

Background: Peptide vaccine-based immunotherapy targeting tumor-associated antigens can elicit CTL responses. However, the expression status of HLA·vaccinated peptide complexes on tumor cells is unknown.
Results: Using a phage display, we isolated mAb D12, which reacted with an HLA-A2·PBF peptide.
Conclusion: We successfully detected the HLA·peptide complex on osteosarcoma cells.
Significance: Assessment of HLA·peptide complexes is important to predict the effect of immunotherapy.
Keywords: Antibody Engineering, Antigen Presentation, Major Histocompatibility Complex (MHC), Phage Display, T Cell Receptor (TCR), HLA-A2, HLA/Peptide Complex, PBF, scFv
Abstract
Osteosarcoma is a rare but highly malignant tumor occurring most frequently in adolescents. The prognosis of non-responders to chemotherapy is still poor, and new treatment modalities are needed. To develop peptide-based immunotherapy, we previously identified autologous cytotoxic T lymphocyte-defined osteosarcoma antigen papillomavirus binding factor (PBF) in the context of HLA-B55 and the cytotoxic T lymphocyte epitope (PBF A2.2) presented by HLA-A2. PBF and HLA class I are expressed in ∼90 and 70% of various sarcomas, respectively. However, the expression status of peptide PBF A2.2 presented by HLA-A2 on osteosarcoma cells has remained unknown because it is difficult to generate a specific probe that reacts with the HLA·peptide complex. For detection and qualification of the HLA-A*02:01·PBF A2.2 peptide complex on osteosarcoma cells, we tried to isolate a single chain variable fragment (scFv) antibody directed to the HLA-*A0201·PBF A2.2 complex using a naïve scFv phage display library. As a result, scFv clone D12 with high affinity (KD = 1.53 × 10−9 m) was isolated. D12 could react with PBF A2.2 peptide-pulsed T2 cells and HLA-A2+PBF+ osteosarcoma cell lines and simultaneously demonstrated that the HLA·peptide complex was expressed on osteosarcoma cells. In conclusion, scFv clone D12 might be useful to select candidate patients for PBF A2.2 peptide-based immunotherapy and develop antibody-based immunotherapy.
Introduction
Osteosarcoma is the most common primary malignant tumor of bone. The survival rate of patients with this disease was under 20% before 1970. Since then, the introduction of neoadjuvant chemotherapy, establishment of guidelines for adequate surgical margins, and development of postexcision reconstruction have raised the 5-year survival rate to 60–70% (1, 2). These advances overshadowed the pioneering adjuvant immunotherapy trials using autologous tumor vaccines for patients with osteosarcoma despite their having some therapeutic efficacy (3–5). However, the survival rate of patients with osteosarcoma has reached a plateau in the last decade (6), which has reignited interest in immunotherapeutic approaches (7–10).
Peptide vaccine-based immunotherapy targeting tumor-associated antigens could elicit CTL2 responses against HLA·peptide complexes. Therefore, expression of targeted HLA·peptide complexes on tumor cells is prerequisite for peptide vaccination. However, the detection and visualization of HLA·peptide complexes is very difficult because (i) it is still hard to establish CTL clones directed to HLA·peptide complexes, (ii) the affinity of soluble natural TCR obtained from CTL clones is not sufficient to visualize HLA·peptide complexes, (iii) generation of anti-HLA·peptide monoclonal antibodies using immunized mice is very difficult, and (iv) the amount of a specific HLA·peptide complex on cells might be too low to detect. Therefore, although generation of anti-HLA·vaccine peptide complex antibodies is very attractive, it remains challenging.
Papillomavirus binding factor (PBF) was first identified as a transcription factor regulating promotor activity on the human papillomavirus type 8 genome (11). We demonstrated that PBF was an osteosarcoma-associated antigen recognized by an autologous cytotoxic T lymphocyte clone (12). Immunohistochemical analysis revealed that 92% of biopsy specimens of osteosarcoma expressed PBF. Moreover, PBF-positive osteosarcoma has a significantly poorer prognosis than that with negative expression of PBF (13). Generally, conventional osteosarcoma is a malignant neoplasm of mesenchymal origin, and there is no specific cause such as viral infection (14). It is suggested that PBF has certain functions not only in transcription of the human papillomavirus genome but also in the cell survival and apoptosis of osteosarcoma (15, 16), innate immunity (17), and adipogenesis (18). Conversely, HLA class I is also expressed in 68% of primary osteosarcoma tissues and correlated with good prognosis (19). These findings suggest that osteosarcoma might be immunogenic for cellular immunity. Next, we identified the CTL epitopes in the context of HLA-A24 and HLA-A2 by a reverse immunology approach (13, 20). We are currently conducting a clinical phase I trial of peptide vaccination therapy for patients with osteosarcoma using these epitopes. Obviously, the expression status of PBF and HLA class I can be assessed by standard immunohistochemistry. However, the expression status of the HLA class I·CTL epitope complex on osteosarcoma cells is still unknown because of the difficulty of visualization of HLA·peptide complexes.
In this study, with the aim to characterize of the expression status of various HLA·peptide complexes on tumor cells, we constructed a naïve single chain variable fragment (scFv) phage display library and generated an artificial scFv antibody that reacts with an HLA·peptide complex using the PBF-derived peptide (PBF A2.2) in the context of HLA-A2 as a prototype antigen. Subsequently, we tried to detect the HLA-A2·PBF A2.2 peptide complex on osteosarcoma cells.
EXPERIMENTAL PROCEDURES
The present study was approved by the Ethics Committee of Sapporo Medical University. The patients, their families, and healthy donors provided informed consent for the use of blood samples and tissue specimens in our research.
Cell Lines
Five human osteosarcoma (OS) cell lines (U2OS, OS2000, KIKU, Saos-2, and HOS) and one bone human malignant fibrous histiocytoma (MFH) cell line (MFH2003) were used. The transporter associated with antigen processing (TAP)-deficient cell line T2 was also used. OS2000, KIKU, and MFH2003 were established in our laboratory (21–23). The other cell lines were kindly donated or purchased from the Japanese Collection of Research Bioresources Cell Bank (Tokyo, Japan) and American Type Culture Collection (Manassas, VA). MFH2003 and OS2000 were cultured with Iscove's modified Dulbecco's Eagle's medium (Invitrogen) containing 10% FBS, and the others were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) containing 10% FBS in a 5% CO2 incubator at 37 °C.
One primary culture of OS cells was used. Fresh specimens taken at surgery from the primary tumor in the left distal femur of a 15-year-old female were minced into small pieces and separately cultured with Iscove's modified Dulbecco's Eagle's medium containing 10% FBS in a 5% CO2 incubator. When the cells grew to confluence, half were trypsinized and passaged. After 30 passages, cultured cells were used for the study. Malignant features of cultured cells were confirmed by cytodiagnosis.
Biotinylated Antigens and Peptides
Biotinylated HLA-A*02:01·peptide complex monomers were constructed by Medical and Biological Laboratories, Co., Ltd. (Nagoya, Japan). Peptides PBF A2.2 (ALPSFQIPV), HIV-A2 (SLYNTVATL), CMV-A2 (NLVPMVATV), Epstein-Barr virus LMP1 (YLQQNWWTL), NY-ESO-1 (SLLMWITQC), WT1 (RMFPNAPYL), human T-lymphotropic virus type I Tax1 (LLFGYPVYV), human T-lymphotropic virus type I Tax2 (QLGAFLTNV), MPT51 (TALGKGISVV), MART-1 (ELAGIGILTV), influenza M1 (GILGFVFTL), gp100M (IMDQVPFSV), tyrosinase (YMDGTMAQV), and gp100 (KTWGQYWQV) were used in the present study.
Construction of scFv Library from Naïve Donors
Construction of the Phagemid Vector pMARXL
We first constructed a phagemid vector based on pMod1 (a kind gift from Dr. Potjamas Pansti, Aarhus University, Aarhus, Denmark) (24). A point mutation at the XhoI site of the ampicillin resistance gene and linker peptide between the cloning sites of variable regions of heavy and light chains (VH and VL, respectively) were introduced into pMod1 as follows. The point mutation was introduced into the XhoI site of the ampicillin resistance gene of pMod1 with a PrimeSTAR Mutagenesis Basal kit using primers AmpR XhoI-mutation sense (5′-TGGGTGCACGAGTGGGTTACATCGAAC-3′) and AmpR XhoI-mutation antisense (5′-CCACTCGTGCACCCAACTGATCTTCAG-3′) according to the manufacturer's protocol. The resultant mutant was digested with XhoI and NotI followed by electrophoresis, cutting, and gel extraction using a QIAEX II Gel Extraction kit (Qiagen, Hilden, Germany). Next, two oligonucleotides containing XhoI-peptide linker-NheI-NotI (sense, 5′-TCGAGGGTGGAGGCGGTTCAGGCGGAGGTGGCTCTGGCGGTGGCGCTAGCGGCAGATCTGATGACGC-3′; antisense, 5′-GGCCGCGTCATCAGATCTGCCGCTAGCGCCACCGCCAGAGCCACCTCCGCCTGAACCGCCTCCACCC-3′) were denatured, annealed, and ligated into the digested vector using DNA Ligation kit version 2 (Takara) followed by transformation of Escherichia coli DH5α. The resultant vector was designated pMARXL (see Fig. 1).
FIGURE 1.

Structure and sequence around the multicloning site of the phagemid vector pMARXL.
scFv phage display libraries were constructed according to the methods described by Pansri et al. (24) and Schofield et al. (25) with some modifications to optimize the experimental conditions. The primers used for the amplification of variable regions are listed in supplemental Table S1.
Source and cDNA Preparation
Peripheral blood mononuclear cells of 31 healthy volunteers and two surgically resected tonsils were used as RNA sources. Peripheral blood mononuclear cells were separately isolated from 50 ml of peripheral blood from each donor followed by total RNA extraction using an RNeasy Mini kit (Qiagen). Total RNA of the tonsils was separately extracted using an RNeasy Maxi kit (Qiagen). mRNA was isolated from each RNA using an Oligotex-dT30 <Super> mRNA Purification kit (Takara, Otsu, Japan). Thirty-one mRNA samples were divided and gathered into six groups (five to six mRNA samples per group). mRNAs of the two tonsils were gathered into a separate group. Then the mRNAs of the seven groups were converted into cDNAs with a First-Strand cDNA Synthesis kit (GE Healthcare). For reverse transcription, the specific primers for the κ and λ light chains and IgM heavy chain were used.
Primary PCR
Amplification of VH and VL was performed with DNA polymerase KODplus (Toyobo) using cDNA and the primers to amplify variable regions of VH and VL including the κ and λ chains (Vk and Vl, respectively). All 5′ primers (14 VH primers, 13 Vk primers, and 15 Vl primers) were used separately for PCR. 3′ primers for VH (four primers), Vk (five primers), and VL (three primers) were mixed in each group and used. Therefore, 294 PCRs were performed separately. The PCR mixture was denatured at 94 °C for 2 min followed by 35 cycles at 94 °C for 15 s, 55 °C for 30 s, and 68 °C for 1 min. Amplicons of VH, Vk, and Vl were electrophoresed (see Fig. 2A). The bands around 350 bp containing VH, Vk, and Vl were cut out, and cDNAs were separately extracted.
FIGURE 2.

Electrophoresis of amplified variable regions after primary PCR. Arrows indicate the adequate amplicons (around 350 bp) containing variable regions.
Secondary PCR
Extracted cDNA was used for secondary PCR to introduce restriction enzyme sites. PCR was performed using of each amplicon as the template and primers. 5′ primers of 14 VH with mixed 3′-primers (four primers), 5′ primers of three Vk with mixed 3′ primers (five primers), and 5′ primers of three Vl with mixed 3′ primers (three primers) were used. The PCR mixture was denatured at 94 °C for 2 min followed by 35 cycles at 94 °C for 15 s, 55 °C for 30 s, and 68 °C for 1 min. Restriction site-introduced amplicons of 14 VH, three Vk, and three Vl were confirmed by electrophoresis (see Fig. 2B). These amplicons were gathered into 17 groups (VH1, VH2, VH3, VH4, VH5, VH6, VH7, Vk1, Vk2, Vk3, Vk4, Vk5, Vk6, Vl1–2, Vl3, Vl4–6, and Vl7–10) and purified with a column (QIAquick PCR Purification kit, Qiagen).
Construction of Initial Library of VH, Vk, and Vl
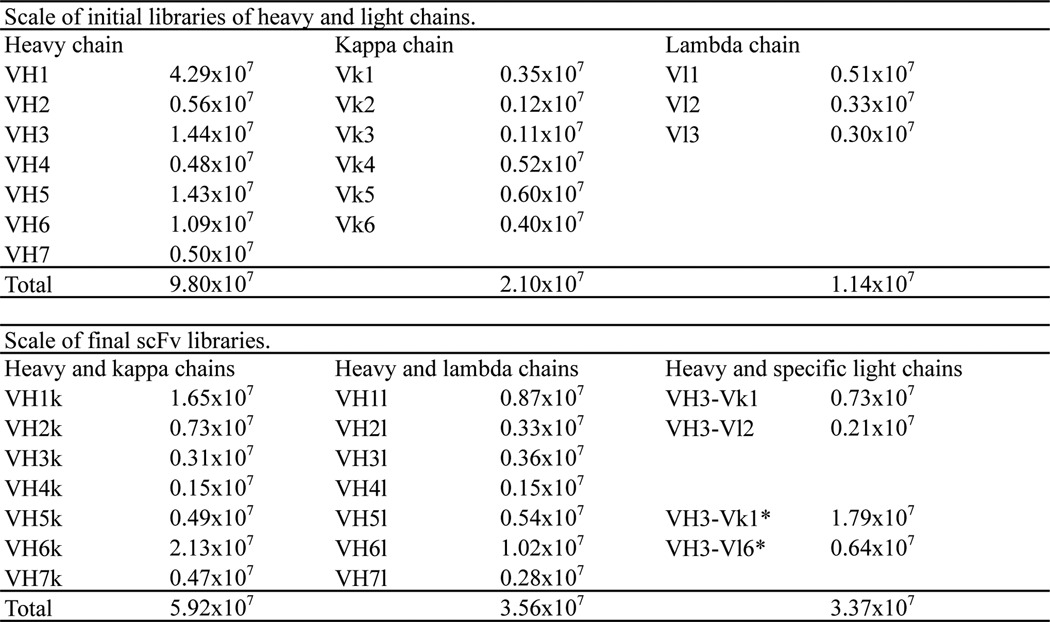
Purified cDNA of VH was digested with SfiI at 50 °C for 2 h followed by purification with the column and digestion with XhoI at 37 °C for 2 h. cDNAs of Vk and Vl were digested with NheI at 37 °C for 2 h followed by purification using the column and digestion with NotI at 37 °C for 2 h. The digested cDNA was electrophoresed, cut, and extracted as with primary PCR products. cDNA of VH was ligated into dephosphorylated pMARXL digested with Sfi1 and XhoI using a Rapid DNA Dephos and Ligation kit (Roche Diagnostics GmbH). All large scale ligation reactions were performed at 16 °C overnight. cDNAs of Vk and Vl were subcloned into pMARXL digested with NheI and NotI. Ligation products were purified with a column and electroporated into DH12S (Invitrogen). These were the initial libraries of VH and VL (Table 1).
TABLE 1.
Scale of constructed libraries
*, prepared as the “additional libraries” described under “Experimental Procedures.”
Construction of the scFv Library
Inserts of VH were prepared from the initial libraries and digested with SfiI and XhoI followed by electrophoresis, cutting, and gel extraction. Vector DNAs coding Vk and Vl were prepared from the initial libraries and digested with SfiI and XhoI followed by dephosphorylation. All κ and λ libraries were combined into one group. Insert cDNAs and vector DNAs were ligated, purified, and electroporated into competent TG1 cells (Lucigen Corp., Middleton, WI). Transformed E. coli were plated on a 1.5% agarose gel of 2× YT containing ampicillin (100 μg/ml) and 2% glucose (2× YTAG) followed by the collection of all colonies into 2× YT liquid. Aliquots of E. coli were divided and frozen with 20% glycerol. In addition, scFv libraries of VH3-Vk1 and VH3-Vl6 were also prepared. These were the main scFv libraries (Table 1).
Construction of Additional scFv Libraries
To construct additional libraries of VH3-VK1 and VH3-Vl6, the primary PCR products of heavy chains (VH3a, VH3b, and VH3c) and light chains (Vk1a, Vk1b, Vk1c, Vk1d, and Vl6) were used independently for secondary PCR to introduce restriction enzyme sites and linker sequences with the primers listed in supplemental Table S1 followed by column purification, electrophoresis, cutting, and gel extraction. cDNAs of VH3 and VL (Vk1 and Vl6, respectively) were assembled by PCR. Equal amounts of cDNAs of heavy and light chains were used in the reaction mixture without primers. The PCR mixture was denatured at 94 °C for 2 min followed by five cycles at 94 °C for 30 s and 68 °C for 1 min. The reaction products were used for pull-through PCR containing the primers PT-BAKSfi (5′-GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC-3′) and PT-FORNot (5′-GAGTCATTCTCGACTTGCGGCCGCAC-3′). The mixture was denatured at 94 °C for 2 min followed by 30 cycles at 94 °C for 15 s, 55 °C for 30 s, and 68 °C for 1 min followed by column purification. Assembled cDNAs of VH and VL were digested with SfiI followed by digestion with NotI. After column purification, digested cDNA was ligated into digested and dephosphorylated pMARXL. After column purification, phagemids were electroporated into competent TG1. Transformed E. coli were plated on 2× YTAG plates followed by the collection of all colonies into 2× YT liquid. Aliquots of E. coli were divided and frozen. The additional libraries were VH3-Vk1 and VH3-Vl6 (Table 1).
Titration
E. coli electroporated with the library phagemid or infected with phages were serially diluted and plated on 2× YTAG plates followed by incubation at 37 °C overnight. The next day, the number of independent colonies was counted, and the titer was calculated.
Preparation of Primary Stocks and Rescue of the Libraries
Glycerol stock of each library was added to 500 ml of 2× YTAG liquid and incubated at 37 °C for 2 h. Cultured liquid containing E. coli was divided into aliquots of 1 ml/tube and cryopreserved with glycerol at −80 °C. These were the primary stocks of the libraries. These primary stocks were rescued as follows. Glycerol stocks of all the libraries were added to 25 ml of 2× YTAG liquid and shaken at 37 °C for 60–90 min until the A600 reached 0.5–1.0 followed by addition of 2.5 × 1010 of M13K07 (Invitrogen). After a 60-min incubation, E. coli were centrifuged. The pellet of E. coli was resuspended in fresh 2× YT containing ampicillin and kanamycin (25 μg/ml) and incubated with shaking at 30 °C overnight.
PEG Precipitation
The overnight culture of E. coli was centrifuged at 1000 × g for 20 min at 4 °C. The supernatant containing phage particles was collected, and volume of 20% polyethylene glycol with 2.5 m NaCl was added followed by incubation on ice for more than 1 h. The phage was pelleted by centrifugation at 12,000 × g for 10 min at 4 °C, and then it was suspended with 4 ml of PBS, filtrated with a 0.45-μm filter, and used for biopanning.
Biopanning with Biotinylated Antigen
Before biopanning, the phage library (0.25 ml) was mixed with PBS containing 4% (w/v) milk (4% PBS-M) in a 1.5-ml tube and incubated with 100 μl of magnetic beads (Dynabeads M-280 Streptavidin, Invitrogen) that were prewashed with PBS with 0.1% Tween 20 (PBS-T) at room temperature for 60 min. After incubation, the phage supernatant was harvested on a magnetic stand. This step can remove nonspecific phages.
Biopanning was performed as follows. 100 μl of the magnetic beads was prewashed with PBS-T and blocked with 2% PBS-M for 1–2 h at room temperature. The phage supernatant was mixed with 0.5 ml of biotinylated antigen (500 nm in the first round and 100 nm in PBS in the second and subsequent ones) and mixed at room temperature for 60 min. After incubation, the phage-antigen mixture was mixed with the magnetic beads followed by additional incubation for 15 min. Then the beads were washed six times with 1 ml of 2% PBS-M with 0.1% Tween 20 and twice with 1 ml of PBS. Specific phage binders were eluted from the magnetic beads by incubation with 1 ml of 100 mm triethylamine for 7 min. The eluted phage aliquot was immediately neutralized with 100 μl of 1 m Tris-HCl (pH 7.4). Next, the resultant phage aliquot was used for phage rescue.
Phage rescue was performed as follows. Half of the phage aliquot after biopanning was added to 10 ml of log-phase TG1 and incubated at 37 °C for 1 h with slow shaking. This step allowed the phage to infect TG1. After incubation, ampicillin (final concentration, 100 μg/ml), glucose (final concentration, 20% (w/v)), and more than 1 × 1010 pfu of helper phage M13K07 was added and incubated for 60 min. Cultured TG1 infected with the phage and M13K07 was centrifuged at 2500 × g for 5 min and resuspended with 25 ml of fresh 2× YT containing ampicillin (100 μg/ml) and kanamycin (25 μg/ml) without glucose followed by incubation in a new flask at 30 °C overnight. The overnight culture of bacteria including the proliferated phage was isolated by PEG precipitation and used for the next round of biopanning.
Preparation of Log-phase TG1
TG1 was plated on agar plates of M9 minimal medium with 1 mm thiamine hydrochloride and 0.1% glucose. After overnight incubation at 37 °C, a single colony was inoculated into 100 ml of 2× YT medium followed by overnight incubation at 37 °C. A 1-ml aliquot of E. coli was inoculated into 100 ml of 2× YT and incubated at 37 °C with fast shaking for 90–120 min until the A600 value reached 0.5–1.0. The resultant E. coli were used as the log-phase TG1 for phage rescue.
Screening Specific Phage Clones Binding to the Biotinylated Antigen
After biopanning, soluble scFv expression of TG1 infected with the phage was induced in a microplate. Half of the phage aliquot after biopanning was added to 10 ml of log-phase TG1 and incubated at 37 °C for 1 h with slow shaking. After incubation, the aliquot of E. coli was diluted, seeded on a 2× YTAG plate, and incubated at 30 °C overnight. The next day, 94 clones were picked up and inoculated independently into wells containing 150 μl of 2× YTAG in a 96-well microculture plate. The plate was incubated at 30 °C with shaking overnight for phage rescue. The following day, 10 μl of cultured E. coli in each well was inoculated into 100 μl of 2× YTAG in a new 96-well microculture plate. The plate was incubated at 30 °C for 5–6 h. After incubation, the plate was centrifuged at 1000 × g for 5 min followed by replacement of the medium with 100 μl of 2× YT containing ampicillin and 1 mm isopropyl 1-thio-β-d-galactopyranoside. Then the plate was incubated at 30 °C overnight. After incubation, it was centrifuged at 1000 × g for 10 min. Then 50 μl of the supernatant was harvested and immediately used for ELISA screening.
For ELISA, 100 μl of biotinylated antigen (5 μg/ml in PBS) was added to each well of a streptavidin-coated 96-well microplate (BioBind Assembly strip, 1 × 8, streptavidin-coated, Thermo Fisher Scientific, Vantaa, Finland) and incubated at room temperature for 1 h. After three washes with 300 μl of PBS-T, each well was blocked with 200 μl of 2% PBS-M containing 5% DMSO for more than 30 min. After blocking, the liquid was discarded, and then 50 μl of 4% PBS-M containing 5% DMSO and 50 μl of culture supernatant containing soluble scFv were added. After a 1.5-h incubation, each well was washed, and 100 μl of an anti-myc-HRP antibody (1:1000 dilution in PBS; Miltenyi Biotec, Gladbach, Germany) was added. After a 1-h incubation, each well was washed, and 100 μl of ABTS Peroxidase Substrate (Kirkegaard and Perry Laboratories, Inc., Gaithersburg, MD) was added. After 20 min, the A405 was assessed using a microplate reader.
Sequence Analysis
Sequences of scFv were analyzed using BigDye Ver1.0 according to the manufacturer's protocol. Sequence primers used in the present study were LMB3 (5′-CAGGAAACAGCTATGAC-3′), Gene III leader (5′-GTGAAAAAATTATTATTCGCA-3′), and FDTSEQ1 (5′-GTCGTCTTTCCAGACGTTAGT-3′) for pMARXL and pFUSE-hIgG1-Fc2 FW(545–572) (5′-CTGAGATCACCGGCGAAGGAGGGCCACC-3′) and pFUSE-hIgG1-Fc2 RV(735–706) (5′-TGGGTTTTGGGGGGAAGAGGAAGACTGACG-3′) for pFX-hIgG1. The sequences were analyzed using the International ImMunoGeneTics Information System website (26).
Generation of scFv-hIgG and scFv Multimer
We constructed a derivative of pFUSE-hIgG-Fc2 (InvivoGen, San Diego, CA) that could express bivalent scFv with a constant region of human IgG to introduce the NotI site into the multicloning site. The mutation was inserted into the NotI site in the 3′-end of the ori sequence using a PrimeSTAR Mutagenesis Basal kit with the primers pFUSE NotI mutation-sense (5′-ATCAGCGCCCGCAATAAAATATCTTTA-3′) and pFUSE NotI mutation-antisense (5′-TATTGCGGGCGCTGATTTAAATGTTAAT-3′) according to the manufacturer's protocol. Next, we introduced a new multicloning site into the mutant vector. The vector was digested with EcoRV and BglII followed by column purification. The digested vector was ligated to annealed double-strand oligonucleotides (EcoRV/BglII pFUSEIghFC2 sense, 5′-ATCGGCCATGGTTTGGTACCTTGCGGCCGCTA-3′; EcoRV/BglII pFUSEIghFC2 antisense, 5′-GATCTAGCGGCCGCAAGGTACCAAACCATGGCCGAT-3′). The derivative of pFUSE-hIgG1-Fc2 was designated pFX-hIgG1. cDNA of VH and VL with a peptide linker of pMARXL was subcloned into pFX-hIgG1 via the NcoI-NotI site. For soluble expression of scFv-hIgG, 4 μg of the plasmid was transfected using Lipofectamine 2000 (Invitrogen) into 293T cells precultured on a 10-cm culture dish in DMEM supplemented with 10% FBS. After 4–5 h, the culture medium was replaced with fresh AIM V (Invitrogen) without serum. The supernatant was harvested and replaced with fresh AIM V at 24, 48, and 72 h after transfection. The collected supernatant was passed through a chromatography column with Protein G at 4 °C. The column was washed with 20 mm sodium phosphate (pH 7.0) and eluted by fraction (1 ml/fraction) with a total of 5 ml of 0.1 m glycine (pH 2.7) followed by immediate neutralization with volume of Tris-HCl (pH 9.0). Fractions containing antibodies were assessed by SDS-PAGE with or without DDT to confirm that oxidized scFv-hIgG formed a dimer protein.
The multimer of scFv-hIgG1 (scFv multimer) was constructed as follows. Briefly, the Bir domain was introduced into the 3′-end of the hIgG1 Fc region by gene synthesis. Soluble expression and purification of the antibody protein were performed as described above. The antibody protein was biotinylated using the BirA enzyme and mixed with phycoerythrin-conjugated streptavidin to achieve a 1:4 molar ratio.
Surface Plasmon Resonance Analysis
Surface plasmon resonance analysis was performed using a ProteOn XPR36 (Bio-Rad) according to the protocol described by Nahshol et al. (27). Briefly, 1 μg/ml biotinylated monomer (HLA-A*02:01·PBF A2.2 peptide or HLA-A*02:01·HIV peptide) in PBS supplemented with 0.005% Tween 20 (PBST) was injected at 30 μl/min at 25 °C to be captured on a neutravidin-immobilized NLC sensor tip. Subsequently, serially diluted scFv-hIgG in PBST was injected at 50 μl/min at 25 °C. All binding sensorgrams were collected and analyzed using ProteOn Manager software (Bio-Rad).
Immunostaining and Flow Cytometry
Before immunostaining, T2 cells (5–10 × 105) were incubated in 200 μl of AIM V with each peptide at 50 μg/ml (PBF A2.2, HIV-A2, and CMV-A2) on a 96-well round bottom microculture plate at 26 °C overnight followed by 2-h incubation at 37 °C. For immunostaining, 5–10 × 106 target cells were seeded in a 96-well microculture plate and incubated with 50–100 μl of 10 μg/ml or 1 mg/ml scFv-hIgG or the supernatant of a hybridoma (BB7.2 or W6/32; purchased from American Type Culture Collection) on ice for 60–90 min. After two washes, the cells were incubated with 100 μl of anti-human IgG conjugated with Qdot655 (1:200 dilution; Invitrogen) or an FITC-conjugated rat anti-mouse IgG antibody (1:200 dilution; Kirkegaard and Perry Laboratories, Inc.) for 40 min. The cells were also stained with 50 μl of the scFv multimer (3 or 10 μg/ml) on ice for 60–90 min. After immunostaining, cells were washed and fixed with 200 μl of PBS with 0.5% formaldehyde and analyzed using FACS Array and CellQuest software (BD Biosciences). The reactivity of the antibodies was evaluated by the percent mean fluorescence intensity (%MFI) increase using the following calculation. %MFI increase = ((MFI of the samples − MFI of the negative control)/(MFI of the negative control)) × 100.
Inhibition Assay of Functional CTL Clone
CTL line 5A9 is an oligoclonal line containing HLA-A*02:01·PBF A2.2 peptide tetramer-positive cells that can recognize peptide PBF A2.2 in the context of HLA-A2 (20). After thawing the cryopreserved stock, cells were cultured in AIM V supplemented with 10% AB human serum and recombinant human IL-2 (200 units/ml; a kind gift from Takeda Pharmaceutical Company, Ltd., Osaka, Japan) in a 96-well microculture plate. On day 27, CTL 5A9 was stained with an anti-CD8-allophycocyanin APC antibody, HLA-A*02:01·PBF A2.2 tetramer-phycoerythrin, and HLA-A*02:01·HIV tetramer-FITC (negative control) (Medical & Biological Laboratories, Co., Ltd.) on ice for 30 min. After staining, the cells were incubated with anti-phycoerythrin magnetic beads (Miltenyi Biotech) followed by magnetic sorting of the tetramer-positive cells according to the manufacturer's protocol. U2OS cells (50,000 cells/well) were seeded in a 96-well flat bottom microculture plate. Next, the cells were preincubated with D12 scFv-hIgG1 or irrelevant D11 scFv-hIgG1 for 1 h at room temperature. Subsequently, sorted tetramer-positive cells (12,000 cells/well) or control medium without cells was added and cocultured for 20 h. The concentration of IFN-γ in the supernatant was analyzed using a human IFN-γ DuoSet ELISA Development kit (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol.
RESULTS
Generation of a Naïve scFv Phage Display Library
We constructed an scFv phage display library with RNA extracted from human peripheral mononuclear cells of 31 naïve donors and two surgically resected tonsils on the new phagemid vector pMARXL (Fig. 1) using a total of 97 primers (supplemental Table S1). All 294 amplicons of VH, Vk, and Vl after primary PCR are shown in Fig. 2. The amplicons were used to introduce restriction enzyme sites by secondary PCR and subcloned into pMARXL. As a result, 16 initial sublibraries and 18 scFv sublibraries containing a total of 1.29 × 108 scFv clones were obtained (Table 1).
Three scFv Clones Recognized HLA-A2·Osteosarcoma Antigen PBF-derived Peptide Complex
We performed biopanning with a biotinylated HLA-A*02:01·PBF A2.2 peptide complex as the antigen and a rescued library phage. The ratios of output phage/input phage were 6.9 × 10−8 in the first round of biopanning, 6.2 × 10−7 in the second round, and 8.2 × 10−5 in the third round (Fig. 3A). The gradual increase of the ratio indicated that adequate enrichment of specific phages was successfully achieved. Considering that the size of the libraries was 1.20 × 108 clones, a 10−20 order of enrichment efficiency was sufficient to isolate specific binders. After three rounds of biopanning, we screened the reactivities of 94 randomly selected scFv clones against the antigen by ELISA. As a result, we obtained three scFv clones (D12, G3, and H7) that were more highly reactive with the HLA-A*02:01·PBF A2.2 peptide complex than with the HLA-A*02:01·HIV peptide complex (Fig. 3B). Sequence analysis revealed that the three scFv clones had identical VH and VL (Fig. 3C).
FIGURE 3.

Three scFv clones recognized HLA-A2·osteosarcoma antigen PBF-derived peptide complex. A, input/output ratios of phage particles. B, ELISA screening of the specific binders of 94 scFv clones reacting with biotinylated HLA-A*02:01·PBF A2.2 peptide complex. C, sequence analysis of complementarity-determining region 3 (CDR3) region of scFv clones.
The scFv Clones Could React with Peptide PBF A2.2 Presented by HLA-A*02:01 on Antigen-presenting Cells with Strong Affinity
Next, we constructed scFv-hIgG possessing bivalent scFv with the human IgG1 constant region and assessed whether the scFv clones could recognize peptide PBF A2.2 presented on the cell surface. D12 scFv-hIgG1 could successfully form an oxidized dimer (Fig. 4A.) The dimeric structures of the other clones were also confirmed (data not shown). We found that the three clones of scFv-hIgG could recognize T2 cells pulsed with peptide PBF A2.2 but not peptide HIV-A2 or peptide CMV-A2 (Fig. 4B). Monovalent scFv could not react with peptide-pulsed T2 cells (data not shown). We selected scFv clone D12 as the representative clone for further analysis because the three clones contained an identical complementarity-determining region sequence and showed the same specificity. Kinetics analysis of protein interaction between scFv D12 and the HLA-A*02:01·PBF A2.2 peptide complex using surface plasmon resonance analysis showed the strong affinity of scFv D12 with the HLA-A*02:01·PBF A2.2 peptide complex (KD = 1.53 × 10−9 m) (Fig. 4C). Simultaneously, D12 scFv-hIgG showed weak binding activity with the HLA-A*02:01·HIV peptide complex (Fig. 4D). Therefore, we assessed the cross-reactivity of D12 scFv-hIgG against the other biotinylated HLA-A*02:01·peptide complexes. We also confirmed that an HLA class I monoclonal antibody (supernatant of hybridoma W6/32) could highly react with all of the immobilized biotinylated complexes (data not shown). As shown in Fig. 4E, scFv D12-hIgG (5 and 0.5 μg/ml) reacted weakly with peptides NY-ESO-1, MPT51, and influenza M1. However, as shown in Fig. 4B, 10 μg/ml scFv D12-hIgG did not react with T2 cells pulsed with peptide HIV-A2 or with peptide CMV. Therefore, we considered that the cross-reactivity of scFv D12 was relatively low.
FIGURE 4.

The scFv clones could react with peptide PBF A2.2 presented by HLA-A*02:01 on antigen-presenting cells with strong affinity. A, soluble fractions of D12 scFv-hIgG1 after purification with Protein G are visualized by SDS-PAGE. The reduced monomer (black arrows) and oxidized dimer of scFv-hIgG (red arrow) of fraction 1 are indicated. B, FACS analysis of scFv-hIgG1. T2 cells were pulsed with the indicated peptides and stained with each scFv-hIgG1 at a concentration of 10 μg/ml. BB7.2 was used to detect expression of HLA-A2 molecules. C and D, surface plasmon resonance analysis. Biotinylated HLA-A*02:01·PBF A2.2 peptide complex (C) or HLA-A*02:01·HIV-A2 peptide complex (D) was immobilized on the sensor tip as the target. Serially diluted D12 scFv-hIgG1 was used as the analyte as described under “Experimental Procedures.” E, ELISA screening of the reactivity of D12 scFv-hIgG against various biotinylated HLA-A*02:01·peptide complexes. RU, response units; EBV, Epstein-Barr virus; HTLV-1, human T-lymphotropic virus type I.
HLA-A2·PBF A2.2 Peptide Complex Was Firmly Expressed on the Surface of Osteosarcoma Cells
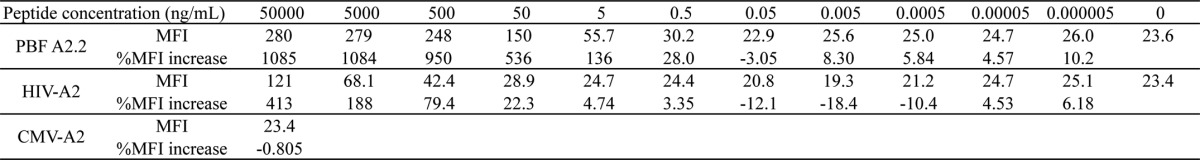
To assess whether scFv could detect peptide PBF A2.2 in the context of HLA-A2 presented on the sarcoma cell surface, we constructed a D12 scFv multimer (Fig. 5A) because bivalent scFv (D12 scFv-hIgG1) failed to react with HLA-A2-positive sarcoma cells (data not shown). The D12 scFv multimer (10 μg/ml) reacted highly with T2 cells pulsed with peptide PBF A2.2 and did not react with the cells pulsed with peptide CMV. Conversely, the multimer reacted with the cells pulsed with peptide HIV-A2 at a concentration of 100 μg/ml. These findings suggested that the low cross-reactivity against peptide HIV-A2 of D12 scFv was enhanced by multimer formation. Therefore, we titrated the reactivity of the multimer by calculation of the MFI against T2 cells pulsed with the peptides at various concentrations between 5 fg/ml and 50 μg/ml (Fig. 5B and Table 2). Although the multimer (3 μg/ml) also reacted with peptide HIV-A2 (pulsed at 50 μg/ml), its reactivity against the peptide was hardly detected at the concentration of 50 ng/ml (Fig. 5B). Conversely, the reactivity against peptide PBF A2.2 was preserved at the lower concentration of 500 pg/ml. The affinity of the multimer against peptide PBF A2.2 was estimated to be 100-fold higher than for peptide HIV-A2. In addition, we considered that a 20.0% MFI increase might be the threshold of the reactivity of the D12 scFv multimer. Next, six sarcoma cell lines and one primary culture of osteosarcoma were stained with the D12 scFv multimer (Fig. 5C). The reactivity of the multimer was graded as follows: strong (≥100% MFI increase), weak (≥20% but <100% MFI increase), and none (<20% MFI increase). In the context of HLA-A2, peptide PBF A2.2 was strongly detected on U2OS (A*02:01), HOS (A*02:11), and Saos-2 (A*02:01); weakly detected on KIKU (A*02:06); and not detected on HLA-A2-negative OS2000 and MFH2003 cells. In addition, peptide PBF A2.2 was also weakly detected on primary cultured osteosarcoma, which highly expressed HLA-A*02:01. These findings suggested that the D12 scFv multimer could react with peptide PBF A2.2 presented by HLA-A2. Simultaneously, the HLA-A2·PBF A2.2 peptide complex was expressed on osteosarcoma cells at various levels.
FIGURE 5.

scFv clone D12 could recognize PBF A2.2 peptide presented on the surface of sarcoma cells. A and B, FACS analysis of reactivity of D12 scFv-hIgG1 (1 mg/ml) and D12 scFv multimer (10 μg/ml) against T2 cells pulsed with the indicated peptides. %MFI increase is indicated. T2 without peptide was used as the negative control. A more than 20.0% MFI increase is indicated by underlining. C, FACS analysis of the reactivity of the D12 scFv multimer (3 μg/ml) with sarcoma cell lines and primary culture cells. Osteosarcoma cell line U2OS (A*02:01/A*3201, PBF+), OS2000 (A*2402, PBF+), KIKU (A*0206/A*2402, PBF+), HOS (A*02:11, PBF+), Saos-2 (A*02:01/A*24:02, PBF+), malignant fibrous histiocytoma cell line MFH2003 (A*2402, PBF−), and primary culture of osteosarcoma (Primary OS; A*02:01, PBF+) were used as target cells. A more than 20.0% MFI increase is indicated by underlining. The expression status of HLA-A2·PBF A2.2 peptide complex was graded as follows: strong (≥100% MFI increase), weak (≥20% but <100% MFI increase), and none (<20% MFI increase).
TABLE 2.
FACS analysis of peptide-pulsed T2 cells
D12 scFv Showed Similar Specificity with Natural CTL Recognizing HLA-A*02:01·PBF A2.2 Peptide Complex
To assess the similarity of specific responses against the HLA-A2·PBF A2.2 peptide complex between natural TCR and scFv, we performed inhibition assay of the CTL-mediated response against PBF A2.2. The frozen oligoclonal CTL line 5A9 was thawed and cultured followed by the selection of tetramer-positive cells using magnetic beads (Fig. 6A). The CTL response against HLA-A*02:01-positive osteosarcoma cell line U2OS was assessed by ELISA (Fig. 6B). Next, U2OS cells were preincubated with D12 scFv-hIgG1 or irrelevant D11 scFv-hIgG1 and cocultured with tetramer-positive CTL 5A9. As a result, partial inhibition (∼20%) of the scFv-mediated CTL response was observed. These findings suggested that scFv had specificity similar to that of functional TCR directed to the cognate antigen.
FIGURE 6.

Clone scFv D12 showed specificity similar to natural CTL recognizing HLA-A*02:01·PBF A2.2 peptide complex. A, tetramer staining of oligoclonal CTL line 5A9 directed to HLA-A*02:01·PBF A2.2 peptide before and after positive selection. B, CTL response against U2OS cells assessed by ELISA. Tetramer-positive cells after enrichment were used as the responder. C, inhibition effect of scFv-hIgG1 on CTL response. scFv D12-hIgG1 and irrelevant scFv D11-hIgG1 were used as blocking antibodies. The inhibition effect on CTL response was calculated as described under “Experimental Procedures.”
DISCUSSION
In the present study, we constructed a naïve scFv phage display library and isolated scFv clone D12, which reacted with the HLA-A*02:01·PBF A2.2 peptide complex with strong affinity. Bivalent and multivalent scFv D12 could react with peptide-pulsed T2 cells and HLA-A2-positive sarcoma cell lines, and D12 scFv could inhibit the natural TCR-mediated CTL response. These findings suggested that the D12 scFv clone had specificity mimicking that of the natural TCR against the HLA-A*02:01·PBF A2.2 peptide complex. Simultaneously, the HLA-A2·PBF A2.2 peptide complex was strongly and naturally expressed on osteosarcoma cells.
Generation of antibodies recognizing HLA·peptide complexes derived from tumor-associated antigens has been reported mainly in the field of melanoma (28). Development of such antibodies against HLA·peptide complexes is significant because assessment of the expression status of HLA·vaccine peptide complexes is important to select candidate patients and predict the efficacy of peptide-based immunotherapy. Moreover, antibodies mimicking TCR with high specificity and affinity could serve as sources of antibody-based therapy and chimeric antigenic receptors. However, the generation of antibodies that react with HLA·peptide complexes is still much more difficult than generation of those that react with the other proteins. This might be because the construction of antibody phage display libraries from naïve donors with sufficient quality and diversity is very hard and requires intensive laboratory work.
Generally, the amount of a single peptide presented by HLA class I molecules is very small. Purbhoo et al. reported that the HLA-A2·NY-ESO-1(157–165) peptide complex presented on melanoma cells ranged from 10 to 50 copies per cell as assessed by soluble high affinity engineered TCR (29). A similarly copy number of the HLA-A*02:01·gp100 peptide complex was barely detected by FACS but could be recognized by CTL (30). In the present study, strong expression levels of the HLA-A*02:01·PBF A2.2 peptide complex were detected on U2OS, HOS, and Saos-2 cells, but only weak levels were detected on KIKU and primary OS. These results suggested that the HLA-A2·PBF A2.2 peptide complex was naturally presented on osteosarcoma cells as in melanoma and might be a good candidate for peptide-based immunotherapy. Obviously, the possibility of the cross-reactivity of scFv clone D12 against the other antigens could not be completely ruled out. Indeed, the D12 scFv multimer mildly reacted with HIV-A2 peptide-pulsed T2 cells and other HLA·peptide complex monomers (NY-ESO-1, MPT51, and influenza M1). Among these peptides, no similar amino acid sequence was observed, excluding the anchor residues of HLA-A2 protein, leucine (Leu) at position 2 and valine (Val) at the C terminus (31). Therefore, the cross-reactivity might not depend on the epitope sequence but on some conformational change of HLA-A*02:01 molecules induced by the binding of peptides into the binding groove of the HLA. However, the reactivity of the multimer against peptide PBF A2.2 was 100-fold higher than for peptide HIV-A2. In addition, the observation that scFv clone D12 could weakly react with primary OS cells highly expressing HLA-A*02:01 suggested that the cross-reactivity of the clone against the other endogenous peptides was very low. The strong expression status of the HLA-A*02:01·PBF A2.2 peptide complex on U2OS cells is a rare condition. However, Michaeli et al. (32) reported that the tyrosinase(369–377) peptide was highly expressed on HLA-A2+ melanoma cells.
For specificity, the ideal probe that reacts with a naturally presented HLA·peptide complex is soluble TCR isolated from a specific CTL clone. We have established several CTL clones that react with tumor-associated antigens. Nevertheless, establishment of CTL clones is still difficult. In contrast, the technique using a scFv phage display library can provide scFv mimicking TCR specificity for a short time. The finding that scFv D12 could partially inhibit the reactivity of a CTL clone directed to the HLA-A*02:01·PBF A2.2 peptide complex suggested that scFv D12 recognized the near part of the HLA·peptide complex docked with TCR of the CTL clone.
Conversely, the advantage of using antibodies for probing HLA·peptide complexes is that they show high affinity on the nanomolar or picomolar order (28). The affinity range of natural TCRs binding to HLA class I·peptides is a KD value of 1–50 × 10−6 m. The affinity of scFv D12 was 3 orders of magnitude greater than that of standard TCRs (33).
The phage display library in the present study was constructed from naïve donors. Therefore, the library could be useful to select specific scFvs that react with the other HLA·tumor-associated antigens. We previously identified various CTL epitopes of tumor-associated antigens and cancer stemlike cell/cancer-initiating cell-associated antigens (12, 13, 20, 34–40). It is also very attractive to identify scFv clones recognizing the CTL epitopes derived from these antigens in the context of HLA-A24 or HLA-A2 for the assessment of the expression status on other tumor cells.
In conclusion, we isolated scFv clone D12, which reacted with the HLA-A*02:01·PBF A2.2 peptide complex presented on osteosarcoma cells. This clone might be useful to detect the HLA·peptide complex on sarcoma cells for selection of patients and prediction of the efficacy of PBF A2.2 peptide-based immunotherapy. In the future, the clone might also be useful for antibody-based immunotherapy and adoptive cell transfer therapy using genetically engineered lymphocytes expressing chimeric antigenic receptors.
Supplementary Material
Acknowledgments
We thank Dr. Hideo Takasu (Dainippon Sumitomo Pharma Co., Ltd., Osaka, Japan) for the kind donation of synthetic peptides and Dr. Shigeru Takamoto (Japanese Red Cross Hokkaido Block Blood Center, Sapporo, Japan) for the kind donation of human sera.
This work was supported by grants from Japan Society for the Promotion of Science Grants-in-aid for Scientific Research (KAKENHI) 21249025 (to N. S.), 20390403 (to T. W.), and 22689041 and 25462344 (to T. Tsukahara); Management Expenses Grants 23-A-10 (to T. W.) and 23-A-44 (to N. S.) from the Government to the National Cancer Center; and Northern Advancement Center for Science and Technology Grant H24-S-5, Takeda Science Foundation Grant 2010-Kenkyu-Shorei, and Suhara Memorial Foundation Grant H24-12 (to T. Tsukahara).

This article contains supplemental Table S1.
- CTL
- cytotoxic T lymphocyte
- PBF
- papillomavirus binding factor
- scFv
- single chain variable fragment
- OS
- osteosarcoma
- MFH
- malignant fibrous histiocytoma
- VH
- variable regions of heavy chains
- VL
- variable regions of light chains
- Vκ
- variable regions of VLκ chains and Vl, variable regions of VLλ chains
- YT
- yeast extract-tryptone
- hIgG
- human IgG
- MFI
- mean fluorescence intensity
- TCR
- T cell receptor.
REFERENCES
- 1. Ferrari S., Palmerini E. (2007) Adjuvant and neoadjuvant combination chemotherapy for osteogenic sarcoma. Curr. Opin. Oncol. 19, 341–346 [DOI] [PubMed] [Google Scholar]
- 2. The Japanese Orthopaedic Association (JOA) Musculoskeletal Tumor Committee (2000) General Rules for Clinical and Pathological Studies on Malignant Bone Tumors, pp. 52–61, 3rd Ed., Kanabara, Tokyo [Google Scholar]
- 3. Southam C. M., Marcove R. C., Levin A. G., Buchsbaum H. J., Miké V. (1972) Proceedings: clinical trial of autogenous tumor vaccine for treatment of osteogenic sarcoma. Proc. Natl. Cancer Conf. 7, 91–100 [PubMed] [Google Scholar]
- 4. Campbell C. J., Cohen J., Enneking W. F. (1975) Editorial: new therapies for osteogenic sarcoma. J. Bone Joint Surg. Am. 57, 143–144 [PubMed] [Google Scholar]
- 5. Kawaguchi S., Wada T., Tsukahara T., Ida K., Torigoe T., Sato N., Yamashita T. (2005) A quest for therapeutic antigens in bone and soft tissue sarcoma. J. Transl. Med. 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewis V. O. (2007) What's new in musculoskeletal oncology. J. Bone Joint Surg. Am. 89, 1399–1407 [DOI] [PubMed] [Google Scholar]
- 7. Meyers P. A., Schwartz C. L., Krailo M., Kleinerman E. S., Betcher D., Bernstein M. L., Conrad E., Ferguson W., Gebhardt M., Goorin A. M., Harris M. B., Healey J., Huvos A., Link M., Montebello J., Nadel H., Nieder M., Sato J., Siegal G., Weiner M., Wells R., Wold L., Womer R., Grier H. (2005) Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J. Clin. Oncol. 23, 2004–2011 [DOI] [PubMed] [Google Scholar]
- 8. Maki R. G. (2006) Future directions for immunotherapeutic intervention against sarcomas. Curr. Opin. Oncol. 18, 363–368 [DOI] [PubMed] [Google Scholar]
- 9. Sato N., Hirohashi Y., Tsukahara T., Kikuchi T., Sahara H., Kamiguchi K., Ichimiya S., Tamura Y., Torigoe T. (2009) Molecular pathological approaches to human tumor immunology. Pathol. Int. 59, 205–217 [DOI] [PubMed] [Google Scholar]
- 10. Tsukahara T., Torigoe T., Tamura Y., Wada T., Kawaguchi S., Tsuruma T., Hirata K., Yamashita T., Sato N. (2008) Antigenic peptide vaccination: provoking immune response and clinical benefit for cancer. Curr. Immunol. Rev. 4, 235–241 [Google Scholar]
- 11. Boeckle S., Pfister H., Steger G. (2002) A new cellular factor recognizes E2 binding sites of papillomaviruses which mediate transcriptional repression by E2. Virology 293, 103–117 [DOI] [PubMed] [Google Scholar]
- 12. Tsukahara T., Nabeta Y., Kawaguchi S., Ikeda H., Sato Y., Shimozawa K., Ida K., Asanuma H., Hirohashi Y., Torigoe T., Hiraga H., Nagoya S., Wada T., Yamashita T., Sato N. (2004) Identification of human autologous cytotoxic T-lymphocyte-defined osteosarcoma gene that encodes a transcriptional regulator, papillomavirus binding factor. Cancer Res. 64, 5442–5448 [DOI] [PubMed] [Google Scholar]
- 13. Tsukahara T., Kawaguchi S., Torigoe T., Kimura S., Murase M., Ichimiya S., Wada T., Kaya M., Nagoya S., Ishii T., Tatezaki S., Yamashita T., Sato N. (2008) Prognostic impact and immunogenicity of a novel osteosarcoma antigen, papillomavirus binding factor, in patients with osteosarcoma. Cancer Sci. 99, 368–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fletcher C. D. M., van den Berg E., Molenaar W. M. (2002) in World Health Organization Classification of Tumors (Fletcher C. D. M., Unni K. K., Mertens F., eds) pp. 120–122, IARC Press, Lyon, France [Google Scholar]
- 15. Sichtig N., Silling S., Steger G. (2007) Papillomavirus binding factor (PBF)-mediated inhibition of cell growth is regulated by 14-3-3β. Arch. Biochem. Biophys. 464, 90–99 [DOI] [PubMed] [Google Scholar]
- 16. Tsukahara T., Kimura S., Ichimiya S., Torigoe T., Kawaguchi S., Wada T., Yamashita T., Sato N. (2009) Scythe/BAT3 regulates apoptotic cell death induced by papillomavirus binding factor in human osteosarcoma. Cancer Sci. 100, 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jordanovski D., Herwartz C., Pawlowski A., Taute S., Frommolt P., Steger G. (2013) The hypoxia-inducible transcription factor ZNF395 is controlled by IκB kinase-signaling and activates genes involved in the innate immune response and cancer. PLoS One 8, e74911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hasegawa R., Tomaru Y., de Hoon M., Suzuki H., Hayashizaki Y., Shin J. W. (2013) Identification of ZNF395 as a novel modulator of adipogenesis. Exp. Cell Res. 319, 68–76 [DOI] [PubMed] [Google Scholar]
- 19. Tsukahara T., Kawaguchi S., Torigoe T., Asanuma H., Nakazawa E., Shimozawa K., Nabeta Y., Kimura S., Kaya M., Nagoya S., Wada T., Yamashita T., Sato N. (2006) Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci. 97, 1374–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsukahara T., Kawaguchi S., Torigoe T., Takahashi A., Murase M., Kano M., Wada T., Kaya M., Nagoya S., Yamashita T., Sato N. (2009) HLA-A*0201-restricted CTL epitope of a novel osteosarcoma antigen, papillomavirus binding factor. J. Transl. Med. 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nabeta Y., Kawaguchi S., Sahara H., Ikeda H., Hirohashi Y., Goroku T., Sato Y., Tsukahara T., Torigoe T., Wada T., Kaya M., Hiraga H., Isu K., Yamawaki S., Ishii S., Yamashita T., Sato N. (2003) Recognition by cellular and humoral autologous immunity in a human osteosarcoma cell line. J. Orthop. Sci. 8, 554–559 [DOI] [PubMed] [Google Scholar]
- 22. Wada T., Uede T., Ishii S., Matsuyama K., Yamawaki S., Kikuchi K. (1988) Monoclonal antibodies that detect different antigenic determinants of the same human osteosarcoma-associated antigen. Cancer Res. 48, 2273–2279 [PubMed] [Google Scholar]
- 23. Tsukahara T., Kawaguchi S., Ida K., Kimura S., Tamura Y., Ikeda T., Torigoe T., Nagoya S., Wada T., Sato N., Yamashita T. (2006) HLA-restricted specific tumor cytolysis by autologous T-lymphocytes infiltrating metastatic bone malignant fibrous histiocytoma of lymph node. J. Orthop. Res. 24, 94–101 [DOI] [PubMed] [Google Scholar]
- 24. Pansri P., Jaruseranee N., Rangnoi K., Kristensen P., Yamabhai M. (2009) A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schofield D. J., Pope A. R., Clementel V., Buckell J., Chapple S. D., Clarke K. F., Conquer J. S., Crofts A. M., Crowther S. R., Dyson M. R., Flack G., Griffin G. J., Hooks Y., Howat W. J., Kolb-Kokocinski A., Kunze S., Martin C. D., Maslen G. L., Mitchell J. N., O'Sullivan M., Perera R. L., Roake W., Shadbolt S. P., Vincent K. J., Warford A., Wilson W. E., Xie J., Young J. L., McCafferty J. (2007) Application of phage display to high throughput antibody generation and characterization. Genome Biol. 8, R254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lefranc M. P. (2003) IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 31, 307–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nahshol O., Bronner V., Notcovich A., Rubrecht L., Laune D., Bravman T. (2008) Parallel kinetic analysis and affinity determination of hundreds of monoclonal antibodies using the ProteOn XPR36. Anal. Biochem. 383, 52–60 [DOI] [PubMed] [Google Scholar]
- 28. Dahan R., Reiter Y. (2012) T-cell-receptor-like antibodies—generation, function and applications. Expert Rev. Mol. Med. 14, e6. [DOI] [PubMed] [Google Scholar]
- 29. Purbhoo M. A., Sutton D. H., Brewer J. E., Mullings R. E., Hill M. E., Mahon T. M., Karbach J., Jäger E., Cameron B. J., Lissin N., Vyas P., Chen J. L., Cerundolo V., Jakobsen B. K. (2006) Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J. Immunol. 176, 7308–7316 [DOI] [PubMed] [Google Scholar]
- 30. Bossi G., Gerry A. B., Paston S. J., Sutton D. H., Hassan N. J., Jakobsen B. K. (2013) Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2, e26840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parker K. C., Bednarek M. A., Coligan J. E. (1994) Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152, 163–175 [PubMed] [Google Scholar]
- 32. Michaeli Y., Denkberg G., Sinik K., Lantzy L., Chih-Sheng C., Beauverd C., Ziv T., Romero P., Reiter Y. (2009) Expression hierarchy of T cell epitopes from melanoma differentiation antigens: unexpected high level presentation of tyrosinase-HLA-A2 complexes revealed by peptide-specific, MHC-restricted, TCR-like antibodies. J. Immunol. 182, 6328–6341 [DOI] [PubMed] [Google Scholar]
- 33. van der Merwe P. A., Davis S. J. (2003) Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 21, 659–684 [DOI] [PubMed] [Google Scholar]
- 34. Hirohashi Y., Torigoe T., Maeda A., Nabeta Y., Kamiguchi K., Sato T., Yoda J., Ikeda H., Hirata K., Yamanaka N., Sato N. (2002) An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin. Cancer Res. 8, 1731–1739 [PubMed] [Google Scholar]
- 35. Sato Y., Nabeta Y., Tsukahara T., Hirohashi Y., Syunsui R., Maeda A., Sahara H., Ikeda H., Torigoe T., Ichimiya S., Wada T., Yamashita T., Hiraga H., Kawai A., Ishii T., Araki N., Myoui A., Matsumoto S., Umeda T., Ishii S., Kawaguchi S., Sato N. (2002) Detection and induction of CTLs specific for SYT-SSX-derived peptides in HLA-A24+ patients with synovial sarcoma. J. Immunol. 169, 1611–1618 [DOI] [PubMed] [Google Scholar]
- 36. Ida K., Kawaguchi S., Sato Y., Tsukahara T., Nabeta Y., Sahara H., Ikeda H., Torigoe T., Ichimiya S., Kamiguchi K., Wada T., Nagoya S., Hiraga H., Kawai A., Ishii T., Araki N., Myoui A., Matsumoto S., Ozaki T., Yoshikawa H., Yamashita T., Sato N. (2004) Crisscross CTL induction by SYT-SSX junction peptide and its HLA-A*2402 anchor substitute. J. Immunol. 173, 1436–1443 [DOI] [PubMed] [Google Scholar]
- 37. Hariu H., Hirohashi Y., Torigoe T., Asanuma H., Hariu M., Tamura Y., Aketa K., Nabeta C., Nakanishi K., Kamiguchi K., Mano Y., Kitamura H., Kobayashi J., Tsukahara T., Shijubo N., Sato N. (2005) Aberrant expression and potency as a cancer immunotherapy target of inhibitor of apoptosis protein family, Livin/ML-IAP in lung cancer. Clin. Cancer Res. 11, 1000–1009 [PubMed] [Google Scholar]
- 38. Honma I., Torigoe T., Hirohashi Y., Kitamura H., Sato E., Masumori N., Tamura Y., Tsukamoto T., Sato N. (2009) Aberrant expression and potency as a cancer immunotherapy target of α-methylacyl-coenzyme A racemase in prostate cancer. J. Transl. Med. 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inoda S., Morita R., Hirohashi Y., Torigoe T., Asanuma H., Nakazawa E., Nakatsugawa M., Tamura Y., Kamiguchi K., Tsuruma T., Terui T., Ishitani K., Hashino S., Wang Q., Greene M. I., Hasegawa T., Hirata K., Asaka M., Sato N. (2011) The feasibility of Cep55/c10orf3 derived peptide vaccine therapy for colorectal carcinoma. Exp. Mol. Pathol. 90, 55–60 [DOI] [PubMed] [Google Scholar]
- 40. Morita R., Nishizawa S., Torigoe T., Takahashi A., Tamura Y., Tsukahara T., Kanaseki T., Sokolovskaya A., Kochin V., Kondo T., Hashino S., Asaka M., Hara I., Hirohashi Y., Sato N. (2014) Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer-initiating cells. Cancer Sci. 105, 389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.