protein in the supernatant was estimated with the BCA protein assay kit from Thermo Scientific (Switzerland) according to the manufacturer's instructions. Proteins diluted in 5× Laemmli buffer (300 mm Tris, pH 6.8, 60% (v/v) glycerol, 18% (w/v) SDS, 3.56 m 2-mercaptoethanol, 0.02% (w/v) bromphenol blue) were separated by 15% SDS-PAGE and transferred onto nitrocellulose membranes (Fisher Scientific). Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences GmbH, Germany) diluted 1:3 in phosphate-buffered saline (PBS) for 30 min and then incubated with the appropriate antibodies. Specific signals were revealed by the LI-COR Odyssey infrared scanner. The level of phosphorylated protein was estimated by measuring the Western blot band intensity using NIH ImageJ and normalized to α-Syn and actin as follows: Ser(P)-129/(α-Syn/actin). Detection of the overexpressed kinases was performed using anti-PLK2 (Santa Cruz Biotechnology) and anti-GRK6 (Santa Cruz Biotechnology).
Subcellular Fractionation
HEK293T cells for subcellular fractionation experiments were plated in 6-well plates. The cells were then transfected with plasmids encoding WT or H50Q α-Syn. Cells were harvested 24 h post-transfection and fractionated as described previously (22). Briefly, the cells were sequentially fractionated using a ProteoExtract® subcellular proteome extraction kit (Calbiochem) to generate the cytosolic, membrane-enriched, and nuclear fractions. The purity of the fractions was further validated by assessing the expression level of housekeeping proteins specific for each fraction (PARP1a for the cytosolic fraction GRP78 membrane fraction and HSP90 and histone H1 for nuclear fractions).
Preparation of Conditioned Medium (CM) and Quantification of α-Syn Secretion
The cells were cultured and transfected as described above. The cell culture medium was replaced with fresh medium containing 2% FBS and 1% penicillin/streptomycin and incubated for 30 h at 37 °C in 5% CO2 starting 48 h post-transfection. The cell culture medium was collected, and the cells were lysed as described above to assess the expression of the endogenous protein. The cell culture medium was sequentially centrifuged at 1000 and 10,000 × g for 5 and 30 min, respectively, to remove debris and dead cells. The supernatant was concentrated 6× using a 10-kDa-cutoff filter (VivaSpin®), and an equal volume of each sample was resolved by 15% polyacrylamide gel electrophoresis followed by Western blot analysis.
Primary Culture of Hippocampal Neurons and Transfection
Hippocampal neurons were prepared from P1 mice pups (Harlan Laboratories) as described previously by Steiner et al. (23) and plated at a density of 350,000 cells/well on polylysine-coated coverslips in growth medium (minimum Eagle's medium with 20 mm glucose, 0.5 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% horse serum).
For neuronal transient transfection, 10-day in vitro neurons were transfected using LipofectamineTM 2000 reagent (Invitrogen) with 0.5 μg of a subcellular localization vector encoding fluorescent YFP fused with the mitochondrial subunit VIII of human cytochrome c oxidase (YFP-mito) (BD Biosciences) in combination with 0.5 μg of empty plasmid, WT α-Syn, or H50Q α-Syn following the manufacturer's instructions. To enhance mitochondrial fragmentation, neurons transfected with YFP-mito were incubated for 2 h with 1 μm staurosporine (Merck4Biosciences, Germany).
Treatment of M17 Cells and Primary Hippocampal Neurons with Recombinant α-Syn
Aggregated (crude) WT α-Syn or H50Q α-Syn was diluted to a final concentration of 10 μm in preheated culture medium. The original culture medium was removed from the cells (M17 or primary hippocampal neurons) and replaced by medium spiked with recombinant α-Syn. Cells were then incubated (37 °C, 5% CO2) for 4 (M17) or 6 days (primary hippocampal neurons) before cell death quantification.
Quantification of Cell Death in Mammalian Cell Lines
The percentage of the cell population permeable to the vital dye propidium iodide (PI) (Invitrogen) was used to quantify cell death in HeLa and M17 cells transfected with either WT α-Syn or H50Q α-Syn or directly treated with extracellular recombinant α-Syn. The supernatant and adherent cells were collected separately at 24, 48, and 72 h post-transfection. After 5 min of centrifugation at 250 × g, cells were resuspended in PBS containing PI at 5 μg/ml. The cells were then analyzed using fluorescence-activated cell sorting (FACS) (Accuri, BD Biosciences). Flow cytometry data were analyzed with FlowJo software (TreeStar).
Quantification of Cell Death in Primary Hippocampal Neurons
The percentage of the cell population permeable to the vital dye SYTOX Green (dead cell stain; Invitrogen) was used to quantify cell death in primary hippocampal neurons. After 6 days of treatment with crude extracellular WT α-Syn or H50Q α-Syn, SYTOX was added to primary hippocampal neurons at a final concentration of 3 nm. The cells were incubated for 30 min at 37 °C before being washed twice with PBS. The total fluorescence of each well was measured by fluorescence top reading with excitation and emission wavelengths of 487 and 519 nm, respectively, using a Tecan Infinite M200 Pro plate reader.
Immunofluorescence and FACS
HeLa cells and M17 cells were fixed in 4% paraformaldehyde in PBS for 20 min at 4 °C at 24, 48, and 72 h post-transfection. After a blocking step with 3% bovine serum albumin in PBS, pH 7.6 with 0.1% Triton X-100 (PBS-T) for 30 min at room temperature, cells were incubated with the mouse monoclonal anti-α-Syn primary antibody for 1 h at room temperature. Cells were rinsed five times in PBS-T and then incubated with the secondary anti-mouse Alexa Fluor 488 antibody. Cells were washed five times in PBS-T and examined by FACS in the Fl-1 channel. Flow cytometry data were analyzed with FlowJo software.
For primary culture, at 48 h post-transfection, neurons were fixed for 15 min at room temperature in a solution (pH 7.4) containing 4% paraformaldehyde and 4% sucrose in PBS. Neurons were then washed three times in MTBS buffer (66 mm NaCl, 100 mm Tris-HCl, pH 7.4) and incubated overnight at 4 °C with primary antibody in 3% bovine serum albumin, 0.3% Triton X-100 in MTBS buffer. The day after, cultures were rinsed three times in MTBS buffer and incubated with the secondary antibody for 2 h at room temperature. After three washes in MTBS buffer, coverslips were mounted on glass slides with DABCO medium and imaged with a Zeiss LSM700 upright confocal microscope.
Transfection of Differentiated SHSY5Y Cells and Preparation of CM
SHSY5Y cells were cultured in 10-cm dishes and differentiated with 10 ml of DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 50 μm retinoic acid in a humidified incubator at 37 °C. To maintain the cells in the differentiated state, the growth medium was renewed every 2 days until the day of experiment. The cells were then electroporated with 10 μg of DNA and kept at 37 °C for 48 h to allow optimal expression of the transgene.
At 48 h post-transfection, the cells were washed two times with prewarmed DMEM and incubated for 18 h with fresh DMEM without FBS. The culture supernatant was collected and centrifuged at 1000 × g for 10 min at 4 °C to pellet cell debris. The supernatant was centrifuged again at 10,000 × g for 15 min at 4 °C, and the CM was collected and processed immediately for analysis by Western blot and enzyme-linked immunosorbent assay (ELISA).
The cells were harvested and lysed in lysis buffer (1% Triton X-100 in cold PBS supplemented with protease inhibitor mixture (Sigma)). After 10 min of incubation on ice, the mixture was centrifuged at 16,000 × g for 10 min at 4 °C. The supernatant (Triton-soluble fraction) was collected in a new, clean tube, and the pellet was resuspended in 1× SDS loading buffer (Triton-insoluble fraction). The concentrations of the samples were determined by BCA protein assay (Thermo Scientific), and equal amounts of protein per sample were resolved by 12% polyacrylamide gel electrophoresis and processed for Western blot analysis.
To assess the membrane integrity, lactate dehydrogenase cytotoxicity assays were performed in cell culture medium using the lactate dehydrogenase cytotoxicity kit (Takara Bio Inc., catalog number MK401) following the manufacturer's instructions. Briefly, 20 μl of crude CM was diluted 5× in PBS and incubated for 30 min at room temperature with equal amounts of catalyst solution (diaphorase/NAD+) in the dark. The enzymatic reaction was quenched with 2 n HCl, and its absorbance was immediately quantified at 490 nm.
Total α-Syn ELISA
ELISAs for the detection of α-Syn in cell culture medium were performed in 96-well plates (BD Biosciences). The plates were coated with the capture antibody, SYN-1, diluted 1:250 in 50 mm sodium carbonate buffer, pH 9.6. The plates were brought to room temperature and washed four times with 200 μl of wash buffer (PBS with 0.5% Tween 20) and then filled with the blocking buffer (SuperBlock T20, Pierce, catalog number 37516) for 1 h at room temperature with gentle shaking (150 rpm). The plates were washed again four times and incubated with 100 μl of samples diluted 1:5 in the blocking solution for 1.5 h together with standards (serial dilutions of recombinant α-Syn monomers from 100 ng/ml stock solution). The plates were washed thoroughly four times with the washing solution and incubated with 100 μl of biotinylated 274 antibody (1 μg/ml) for 1.5 h. Excess detection antibody was removed by washing four times with PBS-T. The plates were further incubated for 1 h with 100 μl of Extravidin peroxidase (Sigma-Aldrich) diluted 1:1500 in the blocking buffer. Finally, the 3,3′,5,5′-tetramethylbenzidine substrate (Sigma-Aldrich) was added to the wells and incubated for 10 min at room temperature with shaking (250 rpm), and the enzymatic reaction was quenched with excess 2 n H2SO4. Absorbance was then read at 450 nm in an ELISA plate reader.
Statistical Analysis
The experiments were repeated three times, yielding the same pattern of results. Statistical analysis was performed using Student's t test. Samples were regarded to have an equal variance unless the F-test returned a p value <0.05. Data were regarded as statistically significant if the p value was <0.05 based on the t test (unpaired, two-tailed distribution). In cases where multiple comparisons were performed, statistical significance was assessed by one-way ANOVA followed by a Tukey-Kramer post hoc test.
RESULTS
The H50Q Mutation Induces Little Perturbation of α-Syn Structure and Oligomeric State in Solution
To determine whether the H50Q mutation affects the biophysical properties of α-Syn, we prepared recombinant WT and H50Q α-Syn and purified both proteins to >98% as assessed by SDS-PAGE, reversed-phase HPLC, and MALDI-TOF (data not shown). Both WT and H50Q α-Syn behaved as unfolded proteins (in phosphate buffer and in the absence of lipid vesicles) as determined by CD spectroscopy (Fig. 1A). Gel filtration chromatography coupled to static light scattering measurements showed that H50Q α-Syn behaved as a monomer and exhibited elution and hydrodynamic properties similar to those of the WT protein. The measured molecular masses of both WT and H50Q α-Syn were also consistent with that of monomers (WT, 13.3 ± 0.27 kDa; H50Q, 13.2 ± 1.05 kDa; Fig. 1B). Furthermore, both proteins co-eluted on the Superdex 200 gel filtration column (WT, VE = 14.04 ml (t = 35.1 min); H50Q, VE = 13.96 ml (t = 34.9 min); Fig. 1B), suggesting that the mutation did not significantly perturb the overall shape or size of the protein.
FIGURE 1.

Biophysical characterization of recombinant WT and H50Q α-Syn. A, CD spectra of recombinant WT (black) and H50Q (red) α-Syn in phosphate buffer. B, gel filtration chromatography with online multiangle static laser light scattering analysis of recombinant WT (black) and H50Q (red) α-Syn. C, 1H/15N HSQC spectra of α-Syn WT (black) andH50Q (red) in aqueous buffer. D, plot of averaged amide chemical shift difference (Δδavg = √(½(ΔδHN2 + (ΔδN/5)2) between the free state H50Q and WT spectra by residue number. The line shows a 3-residue average. E, plot of α-carbon secondary shifts (difference between measured chemical shift and random coil chemical shift) for α-Syn WT (black) and H50Q (red) in aqueous buffer by residue number. The lines show 3-residue averages. F, plot of the ratio of amide cross-peak intensity in paramagnetically labeled samples to the intensity in diamagnetic samples of E20C α-Syn (black) and E20C/H50Q α-Syn (red). The lines show 5-residue averages. deg, degrees; Abs, absorbance.
To further investigate the effect of the H50Q mutation on the conformational properties of α-Syn, we used NMR spectroscopy. Fig. 1C shows an overlay of 1H/15N HSQC spectra for WT and H50Q α-Syn. The corresponding plot of per residue chemical shift differences for amide cross-peaks is shown in Fig. 1D. These data show that structural differences in the free state were limited to the few (±3) residues around the mutation site. Furthermore, secondary structure propensities were evaluated by α-carbon secondary shifts and show little perturbation from the WT protein (Fig. 1E). In addition, the previously shown N- to C-terminal contacts in WT α-Syn were preserved as observed by PRE NMR experiments (Fig. 1F). Specifically, PRE effects in the C-terminal tail of the protein are essentially unaltered in the H50Q mutant. Small local variations in PRE effect in other regions (for example near positions 40, 50, and 80) are estimated to be within the error of the experiment. Together, these data strongly suggest that the H50Q mutation induces little perturbation of α-Syn structure in solution.
WT and H50Q α-Syn Exhibit Similar Interactions with Negatively Charged Lipids
Among the previously characterized PD-linked mutations (A30P, E46K, and A53T), only A30P has been shown to attenuate α-Syn interactions with negatively charged phospholipids in vitro (24, 25), in cell culture (26), and in vivo (27, 28). To investigate whether the H50Q mutation could affect α-Syn membrane binding in vitro, we first performed CD measurements of different mixtures of proteins and artificial liposomes because α-Syn is known to adopt α-helical conformations upon binding to lipid membranes (16). We incubated recombinant WT or H50Q α-Syn with 100-nm-diameter POPG unilamellar vesicles as a membrane model. Analysis of protein/lipid mixtures containing protein:lipid mass ratios of 0.5:1 and 5:1, revealed that the canonical α-helix CD signal of α-Syn increased with increasing concentration of lipid vesicles with no significant difference between the WT protein and the H50Q mutant (Fig. 2, A and B).
FIGURE 2.

In vitro membrane binding analyses. A, CD spectra of recombinant WT α-Syn with increasing concentrations of 100-nm-diameter POPG vesicles. B, CD spectra of recombinant H50Q α-Syn with increasing concentrations of 100-nm-diameter POPG vesicles. C, 1H/15N HSQC spectra of WT α-Syn (black) and H50Q (red) in the presence of 40 mm SDS. D, plot of averaged amide chemical shift difference (Δδavg = √(½(ΔδHN2 + (ΔδN/5)2) between the SDS H50Q and WT spectra by residue number. The line shows a 3-residue average. E, plot of α-carbon secondary shifts (difference between measured chemical shift and random coil chemical shift) for α-Syn WT (black) and H50Q (red) in 40 mm SDS by residue number. The lines show 3-residue averages. F, ratio of amide cross-peak intensities in samples with 3 mm SUVs to the intensity in samples without SUVs for WT α-Syn (black) and H50Q (red) by residue number. deg, degrees.
We then sought to investigate the effect of the H50Q mutation on the lipid- and detergent-bound states of α-Syn in greater detail using NMR techniques. Previous studies on the lipid-bound state of α-Syn have used the anionic detergent SDS as a membrane mimic (18, 29). Fig. 2C shows the 1H/15N HSQC spectra of WT and H50Q α-Syn in the presence of SDS micelles (40 mm SDS) showing no effect of the H50Q mutation on the gross micelle-bound structure of the protein. However, minor changes in NMR resonance positions were observed further out from position 50 (±10 residues) than might be expected for a single amino acid substitution (Fig. 2D). This may indicate some effect on the N-terminal portion of the second micelle-bound helix and the linker region between the two helices. However, analysis of the secondary structure propensities via α-carbon secondary shifts (Fig. 2E) showed no major difference between WT and H50Q. We also investigated the 1H/15N HSQC profiles of both proteins in the presence of SUVs composed of 15:25:60 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS):1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE):1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). At a lipid:protein molar ratio of about 30:1, the binding profile and extent of binding of H50Q α-Syn were similar to those of the WT protein (Fig. 2F). Together, these results indicate that any effects that the H50Q mutation may exert on the interaction of α-Syn with membranes are subtle at most.
The H50Q Mutation Accelerates α-Syn Fibril Formation in Vitro
Previous studies have consistently shown that the PD-linked mutations (A30P, E46K, and A53T) increase α-Syn oligomerization and fibril formation in vitro (7–9). To determine whether the H50Q mutation affects α-Syn aggregation in vitro, we compared the kinetics and extent of fibrillization by WT and H50Q α-Syn at 37 °C under constant agitation over the concentration range of 5–45 μm. ThT binding kinetics performed on aggregation experiments with various initial protein concentrations (from 5 to 45 μm; Fig. 3) showed that H50Q α-Syn had slightly higher fibril formation rates compared with the WT protein except at 45 μm where the differences between WT and H50Q α-Syn were small (Fig. 3, A–D, black curves) The slightly faster aggregation kinetics of H50Q α-Syn were further demonstrated by quantifying the remaining soluble protein at each time point using an SDS-PAGE/Coomassie staining assay after performing sedimentation assays. At all of the protein concentrations we used, H50Q α-Syn precipitated slightly faster than the WT protein (Fig. 3, A–D, red curves). Similar to the ThT binding data, the effect was generally rather small except when the experiment was performed at a starting protein concentration of 20 μm. Apparent fibril growth rates were quantified from the ThT binding kinetics as described under “Experimental Procedures” and showed that H50Q α-Syn fibrillizes faster than the WT protein at all concentrations except at 45 μm; however, differences in fibril growth rates were also not statistically significant (Fig. 3F). Together, these results suggest that H50Q α-Syn slightly accelerates α-Syn fibril formation.
FIGURE 3.

Comparison of WT and H50Q α-Syn fibrillization properties. A, ThT and sedimentation analyses of WT and H50Q α-Syn at a starting concentration of 5 μm. B, C, and D, the same experiments as in A were performed with initial protein concentrations of 10, 20, and 45 μm, respectively. In A–D, black curves correspond to ThT fluorescence kinetics (left ordinate axes), and red curves correspond to quantification of soluble protein content by sedimentation analysis (right ordinate axes). Dashed lines indicate WT α-Syn, and solid lines indicate H50Q α-Syn. Each curve is a representative specimen from at least three independent experiments. E, quantification of apparent fibril growth rates based on ThT fluorescence curves. Error bars represent mean ± S.E. A.U., absorbance units.
To further characterize the effect of the H50Q mutation on α-Syn aggregation, we monitored the misfolding and oligomerization of WT and H50Q α-Syn by CD spectroscopy and AFM. CD spectra taken at different time points during the aggregation experiments at a starting protein concentration of 20 μm revealed that H50Q α-Syn transitioned toward β-sheet-rich species faster than the WT protein (Fig. 4A). After 36 h of incubation, WT α-Syn shifted toward β-structures, but random coil signals remained prevalent, whereas H50Q α-Syn displayed a signal consistent with a predominantly β-sheet conformation. At later time points (≥72h), both proteins showed a fully β-sheet character (Fig. 4A). A similar behavior was observed when using a lower initial protein concentration (10 μm; data not shown).
FIGURE 4.

Effect of the H50Q mutation on α-Syn misfolding and oligomerization. A, CD spectra of WT (black) and H50Q (red) α-Syn taken at different time points of an aggregation experiment done at a starting protein concentration of 20 μm. For CD analysis, samples were diluted to 10 μm with water and analyzed in a 1-mm-optical path cell. B, AFM analysis of freshly prepared α-Syn samples at a low concentration of deposition. Panel a, analysis of freshly prepared WT α-Syn deposited at 0.5 μm on bare mica. Top left panel, high resolution height map (scale bar, 100 nm); bottom left panel, zoomed section of the above picture showing typical monomer and dimer particles (scale bar, 10 nm); top right panel, height distribution histogram; bottom right panel, cross-sectional analysis of WT α-Syn monomer (magenta line) and dimer (green line) particles from the bottom left picture. Panel b, the same analyses were performed on freshly prepared H50Q α-Syn samples. Top left panel, high resolution height map (scale bar, 100 nm); bottom left panel, zoomed section of the above picture showing typical monomer and dimer particles (scale bar, 10 nm); top right panel, height distribution histogram; bottom right panel, cross-sectional analysis of H50Q α-Syn monomer (magenta line) and dimer (green line) particles from the bottom left picture. deg, degrees.
We then used AFM to investigate whether the H50Q mutation could affect the early stages of α-Syn oligomerization during the aggregation process. Because it is negatively charged at physiological pH, α-Syn has an affinity toward positively charged surfaces. α-Syn aggregation was imaged on two surfaces, bare mica and mica treated with APTES. Bare mica has a negatively charged surface in physiological solutions and a low topographical roughness, allowing higher resolution imaging. APTES-functionalized mica has a positively charged surface but a higher surface roughness. Both surfaces were used to assess the aggregation of WT and H50Q α-Syn. At a low concentration of deposition (0.5 μm; i.e. low surface coverage), bare mica was used because it is more suitable for the visualization and statistical analysis of the single monomers. Freshly prepared WT and H50Q α-Syn (initial concentration, 45 μm) diluted to 0.5 μm and deposited on bare mica surfaces displayed a mixture of monomeric and dimeric species as well as some higher order oligomers (Fig. 4B). Monomers and dimers from both α-Syn variants displayed similar average diameters and heights (Fig. 4B), but H50Q α-Syn showed more elongated oligomeric structures than WT α-Syn (Fig. 4B). The height histogram in Fig. 4B shows a first height population (monomers) with an average height of 0.3 nm and a second population composed by dimers with an average height of about 0.9 nm. These dimers were always present for both WT and H50Q α-Syn with a higher proportion of dimers in H50Q compared with WT α-Syn (Fig. 4B). Although samples were freshly filtered through 100-kDa membranes (see “Experimental Procedures”), these dimers quickly re-form after the filtration and might be deposited more efficiently than the monomers on the mica surface. No larger species are detectable in significant proportions, thus excluding the presence of large oligomers and/or protofibrils. Because of the limit of the lateral resolution of the AFM tip, it is not possible to obtain reliable lateral diameter estimation of the particles. Thus, the larger structures seen with H50Q α-Syn (Fig. 4B, panel b) correspond to closely associated monomers and dimers in the x-y plane. Together, these data suggest faster rates of β-sheet-rich oligomer formation for H50Q α-Syn compared with its WT counterpart.
Next, we determined whether the structural properties of mature α-Syn amyloid fibrils could be affected by the H50Q mutation. After 6 days of incubation at 37 °C, the fibrils formed by both proteins were observed by AFM on APTES-functionalized mica surfaces (which are more suitable for a statistical characterization of protofibrils and fibrils) (Fig. 5A, panels a and e). H50Q α-Syn fibrillar structures were composed of mainly two populations characterized by average heights of 4 ± 0.4 and 6.2 ± 0.5 nm, respectively (Fig. 5A, panel c) and an average length of 80 ± 30 nm (Fig. 5A, panel d). Our group and others reported previously that the height of α-Syn mature fibrils is within the 6–9-nm range (30). Thus, it is likely that the first population corresponded to protofilament species, whereas the second one represented a more mature fibril species but not the final state of α-Syn assembly because mature α-Syn fibrils are composed of four protofilaments (31). Conversely, AFM images of WT α-Syn fibrils showed mainly a single population with an average height of 4.8 ± 0.5 nm (Fig. 5A, panels f–h), which is consistent with an abundant presence of protofilaments and a paucity of mature fibrils. WT α-Syn fibrillar structures were also characterized by a broad length distribution (150 ± 70 nm; Fig. 5A, panels g and i). Transmission electron micrographs revealed no major morphological differences between the fibrils formed by WT and H50Q α-Syn at early (24 h) or late time points (120 h) (Fig. 5B). In conclusion, the H50Q mutation enhances the rate of α-Syn oligomerization and fibrillization. The differences in the distribution of protofilaments and fibrillar species between the two proteins may explain why the plateau amplitudes in the ThT binding assay for H50Q and WT α-Syn were different.
FIGURE 5.

Analysis of WT and H50Q α-Syn amyloid fibrils. A, AFM analysis of the morphology of fibrillar structures of H50Q (panels a–d) and WT α-Syn (panels e–i) after 6 days of incubation deposited at high concentration (45 μm) on APTES-functionalized mica. Panel a, overview of H50Q α-Syn fibrillar aggregates. Panel b, details of H50Q protofibrils and fibrils with measurement of their cross-sectional dimensions and comparison with oligomer height. Panels c and d, distribution of the average height and length of H50Q fibrillar structures. Panel e, overview of WT α-Syn fibrillar aggregates. Panel f, details of WT protofibrils and fibrils and their cross-sectional dimensions. Panel g, high resolution scan of a WT protofibril and measurement of its cross-sectional dimensions. Panels h and i, distribution of the average height and length of WT fibrillar structures. B, transmission electron microscopy analysis of negatively stained samples of WT and H50Q α-Syn taken after 24 (top) or 120 h (bottom) of agitation at 37 °C. Scale bars, 200 nm. Av., average.
The H50Q Mutation Does Not Affect α-Syn Interaction with Metals at Sites Other than H50
Because the His-50 residue has been implicated in the binding of Cu(II) and several other metal ions (32), we also investigated how the H50Q mutation affected the binding of several metals to α-Syn using NMR. For Cu(II), we monitored the attenuation of amide cross-peaks (due to increased relaxation when a paramagnetic moiety is bound) when the proteins were incubated with increasing concentrations of CuCl2. Fig. 6A shows a titration of α-Syn H50Q with increasing concentrations of Cu(II). Addition of Cu(II) led to amide cross-peak intensity decreases in several regions of the protein, suggesting copper-protein interactions. The binding profile for α-Syn H50Q was similar to the profile we reported previously for the WT protein (Fig. 6A) (18) with deep minima at the N terminus, residue 20, residue 60, and residue 85 and many minima in the C-terminal tail; the only obvious difference was the expected loss of the His-50 binding site. In our previous report, we examined the loss of copper coordination by the His-50 residue by mutation to alanine; the resultant copper binding profile of α-Syn H50A was very similar to that of α-Syn H50Q (Fig. 6B). For Fe(III) and Zn(II), both proteins were incubated in the presence of a 1 mm concentration of each metal ion, and the difference in amide chemical shifts between metal-free and metal-containing samples was measured. For Fe(III), interaction sites with WT α-Syn were observed in the C-terminal region and at the N terminus to a lesser extent, and strong binding was seen at His-50 (Fig. 6C). All of these sites (except His-50) were retained at similar levels in the H50Q mutant (Fig. 6C). Zn(II) binding sites were stronger within the C-terminal region as well as at a binding site around His-50 that was wider than for Fe(III) (up to residue ∼60 for WT α-Syn) (Fig. 6D). This site was lost in the H50Q mutant, but the binding of Zn(II) to the C-terminal region of H50Q α-Syn was also similar to that of the WT protein (Fig. 6D).
FIGURE 6.

1H/15N HSQC NMR-based measurements of the effect of the H50Q mutation on metal binding. A, plot of the ratio of cross-peak intensities in H50Q α-Syn samples containing 1:1 Cu(II):protein (green), 2:1 Cu(II):protein (red), and previously measured WT α-Syn samples containing 2:1 Cu(II):protein (black) (18) to the intensities in copper-free samples by residue number. B, plot of the intensity ratios for H50Q α-Syn containing 2:1 Cu(II):protein (red) compared with previously measured intensity ratios for H50A α-Syn containing 2:1 Cu(II):protein (blue) (17). C, plot of averaged amide chemical shift difference (Δδavg = √(1/2(ΔδHN2 + (ΔδN/5)2) upon the addition of 1 mm Fe(III) (∼10:1 Fe(III):protein) by residue number for both H50Q (red) and WT (black) α-Syn. D, plot of averaged amide chemical shift difference (Δδavg = √(1/2(ΔδHN2 + (ΔδN/5)2) upon the addition of 1 mm Zn(II) (∼10:1 Zn(II):protein) by residue number for both H50Q (red) and WT (black) α-Syn.
The H50Q Mutation Does Not Affect α-Syn Phosphorylation at Serine or Tyrosine Residues in Vitro or in a Cell-based Assay
Phosphorylation at serine residues (Ser-87 and Ser-129) has emerged as an important molecular switch for the regulation of α-Syn conformation, aggregation, subcellular localization, and clearance (33). To investigate the impact of the H50Q mutation on α-Syn phosphorylation at these residues, we performed an in vitro kinase assay by incubating recombinant WT or H50Q α-Syn with PLK3. As we reported previously (22), MALDI-TOF mass spectrometry and Western blot analysis revealed that PLK3 phosphorylated WT and H50Q α-Syn exclusively at Ser-129 (Fig. 7, A and B). Moreover, the incubation with PLK3 induced a total conversion of both WT and H50Q α-Syn toward the phosphorylated form (Fig. 7A) (22). Using another kinase that phosphorylates α-Syn at Ser-87 and Ser-129 residues, CK1 (15), MALDI-TOF analysis revealed two sites of phosphorylation, Ser-87 and Ser-129. This finding was confirmed by Western blotting using a specific antibody raised against these two residues (Fig. 7, A and C). In a cell-based assay, the co-expression of WT or H50Q α-Syn with PLK2 or GRK6 (Fig. 8A) resulted in extensive phosphorylation of both proteins at Ser-129. Interestingly, the quantification did not reveal any effect of H50Q mutation on the extent of α-Syn phosphorylation (Fig. 8B).
FIGURE 7.

In vitro phosphorylation of WT and H50Q α-Syn by PLK2, CK1, and SYK. A, MALDI-TOF-MS analysis of the phosphorylation reaction after 17-h incubation with PLK3, CK1, and SYK. The analysis demonstrated that WT and H50Q α-Syn are quantitatively phosphorylated at Ser-129 after incubation with PLK3. Double (Ser-87 and Ser-129) and triple phosphorylations (Tyr-39, Tyr-125, and Tyr-133) were observed after α-Syn incubation with CK1 and SYK, respectively. B–D, immunoblots of Ser(P)-129, Ser(P)-87, Tyr(P)-39, Tyr(P)-125, and Tyr(P)-133 α-Syn phosphorylated by PLK3, CK1, and SYK.
FIGURE 8.

Cell-based phosphorylation and degradation of α-Syn. A, PLK2- and GRK6-mediated phosphorylation in cell culture. HEK cells were transfected with 3 μg of plasmid DNA for α-Syn and 1 μg of PLK2 or GRK6 and collected 24 h post-transfection. B, Ser(P)-129/α-Syn ratio shows that WT and H50Q α-Syn exhibit the same extent of phosphorylation. C, WT and H50Q α-Syn are both degraded after PLK2 overexpression. Western blotting and optical density quantification revealed that PLK2 overexpression induces a significant decrease of WT and H50Q α-Syn signal. Error bars represent means ± S.D. An ANOVA test followed by a Tukey-Kramer post hoc test was performed. **, p < 0.01 (presence versus absence of the kinase). A.U., absorbance units.
We then extended our analysis to investigate the effect of H50Q mutation on α-Syn phosphorylation at tyrosine residues. In vitro incubation with SYK induced α-Syn phosphorylation at multiple tyrosine residues (Fig. 7A). Western blot analysis demonstrated that SYK phosphorylated WT and H50Q α-Syn at Tyr-39, Tyr-125, and Tyr-133 (Fig. 7D). The H50Q mutation did not affect SYK-mediated α-Syn phosphorylation at the tyrosine residues.
We recently showed that PLK2 overexpression induces clearance of α-Syn through the autophagy-lysosomal pathway (34). To investigate whether the H50Q mutant could affect PLK2-mediated α-Syn turnover, we co-overexpressed WT α-Syn or H50Q α-Syn with PLK2 in HEK cells. Western blot analysis and optical density quantification showed a similar decrease in α-Syn after PLK2 overexpression, demonstrating that H50Q mutation does not affect PLK2-mediated α-Syn turnover (Fig. 8C). Together, these data indicate that H50Q mutation does not affect α-Syn phosphorylation at serine or tyrosine residues in vitro or in cell culture, nor does it affect PLK2-mediated degradation of α-Syn.
H50Q and WT α-Syn Exhibit Similar Subcellular Localizations in Primary Culture and in Mammalian Cell Lines
The position of histidine 50 close to the short protein loop connecting the two α-helices suggests that the His → Gln mutation may influence α-Syn interaction with membranes, which could alter its subcellular localization (35). To further test this hypothesis in a cellular context, we overexpressed WT or H50Q α-Syn and evaluated their subcellular localization in primary hippocampal neurons. The results shown in Fig. 9 reveal similar cytosolic and membranous localization of the two proteins (Fig. 9A). Similar results were observed in a mammalian cell line where WT and H50Q α-Syn exhibited both cytoplasmic and membranous localizations in HeLa cells, suggesting that the H50Q mutation does not affect α-Syn subcellular localization (Fig. 9B). To rule out the possibility that the cellular imaging analysis is not sufficiently robust to reveal a difference in subcellular localization between WT and H50Q α-Syn, we used a biochemical subcellular fractionation assay that detects differences in membrane binding among α-Syn mutants with different membrane affinities (data not shown). Western blot and optical density analyses showed that both WT and H50Q forms were mainly present in the cytosolic and membrane fractions (Fig. 9C), and no differences were observed in α-Syn protein levels between WT and H50Q in the different cellular fractions (Fig. 9D).
FIGURE 9.

The H50Q mutation does not affect α-Syn subcellular localization. Immunocytochemistry in mouse hippocampal primary neurons (A) and mammalian HeLa cells (B) shows that WT α-Syn and the H50Q mutant exhibit similar cytoplasmic and membranous subcellular localization. Biochemical subcellular fractionation showed that the two forms of α-Syn (WT and H50Q) are enriched in the cytosolic and membrane fractions (C), and the optical density analysis (D) (n = 3) confirmed similar levels of the different α-Syn forms in the cytosolic (black columns) and membrane (hatched columns) fractions. The purity of the fractions was further validated by assessing the expression level of housekeeping proteins specific for each fraction (PARP1a for the cytosolic fraction, GRP78 for the membrane fraction, and HSP90 and histone H1 for nuclear fractions). Error bars represent mean ± S.D. Scale bars correspond to 10 μm.
The H50Q Mutation Enhances α-Syn Secretion
Early studies from several research groups have provided strong evidence for α-Syn cell-to-cell transmission in vivo (36–38) and in cell-based assays (38–41). Those findings suggest that this process is essential for promoting the seeding and spreading of pathological α-Syn in a prion-like manner. To assess the impact of the novel H50Q mutation on α-Syn secretion in neuronal cell lines, we overexpressed WT or H50Q α-Syn in differentiated SHSY5Y cells. WT and H50Q showed the same expression level in the Triton X-100-soluble fraction, and no significant changes in high molecular weight aggregates in the Triton-insoluble fraction were observed (Fig. 10). Interestingly, analysis of the CM derived from the cells expressing the H50Q mutation showed a significant increase in secretion of α-Syn (Fig. 10A) as revealed by Western blot densitometry and sandwich ELISA (Fig 10, B and C). Moreover, greater amounts of high molecular weight aggregates were found in the CM with H50Q mutants than with the WT. To rule out the possibility that the H50Q mutant was toxic to the cells during the time course of our experiment, we evaluated cytotoxicity by monitoring the release of lactate dehydrogenase in the cell culture medium. Our data revealed no significant difference in lactate dehydrogenase (Fig. 10D). It is worth noting that the evaluation of the levels of secreted α-Syn relative to lactate dehydrogenase activity also revealed that H50Q in the conditioned medium is significantly higher compared with the levels α-Syn WT, hence confirming the enhanced secretion of α-Syn H50Q (data not shown). All together, these data suggest that the H50Q mutation causes increased secretion of both monomeric and aggregated α-Syn by an unknown mechanism.
FIGURE 10.

The H50Q mutation enhances the secretion of α-Syn and α-Syn aggregation in differentiated SHSY5Y cells. A, differentiated SHSY5Y cells overexpressing WT α-Syn or H50Q were subfractionated into Triton X-100-soluble (TX-Sol) and -insoluble (TX inSol) fractions. Both fractions as well as CM derived from these cells were analyzed by Western blotting. Similar α-Syn expression levels were obtained in Triton X-100- (left panel) and Triton-insoluble fractions (middle panel). The H50Q mutant in contrast showed elevated monomers and higher molecular weight (HMW) aggregates in the CM (right panel). β-Actin was used to assess the amount of total proteins loaded (bottom panel). B, densitometric analysis of α-Syn monomers released into CM normalized to α-Syn in the Triton X-100-soluble fraction. C, sandwich ELISA of total α-Syn in CM showing a significant increase of H50Q compared with WT (t test; *, p < 0.05). D, lactate dehydrogenase (LDH) assay showing no significant difference between the cytotoxicity of WT and H50Q. Error bars represent mean ± S.D.
The H50Q Mutation Exacerbates Neuronal Toxicity of Extracellular α-Syn
Next, we sought to compare the potential toxicity of WT and H50Q α-Syn overexpressed in HeLa and M17 mammalian cell lines. Using the uptake of the vital dye PI as a marker for membrane disruption and toxicity, we showed that neither WT nor H50Q mutant overexpression induced toxicity at 24, 48, or 72h post-transfection in comparison with the luciferase-overexpressing control cells (Fig. 11, A and B). As a positive control, the overexpression of BAX, a proapoptotic protein, was used to induce time-dependent cell death (Fig. 11, A and B). These data are in accordance with the results using the lactate dehydrogenase release in differentiated neuroblastoma (Fig. 10D).
FIGURE 11.

The H50Q mutation exacerbates neuronal toxicity of extracellular α-Syn. A and B, WT α-Syn or its mutant H50Q does not induce cell death when ectopically expressed in mammalian cell lines. HeLa (A) and M17 cells (B) were transfected with a negative control plasmid coding for luciferase (LUC), a positive control plasmid coding for BAX, or a plasmid encoding WT α-Syn or its mutant H50Q. HeLa (A, panel a) and M17 cells (B, panel a) were then harvested 24, 48, or 72 h post-transfection and stained with PI, a vital dye, to score cell death; cells permeable to PI were counted as dead by FACS analysis. Shown are the means of three independent experiments done in triplicate for each construct in HeLa cells (A, panel a) and in M17 cells (B, panel a). An ANOVA test followed by a Tukey-Kramer post hoc test was performed. **, p < 0.005 (luciferase versus BAX). The relative expressions levels of WT and H50Q α-Syn after overexpression were evaluated by FACS analysis in HeLa (A, panel b) and M17 cell lines (B, panel b). C and D, extracellular WT α-Syn and H50Q crude preparations induce cell death in M17 mammalian cells and primary hippocampal neurons. C, M17 cells were treated for 4 days with Tris (negative control) or with a crude preparation of α-Syn recombinant protein (WT or H50Q) containing a mixture of monomers, oligomers, and protofibrils at a final concentration of 10 μm. The cells were then harvested and stained with the vital dye PI. Cell death was scored on the basis of cell population permeable to PI as measured by FACS analysis. Shown is a representative histogram of three independent experiments done in triplicate for each condition (n = 3). *, p < 0.01. An ANOVA test followed by a Tukey-Kramer post hoc test was performed (Tris versus crude α-Syn WT or H50Q). D, primary hippocampal neurons were treated for 6 days with a crude preparation of α-Syn recombinant protein (WT or H50Q) at a final concentration of 10 μm. Cell death was scored on the basis of cell population permeable to the vital dye SYTOX Green as measured by total fluorescence assessed by fluorescence plate reading. Shown is a representative histogram of three independent experiments done in triplicate for each condition (n = 3). An ANOVA test followed by a Tukey-Kramer post hoc test was performed. **, p < 0.005 (crude WT versus crude H50Q); ***, p < 0.00001 (Tris versus crude α-Syn WT or H50Q). Error bars in A–D represent means ± S.D.
Then, motivated by the enhanced secretion of H50Q α-Syn, we sought to assess the toxicity of extracellular α-Syn by the addition of a crude mixture of aggregated recombinant WT or H50Q α-Syn containing a mixture of monomers, oligomers, and protofibrils at a final concentration of 10 μm to the culture medium of the M17 cells. After 4 days, we assessed cell viability using the PI exclusion method. Moderate but significant toxicity was observed when M17 cells were treated with the crude mixture of aggregated α-Syn, but no difference was observed between WT and H50Q (Fig. 11C). Interestingly, analysis of the uptake of SYTOX Green, a fluorescent vital dye, showed that primary hippocampal neurons exhibited a highly toxic reaction after treatment with the crude mixture of WT or H50Q compared with the Tris-treated neurons (Fig. 11D). Strikingly, the treatment with H50Q crude mixture exhibited significantly more neuronal toxicity than that with WT α-Syn. Together, these data show that aggregated forms of α-Syn and H50Q are toxic in neuronal culture, and in the specified conditions, the H50Q crude mixture is more toxic.
The H50Q Mutation Enhances α-Syn-induced Mitochondrial Fragmentation
α-Syn overexpression can alter mitochondrial morphology and functions in cell culture and in vivo. This deleterious effect mediated by α-Syn seems to be enhanced by the PD-linked mutation A53T and correlated with increased vulnerability of mammalian cell lines (42, 43). To determine whether the H50Q mutation affects mitochondrial morphology, we overexpressed WT or H50Q α-Syn or an empty vector together with YFP-mito in mouse hippocampal primary neurons. Cells were fixed, stained, and analyzed 48 h post-transfection by confocal microscopy. As a positive control, we treated the hippocampal neurons with 1 μm staurosporine for 2 h to enhance mitochondrial fragmentation (44). YFP-mito staining revealed the presence of smooth and filamentous mitochondrial network in the majority of the control cells transfected with the empty vector. After staurosporine treatment, the majority of the neurons exhibited dense, punctate mitochondrial fluorescence, reflecting mitochondrial fragmentation (44) (Fig. 12, A and B). As expected, both WT and H50Q α-Syn overexpression induced mitochondrial fragmentation in the transfected neurons (Fig. 12, A and B). Interestingly, the quantification of α-Syn-positive neurons with fragmented mitochondria revealed that H50Q-overexpressing cells had more neurons with fragmented mitochondria compared with WT α-Syn, suggesting that H50Q mutation could enhance mitochondrial dysfunction (Fig. 12C).
FIGURE 12.
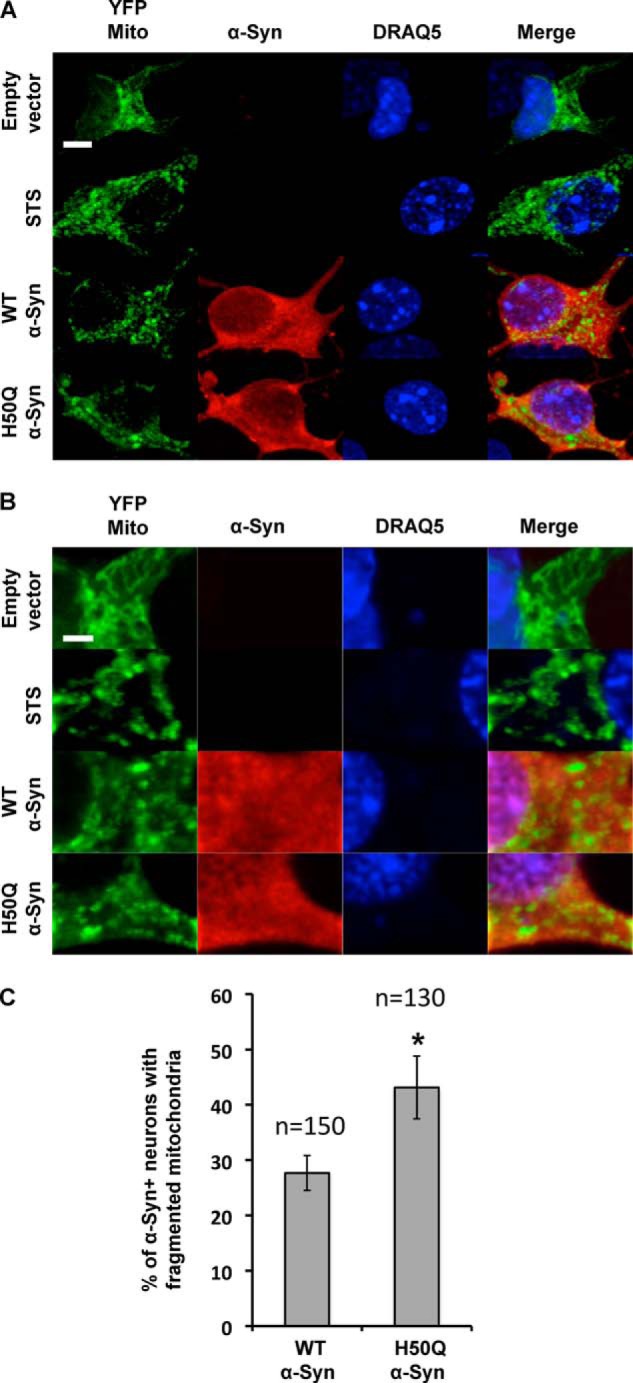
Effect of the H50Q mutation on mitochondrial morphology. A and B, low and high magnification confocal images illustrating the effect of α-Syn overexpression on the mitochondrial morphology in hippocampal primary culture. Overexpression of WT and H50Q α-Syn along with YFP-mito in primary mouse hippocampal neurons shows enhanced mitochondrial fragmentation. As a positive control, treatment with staurosporine (STS) (1 μm) for 2 h induced an increase in the number of neurons that displayed mitochondrial fragmentation. DRAQ5® is a cell-permeable far-red fluorescent DNA dye used to stain the nuclei. Scale bars, 5 μm (A) and 2 μm (B). C, quantification of the α-Syn-positive neurons showing fragmented mitochondria demonstrated that H50Q α-Syn overexpression induced more cells exhibiting mitochondrial fragmentation (n = 3). Error bars represent means ± S.D. An ANOVA test followed by a Tukey-Kramer post hoc test was performed. *, p < 0.005 (H50Q versus WT α-Syn).
DISCUSSION
Studies using different in vitro and in vivo model systems have shown that the PD-linked mutations A30P, E46K, A53T, and G51D affect α-Syn aggregation and toxicity (7–9, 45–47). To gain insight into the mechanisms by which the H50Q mutation contributes to the pathogenesis of PD, we assessed the effect of the His-50 → Gln substitution on α-Syn structure, aggregation, phosphorylation, subcellular localization, secretion, and toxicity.
Our CD spectroscopy and NMR analyses demonstrate that the H50Q mutation does not change the gross structure of the free or membrane-bound state of the protein. In addition, α-Syn H50Q retains the long range intramolecular N- to C-terminal contacts as reported previously for the WT protein (48–50). In comparison, the PD-associated mutations A30P and A53T are reported to have either minor (50) or no (51) effects on such contacts, whereas the E46K mutation enhances such intramolecular interactions (51). The NMR analysis by Ghosh et al. (52) revealed some chemical shift changes in the C-terminal tail of α-Syn that were not seen in this study. These chemical shift changes are likely a result of altered electrostatics in the no-salt buffer used by Ghosh et al. (52) as opposed to the more physiological salt concentration (100 mm NaCl) used here.
The effects of the H50Q mutation on the structure of the lipid-bound protein were assessed in the presence of spheroidal micelles of SDS, a system often used to mimic the lipid-bound state of α-Syn while avoiding the large, NMR-unfriendly size of a protein-vesicle complex. Except for chemical shift perturbations restricted around the mutation site, the HSQC spectra for H50Q and WT α-Syn in the presence of SDS micelles are similar. The secondary structure is also not perturbed by the mutation, which is located near the N-terminal end of the second of the two α-helices in the micelle-bound state of the protein (53). The extended effect of the mutation on amide chemical shifts may be due to the helical nature of the mutation region, which allows the effect of the mutation on chemical shift to propagate further than in a more extended, disordered conformation. It is interesting to note that the chemical shift perturbation region encompasses the N-terminal end of the C-terminal helix as well as the linker region between the two helices. Together, these data demonstrate that the H50Q mutation does not affect the structure of free and membrane-bound α-Syn monomer.
Cu(II) (and other metals) binding to α-Syn has been extensively studied (32, 48) and enhances α-Syn aggregation in vitro (54). NMR studies of WT α-Syn binding to Cu(II) suggested that His-50 forms a major binding site for Cu(II) ions (55). Thus, we expected that the H50Q mutation would affect Cu(II) and possibly other metal binding by α-Syn. However, incubation of Cu(II) with α-Syn H50Q led to localized resonance broadening due to the high dipole moment of the unpaired electrons of the Cu(II) ions. As expected, no broadening was seen near residue 50 due to the loss of the histidine coordination site. Otherwise, the pattern of broadening was similar to that seen for the WT protein with other N- and C-terminal binding sites (for metals such as Fe(III), Mn(II), and Zn(II)) preserved.
Our in vitro aggregation studies of WT and H50Q α-Syn over the concentration range of 5–45 μm revealed that the H50Q mutation accelerates α-Syn oligomerization and fibrillization. This effect is reflected by fact that the H50Q mutants exhibit (i) higher fibril growth rate in a ThT binding assay (Fig. 3), (ii) rapid loss of monomers and precipitation compared with WT α-Syn in the sedimentation assay, and (iii) faster misfolding and formation of oligomers and β-sheet-rich species compared with WT α-Syn as shown by CD and AFM studies. These observations are consistent with a recent report by Ghosh et al. (52) where they showed that H50Q mutation accelerates α-Syn aggregation despite several important differences. Ghosh et al. (52) used a single and much higher protein concentration (300 μm), whereas we showed that the effect of the H50Q mutation persists even at much lower concentrations (5 μm) that are closer to intraneuronal α-Syn concentrations. The fact that we obtained similar results despite performing aggregation experiments under markedly different conditions (such as higher salt concentration and higher agitation speed) compared with those from Ghosh et al. (52) strengthens the conclusion that H50Q enhances α-Syn fibrillization properties. Similarly to Ghosh et al. (52), we confirmed H50Q α-Syn by EM and AFM; our in-depth AFM analyses (using statistical dimensional examination of oligomers and protofilaments) further showed that the H50Q mutation increases oligomer formation and increases the fibril polymerization rate.
To assess the effect of H50Q mutation on the cellular properties of α-Syn, we transiently overexpressed H50Q or WT α-Syn in mammalian cells and compared the subcellular distribution, secretion, and toxicity of the two proteins. In accordance with our previous work, immunocytochemistry and biochemical analysis in mammalian cell lines and primary culture revealed that α-Syn exhibited both cytoplasmic and membranous subcellular localization (22). These analyses revealed that the H50Q mutant exhibits a subcellular localization similar to that of WT α-Syn, which is expected given our observations that the His-to-Gln substitution did not affect α-Syn structure or its interaction with membranes in vitro (Figs. 1 and 2).
Both monomeric and aggregated forms of α-Syn are secreted into cell culture medium (40, 56). Interestingly, we consistently observed higher levels of the H50Q mutant compared with WT α-Syn in the conditioned medium of cells overexpressing WT or H50Q. This increase in extracellular release could be explained by the fact H50Q forms more β-sheet-rich aggregates than WT α-Syn. This hypothesis is supported by recent studies showing that enhancing aggregation increases α-Syn secretion (57).
To determine whether the H50Q mutation influences the toxicity of α-Syn, we first transiently overexpressed this mutant in mammalian cell lines. Our data show that neither H50Q nor WT α-Syn induced cell loss. These data are in accordance with previous work reporting that the overexpression of WT α-Syn or its PD-linked mutant forms in mammalian cell lines does not induce toxicity per se; however, it increases cell vulnerability to various insults (e.g. oxidative stress and dopamine toxicity) (58–60).
The increased release of H50Q α-Syn into the culture medium suggested that the toxic properties of this mutant might at least be partially mediated by its effect on α-Syn toxicity or spreading. Therefore, we compared the extracellular toxicity of H50Q and WT α-Syn in mammalian cell lines and in hippocampal neuron culture. Our results demonstrate that both extracellular aggregated forms of WT and H50Q α-Syn are toxic. Interestingly, exogenous H50Q α-Syn aggregates exhibited more toxicity and induced significantly more cell loss than WT α-Syn. These findings, combined with the increased rate of H50Q secretion, suggest that the enhanced pathogenicity of H50Q could be related to its enhanced oligomerization, secretion, and extracellular toxicity.
The integrity of mitochondria and alterations in the equilibrium between mitochondrial fission and fusion have been linked to the pathogenesis of several neurodegenerative disorders, including PD (61, 62). In our study, the overexpression of human α-Syn induced mitochondrial fragmentation in mouse hippocampal neuronal culture. Interestingly, overexpression of the H50Q mutant form significantly increased the number of cells exhibiting fragmented mitochondria, demonstrating that H50Q mutation may exacerbate mitochondrial dysfunction. A similar effect was observed in neuronal cultures from mice overexpressing another PD-linked mutation, A53T. Together, these data show that the PD-linked mutation H50Q, similar to A53T, exacerbates α-Syn-mediated mitochondrial dysfunctions and suggest that common pathways could mediate the pathogenicity of these mutations.
In conclusion, our results show that the novel PD-linked H50Q mutant does not affect α-Syn structure, membrane interactions, subcellular localization, or phosphorylation by other kinases in vitro or in cell culture. However, substitution of His-50 with Gln accelerates the conversion of α-Syn into β-sheet-rich oligomers, alters the height distribution of the protofilaments and fibrils, and increases α-Syn secretion and extracellular toxicity. These results provide novel insight into the mechanism by which this mutation may contribute to the pathogenesis of PD. Further investigation is required to validate these results and explore the effect of this mutation on α-Syn aggregation, turnover, toxicity, and pathological spreading in in vivo models of synucleinopathies.
Acknowledgments
We thank Nathalie Jordan and Trudy Ramlall for their excellent technical support. The 900 MHz NMR spectrometers were purchased with funds from National Institutes of Health Grant P41GM066354, the Keck Foundation, the New York State Assembly, and the U.S. Department of Defense.
Note Added in Proof
While the present manuscript was under review, an independent study reported that the H50Q mutation in α-Syn, but not other mutations to H50, results in significant conformational changes at the C terminus of the protein (Chi, Y. C., Armstrong, G. S., Jones, D. N., Eisenmesser, E. Z., and Liu, C. W. (2014) Residue histidine 50 plays a key role in protecting α-synuclein from aggregation at physiological pH. J Biol Chem. 289, 15474–15481). These results are in contrast to our work here, where no C-terminal effects of H50Q are detected, and that of Ghosh et. al. (discussed above), who report only very subtle effects. The contrasting results could potentially be explained by metal ion contamination, as this can cause significant spectral changes for the C-terminal region of α-Syn. Chi et. al. also report that the H50Q mutation increases α-Syn aggregation rates in vitro, consistent with our own data described here.
This work was supported, in whole or in part, by National Institutes of Health Grants R37AG019391, CO6RR015495 and P41GM066354, the Keck Foundation, New York State Assembly, the U.S. Department of Defense (to I. D. and D. E.), an ERC Starting Grant 587137 (to H. A. L., A. L. M. M., and B. F.), and EPFL funding (to H. A. L., O. K., A. L. M. M., M. K. M., and F. V.).
- α-Syn
- α-synuclein
- PD
- Parkinson disease
- ThT
- thioflavin T
- POPG
- 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-racemic glycerol)
- PRE
- paramagnetic relaxation enhancement
- HSQC
- heteronuclear single quantum coherence
- SUV
- small unilamellar vesicle
- AFM
- atomic force microscopy
- APTES
- (3-aminopropyl)triethoxysilane
- CM
- conditioned medium
- PI
- propidium iodide
- ANOVA
- analysis of variance
- PLK3
- Polo-like kinase 3
- CK1
- casein kinase 1.
REFERENCES
- 1. Lashuel H. A., Overk C. R., Oueslati A., Masliah E. (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F., Zigmond M. J. (2002) A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 4. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., Iwatsubo T. (1998) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 5. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 6. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 7. Conway K. A., Harper J. D., Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 8. Narhi L., Wood S. J., Steavenson S., Jiang Y., Wu G. M., Anafi D., Kaufman S. A., Martin F., Sitney K., Denis P., Louis J. C., Wypych J., Biere A. L., Citron M. (1999) Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846 [DOI] [PubMed] [Google Scholar]
- 9. Pandey N., Schmidt R. E., Galvin J. E. (2006) The α-synuclein mutation E46K promotes aggregation in cultured cells. Exp. Neurol. 197, 515–520 [DOI] [PubMed] [Google Scholar]
- 10. Appel-Cresswell S., Vilarino-Guell C., Encarnacion M., Sherman H., Yu I., Shah B., Weir D., Thompson C., Szu-Tu C., Trinh J., Aasly J. O., Rajput A., Rajput A. H., Jon Stoessl A., Farrer M. J. (2013) α-Synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813 [DOI] [PubMed] [Google Scholar]
- 11. Proukakis C., Dudzik C. G., Brier T., MacKay D. S., Cooper J. M., Millhauser G. L., Houlden H., Schapira A. H. (2013) A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Proukakis C., Houlden H., Schapira A. H. (2013) Somatic α-synuclein mutations in Parkinson's disease: hypothesis and preliminary data. Mov. Disord. 28, 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dudzik C. G., Walter E. D., Millhauser G. L. (2011) Coordination features and affinity of the Cu2+ site in the α-synuclein protein of Parkinson's disease. Biochemistry 50, 1771–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uversky V. N., Li J., Fink A. L. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 15. Paleologou K. E., Schmid A. W., Rospigliosi C. C., Kim H. Y., Lamberto G. R., Fredenburg R. A., Lansbury P. T., Jr., Fernandez C. O., Eliezer D., Zweckstetter M., Lashuel H. A. (2008) Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J. Biol. Chem. 283, 16895–16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eliezer D., Kutluay E., Bussell R., Jr., Browne G. (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 17. Marley J., Lu M., Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 18. Bussell R., Jr., Eliezer D. (2003) A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 329, 763–778 [DOI] [PubMed] [Google Scholar]
- 19. Hejjaoui M., Butterfield S., Fauvet B., Vercruysse F., Cui J., Dikiy I., Prudent M., Olschewski D., Zhang Y., Eliezer D., Lashuel H. A. (2012) Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies: α-synuclein phosphorylation at tyrosine 125. J. Am. Chem. Soc. 134, 5196–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oueslati A., Paleologou K. E., Schneider B. L., Aebischer P., Lashuel H. A. (2012) Mimicking phosphorylation at serine 87 inhibits the aggregation of human α-synuclein and protects against its toxicity in a rat model of Parkinson's disease. J. Neurosci. 32, 1536–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wigler M., Silverstein S., Lee L. S., Pellicer A., Cheng Y. c., Axel R. (1977) Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell 11, 223–232 [DOI] [PubMed] [Google Scholar]
- 22. Mbefo M. K., Paleologou K. E., Boucharaba A., Oueslati A., Schell H., Fournier M., Olschewski D., Yin G., Zweckstetter M., Masliah E., Kahle P. J., Hirling H., Lashuel H. A. (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Steiner P., Sarria J. C., Glauser L., Magnin S., Catsicas S., Hirling H. (2002) Modulation of receptor cycling by neuron-enriched endosomal protein of 21 kD. J. Cell Biol. 157, 1197–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jo E., Fuller N., Rand R. P., St George-Hyslop P., Fraser P. E. (2002) Defective membrane interactions of familial Parkinson's disease mutant A30P α-synuclein. J. Mol. Biol. 315, 799–807 [DOI] [PubMed] [Google Scholar]
- 25. Wislet-Gendebien S., Visanji N. P., Whitehead S. N., Marsilio D., Hou W., Figeys D., Fraser P. E., Bennett S. A., Tandon A. (2008) Differential regulation of wild-type and mutant α-synuclein binding to synaptic membranes by cytosolic factors. BMC Neurosci. 9, 92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Kim Y. S., Laurine E., Woods W., Lee S. J. (2006) A novel mechanism of interaction between α-synuclein and biological membranes. J. Mol. Biol. 360, 386–397 [DOI] [PubMed] [Google Scholar]
- 27. Jensen P. H., Nielsen M. S., Jakes R., Dotti C. G., Goedert M. (1998) Binding of α-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J. Biol. Chem. 273, 26292–26294 [DOI] [PubMed] [Google Scholar]
- 28. Kuwahara T., Tonegawa R., Ito G., Mitani S., Iwatsubo T. (2012) Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 287, 7098–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ulmer T. S., Bax A., Cole N. B., Nussbaum R. L. (2005) Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 280, 9595–9603 [DOI] [PubMed] [Google Scholar]
- 30. Sweers K., Segers-Nolten I., Bennink M., Subramaniam V. (2012) Structural model for α-synuclein fibrils derived from high resolution imaging and nanomechanical studies using atomic force microscopy. Soft Matter 8, 7215–7222 [Google Scholar]
- 31. Khurana R., Ionescu-Zanetti C., Pope M., Li J., Nielson L., Ramírez-Alvarado M., Regan L., Fink A. L., Carter S. A. (2003) A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 85, 1135–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kowalik-Jankowska T., Rajewska A., Jankowska E., Grzonka Z. (2006) Copper(II) binding by fragments of α-synuclein containing M1-D2- and -H50-residues; a combined potentiometric and spectroscopic study. Dalton Trans. 42, 5068–5076 [DOI] [PubMed] [Google Scholar]
- 33. Oueslati A., Fournier M., Lashuel H. A. (2010) Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145 [DOI] [PubMed] [Google Scholar]
- 34. Oueslati A., Schneider B. L., Aebischer P., Lashuel H. A. (2013) Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–E3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kara E., Lewis P. A., Ling H., Proukakis C., Houlden H., Hardy J. (2013) α-Synuclein mutations cluster around a putative protein loop. Neurosci. Lett. 546, 67–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hansen C., Angot E., Bergström A. L., Steiner J. A., Pieri L., Paul G., Outeiro T. F., Melki R., Kallunki P., Fog K., Li J. Y., Brundin P. (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 121, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kordower J. H., Chu Y., Hauser R. A., Freeman T. B., Olanow C. W. (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506 [DOI] [PubMed] [Google Scholar]
- 38. Desplats P., Lee H. J., Bae E. J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S. J. (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emmanouilidou E., Stefanis L., Vekrellis K. (2010) Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 31, 953–968 [DOI] [PubMed] [Google Scholar]
- 40. Lee H. J., Patel S., Lee S. J. (2005) Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 25, 6016–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee H. J., Suk J. E., Bae E. J., Lee S. J. (2008) Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428 [DOI] [PubMed] [Google Scholar]
- 42. Gui Y. X., Wang X. Y., Kang W. Y., Zhang Y. J., Zhang Y., Zhou Y., Quinn T. J., Liu J., Chen S. D. (2012) Extracellular signal-regulated kinase is involved in α-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol. Aging 33, 2841–2854 [DOI] [PubMed] [Google Scholar]
- 43. Choubey V., Safiulina D., Vaarmann A., Cagalinec M., Wareski P., Kuum M., Zharkovsky A., Kaasik A. (2011) Mutant A53T α-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meuer K., Suppanz I. E., Lingor P., Planchamp V., Göricke B., Fichtner L., Braus G. H., Dietz G. P., Jakobs S., Bähr M., Weishaupt J. H. (2007) Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 14, 651–661 [DOI] [PubMed] [Google Scholar]
- 45. Karpinar D. P., Balija M. B., Kügler S., Opazo F., Rezaei-Ghaleh N., Wender N., Kim H. Y., Taschenberger G., Falkenburger B. H., Heise H., Kumar A., Riedel D., Fichtner L., Voigt A., Braus G. H., Giller K., Becker S., Herzig A., Baldus M., Jäckle H., Eimer S., Schulz J. B., Griesinger C., Zweckstetter M. (2009) Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 28, 3256–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fares M. B., Bouziad N. A., Dikiy I., Mbefo M. K., Jovicic A., Kiely A., Holton J. L., Lee S. J., Gitler A. D., Eliezer D., Lashuel H. A. (2014) The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 10.1093/hmg/ddu165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sung Y. H., Eliezer D. (2007) Residual structure, backbone dynamics, and interactions within the synuclein family. J. Mol. Biol. 372, 689–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dedmon M. M., Lindorff-Larsen K., Christodoulou J., Vendruscolo M., Dobson C. M. (2005) Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127, 476–477 [DOI] [PubMed] [Google Scholar]
- 50. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rospigliosi C. C., McClendon S., Schmid A. W., Ramlall T. F., Barré P., Lashuel H. A., Eliezer D. (2009) E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J. Mol. Biol. 388, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ghosh D., Mondal M., Mohite G. M., Singh P. K., Ranjan P., Anoop A., Ghosh S., Jha N. N., Kumar A., Maji S. K. (2013) The Parkinson's disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 52, 6925–6927 [DOI] [PubMed] [Google Scholar]
- 53. Bussell R., Jr., Ramlall T. F., Eliezer D. (2005) Helix periodicity, topology, and dynamics of membrane-associated α-synuclein. Protein Sci. 14, 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang H., Griggs A., Rochet J. C., Stanciu L. A. (2013) In vitro study of α-synuclein protofibrils by cryo-EM suggests a Cu2+-dependent aggregation pathway. Biophys. J. 104, 2706–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sung Y. H., Rospigliosi C., Eliezer D. (2006) NMR mapping of copper binding sites in α-synuclein. Biochim. Biophys. Acta 1764, 5–12 [DOI] [PubMed] [Google Scholar]
- 56. Danzer K. M., Ruf W. P., Putcha P., Joyner D., Hashimoto T., Glabe C., Hyman B. T., McLean P. J. (2011) Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee H. J., Baek S. M., Ho D. H., Suk J. E., Cho E. D., Lee S. J. (2011) Dopamine promotes formation and secretion of non-fibrillar α-synuclein oligomers. Exp. Mol. Med. 43, 216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee M., Hyun D., Halliwell B., Jenner P. (2001) Effect of the overexpression of wild-type or mutant α-synuclein on cell susceptibility to insult. J. Neurochem. 76, 998–1009 [DOI] [PubMed] [Google Scholar]
- 59. Ko L. W., Ko H. H., Lin W. L., Kulathingal J. G., Yen S. H. (2008) Aggregates assembled from overexpression of wild-type α-synuclein are not toxic to human neuronal cells. J. Neuropathol. Exp. Neurol. 67, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tabrizi S. J., Orth M., Wilkinson J. M., Taanman J. W., Warner T. T., Cooper J. M., Schapira A. H. (2000) Expression of mutant α-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 9, 2683–2689 [DOI] [PubMed] [Google Scholar]
- 61. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 62. Knott A. B., Bossy-Wetzel E. (2008) Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann. N.Y. Acad. Sci. 1147, 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]